

Abstract
Abasic (AP) sites arise in DNA through spontaneous base loss and enzymatic removal of damaged bases. APN1 encodes the major AP-endonuclease of Saccharomyces cerevisiae. Human HAP1 (REF1) encodes the major AP endonuclease which, in addition to its role in DNA repair, functions as a redox regulatory protein. We identify APN2, the yeast homolog of HAP1 and provide evidence that Apn1 and Apn2 represent alternate pathways for repairing AP sites. The apn1Δ apn2Δ strain displays a highly elevated level of MMS-induced mutagenesis, which is dependent on the REV3, REV7, and REV1 genes. Our findings indicate that AP sites are highly cytotoxic and mutagenic in eukaryotes, and that the REV3, REV7-encoded DNA polymerase ζ mediates the mutagenic bypass of AP sites.
Keywords: APN2, APN1, base excision repair, mutagenic bypass, yeast
Abasic (AP) sites arise in DNA at a significant rate by spontaneous hydrolysis of the N-glycosylic bond. Extrapolating from the rate of depurination occurring in vitro under physiological conditions, it has been estimated that as many as 104 purines are lost spontaneously in a human cell per day (Lindahl and Nyberg 1972). AP sites are also formed in DNA as intermediates in base excision repair (BER).
Two major class II AP Endos have been identified in Escherichia coli. exonuclease III, the product of the xth gene, is a constitutive and abundant enzyme representing ∼90% of the total AP activity, and endonuclease IV, encoded by the nfo gene, is an inducible activity representing 10%–50% of the activity in uninduced or induced cells, respectively. Both enzymes also possess a 3′-phosphatase, and a 3′-phosphodiesterase activity that can remove 3′-terminal groups formed in DNA by free radical attack or by the action of β-lyase activity associated with some of the DNA glycosylases (Demple and Harrison 1994; Seeberg et al. 1995; Wallace 1997).
Homologs of E. coli exonuclease III and endonuclease IV (Exo III, Endo IV) have been identified in eukaryotes. In Saccharomyces cerevisiae, APN1 encodes the major AP Endo, and it shares extensive homology with Endo IV (Popoff et al. 1990). Like its E. coli counterpart, Apn1 is a class II AP endonuclease, and it has 3′-diesterase, and 3′-phosphatase activities (Johnson and Demple 1988a,b). A homolog of E. coli Exo III has not yet been identified in yeast. In contrast to yeast, Exo III homologs have been identified from mammalian and other sources. The enzyme from humans, variously known as HAP1, APE, Ref-1, or Apex, has a robust class II AP endonuclease activity, but its 3′-phosphodiesterase and 3′-phosphatase activities are relatively weak (Chen et al. 1991; Winters et al. 1994). Interestingly, HAP1 also functions as a regulatory protein, stimulating the DNA-binding activity of AP-1 proteins, such as c-Jun and c-Fos, by a redox-dependent mechanism (Xanthoudakis and Curran 1992). The stimulatory effect of HAP1 on Jun and Fos is mediated through reduction of a conserved cysteine residue present in the DNA-binding domain of each protein (Abate et al. 1990; Xanthoudakis et al. 1992). HAP1 also stimulates the DNA-binding activity of other redox regulated transcription factors, including p53 (Xanthoudakis et al. 1992; Jayaraman et al. 1997). The DNA repair and redox activities of HAP1 reside in distinct regions of the protein and can function independently of one another (Walker et al. 1993; Xanthoudakis et al. 1994). HAP1 is essential for the early embryonic development of mice, as mice lacking a functional HAP1 gene die shortly after implantation (Xanthoudakis et al. 1996). Because sequences containing both the DNA repair and redox regulatory activities have been disrupted in these mutant mice, it is not known whether one or both of these activities are necessary for embryonic development.
Here we identify a homolog of Exo III/HAP1 in S. cerevisiae, which we have named APN2, and examine the effects of apn1Δ and apn2Δ mutations on viability, mutagenesis, and the repair of AP sites.
Results
APN2 is a member of the Exo III family of AP endonucleases
The S. cerevisiae APN2-encoded protein is homologous to E. coli Exo III (Xth), human HAP1, and to the other related enzymes from Drosophila, Arabidopsis, and other bacterial species (Fig. 1). The HAP1 protein from residues 62–318 retains all of the DNA repair activities (Walker et al. 1993; Xanthoudakis et al. 1994; Izumi and Mitra 1998). This region of ∼250 residues is highly conserved among HAP1, Apn2, Exo III, and other members of the family (Fig. 1A,B). In this region, Apn2 shares 33% similar and 21% identical residues with HAP1, and 26% similar and 19% identical residues with the E. coli Xth protein.
Figure 1.


Apn2 is a member of the Exo III/HAP1 family of proteins. (A) Alignment of human HAP1, S. cerevisiae Apn2, and E. coli Exo III (Xth) proteins. Identical and highly conserved residues are highlighted. Amino acid positions are indicated by numbers in parentheses. The conserved residues thought to be involved in catalytic activity (Glu-96, Asp-283, and His-309 in HAP1 protein) are indicated by arrows. The Cys-65 residue in HAP1 essential for the redox activity is circled and indicated by an asterisk. (B) Schematic alignment of Exo III-type nucleases from various organisms. Boxes represent primary amino acid sequence. Narrow regions indicate unique sequences, whereas larger, shaded, or stippled regions indicate regions of homology among the proteins. The different shades within the boxes indicate independent domains; spaces indicate gaps that were introduced to optimize alignment of the proteins. Protein lengths are indicated by numbers at right. Positions of the highly conserved amino acid sequences QE(T/L/I)K, R(L/I)D, and SDH(C/A)P are indicated. Positions of the redox cysteine residue in human HAP1, and of cysteine residues present at the corresponding position in Drosophila RRP and Arabidopsis thaliana ARP proteins are indicated by the letter C within the alignments.
From the x-ray crystal structure of Exo III, residues Asp-229 and His-259 have been proposed to mediate catalysis, and Glu-34 to bind a metal ion and to facilitate catalysis (Mol et al. 1995). HAP1 shares considerable structural similarity to Exo III (Gorman et al. 1997). Mutational inactivation of the corresponding HAP1 active site residues Asp-283, His-309, and Glu-96 to alanine has indicated a critical role of these residues in catalysis (Barzilay et al. 1995). These residues and the sequences flanking them have been very highly conserved in Apn2 and in the other members of this nuclease family (Fig. 1A,B). Apn2 and its counterpart from Schizosaccharomyces pombe have a carboxy-terminal domain that is absent in the other homologs (Fig. 1B). This region of the protein may be involved in species-specific protein–protein interactions.
The amino-terminal 61 residues of HAP1 are absent in Exo III, and this region is not conserved in Apn2 (Fig. 1A). This amino-terminal region of HAP1 is important for redox activity but not for DNA repair functions. The Cys-65 residue present in HAP1 is essential for redox activity, as replacement of this residue with alanine inhibits the ability of HAP1 to stimulate Jun DNA binding (Walker et al. 1993; Xanthoudakis et al. 1994). This cysteine residue is not conserved in Exo III or Apn2. Exo III has no redox activity (Walker et al. 1993), and Apn2 is likely to be devoid of this activity.
APN2 is not essential for growth or survival
As previously reported (Ramotar et al. 1991), deletion of APN1 has no effect on the growth or viability of yeast cells. We have observed no effect of the apn2Δ mutation on viability or growth, and the apn1Δ apn2Δ double mutant also grows normally (data not shown). Because AP sites are noncoding, we determined whether spontaneous mutations arise at a much higher rate in the strains deleted for both of the APN genes. Deletion of APN1 increased the rate of can1r mutations 3.5-fold, a result similar to that reported previously (Ramotar et al. 1991). The rate of spontaneous can1r mutations in the apn2Δ strain was the same as in the wild-type strain, and the apn1Δ apn2Δ strain exhibited the same level of spontaneous mutability as the apn1Δ strain (data not shown).
The apn2Δ mutation enhances the sensitivity of the apn1Δ strain to alkylation damage
To determine the relative contribution of the APN1 and APN2 genes to the repair of AP sites, we tested the sensitivity of the apn1Δ, apn2Δ, and apn1Δ apn2Δ mutant strains to the alkylating agent MMS. MMS alkylates the bases in DNA, particularly adenine at the N3 position (3MeA) and guanine at the N7 position (7MeG). The alkylated bases are removed by the action of N-methylpurine DNA glycosylases (MPG), which act on 3MeA and 7MeG and also on a variety of other alkylated bases (Roy et al. 1994; Bjoras et al. 1995). The AP sites resulting from DNA glycosylase action would be acted on by the Apn1 or Apn2 proteins. As shown in Figure 2A, and as reported previously (Ramotar et al. 1991), the apn1Δ mutant strain exhibits moderate sensitivity to MMS. The MMS sensitivity of the apn2Δ strain is similar to that of the wild-type strain. The apn1Δ apn2Δ strain, however, displays an extremely enhanced MMS sensitivity, suggesting that Apn1 and Apn2 compete for the repair of AP sites, and in the absence of both AP endonucleases, AP sites remain in DNA and result in cell lethality.
Figure 2.

Enhanced MMS sensitivity and MMS mutagenesis in the apn1Δ apn2Δ strain. (A) MMS sensitivity of various yeast strains. Cells grown overnight in YPD medium were treated with MMS at the concentrations indicated for a 20-min period. Appropriate dilutions were spread onto YPD plates. Each curve represents the average of two to three experiments for each strain. (•) EMY74.7, wild type (APN1 APN2); (□) YRP190, apn1Δ; (▵) YRP263, apn2Δ; (○) YRP269, apn1Δ apn2Δ. (B) MMS-induced mutations at the CAN1 locus. Cells grown overnight in YPD medium were treated with MMS at the concentrations indicated for a 20-min period. Appropriate dilutions were spread onto YPD plates for viability determinations and onto synthetic complete medium lacking arginine and containing canavanine for the determination of CAN1S to can1r mutagenesis. Each curve represents the average of two to three experiments for each strain. (•) EMY74.7, wild type (APN1 APN2); (□) YRP190, apn1Δ; (▵) YRP263, apn2Δ; (○) YRP269, apn1Δ apn2Δ.
To confirm that the extreme MMS-sensitive phenotype of the apn1Δ apn2Δ strain was, in fact, attributable to deletions of these two genes, we constructed low copy CEN vectors carrying the wild-type APN1 or the APN2 gene, and transformed these plasmids into the apn1Δ apn2Δ strain to test for complementation. As expected, introduction of the APN1 plasmid into the apn1Δ apn2Δ strain restored MMS survival to wild-type levels, whereas introduction of the APN2 plasmid conferred MMS resistance to the level equivalent to that seen in the apn1Δ mutant strain (data not shown).
Enhanced MMS-induced mutagenesis in the apn1Δ apn2Δ strain
Because AP sites are noncoding, they would present a block to the DNA replicational machinery. Replication through such sites could occur by a mutagenic process in which DNA polymerase misincorporates a nucleotide opposite such a lesion, or these sites may be bypassed by error-free postreplicative repair processes in which the gap left opposite the AP site is filled in either by a ‘copy choice’ type of DNA synthesis in which the undamaged sister duplex is used as the template for copying the missing information (Higgins et al. 1976) or by a recombinational mechanism. To evaluate the extent to which AP sites are mutagenic in eukaryotes, we determined the rate of CAN1S to can1r mutations in MMS-treated wild-type, apn1Δ, apn2Δ, and apn1Δ apn2Δ strains (Fig. 2B). The incidence of MMS-induced can1r mutations was the same in the wild-type and apn2Δ strains, and was elevated only slightly in the apn1Δ strain. In contrast, the frequency of MMS-induced can1r mutations rose sharply in the apn1Δ apn2Δ strain. For instance, whereas treatment with 0.1% MMS produced 250–500 can1r mutants per 107 viable cells in the wild-type, apn1Δ, and apn2Δ strains, the can1r mutation frequency rose to 19,000 per 107 cells in the apn1Δ apn2Δ strain (Fig. 2B). Introduction of the APN1 or the APN2 gene carried on a CEN plasmid into the apn1Δ apn2Δ strain lowered MMS mutagenesis to levels seen in the wild-type and the apn1Δ strain, respectively (data not shown). These findings provide the first clear evidence of the high mutagenic potential of unrepaired AP sites in eukaryotes.
Defective repair of MMS-induced DNA damage in the apn1Δ apn2Δ mutant strain
The greatly enhanced MMS sensitivity of the apn1Δ apn2Δ mutant strain suggested that both Apn1 and Apn2 function in the removal of AP sites from DNA. To directly assess the role of these nucleases in the repair of AP sites, we examined the level of AP sites in MMS-treated wild-type, apn1Δ, apn2Δ, and apn1Δ apn2Δ mutant strains by sedimentation of DNA in alkaline sucrose gradients. Because NaOH hydrolyzes the phosphodiester bond near each AP site, the size reduction in DNA observed on sedimentation in alkaline sucrose gradients is a good measure of the AP sites present. Yeast cells were treated with 0.1% MMS for 20 min, and the sedimentation profile of DNA was evaluated immediately following this treatment, and following incubation of these cells for an additional 1 or 2 hr to allow time for DNA repair to occur (Fig. 3). In all of the strains, DNA from MMS-treated cells sediments toward the top of the gradient, indicating the presence of AP sites. The sedimentation pattern of DNA obtained from wild-type or apn2Δ cells that had been incubated for 1 hr following MMS treatment was the same as that of DNA from untreated control cells, indicating proficient repair of AP sites in both these strains during the 1-hr period (Fig. 3A,C). As previously reported (Ramotar et al. 1991), removal of AP sites was less efficient in the apn1Δ strain. DNA obtained from MMS-treated apn1Δ cells that were allowed a 1-hr repair period sedimented at a slower rate than DNA from untreated cells. The size of DNA increased in cells that had been incubated for 2 hr after MMS treatment, but even after this longer repair period, DNA was not fully restored to normal size (Fig. 3B). The removal of AP sites was severely impaired in the apn1Δ apn2Δ mutant strain, as DNA from MMS-treated apn1Δ apn2Δ cells allowed a 2-hr repair period sedimented only slightly faster than DNA from cells that were treated with MMS but allowed no repair period (Fig. 3D).
Figure 3.
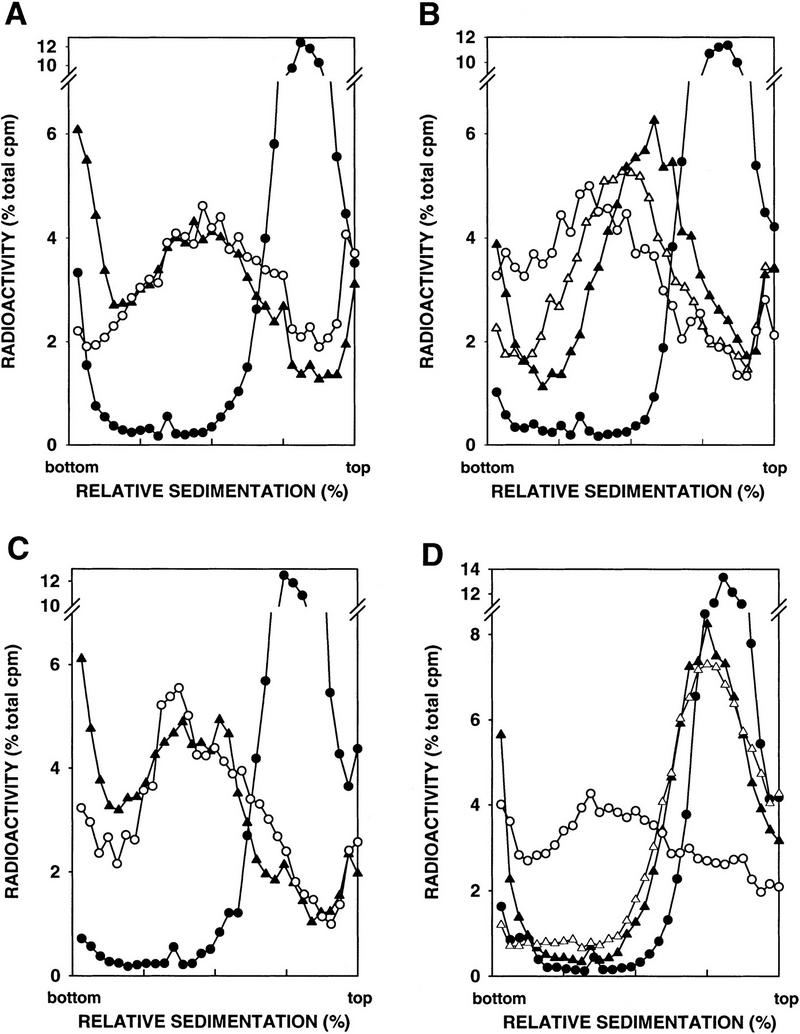
Alkaline sucrose gradient analysis of DNA from cells treated with 0.1% MMS for 20 min. (A) YRP276, wild type (APN1 APN2). (B) YRP210, apn1Δ. (C) YRP291, apn2Δ. (D) YRP292, apn1Δ apn2Δ. (○) Untreated cells; (•) cells treated with MMS for 20 min; (▴) cells treated with MMS for 20 min, and then given a 1-hr repair period; (▵) cells treated with MMS for 20 min, and then given a 2-hr repair period.
Requirement of REV genes in mutagenic bypass of AP sites
Recent in-vitro studies with mammalian DNA polymerase δ (Pol δ) and proliferating cell nuclear antigen (PCNA) have indicated that PCNA allows Pol δ to replicate past an AP site present in the template strand, resulting in the formation of full-length product (Mozzherin et al. 1997). As expected from the A-rule (Strauss 1991), fully extended primers incorporated predominantly dAMP opposite the template lesion. Extensive genetic studies in yeast, however, have provided evidence for the involvement of REV3, REV7, and REV1 genes in mutagenesis induced by UV light (Lawrence and Hinkle 1996). More recent biochemical studies have indicated that Rev3 and Rev7 consitute two subunits of DNA Pol ζ, which has the ability to replicate past a thymine cis–syn cyclobutane dimer (Nelson et al. 1996b). Rev1 has a deoxycytidyl transferase activity, and it transfers a dCMP residue to the 3′ end of a primer in a template-dependent reaction. Interestingly, the enzyme efficiently inserts dCMP opposite an AP site, producing a terminus that can be extended by Pol ζ (Nelson et al. 1996a).
The availablity of the apn1Δ apn2Δ strain has provided us the opportunity to test whether mutagenic bypass of AP sites is mediated by the Rev proteins. To examine this, we incorporated the rev3Δ, rev7Δ, and rev1Δ mutations in the apn1Δ apn2Δ strain, and examined the incidence of MMS-induced mutations. As shown in Figure 4, the rev3Δ, rev7Δ, or rev1Δ mutations did not enhance the MMS sensitivity of apn1Δ apn2Δ strain, but, interestingly, no MMS-induced can1r mutations were recovered in any of these apn1Δ apn2Δ revΔ mutant strains. These results indicate that mutagenic bypass of AP sites is controlled by Pol ζ and its accessory factors.
Figure 4.

Requirement of REV genes for MMS-induced mutagenesis in the apn1Δ apn2Δ strains. Methods are as described in the legend to Fig. 2. (A) MMS sensitivity of yeast strains. (•) EMY74.7, wild type (APN1 APN2); (○) YRP269, apn1Δ apn2Δ; (▵) YREV1.15, rev1Δ; (█) YREV3.15, rev3Δ; (▾) YREV7.1, rev7Δ; (▴) YREV1.13, apn1Δ apn2Δ rev1Δ; (□) YREV3.40, apn1Δ apn2Δ rev3Δ; (▿) YREV7.4, apn1Δ apn2Δ rev7Δ. (B) MMS-induced mutations of CAN1S to can1r. (○) YRP269, apn1Δ apn2Δ; (▴) YREV1.13, apn1Δ apn2Δ rev1Δ; (□) YREV3.40, apn1Δ apn2Δ rev3Δ; (▿) YREV7.4, apn1Δ apn2Δ rev7Δ.
Discussion
AP sites are one of the most common lesions in DNA. To define the pathways for the repair of AP sites in eukaryotes, here we identify yeast APN2, a homolog of E. coli Exo III and human HAP1. Deletion of APN2 has no effect on cell viability or growth, and the apn1Δ apn2Δ mutant also grows normally. Sensitivity to MMS is not affected by the apn2Δ mutation, but the apn1Δ apn2Δ strain displays a much higher level of sensitivity to MMS than the apn1Δ strain, which is only moderately sensitive to this DNA-damaging agent. Consistent with this observation, the apn1Δ apn2Δ mutant is highly deficient in the removal of AP sites, as determined by alkaline sucrose gradient sedimentation of DNA obtained from MMS-treated cells. These results provide strong evidence for a role of Apn2 in the removal of AP sites, and they indicate that Apn1 and Apn2 represent alternate ways of removing AP sites.
The identification of Apn2 and the demonstration of its involvement in the repair of AP sites in yeast attests to the high degree of conservation of pathways for repairing AP sites among prokaryotes and eukaryotes. Therefore, Exo III in E. coli, Apn2 in S. cerevisiae, and HAP1 in humans, all function in the removal of AP sites. Even though no Endo IV/Apn1 homolog has yet been indentified in humans and higher eukaryotes, such an activity is likely to be present in there as well. It is of some interest to note that although the enzymes for removing AP sites, as well as many of the DNA N-glycosylases, have been conserved between prokaryotes and eukaryotes, no such conservation exists among the components of the nucleotide excision repair machinery.
Apn1 accounts for >90% of the class II AP endonuclease/3′-diesterase activity in wild-type yeast cell extracts, suggesting that the activity encoded by APN2 is relatively minor. This is consistent with the observation that deletion of APN1 increases the rate of spontaneous mutations as well as MMS sensitivity, but deletion of APN2 has no effect on these phenotypes. In the absence of Apn1, however, cells rely on the repair function of Apn2, as deletion of both the APN1 and APN2 genes elicits a large increase in MMS sensitivity and confers a severe reduction in the ability to remove AP sites. A class II AP endonuclease and 3′-phosphodiesterase activity has been recently partially purified from apn1Δ S. cerevisiae cells. Unlike Apn1, but similar to Exo III, this activity is strongly dependent on Mg2+ (Sander and Ramotar 1997). APN2 may encode this activity.
The high level of deficiency in the removal of AP sites in the apn1Δ apn2Δ strain would predict that spontaneously arising AP sites would accumulate in this strain. It is therefore somewhat surprising that simultaneous inactivation of the APN1 and APN2 genes has no effect on cell viability or growth, and spontaneous mutability is increased only a few fold in the apn1Δ apn2Δ strain. On infliction of additional DNA damage, however, as on treatment with MMS, apn1Δ apn2Δ cells suffer a drastic reduction in survival, and they display a sharp rise in mutagenesis. We interpret these observations as follows. In normally growing apn1Δ apn2Δ cells, the AP sites resulting from spontaneous base loss can be efficiently handled by postreplicative bypass processes, and a majority of these act in an error-free manner. In MMS-treated cells, however, the rising level of AP sites is handled by error-free as well as mutagenic bypass processes, resulting in a steep rise in the incidence of mutations in the surviving apn1Δ apn2Δ cells and cell death occurs if all the bypass processes are saturated.
Even though in vitro studies have indicated that mammalian DNA Pol δ, in concert with PCNA, can efficiently bypass AP sites (Mozzherin et al. 1997), our genetic studies provide evidence for the requirement of REV3 and REV7 encoded DNA Pol ζ and of Rev1 protein in mutagenic bypass of AP sites. Deletion of any of these REV genes in the apn1Δ apn2Δ strain, however, causes no significant enhancement of MMS sensitivity, suggesting that mutgenic bypass by the REV3 system makes only a minor contribution to cell survival and that a majority of AP sites are bypassed in an error-free manner. Because AP sites would represent one of the most frequently formed DNA lesions in mammalian cells (Lindahl and Nyberg 1972), however, DNA Pol ζ-mediated mutagenic bypass of AP sites may make a significant contribution to mutagenesis, and to tumorigenesis and carcinogenesis. The identification of REV3 and REV7 counterparts in humans (Lawrence and Hinkle 1996; Gibbs et al. 1998) supports such a possiblity.
In contrast to the absence of any effect on cell viability of the apn2Δ mutation either alone or when combined with the apn1Δ mutation, mice lacking a functional HAP1 (REF1) gene die during embryonic development, following blastocyst formation (Xanthoudakis et al. 1996). This lethality may arise from a deficiency in the repair of AP sites or of oxidative damage, or from the inactivation of the redox regulatory function, or from the combined effects of these deficiencies. The redox activity of HAP1 protein resides in the amino-terminal domain, and cysteine 65 is essential for this function. Because neither the amino-terminal sequence nor the cysteine 65 residue have been conserved in Apn2, this protein may have no role in redox regulation. Our observations with the yeast apn1Δ apn2Δ mutant may suggest that embryonic lethality of hap1 −/− mice results from a deficiency in the redox regulatory function and not from the inactivation of the DNA repair function of HAP1.
Materials and methods
Identification of APN2
To identify the yeast homolog of the human AP endonuclease HAP1, the HAP1 protein sequence (GenBank accession no. M92444) was used to search for homologs in the S. cerevisiae nonredundant protein data set at the Saccharomyces Genome Database using the BLAST program (Altschul et al. 1990). The S. cerevisiae open reading frame (ORF) designated YBL019W (accession no. Z35780) showed homology to HAP1-encoded protein, and we have named this gene APN2.
Generation of yeast null mutations
Deletion mutations were generated in yeast by the gene replacment method (Rothstein 1991) using the URA3 geneblaster. To construct a genomic deletion mutation of APN1, a 1.9-kb PCR product containing the APN1 gene was cloned into pUC19, generating the plasmid pPM750. The region of APN1 from the BglII site at nucleotide position +25 to the EcoRV site at postion +1057 within the APN1 ORF was replaced with the URA3 geneblaster fragment. The resulting plasmid, pPM753, when cut with PvuII results in a linear DNA fragment, incorporation of which in the yeast genome deletes APN1 from nucleotide +30 to +1060 of the 1104-bp APN1 ORF. To construct the apn2 deletion generating plasmid, a 2.54-kb PCR product encompassing the APN2 gene was generated and cloned into pUC19. The 1.37-kb BglII fragment within the APN2 ORF was excised and replaced with the URA3 geneblaster fragment. The apn2Δ-generating plasmid pPM838, when digested with BamHI and SalI, releases a linear DNA fragment, incorporation of which in the yeast genome deletes APN2 from nucleotide +135 to +1505 of the 1563-bp APN2 ORF. Linear deletion generating DNA was introduced into yeast strains by the lithium acetate method (Ito et al. 1983). Generation of genomic deletions was confirmed by PCR of genomic DNA. The URA3 geneblaster fragment contains the URA3 gene flanked by two direct repeats of the Salmonella HisG sequence. These repeats allow for recombination in yeast to excise the URA3 gene (Alani et al. 1987). Loss of the URA3 gene was selected for by plating cells on medium containing 5-fluoro-orotic acid (5-FOA).
Spontaneous forward mutation of CAN1S
To measure the rates of spontaneous foward mutation at the CAN1S locus, for each strain, 11 independent cultures were grown in 500 μl of YPD, each starting from ∼10 canavanine-sensitive cells. Cells were grown for 3 days at 30°C and subsequently plated onto synthetic complete (SC) medium lacking arginine but containing canavanine. Plates were incubated for 4–5 days before counting canavanine-resistant colonies. Rates of spontaneous forward mutation were determined by the method of the median (Lea and Coulson 1949). Each experiment was performed in duplicate.
MMS sensitivity and mutagenesis
All of the strains used for these experiments were derived from DBY747 that had been made trp1Δ, resulting in the strain EMY74.7 (MATa his3-Δ1 leu2-3,-112 trp1Δ ura3-52). For determining sensitivity to MMS and for measuring the rate of MMS-induced forward mutations at the CAN1S locus, cells were grown overnight in YPD medium, sonicated to disperse clumps, washed, and resuspended in 0.05 m KPO4 buffer at pH 7.0. Appropriate dilutions of MMS were added to 1-ml suspensions of cells adjusted to 1.5 × 108 cells per ml. Samples were incubated in the presence of MMS with vigorous shaking at 30°C for 20 min. The reaction was terminated by the addition of 1 ml of 10% sodium thiosulfate. Appropriate dilutions of cells were plated on YPD for viability determinations and on synthetic complete medium lacking arginine but containing canavanine for determining the frequency of can1r mutations. Plates were incubated at 30°C and counted after 3 and 4–5 days for viability and mutagenesis determinations, respectively.
Alkaline sucrose gradients
All strains for alkaline sucrose gradient sedimentation were derived from EMY74.7. ρ0 derivatives lacking mitochondrial DNA were obtained by ethidium bromide mutagenesis, resulting in the following isogenic strains used in these experiments: YRP276, his3-Δ1 leu2-3,-112 trp1Δ ura3-52 ρ0; YRP210, his3-Δ1 leu2-3,-112 trp1Δ ura3-52 ρ0 apn1Δ; YRP291, his3-Δ1 leu2-3,-112 trp1Δ ura3-52 ρ0 apn2Δ; YRP292, his3-Δ1 leu2-3,-112 trp1Δ ura3-52 ρ0 apn1Δ apn2Δ. Strains grown overnight at 30°C in SC medium containing 10 μg/ml of uracil and 10 μCi/ml [3H]uracil were collected by filtration, washed, and suspended in the same volume of SC containing 30 μg/ml of uracil. Cells were then incubated for 1 hr at 30°C to allow for depletion of the intracellular pool of [3H]uracil to occur. Cells were collected by filtration, washed, and suspended in 0.05 m KPO4 buffer at pH 7.0, and incubated for 20 min with MMS at a final concentration of 0.1%. Cells were collected by filtration, washed, and resuspended in 3 ml of water. One milliliter of cells was used for the 0-hr sample and the other 2 ml of cells was diluted to 20 ml with YPD medium and incubated for an additional 1 or 2 hr to allow for repair to occur. Conversion of cells to spheroplasts, alkaline sucrose gradient sedimentation, and processing of samples were performed as described (Torres-Ramos et al. 1996), except that acid precipitation of alkali-hydrolyzed samples was carried out with 1 n HCl and 0.1 m sodium pyrophosphate rather than TCA.
Acknowledgments
This work was supported by grants GM19261, CA41261, and CA53791 from the National Institutes of Health, ES08547 from the National Institute of Environmental Health Sciences, and grant DEFGO3–93ER61706 from the Department of Energy.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lprakash@scms.utmb.edu; FAX (409) 747-8608.
References
- Abate C, Patel L, Rauscher FJ, III, Curran T. Redox regulation of Fos and Jun DNA-binding activity in vitro. Science. 1990;249:1157–1161. doi: 10.1126/science.2118682. [DOI] [PubMed] [Google Scholar]
- Alani E, Cao L, Kleckner N. A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted genes. Genetics. 1987;116:541–545. doi: 10.1534/genetics.112.541.test. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman J. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Barzilay G, Mol CD, Robson CN, Walker LJ, Cunningham RP, Tainer JA, Hickson ID. Identification of critical active-site residues in the multifuncitonal human DNA repair enzyme HAP1. Nat Struct Biol. 1995;2:561–568. doi: 10.1038/nsb0795-561. [DOI] [PubMed] [Google Scholar]
- Bjoras M, Klungland A, Johansen RF, Seeberg E. Purification and properties of the alkylation repair DNA glycosylase encoded MAG gene from Saccharomyces cerevisiae. Biochemistry. 1995;34:4577–4582. doi: 10.1021/bi00014a010. [DOI] [PubMed] [Google Scholar]
- Chen DS, Herman T, Demple B. Two distinct human DNA diesterases that hydrolyze 3′-blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Res. 1991;19:5907–5914. doi: 10.1093/nar/19.21.5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demple B, Harrison L. Repair of oxidative damage to DNA: Enzymology and biology. Annu Rev Biochem. 1994;63:915–948. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- Gibbs PEM, McGregor WG, Maher VM, Nisson P, Lawrence CW. A human homolog of the Saccharomyces cerevisiae REV3 gene, which encodes the catalytic subunit of DNA polymerase ζ. Proc Natl Acad Sci. 1998;95:6876–6880. doi: 10.1073/pnas.95.12.6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman MA, Morera S, Rothwell DG, de la Fortelle E, Mol CD, Tainer JA, Hickson ID, Freemont PS. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 1997;16:6548–6558. doi: 10.1093/emboj/16.21.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins NP, Kato K, Strauss B. A model for replication repair in mammalian cells. J Mol Biol. 1976;101:417–425. doi: 10.1016/0022-2836(76)90156-x. [DOI] [PubMed] [Google Scholar]
- Ito H, Faukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi T, Mitra S. Deletion analysis of human AP-endonuclease: Minimum sequence required for the endonuclease activity. Carcinogenesis. 1998;19:525–527. doi: 10.1093/carcin/19.3.525. [DOI] [PubMed] [Google Scholar]
- Jayaraman L, Murthy KGK, Zhu C, Curran T, Xanthoudakis S, Prives C. Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes & Dev. 1997;11:558–570. doi: 10.1101/gad.11.5.558. [DOI] [PubMed] [Google Scholar]
- Johnson AW, Demple B. Yeast DNA 3′-repair diesterase is the major cellular apurinic/apyrimidinic endonuclease: Substrate specificity and kinetics. J Biol Chem. 1988a;263:18017–18022. [PubMed] [Google Scholar]
- Johnson AW, Demple B. Yeast DNA diesterase for 3′-fragments of deoxyribose: Purification and physical properties of a repair enzyme for oxidative DNA damage. J Biol Chem. 1988b;263:18009–18016. [PubMed] [Google Scholar]
- Lawrence CW, Hinkle DC. DNA polymerase ζ and the control of DNA damage induced mutagenesis in eukaryotes. Cancer Surv. 1996;28:21–31. [PubMed] [Google Scholar]
- Lea DE, Coulson CA. The distribution of the numbers of mutants in bacterial populations. J Genet. 1949;49:264–285. doi: 10.1007/BF02986080. [DOI] [PubMed] [Google Scholar]
- Lindahl T, Nyberg B. Rate of depurination of native deoxyribonucleic acid. Biochemistry. 1972;11:3610–3617. doi: 10.1021/bi00769a018. [DOI] [PubMed] [Google Scholar]
- Mol CD, Kuo C-F, Thayer MM, Cunningham RP, Tainer JA. Structure and function of the multifunctional DNA-repair enzyme exonuclease III. Nature. 1995;374:381–386. doi: 10.1038/374381a0. [DOI] [PubMed] [Google Scholar]
- Mozzherin DJ, Shibutani S, Tan C-K, Downey KM, Fisher PA. Proliferating cell nuclear antigen promotes DNA synthesis past template lesions by mammalian DNA polymerase δ. Proc Natl Acad Sci. 1997;94:6126–6131. doi: 10.1073/pnas.94.12.6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JR, Lawrence CW, Hinkle DC. Deoxycytidyl transferase activity of yeast REV1 protein. Nature. 1996a;382:729–731. doi: 10.1038/382729a0. [DOI] [PubMed] [Google Scholar]
- ————— Thymine-thymine dimer bypass by yeast DNA polymerase ζ. Science. 1996b;272:1646–1649. doi: 10.1126/science.272.5268.1646. [DOI] [PubMed] [Google Scholar]
- Popoff SC, Spira AI, Johnson AW, Demple B. Yeast structural genes (APN1) for the major apurinic endonuclease: Homology to Escherichia coli endonuclease IV. Proc Natl Acad Sci. 1990;87:4193–4197. doi: 10.1073/pnas.87.11.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramotar D, Popoff SC, Gralla EB, Demple B. Cellular role of yeast Apn1 apurinic endonuclease/3′-diesterase: Repair of oxidative and alkylation DNA damage and control of spontaneous mutation. Mol Cell Biol. 1991;11:4537–4544. doi: 10.1128/mcb.11.9.4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein R. Targeting, disruption, replacement, and allele rescue: Integrative DNA transformation in yeast. Methods Enzymol. 1991;194:281–301. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- Roy R, Brooks C, Mitra S. Purification and biochemical characterization of recombinant N-methylpurine-DNA glycosylase of the mouse. Biochemistry. 1994;33:15131–15140. doi: 10.1021/bi00254a024. [DOI] [PubMed] [Google Scholar]
- Sander M, Ramotar D. Partial purification of PdeI from Saccharomyces cerevisiae: Enzymatic redundancy for the repair of 3′-terminal DNA lesions and abasic sites in yeast. Biochemistry. 1997;36:6100–6106. doi: 10.1021/bi970048y. [DOI] [PubMed] [Google Scholar]
- Seeberg E, Eide L, Bjorås M. The base excision repair pathway. Trends Biochem Sci. 1995;20:391–396. doi: 10.1016/s0968-0004(00)89086-6. [DOI] [PubMed] [Google Scholar]
- Strauss BS. The ‘A-rule’ of mutagen specificity: A consequence of DNA polymerase bypass of non-instructional lesions? BioEssays. 1991;13:79–84. doi: 10.1002/bies.950130206. [DOI] [PubMed] [Google Scholar]
- Torres-Ramos CA, Yoder BL, Burgers PMJ, Prakash S, Prakash L. Requirement of proliferating cell nuclear antigen in RAD6-dependent postreplicational DNA repair. Proc Natl Acad Sci. 1996;93:9676–9681. doi: 10.1073/pnas.93.18.9676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LJ, Robson CN, Black E, Gillespie D, Hickson ID. Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol Cell Biol. 1993;13:5370–5376. doi: 10.1128/mcb.13.9.5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace S. Oxidative damage to DNA and its repair. In: Scandalios JG, editor. Oxidative stress defenses. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 49–90. [Google Scholar]
- Winters TA, Henner WD, Russell PS, McCullough A, Jorgensen TJ. Removal of 3′-phosphoglycolate from DNA strand-break damage in an oligonucleotide substrate by recombinant human apurinic/apyrimidinic endonuclease 1. Nucleic Acids Res. 1994;22:1866–1873. doi: 10.1093/nar/22.10.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S, Curran T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992;11:653–665. doi: 10.1002/j.1460-2075.1992.tb05097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S, Miao G, Wang F, Pan Y-CE, Curran T. Redox activation of Fos-Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992;11:3323–3335. doi: 10.1002/j.1460-2075.1992.tb05411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S, Miao GG, Curran T. The redox and DNA-repair activities of Ref-1 are encoded by nonoverlapping domains. Proc Natl Acad Sci. 1994;91:23–27. doi: 10.1073/pnas.91.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci. 1996;93:8919–8923. doi: 10.1073/pnas.93.17.8919. [DOI] [PMC free article] [PubMed] [Google Scholar]