

Abstract
The anti-σ factor FlgM of Salmonella typhimurium inhibits transcription of class 3 flagellar genes through a direct interaction with the flagellar-specific σ factor, σ28. FlgM is believed to prevent RNA polymerase (RNAP) holoenzyme formation by sequestering free σ28. We have analyzed FlgM-mediated inhibition of σ28 activity in vitro. FlgM is able to inhibit σ28 activity even when σ28 is first allowed to associate with core RNAP. Surface plasmon resonance (SPR) was used to evaluate the interaction between FlgM and both σ28 and σ28 holoenzyme (Eσ28). The Kd of the σ28–FlgM complex is ∼2 × 10−10 m; missense mutations in FlgM that cause a defect in σ28 inhibition in vivo increase the Kd of this interaction by 4- to 10-fold. SPR measurements of Eσ28 dissociation in the presence of FlgM indicate that FlgM destabilizes Eσ28, presumably via an interaction with the σ subunit. Our data provide the first direct evidence of an interaction between FlgM and Eσ28. We propose that this secondary activity of FlgM, which we term holoenzyme destabilization, enhances the sensitivity of the cell to changes in FlgM levels during flagellar biogenesis.
Keywords: σ-factors, RNA polymerase, transcription, Salmonella typhimurium
Bacterial core RNA polymerase (RNAP), a multimeric enzyme with a molecular composition of α2ββ′, has the ability to transcribe RNA processively from a nick or bubble in double-stranded DNA, but is unable to initiate transcription specifically at a promoter (Burgess and Travers 1969). Promoter specificity is conferred by one of a number of dissociable σ subunits, which contain the determinants necessary for promoter recognition and localized denaturation of the DNA (Helmann and Chamberlin 1988). The interaction of σ with core RNAP to form RNAP holoenzyme is mediated by at least one domain of σ, located in conserved σ regions 2.1/2.2 (Lesley and Burgess 1989; Shuler et al. 1995; Tintut and Gralla 1995; Severinova et al. 1996; Cliften et al. 1997; Joo et al. 1997; Léonetti et al. 1998; Owens et al. 1998) and may also involve conserved σ regions 3 (Zhou et al. 1992; Joo et al. 1998; Cliften et al. 1997; Owens et al. 1998). The holoenzyme complex is extremely stable, with a Kd of ∼5 × 10−10 m(Gill et al. 1991).
Escherichia coli contains seven species of σ subunits (Ishihama 1997), which share significant sequence similarity (Lonetto et al. 1992). The majority of bacterial genes are transcribed by RNAP holoenzyme containing the vegetative σ factor, σ70 (Harris et al. 1978; Osawa and Yura 1981). Subsets of genes whose products are required only during certain developmental stages or under specific conditions are often coordinately transcribed by RNAP holoenzyme containing an alternative σ factor. Pools of alternative σ factors may be synthesized before they are needed and maintained in an inactive state until the cell receives a signal that they are required. This strategy is often employed in situations that demand a rapid and coordinated shift in the focus of transcription, such as during flagellar synthesis in E. coli, S. typhimurium, and other species (Ohnishi et al. 1990; Helmann 1991; Liu and Matsumura 1995). The sequestering of σ factors from core RNAP is carried out by a family of negative regulatory proteins known as anti-σ factors (for review, see Brown and Hughes 1995; Hughes and Matthai 1998). Anti-σ factors bind directly to free σ subunits and appear to inhibit an early step in the transcription cycle prior to the stable association of RNAP holoenzyme with promoter DNA. Anti-σ factors are themselves negatively regulated by a variety of mechanisms, including export of the anti-σ factor out of the cell, and competitive binding by anti-anti-σ factors.
Studies of the interaction between the anti-σ factor FlgM and the flagellar-specific σ28 of Salmonella typhimurium led to the proposal that FlgM prevents holoenzyme formation by binding to the amino-terminal conserved σ regions 2.1/2.2 of σ28, thereby masking important core RNAP-binding residues (Ohnishi et al. 1992). The discovery that the minimal FlgM-binding domain of σ28 was contained in the carboxy-terminal −35 promoter-binding domain, comprised by regions 4.1/4.2, led to revision of this model (Kutsukake et al. 1994). It was proposed that FlgM bound to σ28 at regions 4.1/4.2 interfered with holoenzyme formation through an allosteric mechanism transmitted from regions 4.1/4.2 to regions 2.1/2.2. Other σ/anti-σ factor pairs appear to interact in a similar manner. It has been demonstrated that a carboxy-terminal fragment of σ70 containing region 4.2 is the minimal epitope capable of binding the phage T4 anti-σ70 factor (Severinova et al. 1996; Adelman et al. 1997; Colland et al. 1998). Likewise, regions 4.1/4.2 of the Bacillus subtilis sporulation-specific σ factor σF fused to maltose-binding protein (MBP) was sufficient to mediate binding to the anti-σ factor SpoIIAB (Decatur and Losick 1996). Genetic analysis of σ28 and σF have identified single amino acid substitutions that allow these σ factors to escape negative regulation by their cognate anti-σ factors, presumably by altering important anti-σ contact points. The majority of these mutations map to regions 4.1/4.2 (Kutsukake et al. 1994; Decatur and Losick 1996; M. Chadsey and K. Hughes, unpubl.).
Despite the emerging similarity among different σ/anti-σ factor systems, little progress has been made in understanding how anti-σ factor binding affects the σ–core RNAP interaction. It is clear that this process is more complicated than the results above would suggest. The same genetic selections that yielded the region 4.1/4.2 substitution mutants of σ28 and σF also produced mutants in the putative core RNAP-binding domains in regions 2.1 and 3.1, and MBP fusions to each of these regions of σF also showed some affinity for SpoIIAB (Decatur and Losick 1996). Some of the altered residues may represent additional anti-σ factor contact points. Alternatively, the substitutions may interfere with an allosteric signal propagating from anti-σ factor bound at the carboxyl terminus. A third possibility is that the substitutions increase the affinity of the σ factors for core RNAP, thus enabling core RNAP to compete more effectively with the anti-σ28 factor for the σ factors.
A major question raised by these analyses is whether the sites recognized by anti-σ factors on free σ are still accessible on holoenzyme. If this is the case, then anti-σ factors could regulate transcription at two levels: (1) By binding to and sequestering free σ from core RNAP (Benson and Haldenwang 1993; Decatur and Losick 1996; Gamer et al. 1996; Gorham et al. 1996; Xie et al. 1996), and (2) by binding to holoenzyme via the σ subunit to prevent transcription directly. We have addressed this question in our analysis of the interaction between σ28 and FlgM in S. typhimurium. During flagellar biogenesis, FlgM regulates the transcription of σ28-dependent genes (Gillen and Hughes 1991), which encode the subunits of the flagellar filament. The genes encoding σ28 and FlgM (fliA and flgM, respectively) are both expressed early in flagellar biogenesis (Kutsukake et al. 1990; Ohnishi et al. 1990; Gillen and Hughes 1993). FlgM checks σ28 activity while the membrane-associated flagellar substructure, the hook/basal body (HBB), is being assembled. Upon completion of the HBB, FlgM is secreted out of the cell through the nascent flagella, thereby releasing σ28 from inhibition (Hughes et al. 1993; Kutsukake 1994). The purpose of our study was to examine in vitro the mechanism of FlgM-mediated inhibition of σ28-dependent transcription. The data presented here suggest that, in addition to sequestering free σ28 from core RNAP, FlgM employs a novel mechanism to regulate σ28 holoenzyme (Eσ28) activity.
Results
Mutations in the carboxy-terminal half of FlgM disrupt anti-σ factor activity
To better understand the mechanism by which FlgM prevents σ28-dependent transcription, we isolated flgM mutants defective in this activity (Hughes et al. 1993; Daughdrill et al. 1997). Two of these, missense mutants with single amino acid substitutions in the carboxy-terminal σ28-binding domain of FlgM (Iyoda and Kutsukake 1995; Daughdrill et al. 1997), were chosen for further characterization in vitro to determine whether their in vivo phenotype was in fact attributable to a decrease in their ability to inhibit the σ28 transcription machinery (σ28 and/or Eσ28). Strains TH2781 and TH3472 express mutant alleles with a Leu to Ser change at residue 66 (FlgM*L66S), and an Ile to Thr substitution at residue 82 (FlgM*I82T), respectively (* is used to designate mutants defective for σ28 inhibition). The mutant flgM* alleles were cloned into amino-terminal histidine-tag fusion vectors as described in Materials and Methods. The purified mutant proteins, His–FlgM*L66S and His–FlgM*I82T, were then compared with His–FlgM for their ability to inhibit σ28-dependent transcription from the fliC promoter in vitro. Reconstituted Eσ28 specifically transcribed the σ28-dependent fliC promoter and reached a near-maximal rate of transcription at a 1:1 ratio between σ28 and core RNAP (Fig. 1A). When Eσ28 was challenged with increasing amounts of His–FlgM, transcription from this promoter rapidly declined, and reached background (σ-independent) levels at a 4:1 ratio of FlgM to σ28 (Fig. 1B). Both FlgM* mutants demonstrated reduced ability to inhibit σ28-dependent transcription; even at a 10:1 ratio of FlgM* to σ28, transcription was not completely abolished. In vivo assays of β-galactosidase expression from σ28-dependent promoters in the presence of the flgM* alleles were consistent with the results of the in vitro transciption assays; strains expressing the mutant FlgM*L66S and FlgM*I82T proteins had three- and six-fold higher levels of β-galactosidase activity, respectively, compared with isogenic flgM+ strains (data not shown). Thus, the simplest explanation for the effect of the substitution mutations in vivo and in vitro is that they fail to bind to σ28, or inhibit its activity, as efficiently as wild-type FlgM. However, neither assay revealed whether the target of FlgM-mediated inhibition was σ28, Eσ28, or both.
Figure 1.
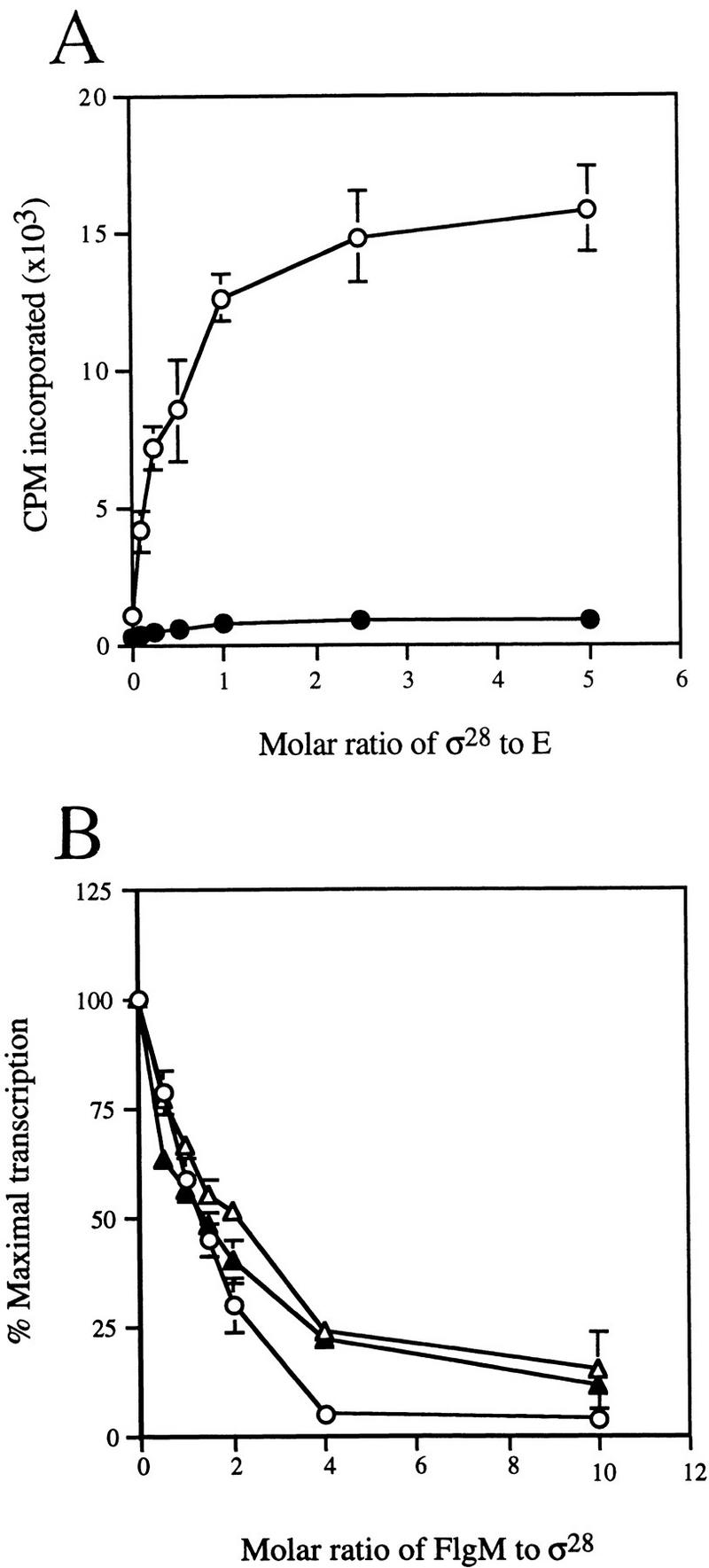
Effect of FlgM on σ28-dependent transcription in vitro. (A) Transcription from a σ28-dependent promoter by reconstituted Eσ28. Reaction mixtures contained 30 nm His-core RNAP (E) and 3 nm template DNA [either the σ28-dependent fliC promoter (○), or the σ70-dependent tac promoter (•)]. The molar ratio of σ28 to core RNAP in each reaction is indicated on the x axis. (B) Inhibition of σ28-dependent transcription by FlgM and mutant FlgM* proteins. Purified His–FlgM proteins were incubated with σ28 and core RNAP prior to the initiation of transcription. Reaction mixtures contained 30 nm core RNAP and 15 nm σ28. (○) FlgM; (▵) FlgM*L66S; (▴) FlgM*I82T. The molar ratio of His–FlgM to σ28 in each reaction is indicated on the x axis. Error bars in A and B indicate s.d. (assays performed in triplicate).
The FlgM*L66S mutant is defective for binding to GST–σ28
Cross-linking of purified FlgM and σ28 in vitro suggested that the two proteins could interact directly (Ohnishi et al. 1992). Copurification of FlgM with a GST–σ28 fusion protein from a crude cell extract demonstrated that the σ28–FlgM complex was sufficiently stable to be isolated without cross-linking (Jishage and Ishihama 1998). The results of a similar GST–σ28 assay for FlgM–σ28 complex formation are shown in Figure 2A. When crude extracts of S. typhimurium cells expressing GST–σ28 were passed over a glutathione–Sepharose column, the GST–σ28 fusion protein, bands corresponding to the α (not visible by Coomassie staining), β, and β′ subunits of RNAP, and the FlgM protein were all retained (lane 2. The presence of α and the identity of FlgM were confirmed by Western analysis; data not shown). Neither FlgM nor the RNAP subunits bound to the unfused GST protein (lane 1). These data confirmed that FlgM is able to bind σ28, and they did not exclude the possibility that FlgM is also capable of binding Eσ28.
Figure 2.
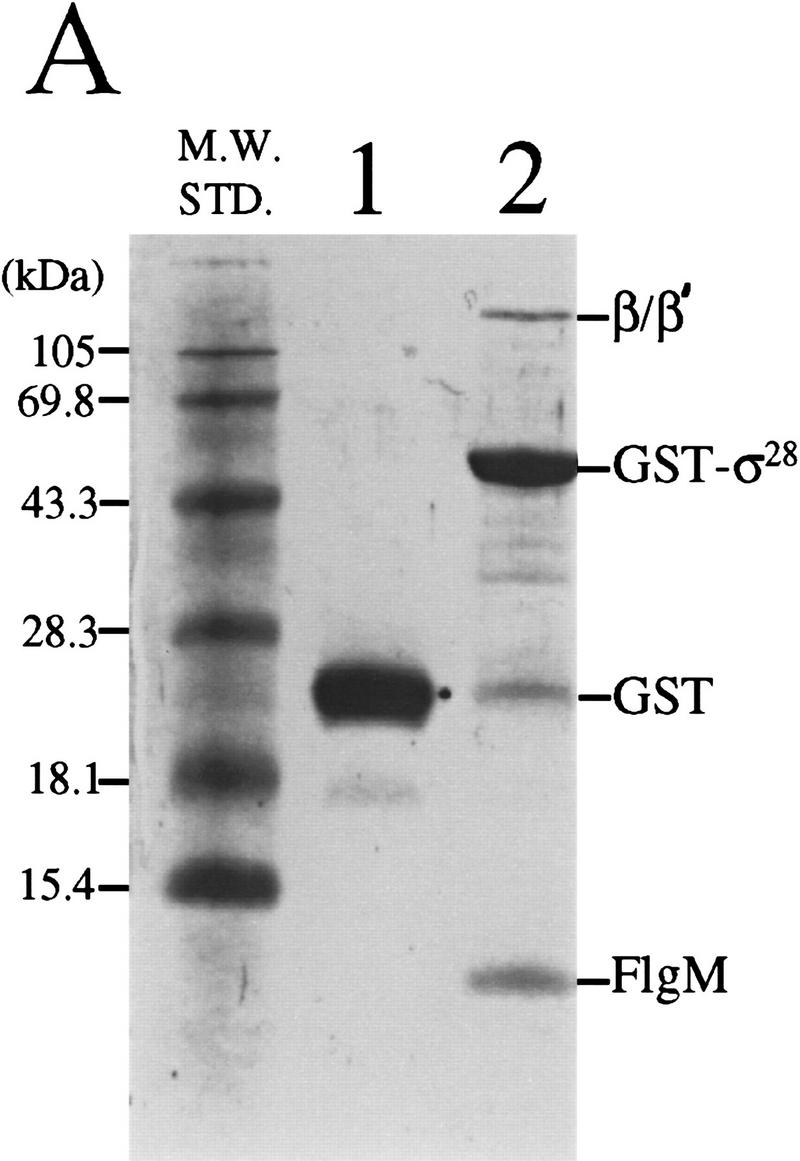
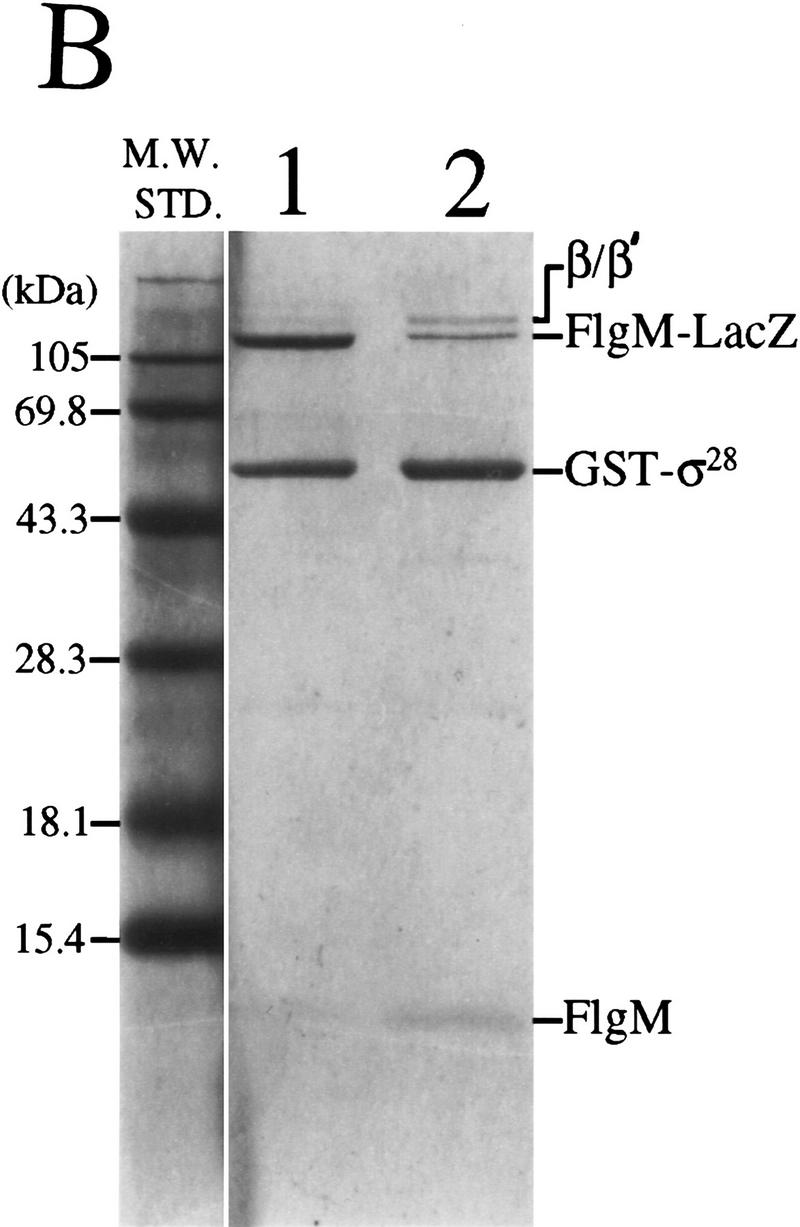
Binding of FlgM and mutant FlgM*L66S proteins to GST–σ28 in crude cell extracts. (A) FlgM associates with GST–σ28. Crude cell extracts from S. typhimurium strain LT2 expressing either GST alone or a GST–σ28 fusion were passed over glutathione–Sepharose affinity columns. Proteins that remained on the column following washing were eluted, separated by SDS-PAGE, and stained with Coomassie blue. (Lane 1) GST alone; (lane 2) GST–σ28 fusion. (B) FlgM*L66S mutation weakens the FlgM–σ28 interaction. Assays were performed as above except that crude cell extracts were prepared from strains expressing FlgM–LacZ (lane 1) and FlgM*L66S–LacZ (lane 2).
The GST–σ28-based assay for σ28–FlgM complex formation was employed to assess the relative affinities of FlgM and FlgM*L66S peptides for GST–σ28. The GST–σ28 expression vector (pKH486) was introduced into merodiploid S. typhimurium flgM strains expressing FlgM–LacZ fusion peptides in addition to native FlgM. Cell extracts from pKH486/TH2822 (expressing FlgM and FlgM–LacZ) and pKH486/TH2825 (expressing FlgM and FlgM*L66S–LacZ) were analyzed as above (Fig. 2B). A comparison of lane 2 with lane 1 reveals that GST–σ28 bound significantly less of the FlgM*L66S–LacZ fusion (and consequently more of the native FlgM protein and the β/β′ subunits of core RNAP) than it did of the FlgM+–LacZ fusion. This result can be attributed to a weaker affinity of the FlgM*L66S–LacZ fusion for GST–σ28, as the intracellular level of the mutant LacZ fusion protein in TH2825 was determined by quantitative Western analysis to be at least as great as that of FlgM+–LacZ in TH2822 (data not shown). These data suggested that the defect in the ability of FlgM*L66S to inhibit σ28 is attributable to a decreased affinity of that mutant for σ28.
FlgM-mediated inhibition of σ28 activity occurs prior to DNA binding
The filter-binding technique (Hinkle and Chamberlin 1972) was employed to test the effect of FlgM on promoter binding by Eσ28. In these assays, specific binding of promoter DNA is tested by challenge of protein–DNA complexes with nonspecific competitor DNA. Neither σ28 nor FlgM alone was able to bind DNA containing the fliC promoter in this assay (Fig. 3A, lanes 2,8). Only RNAP containing σ28 bound specifically to the fliC promoter (Fig. 3 cf. A, lanes 4 and 6 with B, lanes 1 and 4). Specific binding of Eσ28 to the fliC promoter was inhibited by FlgM regardless of the order of addition of proteins to the reaction mixture (Fig. 3B, lanes 2,3). In these two lanes, it appeared that σ28 that had first been incubated with core RNAP for a period of time sufficient to allow holoenzyme formation (as evidenced by the data in Fig. 1) was as susceptible to FlgM as free σ28. However, once Eσ28 formed a complex with the promoter, subsequent incubation with FlgM could not reverse this process (data not shown), which suggested that FlgM is not able to interact with Eσ28 once it is bound to DNA.
Figure 3.
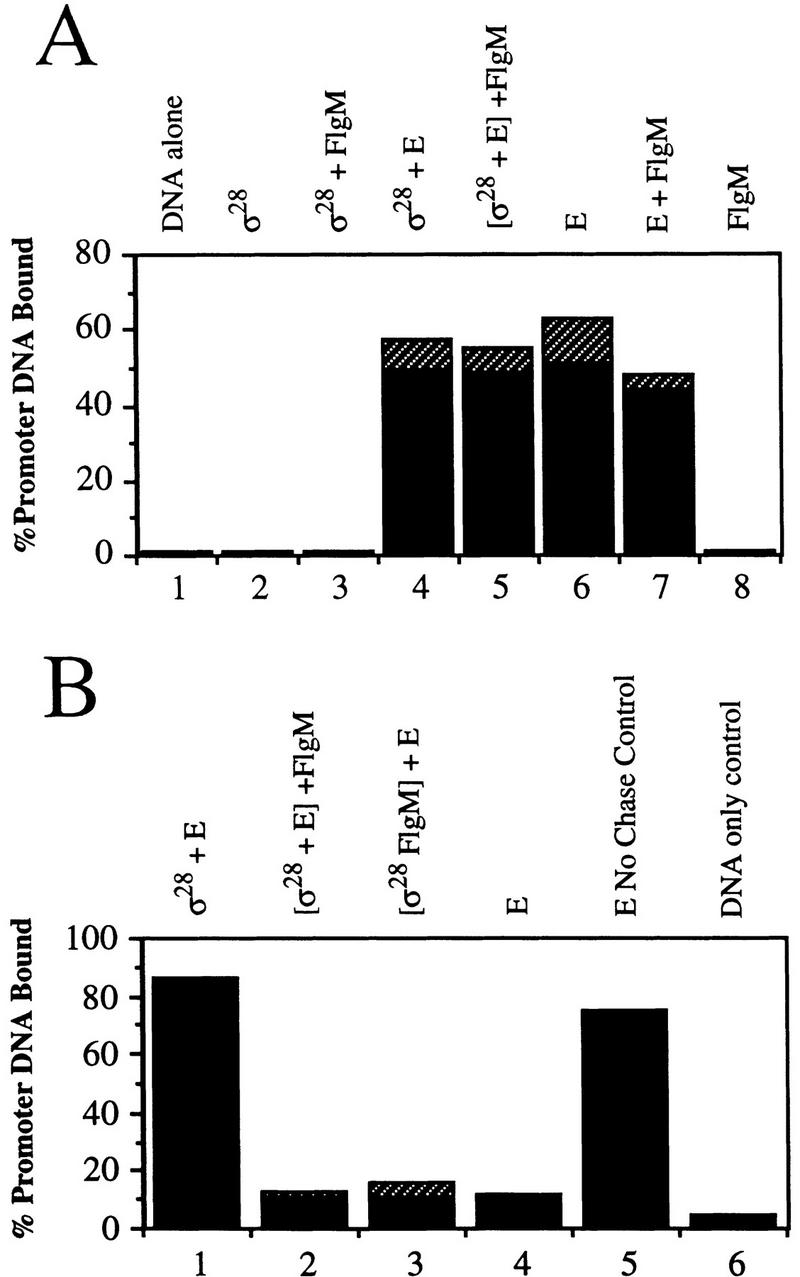
FlgM interferes with specific binding of Eσ28 to the fliC promoter. Filter binding assays were used to evaluate the effect of FlgM on promoter binding by Eσ28. Proteins indicated above each lane were incubated together for 10 min prior to the addition of labeled promoter DNA. In lane 5 (A) and lanes 2 and 3 (B), proteins enclosed in brackets were preincubated for 10 min before the addition of the third protein; reactions were incubated a further 5 min before DNA was added. The hatched bars indicate s.d. (assays performed in triplicate). (A) Noncompeted binding of the fliC promoter by σ28, core RNAP (E), and FlgM. (B) The effect of FlgM on specific binding of Eσ28 to the fliC promoter. Samples were chased with unlabeled sheared salmon sperm DNA (a competitor for nonspecific DNA binding) prior to sampling, so promoter DNA retained on filters represents only that bound specifically by protein.
FlgM comigrates with Eσ28 in native PAGE
Resolution of protein complexes on native gels was employed to test for the existence of a ternary FlgM–Eσ28 complex. FlgM, σ28, and core RNAP were electrophoresed either by themselves, or in different combinations on a native gel (Fig. 4A, lanes 1–7). Protein complexes from the native gel were analyzed by SDS-PAGE to identify their constituents (Fig. 4B, lanes 10–14). When σ28 and FlgM were mixed, a novel band appeared on the native gel (Fig. 4A, lane 3) which proved to be a heterocomplex (Fig. 4B, lane 14). The possibility that FlgM was comigrating with σ28 by virtue of a nonspecific interaction was tested by mixing FlgM with BSA, which has a predicted pI similar to that of σ28 (4.97 and 4.91, respectively). No FlgM was found associated with BSA (data not shown). When core RNAP was mixed with σ28, FlgM, or both proteins together, no novel bands appeared, though the appearance of the RNAP complex in the presence of σ28 was changed, possibly resulting from the formation of Eσ28 (Fig. 4A, lanes 4–7). SDS-PAGE analysis of these core complexes revealed the presence of comigrating proteins (Fig. 4B, lanes 9–13). As expected, σ28 associated with core RNAP (Fig. 4B, lane 11). Although FlgM by itself did not associate with core RNAP (Fig. 4B, lane 10), separation of the RNAP complex in Figure 4A (lane 6; σ28 and FlgM added) revealed that a small amount (visible by Coomassie staining) was found to comigrate with the σ28–core RNAP complex (Fig. 4B, lane 12). Changing the order in which the proteins were added to the reaction mixture did not alter the outcome of this experiment (Fig. 4B, lane 13). The comigration of FlgM with the σ28–core RNAP complex was evidence for the existence of a ternary complex, but to adequately test this possibility, it would be necessary to use an assay that would not allow FlgM, σ28, and core RNAP to exist in dyamic equilibrium (i.e., where σ28 could be rapidly alternating between a complex with core RNAP and a complex with FlgM).
Figure 4.


FlgM associates with Eσ28 in vitro. A combination of native PAGE (A) and denaturing SDS-PAGE (B) was used to detect complexes formed by FlgM, σ28, and core RNAP. (A) Proteins shown at the top were incubated together for 15 min and electrophoresed on a native polyacrylamide gel. In lane 6, proteins enclosed in brackets were preincubated for 10 min before the addition of the third protein and a further 5-min incubation. Protein complexes were visualized by staining with Coomassie blue. (B) Bands corresponding to core RNAP or the σ28–FlgM complex in A were excised and separated by SDS-PAGE to identify the proteins contained within them. (Lane 8) σ28 and FlgM standards; (lanes 9–12), the core RNAP bands from lanes 4, 7, 5, and 6, respectively; (lane 13) the core RNAP band from a separate native gel in which the order of protein addition is indicated at the top of the gel; (lane 14), the σ28–FlgM band from lane 3.
Kinetic analysis of the FlgM–Eσ28, FlgM–σ28, and σ28–core RNAP interactions
The surface plasmon resonance (SPR)-based system (Biacore) was used to evaluate the interaction of FlgM with Eσ28 under nonequilibrium conditions. SPR enables the measurement of molecular interactions in real time by following the specific adsorption and desorption of a protein in solution (the analyte) to its binding partner (the ligand) immobilized on a sensor surface (Karlsson et al. 1991). The association rate constant (ka) can be determined by analysis of a collection of binding curves in which the concentration of analyte flowing over a fixed concentration of ligand is varied. The dissociation rate constant (kd) can likewise be calculated from the subsequent decrease in signal that occurs after the analyte pulse has ended, and the continuous flow of buffer removes analyte from the sensor surface as it dissociates from bound ligand.
This latter feature of the SPR system enabled us to test one model for FlgM inhibition of Eσ28 that was not easily evaluated by other methods, that is, that FlgM binds to Eσ28 and increases the rate of dissociation of σ28 from core RNAP. We reasoned that if FlgM possessed the ability to destabilize holoenzyme, then the dissociation rate of the Eσ28 complex would increase in the presence of FlgM. If, however, the apparent susceptibility of Eσ28 to FlgM demonstrated by the filter binding assays was simply attributable to the ability of FlgM to compete with core RNAP for spontaneously dissociated σ28, then the dissociation rate of the Eσ28 complex would be unaffected by FlgM. For these experiments, σ28 (28 kD) was used as the ligand and core RNAP (380 kD) as the analyte, as this arrangement would result in the largest change in signal per molecule of analyte released. Controls to ensure that the measurements would not be skewed by analyte rebinding during the dissociation phase were performed (see Materials and Methods).
The effect of FlgM on the rate of dissociation of Eσ28 is shown in Figure 5A. The results indicate that FlgM significantly increases the rate of Eσ28 dissociation, increasing the kd of the complex approximately fourfold. Reducing the FlgM concentration from 250 nm (used above) to 50 nm diminished this effect (such that the kd was increased only 2.5-fold), and the effect was not completely abolished until the concentration of FlgM was decreased to 10 nm (data not shown). A FlgM-induced increase in the kd of holoenzyme was not observed when His–σ70 was substituted as the ligand (Fig. 5B), demonstrating specificity for Eσ28 (the sharp drop in signal immediately following the end of analyte injection was caused by the difference in refractive index between the analyte buffer and the running buffer in this experiment and did not affect the interpretation of the data.) The fact that FlgM did not appear to affect Eσ70 suggests that the sites on Eσ28 with which FlgM interacts are completely or partially contained on the σ28 subunit. In a separate experiment, the ability of core RNAP to bind to FlgM was tested by injection of a 125 nm solution of core RNAP over a dense FlgM ligand surface; no specific binding was observed (data not shown).
Figure 5.

SPR analysis of the effect of FlgM proteins on the rate of dissociation of RNAP holoenzyme. An SPR-based assay was used to evaluate the effect of FlgM on the stability of Eσ28. The amount of protein associated with the chip surface was measured in RU; the change in RU during the course of the experiments is plotted as a function of time (seconds). All curves have been normalized relative to the start of analyte injection. The horizontal bar at the top of each panel defines the period during which core RNAP (the analyte) was injected (association phase). The dissociation rate constants (kd ) for Eσ28 in the presence of FlgM proteins or in HBS buffer are displayed next to the curves. (A) FlgM increases the rate of Eσ28 dissociation, and FlgM* mutants are defective for this activity. A 50 nm solution of purified core RNAP in HBS buffer was injected over a surface of immobilized σ28 to generate a layer of Eσ28. Dissociation of Eσ28 was carried out in HBS buffer or in HBS buffer containing 250 nm His–FlgM, 250 nm His–FlgM*L66S, or 250 nm His–FlgM*I82T. (B) Eσ70 is not destabilized by FlgM. These experiments were carried out in the same manner as above, except that His–σ70 instead of σ28 served as the immobilized ligand, and the amount of His–FlgM during dissociation was 50 nm.
SPR was used to compare σ28 binding by the FlgM*L66S and FlgM*I82T missense mutants with that of wild-type FlgM. In the first set of experiments, His–FlgM proteins were used as the ligand, and the analyte was native σ28. The ka and kd for each interaction, the calculated dissociation constant (Kd), and the relative affinity of the mutant FlgM* variants for σ28 are reported in Table 1. The ∼3.6-fold weaker affinities of the mutant FlgM* proteins for σ28 in these experiments were reproducible; independent preparations of FlgM proteins and native σ28 gave essentially identical results. In the second set of experiments, the identities of the ligand and analytes were reversed; His–σ28 was used as the ligand, and wild-type and mutant His–FlgM proteins as analytes. The results of these experiments paralleled those from the first set of assays, though the σ28 binding defect of the mutants was more pronounced here, mainly because of an increase in the kd (this discrepancy might be attributed to the presence of the histidine tag on σ28, or to the different ligand capture method used in these experiments). Also presented in Table 1 are the ka , kd, and the calculated Kd obtained from a kinetic analysis of the σ28–core RNAP interaction. Although the calculated Kd for the Eσ28 complex is similar to that measured for Eσ70 using fluorescent spectroscopy (Gill et al. 1991), the accuracy of this estimate should be confirmed using an alternative technique before it is accepted. The SPR technique is used most reliably to derive relative kinetic measurements, as was done to test the effect of FlgM on Eσ28 dissociation. In a relative context, however, it is worth pointing out that the affinity of σ28 for core in these experiments was similar to that of σ28 for FlgM. This may have important consequences on the ability of FlgM and core RNAP to compete for σ28 in vivo (see Discussion).
Table 1.
Analysis of binding of FlgM proteins and core RNAP to σ28
| Ligand
|
Analyte
|
ka (105/m per sec)
|
kd (10−4/sec)
|
Kd (pM)
|
Relative affinity (Kd*/Kd WT)
|
|---|---|---|---|---|---|
| His–FlgM | σ28 | 8.9 | 1.6 | 180 | — |
| (5.0) | (1.0) | (200) | — | ||
| His–FlgM*L66S | σ28 | 2.4 | 1.5 | 630 | 3.5× |
| (2.3) | (1.6) | (720) | (3.6×) | ||
| His–FlgM*I82T | σ28 | 5.6 | 3.7 | 660 | 3.7× |
| (3.4) | (2.5) | (740) | (3.7×) | ||
| His–σ28 | His–FlgM | 5.5 | 1.6 | 290 | — |
| His–σ28 | His–FlgM*L66S | 2.1 | 8.5 | 4000 | 13.8× |
| His–σ28 | His–FlgM*I82T | 5.5 | 16 | 3000 | 10.3× |
| His–σ28 | core RNAP | 4.8 | 3.9 | 800 |
His–FlgM proteins (wild type and mutant) were prepared in parallel. Data set in parentheses was generated with an independently purified series of His–FlgM proteins and native σ28.
The ability of mutant FlgM* proteins to increase the kd of Eσ28 was tested to determine whether their σ28 binding defects also affected this interaction. Figure 5A shows that at 250 nm, neither of the FlgM* proteins was able to significantly increase the dissociation of Eσ28 above the rate in the control experiment, providing further evidence that destabilization of Eσ28 by FlgM appears to involve specific contacts between FlgM and σ28.
Discussion
SPR experiments designed to test the effect of FlgM on Eσ28 stability revealed that FlgM increases the rate of dissociation of the complex, presumably through an interaction with the σ28 subunit. Though this effect could be caused by the ability of FlgM to interfere with rebinding of core RNAP to σ28 during dissociation, extensive controls indicated this was unlikely to be the case here (see Materials and Methods). The finding that FlgM is able to increase the dissociation rate of Eσ28 in vitro, an activity we term holoenzyme destabilization, allows us to speculate about the nature of the σ28–FlgM interaction, and about the behavior of this σ subunit when it is bound to core RNAP.
The σ28–FlgM complex may involve multiple contacts between FlgM and σ28 at regions 2.1, 3.1, and 4.1/4.2, analogous to the interaction between σF and SpoIIAB (Kutsukake et al. 1994; Decatur and Losick 1996; M. Chadsey and K. Hughes, unpubl.). Multiple interactions between σ28 and FlgM could account for the extreme stability of the complex, which must be dissolved in 6 m guanidine HCl to achieve complete dissociation (see Materials and Methods). The region of FlgM identified as the σ28-binding domain is extensive (between 25 and 57 contiguous residues at the carboxyl terminus; Iyoda and Kutsukake 1995; Daughdrill et al. 1997, 1998), and could conceivably make contacts with noncontiguous domains on σ28. This entire region, normally unstructured, is known to undergo a conformational change when it binds σ28, as upon complex formation it becomes constrained (Daughdrill et al. 1997, 1998). The substitution at FlgM residue 66 is part of a contiguous 14 residue region that appears to adopt a helical structure upon σ28 binding (Daughdrill et al. 1998). Unlike the I82T mutation, L66S appears to affect the ka of the σ28–FlgM interaction, which may imply that substitution of a polar residue for a hydrophobic one at this position perturbs the folding of FlgM that occurs upon binding of σ28. The observation that the FlgM missense mutants that are defective for free σ28 binding are also defective for the interaction with Eσ28 suggests that the site(s) on free σ28 that contacts the altered FlgM residues also mediates the interaction between FlgM and Eσ28.
If, as is likely, the emerging concept of the σ–core RNAP interaction can be generalized to σ28, then both regions 2 and 3 of σ28 may participate in core RNAP binding. A multipartite interaction between σ28 and core RNAP means that the Eσ28 complex might breathe. A breathing complex could be viewed as one in which σ regions 2 and 3 are alternately exposed as they temporarily lose contact with core RNAP, making them available to competing binding partners such as FlgM (Fig. 6, pathway I). Alternatively, breathing could be imagined as an allosteric process in which the conformation of σ alternates between a tightly bound conformation (bound at both regions 2 and 3) and a weakly bound conformation (bound at region 2 or 3; Fig. 6, pathway II). This alternative scenario does not assume that σ regions normally buried in the σ28–core RNAP interface are exposed by breathing.
Figure 6.

Possible mechanisms for holoenzyme destabilization by FlgM. Conserved σ regions 2, 3, and 4 are numbered. σ28 regions interacting with core RNAP (E) are shaded dark gray; σ28 regions interacting with FlgM are shaded light gray. Free FlgM is depicted as an unstructured molecule; σ28-bound FlgM is depicted in a constrained conformation. For simplicity, only the carboxy-terminal σ28-binding domain of FlgM is shown participating in the interaction with σ28. (I) Competitive displacement model. Eσ28 breathing alternately exposes σ regions 2 and 3. FlgM partially bound to σ28 can compete with E for rebinding to regions 2 and 3 to effect dissociation. For simplicity, FlgM is only shown interacting with one of the two partially bound forms of σ28. (II) Allosteric displacement model. Binding of FlgM to exposed region 4 stabilizes a σ28 conformation that is incompatible with E, resulting in the release of the σ28–FlgM complex.
It is not clear how FlgM is able to destabilize Eσ28. We can envision two models, which our data do not allow us to distinguish at this point (Fig 6). The competitive displacement model (pathway I) proposes that destabilization of Eσ28 results from the displacement of core RNAP from σ as FlgM competes for binding to transiently exposed σ regions 2 and 3. FlgM may initiate this process by interacting with its unique binding site in regions 4.1/4.2. Establishing contact at this site would increase the probability of subsequent interactions with regions 2.1 and 3.1. It is reasonable to expect regions 4.1/4.2 to be accessible on Eσ28, as they must be able to interact with the −35 promoter determinants. Alternatively, FlgM may bind first to one of the domains it shares with core RNAP (these two possibilities are not differentiated in Fig. 6). The allosteric displacement model (pathway II) postulates that FlgM interacts with Eσ28 at exposed σ regions 4.1/4.2 to stabilize a FlgM-bound conformation of σ28 that cannot interact efficiently with core RNAP. Additional contacts between FlgM and σ28 are proposed to form after dissociation of the σ subunit from Eσ28.
Both models invoke a ternary complex between FlgM and Eσ28. Attempts to demonstrate similar complexes in other anti-σ/σ factor systems have been largely unsuccessful, with the exception of the copurification of the phage T4 protein AsiA with Eσ70 (Stevens 1976; Orsini et al. 1993). The relative stability of the AsiA–Eσ70 complex can be attributed to the positive regulatory role of σ70-bound AsiA (in conjuction with the phage T4 protein MotA) at σ70-dependent middle phage promoters (Ouhammouch et al. 1994, 1995). Often, the conclusion drawn from these negative results is that core RNAP binding and anti-σ factor binding are mutually exclusive. We submit that, at least in the case of Eσ28 and FlgM, it is only the unstable nature of the ternary complex that has prevented its isolation. Detection of an Eσ28–FlgM complex in the native gel assays was probably attributable to the extremely high concentration (>1 μm σ28 and FlgM; 100 nm core RNAP) of the proteins in the samples.
The SPR experiments revealed that in the presence of FlgM, the half-life of the Eσ28 complex decreased approximately fourfold to 4 min. However, because the signal generated by FlgM binding to Eσ28 was insignificant compared to the drop in signal caused by Eσ28 dissociation, we were unable estimate the half-life of the putative FlgM–Eσ28 complex itself. It is possible that the half-life of this complex is significant on the time scale required for Eσ28 to initiate transcription at a promoter (Record 1996). If this is the case, then the primary mechanism of Eσ28 inhibition by FlgM may be steric interference with promoter recognition by the −35 promoter-binding domain in regions 4.1/4.2, analagous to the proposed effect of σ70-bound AsiA at phage T4 early promoters (Severinova et al. 1996, 1998; Adelman et al. 1997; Colland et al. 1998).
In our SPR experiments, FlgM had to be present at a concentration at least two orders of magnitude higher than the Kd of the σ28–FlgM complex (which was estimated to be ∼200 pm) to have an effect on the dissociation rate of Eσ28. This observation is consistent with both of the proposed models. In the competitive displacement model, a high concentration of FlgM might be necessary to compensate for the fact that some of its binding determinants on σ28 are masked by core RNAP. In the allosteric displacement model, the high concentration of FlgM might be required to overcome a kinetically unfavorable interaction with the core RNAP-associated conformation of σ28.
Preliminary measurements of FlgM in exponentially growing S. typhimurium indicate that the concentration of FlgM is within the range required for it to have an impact on Eσ28 activity. FlgM is present at ∼400 nm in wild-type cells, and is at least twice that in a mutant strain defective for FlgM export (assuming a cell size of 0.5 μm × 2 μm; data not shown). Two predictions can be made on the basis of these measurements. First, at these FlgM concentrations, there should be essentially no free σ28 in the cell; all σ28 should be either involved in a potentially active complex with core RNAP or sequestered by FlgM (the estimated Kd of the σ28–FlgM interaction is of the same magnitude as the estimated Kd of the σ28–core RNAP interaction; see Table 1). Second, relatively small changes in the concentration of FlgM when it is already approximately three orders of magnitude over the Kd of the σ28–FlgM complex will not affect the efficiency with which that interaction occurs, but they could affect the ability of FlgM to interact with Eσ28. Therefore, at least while FlgM levels are high, as they are believed to be prior to the completion of the HBB substructure (Hughes et al. 1993; Kutsukake 1994), FlgM-mediated inhibition of Eσ28 may be an important regulatory mechanism. There is a 10- to 15-min interval between the onset of expression of the genes encoding σ28 and FlgM, and the appearance of σ28-dependent gene products inside the cell (J. Karlinsey and K. Hughes, unpubl.). Assuming a 4-min half-life of Eσ28 in the presence of a high concentration of FlgM (>250 nm), this interval should be sufficient for Eσ28 levels to be significantly reduced by FlgM.
It appears that small changes in the affinity or concentration of FlgM are sufficient for FlgM to affect σ28 activity in vivo. S. typhimurium mutants expressing FlgM* proteins with only a 4- to10-fold higher Kd for the interaction with σ28 could be easily distinguished from their flgM+ parent strain on the basis of σ28-dependent gene expression (Daughdrill et al. 1997). In our SPR experiments, the effect of these mutations on the ability of FlgM to destabilize Eσ28 was significant. The level of FlgM in a S. typhimurium basal body mutant that is unable to export FlgM (and therefore does not express σ28-dependent genes) is only twofold higher than that of an export-competent strain that expresses σ28-dependent genes normally (Karlinsey et al. 1998). These observations are consistent with intracellular FlgM levels being close to the threshold concentration for σ28 inhibition.
Our data suggest that in addition to simply sequestering free σ28 from core RNAP, FlgM is capable of negatively regulating Eσ28 itself. We propose that regulation at the level of Eσ28 serves to enhance the sensitivity of the cell to changes in FlgM levels during assembly of the flagellar organelle. The ability to finely tune the expression of σ28-dependent genes during this process may be critical for efficient assembly. Recent work in our lab suggests that the level of FlgM prior to HBB completion may be more dynamic and responsive to intermediate stages in HBB development than was previously thought (Karlinsey et al. 1998). Our revised model is that FlgM levels are highest while the basal body substructure is being assembled, then decrease slightly during hook assembly as a prelude to dropping to their lowest level once the hook is completed and the HBB becomes competent for export. One purpose of the slight decrease in FlgM levels could be to ease FlgM inhibition of Eσ28 activity just prior to completion of the HBB. A low level of σ28-dependent expression at this stage would enable the cell to synthesize a small pool of filament subunits for polymerization the moment the HBB was completed. This strategy would prevent there being a lag in flagellar biogenesis while maximal expression of flagellin was getting underway. Once export of FlgM through the completed HBB had commenced, the level of intracellular FlgM might reach a point where even sequestration of free σ28 from core RNAP is no longer efficient; during maturation of the filament, σ28-dependent expression of flagellin would be expected to reach its highest rate.
It has been proposed that as the flagella increase in length, export of FlgM should become increasingly difficult (Hughes et al. 1993). This, together with the fact that flgM transcription is positively regulated by Eσ28 (Gillen and Hughes 1993) and, therefore, is expected to be maximal during filament polymerization, could mean that the intracellular concentration of FlgM gradually returns to a high level as the flagella reach their mature length. FlgM inhibition of Eσ28 may play a homeostatic role once flagellar biogenesis is complete. Presumably at this stage there would be a large pool of active Eσ28. Dismantling of Eσ28 might play an important role in shifting the focus of cellular transcription away from flagellar promoters.
Materials and methods
Bacterial strains
S. typhimurium strains used were isogenic with wild-type strain LT2 (John Roth, University of Utah, Salt Lake City). flgM mutants TH2781 and TH3472 were isolated as described (Daughdrill et al. 1997). FlgM–LacZ fusion strains TH2822 {DUP1114[(flgM5208)*MudB*(purB1879)]} and TH2825 {DUP1116[(flgM*5224 (L66S)flgM5208)*MudB*(purB1879)]} were constructed by standard genetic procedures (Davis et al. 1980). The strain TH3920 {fliC5050::MudA DEL1141[(flgA5211)* Mud Cm(flgN5220)] vh2− fljBenx} was used to assay the activity of the His-tagged FlgM proteins in vivo. TH3656 {leuBCD485 trp::[spcR PlacT7RNAPlacI]} (Stanley Maloy, University of Illinois, Urbana) was used for the expression of His–core RNAP. E. coli strain DH5α was used for cloning, and E. coli strain BL21(DE3) lysogenic for bacteriophage DE3 (Novagen) was used for the expression of His–σ28, His–σ70, and His–FlgM proteins. Cells were transformed by standard procedures.
Plasmid constructions
The His–σ28 fusion expression vector pKH445 was derived as follows: A 764-bp EcoRI fragment from pMS531 (Starnbach and Lory 1988) was cloned into the BamHI site of the vector pET15b (Novagen) to create pKH441 (both insert and vector were treated with Klenow prior to ligation). pKH441 was linearized with XhoI, partially filled in with Klenow, dTTP, dCTP, and dGTP, blunted with mung bean nuclease, and religated to create pKH445. The GST–σ28 fusion expression vector pKH486 was made by ligation of the EcoRI fragment from pMS531 into the EcoRI site of pGEX-3X (Pharmacia).
The FlgM expression vector pMC64 was derived as follows: A 326-bp DNA fragment containing the flgM gene was amplified from the LT2 chromosome by PCR, with primers EcoRI–BspHI–FlgM (GAATTCATGAGCATTGACCGTACC) and FlgMend (GTATTTCTGACAAACGAGTC) such that a BspHI site was introduced at the start codon of the flgM gene. This fragment was cloned into the SmaI site of BluescriptII vector KS+ (Stratagene) to create pMC56. A BspHI (Klenow-treated–HindIII fragment from pMC56 was cloned into pTrc99A (Pharmacia) digested with NcoI, blunted with mung bean nuclease, and digested with HindIII.
pMC96 is a derivative of pMC64 designed to co-express FlgM and σ28 and was derived as follows: The 764-bp EcoRI fragment from pMS531 was cloned into pTrc99A digested with NcoI to create the σ28 expression vector pMC61 (both insert and vector were treated with Klenow prior to ligation). An SspI fragment carrying the σ28 expression cassette from pMC61 was cloned into the unique NsiI site of pMC64 to create pMC96.
The His–FlgM+ expression vector pJK302 was constructed by cloning a BspHI (Klenow-treated)–HindIII fragment from pMC56 into pET28c (Novagen) digested with NheI, treated with Klenow, and digested with HindIII. The His–FlgM*I82T expression vector pJK306 was constructed as follows: flgM*5436(I82T) was PCR amplified from the chromosome of TH3472 with primers EcoRI–BspHI–FlgM and FlgMend. The PCR fragment was treated with Klenow, digested with EcoRI, and ligated into BluescriptII vector SK− digested with EcoRI and EcoRV to create pJK300. A BspHI (Klenow-treated)–HindIII fragment from pJK300 was cloned into pET28c as in the construction of pJK302 to create pJK306. The His–FlgM*L66S expression vector pJK314 was constructed as follows: flgM*5224(L66S) was cloned by overlap extension mutagenesis as described (White 1993). The first round of PCR used pMC56 as a template for two sets of reactions. Set A primers: M5′sense (GGAATTCCATATGAGCATTGACCGT) and L66Santisense (GCCGTTTTGCTTGCTTCGAC); set B primers L66Ssense (GTCGAAGCAAGCAAAACGGC) and FlgMend. PCR fragments generated from sets A and B were gel purified, pooled, and used as templates for a second round of PCR with primers M5′sense and FlgMend, generating a 340-bp fragment containing the flgM*5224 allele with an NdeI site at the start codon. This fragment was treated with Klenow and cloned into BluescriptII vector SK− digested with SmaI to create pJK310. An NdeI–BamHI fragment from pJK310 was cloned into pET28c digested with NdeI and BamHI to create pJK314.
The phage P22 integration vectors pJK374 (His–FlgM+), pJK376 (His–FlgM*I82T), and pJK378 (His–FlgM*L66S) were constructed by subcloning the NcoI–HindIII fragments from pJK302, pJK306, and pJK314, respectively, into pBAD24 (Guzman et al. 1995) digested with NcoI and HindIII to create pJK368, pJK369, and pJK370. BspEI (Klenow-treated)–SspI fragments from these plasmids were cloned into the EcoRI site of the phage P22 integration vector pMC16 (Chadsey 1998; details of construction available on request) to create pJK374, pJK376, and pJK378, respectively.
pJK127 was made by cloning of a 56-bp HindIII–EcoRI fliC promoter fragment from M13mp19-PfliC (A. Dombroski, University of Texas, Houston) into BluescriptII vector SK− digested with the same enzymes. A 211-bp BssHII fragment from pJK127 served as the DNA fragment for the DNA filter binding assays. pJK128 was made by cloning of a 141-bp HindIII–BssHII fragment from pJK127 into pKK233-2 (Pharmacia) digested with EcoRI and HindIII (both insert and vector were treated with Klenow prior to ligation).
In vivo assay of His–FlgM+ and His–FlgM* proteins
To verify that the His-tagged derivatives of FlgM+, FlgM*L66S, and FlgM*I82T were active in vivo, the genes expressing these fusion proteins (under the control of the arabinose-inducible PBAD promoter) were introduced into the chromosome in single copy as follows: Plasmids pJK374 (PBADhis–flgM+), pJK378 [PBADhis–flgM* 5224(L66S)], and pJK376 [PBADhis–flgM* 5436(I82T)] were recombined onto the chromosome of S. typhimurium phage P22 essentially as described (Youderian et al. 1983), except that mitomycin C treatment instead of UV irradiation was used to stimulate recombination between the plasmids and phage P22, to generate recombinant phage P22(JK374), P22(JK378), and P22(JK376). Though these phage have a partial deletion of the immI region, they can be propagated and lysogenized normally. Phage Φ374, Φ378, and Φ376 were lysogenized into strain TH3920 to introduce the PBADhis–flgM alleles into the S. typhimurium chromosome at the ataA locus. TH3920 is deleted for the flgM locus, and contains a transcriptional fusion of lacZ to the fliC gene, which serves as a reporter for σ28-dependent transcription. To assay His–FlgM-mediated inhibition of σ28-dependent LacZ expression, the lysogens were grown to mid-log stage in the presence of arabinose, and assayed for β-galactosidase activity in triplicate as described (Davis et al. 1980). The results of these assays demonstrate that the His-tagged FlgM proteins are active in vivo, and that the His–FlgM* proteins retain their mutant phenotype (Fig. 7).
Figure 7.
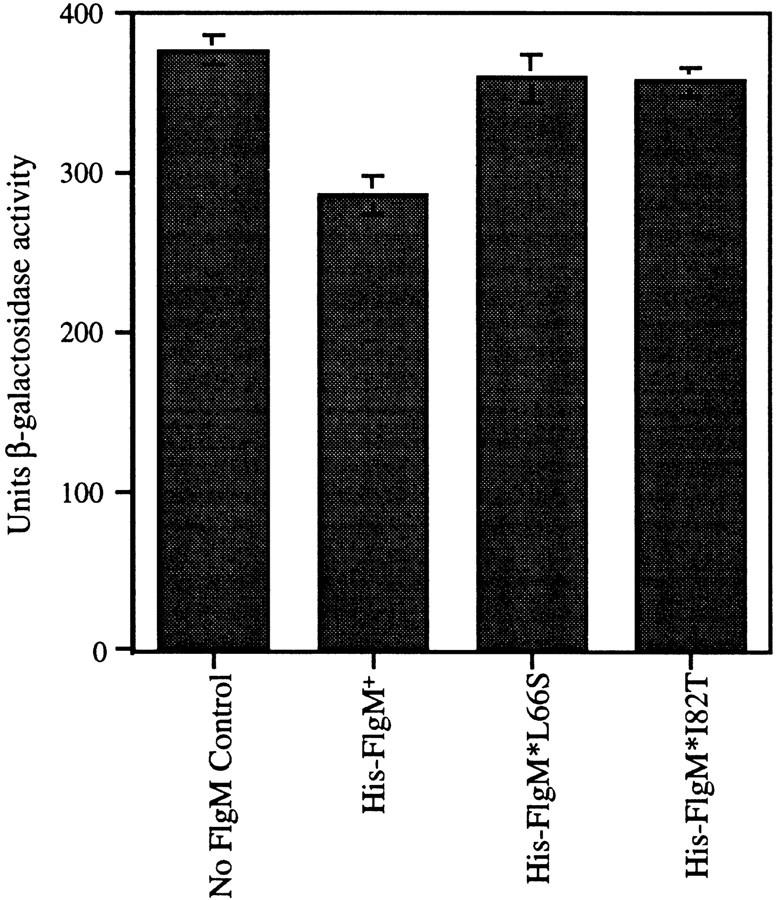
The His–FlgM fusion proteins are active in vivo and the His–FlgM* mutants are defective for σ28 inhibition. β-Galactosidase assays demonstrating inhibition of σ28-dependent lacZ expression by the His–FlgM proteins (both wild type and mutant) in vivo. The His–FlgM proteins are being expressed from the arabinose-inducible PBAD promoter from the S. typhimurium chromosome. The ability of the His–FlgM proteins to inhibit σ28-dependent expression of lacZ was measured in strains grown to mid-log phase in the presence of 1.3 μm arabinose. β-Galactosidase activities are expressed as nmole/min per OD650/ml (Davis et al. 1980).
Expression of proteins for purification
For the production of His–σ28, pKH445 was transformed into bacterial strain BL21(DE3). Cells were grown at 37°C in 1-liter batches in LB broth plus 50 μg/ml carbenicillin to A600 of 0.8. The fusion protein was induced by the addition of 1 mm IPTG for 1 hr. For the production of native FlgM, plasmid pMC64 was transformed into S. typhimurium strain LT2, and the cells were grown and induced as described above except that IPTG induction was continued for 2 hr. For the production of His–FlgM proteins, pJK302, pJK306, and pJK314 were transformed into bacterial strain BL21(DE3). Cells were grown at 37°C in 1-liter batches in LB broth plus 50 μg/ml kanamycin to A600 of 0.8. The fusion proteins were induced by the addition of 1 mm IPTG for 2 hr. For the production of His–σ70, E. coli strain BL21(DE3) containing pQETσ70 (A. Dombroski) was grown and induced as described (Wilson and Dombroski 1997). For the production of His–core RNA polymerase, pT7his6flα (R. Gourse, University of Wisconsin, Madison) was transformed into TH3656. Cells were grown at 37°C in 1-liter batches in LB broth plus 50 μg/ml carbenicillin and 0.1 mm IPTG to A600 of 0.8.
Purification of native σ28 and derivatives
All steps were carried out at 4°C. σ28–FlgM complex was purified as described (Daughdrill et al. 1997) and dissociated by the addition of guanidine-HCl to 6 m. Denatured proteins were separated at room temperature over a Pharmacia Superdex 75 HR 10/30 FPLC column equilibrated with denaturing FPLC buffer (6 m guanidine-HCl, 50 mm Tris-HCl at pH 7.9, 150 mm NaCl, 0.1 mm EDTA, 1 mm DTT). σ28 eluted close to the void volume, whereas FlgM did not elute from the column. Fractions containing FlgM-free σ28 were pooled, concentrated, then renatured by a rapid 50× dilution into dilution buffer (20% glycerol, 50 mm Tris-HCl at pH 7.9, 150 mm NaCl, 0.1 mm EDTA, 1 mm DTT; Hagar and Burgess 1980) plus 0.01% Triton X-100 at room temperature. Renatured pure σ28 was run a second time over the Superdex 75 column equilibrated with TGED (10 mm Tris-HCl at pH 7.9, 0.1 mm EDTA, 0.1 mm DTT, 5% glycerol; Lowe et al. 1979) containing 500 mm NaCl (TGED-500) plus 0.01% Triton X-100 to isolate monomeric σ28. The His–σ28 fusion protein was purified according to Novagen’s pET-His protocols with 6 m guanidine-HCl in all buffers. Proteins were stored in storage TGED-500 (containing 50% glycerol instead of 5%) plus 0.01% Triton X-100 at −20°C or −80°C.
Purification of FlgM and derivatives
All steps were carried out at 4°C. For native FlgM purification, cell pellets were resuspended in AMK buffer (Ohnishi et al. 1992) and lysed by French press at 20,000 psi. Total soluble protein was dialyzed into buffer A (10 mm NaOAc at pH 5.2, 0.1 mm EDTA), bound to a CM-52 (Whatman) column, and eluted with a 0–500 mm linear NaCl gradient in buffer A. FlgM-containing fractions were loaded onto a Pharmacia FPLC Mono S column in buffer A and eluted with a 0–250 mm linear NaCl gradient in buffer A. FlgM-containing fractions were pooled and loaded onto a FPLC phenyl–Sepharose column in buffer B [1 m (NH4)2SO4, 20 mm Tris-HCl at pH 7.2, 1 mm EDTA), and eluted with a 0–100% linear gradient of buffer C (20 mm Tris-HCl at pH 7.2, 1 mm EDTA). FlgM fractions were pooled and dialyzed into TGED storage buffer containing 100 mm NaCl (TGED-100). FlgM was homogeneous as determined by Coomassie-stained SDS-PAGE analysis. His–FlgM proteins were purified according to Novagen’s pET–His protocols. The binding buffer and initial washes contained 6 m guanidine HCl; subsequent steps were performed with nondenaturing buffers. Proteins were stored in TGED-100 at 4°C or storage TGED-100 at −20°C.
Purification of His–σ70
His–σ70 was purified as described (Wilson and Dombroski 1997) and stored in storage TGED-500 plus 0.01% Triton X-100 at −80°C.
Purification of core RNAP
Core RNAP was purified from 50 grams of S. typhimurium strain LT2 as described (Thompson et al. 1992). His–core RNAP from 28 grams of S. typhimurium TH3656 was purified by the same protocol with an extra passage over the Biorex 70 column to completely separate His–core RNAP from contaminating σ subunits. His–core RNAP was separated from native core RNAP by passage over a HisBind (Novagen) column under nondenaturing conditions according to Novagen’s pET–His protocols. Proteins were stored in storage TGED-100 at −20°C.
Protein concentration determinations
The concentration of purified proteins was measured by use of the standard Bradford procedure (Bradford 1976) or determined by adsorbance readings at 280 nm corrected for light scattering. Molar extinction coefficients were calculated on the basis of the method of Gill and von Hippel (1989) and are reported in units of m 1/cm: σ28 and His–σ28, 27310; FlgM and His–FlgM, 1280; core RNAP, 198500; His–σ70, 39400.
In vitro transcription assays
Spot transcription assays were performed essentially as described (Thompson et al. 1992) except that the reaction buffer contained 150 mm NaCl, 0.05% BSA, and 0.01% Triton X-100, and the final wash of the DEAE–cellulose disks in ether was omitted. Reactions (100 μl) contained 0.3 pmole of linear DNA promoter template and 3 pmoles of native or His–core RNAP. Linear templates were PCR amplified from pJK128 with primers 128/233 (CTCATCCGCCAAAACAGCC) and 128 (GATCTTCCCCATCGGTGATGT) to generate a 271-bp fliC promoter fragment and from pKK233-2 with primers 128/233 and 233 (GCGCCGACATATAAACGG) to generate a 158-bp tac promoter fragment. Proteins were added to the transcription assay buffer containing NTPs on ice, allowed to equilibrate at 37°C for 10 min, and initiated by the addition of template.
Copurification of GST–σ28 and σ28-associated proteins
pKH486 or pGEX-3X plasmid DNA was electroporated into LT2, TH2822, and TH2825 cells. Cells were grown in LB plus 100 μg/ml ampicillin at 37°C to A600 of 0.4–0.6, then allowed to grow for an additional hour without induction. The cells were lysed by passage through a French press twice at 20,000 psi. The GST–σ28 fusion protein and other complexed proteins were affinity purified over a Sepharose–glutathione column (Sigma) as described (Smith and Johnson 1988). Column eluants were electrophoresed on 10% tricine/SDS-polyacrylamide gels (Schagger and Jagow 1987). Protein bands were identified by Western analysis or by comparison with purified protein standards.
Filter binding assays
DNA filter binding assays were performed as described (Hinkle and Chamberlin 1972). The DNA template, a 211-bp BssHII fragment from pJK127 containing the fliC promoter was end-labeled with [α-32P]dCTP (NEN) in a Klenow reaction. DNA binding reactions were carried out in 50 μl of TXN buffer (50 mm Tris-HCl at pH 7.8, 50 mm NaCl, 0.1 mm EDTA, 0.1 mm DTT, 3 mm MgOAc, 25 μg/ml BSA) at 37°C. The concentration of core RNAP was 30 nm. The concentrations of σ28 and FlgM were both 60 nm. Proteins were incubated together for 10 or 15 min, followed by the addition of 5 μl of prewarmed radiolabeled fliC promoter DNA to a final concentration of 6 nm, and incubated for 5 min (samples chased with 1 μg of sheared salmon sperm DNA were incubated an additional 5 min after the addition of nonspecific competitor DNA). Samples were diluted to 1 ml in TXN buffer and filtered through a 13-mm BA83 nitrocellulose filter (Schleicher & Schuell) prewetted with 1 ml of TXN buffer on a Millipore sampling manifold. The filters were dried and counted by liquid scintillation.
Native PAGE analysis of Eσ28-associated proteins
Reactions (10 μl) containing (where indicated) 5 μg of σ28, 5 μg of FlgM, and/or 1 μg of core RNAP in modified TXN buffer containing 100 mm NaCl and lacking BSA, were incubated for 15 min at 37°C. Native sample buffer (Novex) was added to 1×, and samples were electrophoresed on a 4%–12% native Tris–glycine gel (Novex) according to the manufacturer’s protocol. Excised gel fragments were equilibrated in 1× Laemmli sample buffer (Laemmli and Favre 1973) for 15 min prior to electrophoresis on a 16.5% tricine/SDS-polyacrylamide gel (Schagger and Jagow 1987).
SPR assays
SPR experiments were carried out with a Biacore 2000 (Biacore). Kinetic constants ka and kd were calculated using the BIAevaluation software version 2.1 (Biacore); models assumed a 1:1 interaction.
Eσ dissociation
Measurements of the kd of Eσ28 and Eσ70 in the presence and absence of FlgM proteins were carried out in HBS buffer (Biacore) at a flow rate of 10 μl/min. Native σ28 or His–σ70 (25 μg/ml in 10 mm acetate at pH 4.0 and pH 4.2, respectively) were coupled to the CM5 sensor chip via standard NHS/EDC activation chemistry (Biacore). Core RNAP in HBS buffer was injected over the ligand surface at 10 μl/min for 2.5 min, followed by injection of HBS buffer, or HBS buffer containing His–FlgM protein. As a control for nonspecific binding, analyte was injected over an adjacent flowcell lacking immobilized ligand; the signal from blank runs was subtracted to correct for noise. The σ surface was cleaned of noncovalently bound protein by a 1-min injection of denaturing FPLC buffer, followed by a 3-min injection of dilution buffer (see section describing purification of native σ28). The immobilized σ factor could be repeatedly regenerated by this procedure, recovering an identical level of analyte binding capacity for over 20 regeneration cycles.
Controls were performed to optimize the level of immobilized ligand (ligand density) and the analyte concentration. This was necessary to eliminate the possibility of analyte rebinding during the dissociation phase of our experiments. If rebinding were allowed to occur, then the observed kd of Eσ28 would be slower than the true kd. In addition, an increase in the apparent kd in the presence of FlgM might then be attributed to the competitive binding of FlgM to ligand sites as they became available, rather than to active dissociation of Eσ28 by FlgM.
Different ligand densities were tested to identify one that could generate a significant binding response curve upon analyte injection, but was not so high that the rates of binding and dissociation were influenced by mass-transport limited kinetics instead of by interaction-limited kinetics. A 50 nm solution of core RNAP was injected over three different levels of ligand [900, 300, and 100 resonance units (RU) of immobilized σ28]. Although the 900 RU ligand surface was somewhat limited by mass transport, as judged by the shape of the binding curve during the initial phase of analyte injection (and was therefore excluded from subsequent experiments), the two lower-density ligand surfaces were not. The finding that dissociation of analyte from the 300 and 100 RU ligand surfaces occurred at nearly the same rate (data not shown) was another indication that our measurements were not skewed by mass-transport effects at these ligand densities. The flow rate was increased from the standard 10 μl/min to 50 μl/min to evaluate the effect of flow rate on analyte dissociation. The kd of Eσ28 was not altered by an increase in flow rate, confirming that dissociation at 10 μl/min was not mass transport limited. Various concentrations of analyte were tested to determine the concentration at which the ligand surface would quickly become saturated. It was important that the ligand surface be saturated for our experiments so that, during the early phase of analyte dissociation, a molecule of core RNAP dissociating from the ligand surface would be unlikely to rebind to an unoccupied molecule of σ28 before flowing out of the sensor chamber. Analyte binding reached a maximum at concentrations of 50 nm and above (data not shown) and, therefore, 50 nm core RNAP was used in all experiments.
As a final test of the experimental conditions, the dissociation of core RNAP was measured in the presence of soluble σ28 (50 nm in HBS buffer) as a competitor for rebinding of core RNAP to immobilized σ28. The presence of competitor did not increase the rate of dissociation of Eσ28 (data not shown), therefore we concluded that analyte rebinding was not occurring under our conditions. All of the Eσ28 dissociation experiments described in Results were performed with both the 300- and 100-RU ligand surfaces; the data depicted in Figure 5A are from the 100-RU experiments only. To obtain the kd values reported in Figure 5, we analyzed the dissociation curves from 210 sec to 330 sec.
Kinetic analysis of FlgM–σ28 and core RNAP–σ28interactions
To prepare the sensor chip for the measurements of σ28–FlgM interactions in experiments where His–FlgM proteins were used as the ligand, 20 μl of a 500 μm NiCl2 solution was passed through the flowcell of a sensor NTA chip at 20 μl/min to prime the surface for capture of the His-tagged ligand. Ligand was applied to the primed flowcell surface by injection of 15 μl of a 50 nm solution of the His–FlgM protein (FlgM+, FlgM*L66S, or FlgM*I82T) in Eluent buffer (Biacore) at 10 μl/min. Typically, 60–80 RU of ligand were captured during these injections. A fresh ligand surface was applied to the chip before each analyte injection. Analyte was serially diluted into Eluent buffer to prepare 100-μl samples ranging in concentration from 0.625 to 40 nm, and injected at 20 μl/min. Rate constants for each σ28–His-FlgM protein pair were derived by analyzing the data from six or seven analyte injections. As a control for nonspecific binding, analyte was injected over an adjacent Ni2+-primed flowcell lacking immobilized ligand; the signal from these blank runs was subtracted to correct for noise. After each experiment, the NTA chip surface was regenerated by injection of 60 μl of regeneration solution (Biacore), followed by 20 μl of 50 mm NaOH at 20 μl/min. This treatment stripped the flow cell of Ni2+ and any nonspecifically bound protein.
For measurement of the σ28–FlgM and σ28–core RNAP interactions in experiments where His–σ28 was used as the ligand, ∼200 RU of His–σ28 was coupled to the second flow cell of a CM5 chip (the first flowcell served as a blank control for nonspecific analyte binding), prepared and regenerated as described in the description of Eσ28 dissociation. Analyte samples (50 ml) were prepared by serial dilution into HBS buffer (concentration range for His–FlgM+ was from 500 to 15.6 nm; for core RNAP, from 80 to 1.25 nm), and injected at 10 μl/min. Data were corrected for noise and analyzed as above. Kd values reported in Table 1 were calculated from the equation Kd = kd/ka.
Acknowledgments
We are grateful for the expert technical advice from Joanne Bruno, Biacore, and Robert Karlsson, Biacore AB, and to Alexander Rudensky, for generously allowing us the use of his Biacore 2000 unit. Our sincere thanks go to Baldomera Olivera, for his suggestion of the GST–σ28 fusion/FlgM–LacZ binding assay, to Richard Burgess and Nancy Thompson, Alicia Dombroski and Christina Wilson, and Richard Gourse for reagents and suggestions, and to Jim Champoux, Kevin Plaxco, Oliver Nanassy, and Richard Burgess for critically reading this manuscript. This work was supported by National Institutes of Health (NIH) Research grants GM43149 and GM56141, and National Sciene Foundation Research grant MCB-9318890. M.C. was supported in part by NIH predoctoral training grant T32 GM07270. K.T.H. is a recipient of a faculty research award from the American Cancer Society.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL hughes@u.washington.edu; FAX (206) 543-8297.
References
- Adelman K, Orsini G, Kolb A, Graziani L, Brody EN. The interaction between the AsiA protein of bacteriophage T4 and the σ70 subunit of Escherichia coli RNA polymerase. J Biol Chem. 1997;272:27435–27443. doi: 10.1074/jbc.272.43.27435. [DOI] [PubMed] [Google Scholar]
- Benson AK, Haldenwang WG. Bacillus subtilis σB is regulated by a binding protein (RsbW) that blocks its association with core RNA polymerase. Proc Natl Acad Sci. 1993;90:2330–2334. doi: 10.1073/pnas.90.6.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Brown KL, Hughes KT. The role of anti-sigma factors in gene regulation. Mol Microbiol. 1995;16:397–404. doi: 10.1111/j.1365-2958.1995.tb02405.x. [DOI] [PubMed] [Google Scholar]
- Burgess RR, Travers AA. Factor stimulating transcription by RNA polymerase. Nature. 1969;221:43–46. doi: 10.1038/221043a0. [DOI] [PubMed] [Google Scholar]
- Chadsey M. “Regulation of the flagellar specific sigma factor, σ28, of Salmonella typhimurium by the anti-sigma factor, FlgM.” Ph.D. dissertation. Seattle, WA: University of Washington; 1998. [Google Scholar]
- Cliften PF, Park JY, Davis BP, Jang S-H, Jaehning JA. Identification of three regions essential for interaction between a σ-like factor and core RNA polymerase. Genes & Dev. 1997;11:2897–2909. doi: 10.1101/gad.11.21.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colland F, Orsini G, Brody EN, Buc H, Kolb A. The bacteriophage T4 AsiA protein: A molecular switch for sigma 70-dependent promoters. Mol Microbiol. 1998;27:819–829. doi: 10.1046/j.1365-2958.1998.00729.x. [DOI] [PubMed] [Google Scholar]
- Daughdrill GW, Chadsey MS, Karlinsey JE, Hughes KT, Dahlquist FW. The C-terminal half of the anti-sigma factor, FlgM, becomes structured when bound to its target, σ28. Nature Struct Biol. 1997;4:285–291. doi: 10.1038/nsb0497-285. [DOI] [PubMed] [Google Scholar]
- Daughdrill GW, Hanely LJ, Dahlquist FW. The C-terminal half of the anti-sigma factor FlgM contains a dynamic equilibrium solution structure favoring helical conformations. Biochemistry. 1998;37:1076–1082. doi: 10.1021/bi971952t. [DOI] [PubMed] [Google Scholar]
- Davis RW, Botstein D, Roth JR. A manual for genetic engineering: Advanced bacterial genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1980. [Google Scholar]
- Decatur AL, Losick R. Three sites of contact between the Bacillus subtilis transcription factor σF and its antisigma factor SpoIIAB. Genes & Dev. 1996;10:2348–2358. doi: 10.1101/gad.10.18.2348. [DOI] [PubMed] [Google Scholar]
- Gamer J, Multhaup G, Tomoyasu T, McCarty JS, Rudiger S, Schonfeld HJ, Schirra C, Bujard H, Bukau B. A cycle of binding and release of the DnaK, DnaJ and GrpE chaperones regulates activity of the Escherichia coli heat shock transcription factor σ32. EMBO J. 1996;15:607–617. [PMC free article] [PubMed] [Google Scholar]
- Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- Gill SC, Weitzel SE, von Hippel PH. Escherichia coli σ70 and NusA proteins. I. Binding interactions with core RNA polymerase in solution and within the transcription complex. J Mol Biol. 1991;220:307–324. doi: 10.1016/0022-2836(91)90015-x. [DOI] [PubMed] [Google Scholar]
- Gillen KL, Hughes KT. Negative regulatory loci coupling flagellin synthesis to flagellar assembly in Salmonella typhiumurium. J Bacteriol. 1991;173:2301–2310. doi: 10.1128/jb.173.7.2301-2310.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Transcription from two promoters and autoregulation contribute to the control of expression of the Salmonella typhimurium flagellar regulatory gene flgM. J Bacteriol. 1993;175:7006–7015. doi: 10.1128/jb.175.21.7006-7015.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorham HC, McGowan SJ, Robson PRH, Hodgson DA. Light-induced carotenogenesis in Myxococcus xanthus: Light-dependent membrane sequestration of ECF sigma factor CarQ by anti-sigma factor CarR. Mol Microbiol. 1996;19:171–186. doi: 10.1046/j.1365-2958.1996.360888.x. [DOI] [PubMed] [Google Scholar]
- Guzman L-M, Belin D, Carson M, Beckwith J. Tight regulation, modulation and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar DA, Burgess RR. Elution of proteins from sodium dodecyl sulfate-polyacrylamide gels, removal of sodium dodecyl sulfate, and renaturation of enzymatic activity: Results with sigma subunit of Escherichia coli RNA polymerase, wheat germ DNA topoisomerase, and other enzymes. Anal Biochem. 1980;109:76–86. doi: 10.1016/0003-2697(80)90013-5. [DOI] [PubMed] [Google Scholar]
- Harris JD, Heilig JS, Martinez II, Calendar R, Isaksson LA. Temperature-sensitive Escherichia coli mutant producing a temperature-sensitive σ subunit of DNA-dependent RNA polymerase. Proc Natl Acad Sci. 1978;75:6617–6681. doi: 10.1073/pnas.75.12.6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmann JD. Alternative sigma factors and the regulation of flagellar gene expression. Mol Microbiol. 1991;5:2875–2882. doi: 10.1111/j.1365-2958.1991.tb01847.x. [DOI] [PubMed] [Google Scholar]
- Helmann JD, Chamberlin MJ. Structure and function of bacterial sigma factors. Annu Rev Biochem. 1988;57:839–872. doi: 10.1146/annurev.bi.57.070188.004203. [DOI] [PubMed] [Google Scholar]
- Hinkle DC, Chamberlin MJ. Studies of the binding of Escherichia coli RNA polymerase to DNA. I. The role of sigma subunit in site selection. J Mol Biol. 1972;70:157–195. doi: 10.1016/0022-2836(72)90531-1. [DOI] [PubMed] [Google Scholar]
- Hughes KT, Gillen KL, Semon MJ, Karlinsey JE. Sensing structural intermediates in bacterial flagellar assembly by export of a negative regulator. Science. 1993;262:1277–1280. doi: 10.1126/science.8235660. [DOI] [PubMed] [Google Scholar]
- Hughes KT, Matthai K. The Anti-sigma factors. Annu Rev Microbiol. 1998;52:231–286. doi: 10.1146/annurev.micro.52.1.231. [DOI] [PubMed] [Google Scholar]
- Ishihama A. Promoter selectivity control of RNA polymerase. In: Eckstein F, Lilley DMJ, editors. Nucleic acids and molecular biology. Berlin: Springer-Verlag; 1997. pp. 53–70. [Google Scholar]
- Iyoda S, Kutsukake K. Molecular dissection of the flagellum-specific anti-sigma factor, FlgM, of Salmonella typhimurium. Mol Gen Genet. 1995;249:417–424. doi: 10.1007/BF00287103. [DOI] [PubMed] [Google Scholar]
- Jishage M, Ishihama A. A stationary phase protein in Escherichia coli with binding activity to the major σ subunit of RNA polymerase. Proc Natl Acad Sci. 1998;95:4953–4958. doi: 10.1073/pnas.95.9.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo DM, Ng N, Calendar R. A σ32 mutant with a single amino acid change in the highly conserved region 2.2 exhibits reduced core RNA polymerase affinity. Proc Natl Acad Sci. 1997;94:4907–4912. doi: 10.1073/pnas.94.10.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo DM, Nolte A, Calendar R, Zhou YN, Jin DJ. Multiple regions on the Escherichia coli heat shock transcription factor σ32 determine core RNA polymerase binding specificity. J Bacteriol. 1998;180:1095–1102. doi: 10.1128/jb.180.5.1095-1102.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlinsey, J.E., H.-C.T. Tsui, M. Winkler, and K.T. Hughes. 1998. Flk couples FlgM translation to flagellar ring assembly in Salmonella typhimurium. J. Bacteriol. 180: (in press). [DOI] [PMC free article] [PubMed]
- Karlsson R, Michaelsson A, Mattsson L. Kinetic analysis of monoclonal antibody-antigen interactions with a new biosensor based analytical system. J Immunol Methods. 1991;45:229–240. doi: 10.1016/0022-1759(91)90331-9. [DOI] [PubMed] [Google Scholar]
- Kundu TK, Kusano S, Ishihama A. Promoter selectivity of Escherichia coli RNA polymerase σF holoenzyme involved in transcription of flagellar and chemotaxis genes. J Bacteriol. 1997;179:4264–4269. doi: 10.1128/jb.179.13.4264-4269.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsukake K. Excretion of the anti-sigma factor through a flagellar substructure couples flagellar gene expression with flagellar assembly in Salmonella typhimurium. Mol Gen Genet. 1994;243:605–612. doi: 10.1007/BF00279569. [DOI] [PubMed] [Google Scholar]
- Kutsukake K, Ohya Y, Iino T. Transcriptional analysis of the flagellar regulon of Salmonella typhimurium. J Bacteriol. 1990;172:741–747. doi: 10.1128/jb.172.2.741-747.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutsukake K, Iyoda S, Ohnishi K, Iino T. Genetic and molecular analyses of the interaction between the flagellum-specific sigma and anti-sigma factors in Salmonella typhimurium. EMBO J. 1994;13:4568–4576. doi: 10.1002/j.1460-2075.1994.tb06778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK, Favre M. Maturation of the head of bacteriophage T4. I. DNA packaging events. J Mol Biol. 1973;80:575–599. doi: 10.1016/0022-2836(73)90198-8. [DOI] [PubMed] [Google Scholar]
- Léonetti JP, Wong K, Geiduschek EP. Core-sigma interaction: Probing the interaction of the bacteriophage T4 gene 55 promoter recognition proteins with E. coli RNA polymerase. EMBO J. 1998;17:1467–1475. doi: 10.1093/emboj/17.5.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesley SA, Burgess RR. Characterization of the Escherichia coli transcription factor σ70: Localization of a region involved in the interaction with core RNA polymerase. Biochemistry. 1989;28:7728–7734. doi: 10.1021/bi00445a031. [DOI] [PubMed] [Google Scholar]
- Liu X, Matsumura P. An alternative sigma factor controls transcription of flagellar class-III operons in Escherichia coli: Gene sequence, overproduction, purification and characterization. Gene. 1995;164:81–84. doi: 10.1016/0378-1119(95)00480-t. [DOI] [PubMed] [Google Scholar]
- Lonetto M, Gribskov M, Gross CA. The σ70 family: Sequence conservation and evolutionary relationships. J Bacteriol. 1992;174:3843–3849. doi: 10.1128/jb.174.12.3843-3849.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe PA, Hagar DA, Burgess RR. Purification and properties of the σ subunit of Escherichia coli DNA-dependent RNA polymerase. Biochemistry. 1979;18:1344–1352. doi: 10.1021/bi00574a034. [DOI] [PubMed] [Google Scholar]
- Ohnishi K, Kutsukake K, Suzuki H, Iino T. Gene fliA encodes an alternative sigma factor specific for flagellar operons in Salmonella typhimurium. Mol Gen Genet. 1990;221:139–147. doi: 10.1007/BF00261713. [DOI] [PubMed] [Google Scholar]
- ————— A novel transcriptional regulation mechanism in the flagellar regulon of Salmonella typhimurium: An antisigma factor inhibits the activity of the flagellum-specific sigma factor, σF. Mol Microbiol. 1992;6:3149–3157. doi: 10.1111/j.1365-2958.1992.tb01771.x. [DOI] [PubMed] [Google Scholar]
- Orsini G, Ouhammouch M, Le C-JP, Brody EN. The asiA gene of bacteriophage T4 codes for the anti-σ70 protein. J Bacteriol. 1993;175:85–93. doi: 10.1128/jb.175.1.85-93.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa T, Yura T. Effects of reduced amount of RNA polymerase sigma factor on gene expression and growth of Escherichia coli: Studies of the rpoD40 (amber) mutation. Mol Gen Genet. 1981;184:166–173. doi: 10.1007/BF00272900. [DOI] [PubMed] [Google Scholar]
- Ouhammouch M, Orsini G, Brody EN. The asiA gene product of bacteriophage T4 is required for middle mode RNA synthesis. J Bacteriol. 1994;176:3956–3965. doi: 10.1128/jb.176.13.3956-3965.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouhammouch M, Adelman K, Harvey SR, Orsini G, Brody EN. Bacteriophage T4 MotA and AsiA proteins suffice to direct Escherichia coli RNA polymerase to initiate transcription at T4 middle promoters. EMBO J. 1995;92:1451–1455. doi: 10.1073/pnas.92.5.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens JT, Miyake R, Murakami K, Chmura AJ, Fujita N, Ishihama A, Meares CF. Mapping the σ70 subunit contact sites on Escherichia coli RNA polymerase with a σ70-conjugated chemical protease. Proc Natl Acad Sci. 1998;95:6021–6026. doi: 10.1073/pnas.95.11.6021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Record T. Escherichia coli RNA polymerase (Eσ70), promoters, and the kinetics of the steps of transcription intitiation. In: Neidhardt FC, editor. Escherichia coli and Salmonella typhimurium. Washington, DC.: ASM Press; 1996. pp. 792–821. [Google Scholar]
- Schagger H, Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- Severinova E, Severinov K, Fenyo D, Marr M, Brody EN, Roberts JW, Chait BT, Darst SA. Domain organization of the Escherichia coli RNA polymerase σ70 subunit. J Mol Biol. 1996;263:637–647. doi: 10.1006/jmbi.1996.0604. [DOI] [PubMed] [Google Scholar]
- Severinova EK, Severinov K, Darst SA. Inhibition of Escherichia coli RNA polymerase by bacteriophage T4 AsiA. J Mol Biol. 1998;279:9–18. doi: 10.1006/jmbi.1998.1742. [DOI] [PubMed] [Google Scholar]
- Shuler MF, Tatti KM, Wade KH, Moran CP., Jr A single amino acid substitution in σE affects its ability to bind core RNA polymerase. J Bacteriol. 1995;177:3687–3694. doi: 10.1128/jb.177.13.3687-3694.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DB, Johnson KS. Single step purification of polypeptides expressed in Escherichia coli as fusions with glutathione-S transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- Starnbach MN, Lory S. The fliA (rpoF) gene of Pseudomonas aeruginosa encodes an alternative sigma factor required for flagellin synthesis. Mol Microbiol. 1988;6:459–469. doi: 10.1111/j.1365-2958.1992.tb01490.x. [DOI] [PubMed] [Google Scholar]
- Stevens A. A salt-promoted inhibitor of RNA polymerase isolated from T4 phage-infected E. coli. In: Losick R, Chamberlin M, editors. RNA polymerase. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1976. pp. 617–627. [Google Scholar]
- Thompson NE, Hager DA, Burgess RR. Isolation and characterization of a polyol-responsive monoclonal antibody useful for gentle purification of Escherichia coli RNA polymerase. Biochemistry. 1992;31:7003–7008. doi: 10.1021/bi00145a019. [DOI] [PubMed] [Google Scholar]
- Tintut Y, Gralla JD. PCR mutagenesis identifies a polymerase-binding sequence of sigma 54 that includes a sigma 70 homology region. J Bacteriol. 1995;177:5818–5825. doi: 10.1128/jb.177.20.5818-5825.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White BA. Methods in Molecular Biology. Totowa, N.J: Humana Press; 1993. PCR protocols: Current methods and applications; p. 392. [Google Scholar]
- Wilson C, Dombroski AJ. Region 1 of σ70 is required for efficient isomerization and initiation of transcription by Escherichia coli RNA polymerase. J Mol Biol. 1997;267:60–74. doi: 10.1006/jmbi.1997.0875. [DOI] [PubMed] [Google Scholar]
- Xie ZD, Hershberger CD, Shankar S, Ye RW, Chakrabarty AM. Sigma factor-anti-sigma factor interaction in alginate synthesis: Inhibition of AlgT by MucA. J Bacteriol. 1996;178:4990–4996. doi: 10.1128/jb.178.16.4990-4996.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youderian P, Vershon A, Bouvier S, Sauer R, Susskind M. Changing the DNA-binding specificity of a repressor. Cell. 1983;35:777–783. doi: 10.1016/0092-8674(83)90110-1. [DOI] [PubMed] [Google Scholar]
- Zhou YN, Walter WA, Gross CA. A mutant σ32 with a small deletion in conserved region 3 of sigma has reduced affinity for core RNA polymerase. J Bacteriol. 1992;174:5005–5012. doi: 10.1128/jb.174.15.5005-5012.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]