

Non-technical summary
A clinical trait of critically ill patients following trauma, surgical complications and/or sepsis is the presence of a marked skeletal muscle wasting, which severely compromises muscle function, especially the respiratory muscles, and clinical outcomes such as morbis, mortality and the length of hospitalization. However, the molecular mechanisms responsible for this are largely unresolved. In this paper we provide novel and interesting translational data that show how the muscle mass is regulated during critical illness. The present observations also show that the muscle signalling pathways are not only important for the size of muscle mass, but could also play a significant role in the whole body glucose control. This is extremely important and relevant to the clinical setting as uncontrolled blood glucose concentrations in critically ill patients impact on clinical outcome. From a clinical perspective, the present data suggest therapeutic strategies to preserve muscle mass and metabolic function in critical illness.
Abstract
Abstract
Critically ill patients experience marked skeletal muscle atrophy, but the molecular mechanisms responsible for this are largely unresolved. Therefore, we investigated key genes and proteins, identified from cell and animal studies to control protein synthesis and breakdown, in vastus lateralis biopsy samples obtained from 10 patients and 10 age- and sex-matched healthy controls. Muscle cytokines IL-6 and TNF-α mRNA were higher in patients than in controls (6.5-fold; P < 0.001 and 2-fold; P < 0.01). From the perspective of muscle protein breakdown, muscle-specific E3-ligases (MAFbx and MuRF1) were higher in patients at mRNA (4.5-fold; P < 0.05 and 2.5-fold; P < 0.05) and protein (5-fold; P < 0.001 and 4.5-fold; P < 0.001) level. Furthermore, 20S proteasome mRNA and protein were higher in patients (5-fold; P < 0.001 and 2.5-fold; P < 0.01). Cathepsin-L mRNA was 2-fold higher (P < 0.01), whilst calpain-3 mRNA (2-fold; P < 0.01) and protein (4-fold; P < 0.01) were lower in patients. Another novel observation was the 3-fold (P < 0.05) and 8.5-fold (P < 0.001) higher expression of myostatin mRNA and protein in patients. Widespread dephosphorylation (inactivation) of proteins regulating translation initiation factor activation and protein synthesis (Akt1, GSK3α,β, mTOR, p70S6K and 4E-BP1) was observed in patients, which was paralleled by increases in their mRNAs. Finally, PDK4 mRNA and protein was 2-fold (P < 0.05) and 2.6-fold (P < 0.01), respectively, higher in patients. In conclusion, we showed comprehensive alterations in molecular events thought to reduce muscle mass and carbohydrate (CHO) oxidation in critically ill patients. Nevertheless, these catabolic events were matched by a cellular programme of anabolic restoration at the transcriptional level. This shows a high molecular plasticity in the muscle of patients, and strategies to preserve muscle mass and metabolic function should focus on maintaining Akt phosphorylation and inhibiting myostatin expression.
Introduction
A clinical trait of critically ill patients following trauma, surgical complications and/or sepsis is the presence of a marked skeletal muscle wasting, which severely compromises muscle function, especially the respiratory muscles, and clinical outcomes such as morbidity and mortality (Gamrin et al. 1997; de Jonghe et al. 2007, 2009; Ali et al. 2008). In quantitative terms, this can amount to an up to 2% loss of muscle mass per day (Gamrin et al. 1997), or an up to 4% reduction in muscle fibre cross-sectional area per day (Helliwell et al. 1991).
Whilst changes in whole body or lower limb muscle protein synthesis have been documented in critically ill patients (Essén et al. 1998; Tjader et al. 2004), our understanding of the molecular regulation of protein turnover in these patients is relatively limited and has been acquired mainly from cell-based and animal studies.
Animal studies suggest that the ubiquitin proteasome system is the main contributor to protein breakdown in muscle wasting conditions (Wray et al. 2003). Two muscle-specific E3-ligases belonging to the ubiquitin–proteasome complex, muscle RING-finger 1 (MuRF1) and muscle atrophy F-box (MAFbx), have been identified as key-regulators of 26S proteasome-mediated protein breakdown in cell-based studies and animal models of muscle wasting (Bodine et al. 2001; Gomes et al. 2001). However, confirmation of the involvement of these two proteolytic markers in critically ill patients is not yet available.
Critically ill patients presenting with systemic inflammation/infection are difficult to wean from mechanical ventilation due to muscle diaphragm wasting. Using a septic animal model, Supinski et al. (2009) recently linked this respiratory muscle wasting to a specific increase in the activity of one of the members of the calpain proteolytic system, calpain-1. Of this system, calpain-1, -2 and -3 are highly expressed in muscle. While increased RNA expression and activity of calpain-1 and -2 is well documented in animal models of muscle wasting (Bartoli & Richard, 2005), the role of the muscle-specific calpain, calpain-3 (or p94), in wasting beyond muscular dystrophies (Richard et al. 1995) is, however, relatively unknown.
Myostatin, a transforming growth factor-β (TGF-β) protein secreted by muscle, appears to be a common feature of human muscle wasting in AIDS (Gonzalez-Cadavid et al. 1998) and chronic muscle disuse (Reardon et al. 2001). Myostatin inhibits proliferation of satellite cells, muscle growth and development (Lee, 2004). More recently, it has been shown that over-expression of myostatin in myoblasts induces muscle wasting by directly inhibiting phosphorylation of Akt (v-akt murine thymoma oncogene homologue 1), and thereby increasing the activity of FOXO transcription factors and the expression of downstream gene targets MuRF1 and MAFbx. Furthermore, myostatin can also inhibit protein synthesis via the Akt/mammalian target of rapamycin (mTOR)/p70S6k signalling pathway (Trendelenburg et al. 2009). Finally, myostatin has been shown to inhibit muscle carbohydrate oxidation (McPherron & Lee, 2002), which is a hallmark of skeletal muscle insulin resistance also present in critical illness (Robinson & van Soeren, 2004). Hyperglycaemia and insulin resistance are common in critically ill patients, even if they have not previously had diabetes. However, the mechanism underlying this inhibition has yet to be elucidated. Irrespective of this point, a role for myostatin in the muscle wasting and insulin resistance in critically ill patients has yet to be demonstrated.
The aim of the present study therefore was to describe for the first time in skeletal muscle of critically ill patients simultaneous changes in anabolic and catabolic pathways thought, from cell- and animal-based research, to be central to the molecular regulation of muscle mass and carbohydrate metabolism.
Methods
Subjects
Twenty volunteers (n = 10 critically ill patients and n = 10 age- and sex-matched, sedentary, healthy controls) participated in the present study, which was approved by the National Research Ethics Committee of the UK and was performed in accordance with the World Medical Association Declaration of Helsinki. The study was performed with the consent or assent from relatives followed by full consent once the patient was capable of giving it. The healthy volunteers also gave their written informed consent. The patient volunteers are described in Table 1, which also contains information about the drugs administered to patients on an individual basis over the course of a stay in the intensive care ward, but not all might have been necessarily given during the first day of admittance when the muscle biopsy was taken. Patients were admitted with various diseases, such as pneumococcal meningitis, burns (35%), postoperative myocardial infarction, respiratory failure, sepsis, multiple cerebral infarcts, hypoxic brain damage following ventricular fibrillation (VF) arrest, oesophagectomy with postoperative leak, and abdominal aortic aneurysm. Patients who had neuromuscular disease, chronic obstructive pulmonary disease (COPD) or chronic heart failure were not included. The APACHE II (Acute Physiology and Chronic Health Evaluation II; Knaus et al. 1985), one of several intensive care unit (ICU) scoring systems, was used to classify the severity of conditions experienced by the patient group. After patient admission to ICU, an integer score from 0 to 71 was computed based on several measurements; higher scores imply a more severe disease and a higher risk of death.
Table 1.
Patients with critical illness – clinical and demographic details
| Diagnosis | No. of systems involved | APACHE II at admission | APACHE II at biopsy | Age | Sex | Medication |
|---|---|---|---|---|---|---|
| Pneumococcal meningitis | CNS, CVS, respiratory | 19 | 10 | 63 | M | Aciclovir, alfentanil, ampicillin, atracurium, ceftriaxone, gentamicin, haloperidol, insulin |
| 35% burns, respiratory failure, sepsis | Respiratory, renal, CVS, skin | 20 | 15 | 71 | M | Adenosine, amiodarone, amphotericin, benzylpenicillin, ceftazidime, ciprofloxacin, erythromycin |
| Pneumonia | Respiratory, CVS | 14 | 13 | 85 | M | Alfentanil, carbimazole, co-amoxiclav, hydrocortisone, propofol, insulin |
| Hypotension | CVS, GCS | 18 | 11 | 77 | F | Amiodarone, atenolol, buprenorphine, digoxin, cyclizine, furosemide (frusemide), insulin |
| Postoperative myocardial infarction | CVS, respiratory, renal | 22 | 19 | 73 | M | Adrenaline, alfentanil, atracurium, isoprenaline, metoclopramide, insulin |
| Multiple cerebral infarcts, sepsis | Brain, CVS, respiratory | 17 | 16 | 34 | M | Aciclovir, amlodipine, atracurium, dexamethasone, erythromycin, fentanyl, metronidazole |
| Hypotension | CVS | 14 | 12 | 77 | M | Carvedilal, cefradine, dexamethasone, esomeprazole, felodopine, morphine, |
| Hypoxic brain damage following VF arrest | Brain, CVS, respiratory | 27 | 21 | 62 | M | Adrenaline, alfentanil, amiodarone, atenelol, enoxoparine, esmolol, insulin |
| Oesophagectomy with postoperative leak | Respiratory, CVS | 10 | 8 | 75 | M | Alfentanil, amitryptylline, amoxicillin, gentamicin, hydrocortisone, oxycodone |
| aAAA repair | Respiratory, renal | 15 | 16 | 71 | M | Amlodipine, bendrofluazide, bispoprolol, ciprofloxacin, clonidine, frusemide, lorazepam, insulin |
AAA, abdominal aortic aneurysm. APACHE II (‘Acute Physiology and Chronic Health Evaluation II’) is a severity of disease classification system. CVS – cardiovascular system; GCS – Glasgow Coma Scale.
Experimental procedure
Muscle biopsy samples were obtained from the vastus lateralis muscle of patients within 6–8 hours of admission to the Intensive Care Unit using the Bergstrom percutaneous needle biopsy technique (Bergstrom & Hultman, 1966). Muscle biopsies were obtained from the age- and sex-matched control volunteers whilst in a resting state the morning after an overnight fast. Tissue was immediately snap-frozen in liquid nitrogen and then stored in liquid nitrogen until mRNA and protein expression measurements were made.
Quantitative RT–PCR
Total RNA was isolated from snap frozen muscle using TriReagent (Ambion) according to the manufacturer's protocol. First strand cDNA was then synthesized from 1 μg RNA sample using random primers (Promega) and PowerScript Reverse Transcriptase (BD Biosciences). Additional reactions were performed, in which the reverse transcriptase was omitted to allow for assessment of genomic DNA contamination.
TaqMan PCR was carried out using an ABI prism 7000 sequence detector (Applied Biosystems, USA), with 2 μl of cDNA, 18 μm of each primer, 5 μm probe, and Universal TaqMan 2× PCR Mastermix (Eurogentec) in a 25 μl final volume. Each sample was run in duplicate. Primers and MGB TaqMan probes (Applied Biosystems) were designed such that probes spanned over exon–exon boundaries to avoid genomic amplification. Hydroxymethylbilane synthase (HMBS) was used as internal control, and all genes of interest were labelled with the fluorescent reporter FAM. The thermal cycling conditions used were: 2 min at 50°C, 10 min at 95°C, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. Ct values of the target gene were normalized to Ct values of the house-keeping gene in patient and control volunteers, and the final results were calculated according to the 2−ΔΔCt method. The average healthy volunteer value was used as calibrator and was set at 1.
Western blotting
Each muscle sample was homogenized in Tris buffer (50 mm Tris, 1 mm EDTA, pH 7.5) supplemented with protease and phosphatase inhibitors (Sigma). After homogenization, each muscle extract was centrifuged for 15 min at 10,000 g. The supernatant was collected and stored at –80°C for further protein immunoblotting according to Constantin et al. (2007). Protein concentration was measured using the Bradford assay. Protein samples were run on a 4–12% Bis-Tris acrylamide gel (Invitrogen, UK) for 2 h at constant 200 V and transferred onto a polyvinylidene difluoride membrane (PVDF) overnight at constant 100 mA, in ice-cold buffers (4°C). The protein transfer was checked with Ponceau S red staining, before blocking the membrane in BSA/Tris-buffered saline (TBS)/Tween for 1 h at room temperature. Membranes were probed with the primary antibody overnight at 4°C.
The antibodies used in this study (phosphorylated Akt (serine473; PAkt) and total Akt (Akt), phosphorylated mTOR (Ser2448 PmTOR) and total mTOR (mTOR), phosphorylated eukaryotic translation factor 4E-binding protein 1 (4E-BP1) (threonine37/46; P4E-BP1), phosphorylated p70 ribosomal S6 kinase (threonine389 Pp70S6K) (Pp70S6K) and total p70D S6K (p70S6K), phosphorylated glycogen synthase kinase (GSK) 3α (serine21; PGSK3α), phosphorylated GSK3β (serine9; PGSK3β) and total GSK3β (GSK3β) were obtained from Cell Signalling Technology (Danvers, MA, USA). Myostatin was obtained from Novus Biologicals (Littleton, CO, USA). MuRF1 and MAFbx, produced in-house by Pfizer Inc. (USA), and pyruvate dehydrogenase kinase isozyme 4 (PDK4), produced in-house by AstraZeneca (UK), were provided to us as gifts. Membranes were washed in Tris buffered saline – Tween (TBS-T) and incubated with the appropriate secondary antibody, either horseradish peroxidase-linked anti-mouse IgG (Dako, UK), or anti-rabbit IgG (GE Healthcare, Amersham, UK). The membranes were washed again in the same buffer, incubated with enhanced chemiluminescence detection (Pierce, UK) and exposed to X-ray film.
Densitometry
Blots were scanned using a Duo Scan T1200 Agfa scanner. Bands were identified using the Quantity One programme from Bio-Rad, and the optical density volume was adjusted by subtracting the local background. All values were normalized to actin placebo for the cytosolic proteins and lamin A/C for the nuclear proteins.
Statistics
Data in text, tables and figures are expressed as mean ± SEM, with n = 10 for patients and n = 10 for controls. Differences between patients and healthy volunteers were determined using the Mann–Whitney U test. Significance was set at the P < 0.05 level of confidence.
Results
Clinical and demographic data
The patient group was age- and sex-matched with a healthy control volunteer group (mean age 68.8 ± 4.4 and 67.0 ± 1.5 years, respectively, and mean BMI 26.8 ± 0.6 and 27.3 ± 0.8 kg m−2, respectively). The clinical and demographic details, and pharmacological treatment data relating to the critically ill patients are presented in Table 1. Their APACHE scores at admission and at the time of muscle biopsy sampling were 17.6 ± 4.8 and 14.1 ± 4.1, respectively (P = 0.009). At the point of muscle biopsy sampling, the mean (95% confidence interval) blood haematocrit, serum creatinine, white blood cell count values and heart rate in the patient group was: 35.9% (29.0–42.8), 96.6 (70.2–123.0) μmol l−1, 18.8 (14.7–22.8) cells μl−1 and 105 (78–132) beats min−1, respectively.
Muscle pro-inflammatory cytokines
mRNA expression levels of the two pro-inflammatory cytokines, interleukin 6 (IL-6) and tumour necrosis factor α(TNF-α), were higher in patients than in controls. Specifically, IL-6 mRNA expression was 6.5-fold higher in patients than in controls (P < 0.001), whilst TNF-α mRNA expression was 2-fold higher (P < 0.01; Fig. 1).
Figure 1.
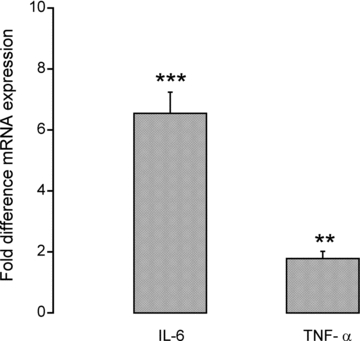
Fold difference in muscle mRNA expression of IL-6 and TNF-α in critically ill patients (n = 10). Values are expressed as 2−ΔΔCt normalized to endogenous HMBS. Muscle biopsies from healthy, age- and sex-matched controls (n = 10) were used as a calibrator with a value of 1. **, ***Significantly different from control (P < 0.01, P < 0.001, respectively). Data represent mean ± SEM.
Muscle protein breakdown
Higher 20S proteasome mRNA expression was identified in patients compared to controls (5-fold, P < 0.001, Fig. 2A). Expression of MAFbx and MuRF1 mRNA was also higher in patients (4.5-fold, P < 0.05 and 2.5-fold, P < 0.05, respectively, Fig. 2A). Furthermore, these mRNA changes observed in patients were matched by increases in 20S proteasome (2.5-fold; P < 0.01), MAFbx (5-fold; P < 0.001) and MuRF1 (4.5-fold; P < 0.001) protein expression (Fig. 2B). Typical Western blots for 20S proteasome, MAFbx and MuRF1 are presented in Fig. 2C. Cathepsin-L mRNA expression (a marker of the lysosomal-mediated protein degradation (Deval et al. 2001) was 2-fold higher in patients than in controls (P < 0.01; Fig. 3A). Calpain-3 mRNA and protein expression was significantly lower in patients (1.3-fold, P < 0.01; 4-fold, P < 0.01; Fig. 3A and B). A typical Western blot for calpain-3 from controls and patients is presented in Fig. 3C.
Figure 2.
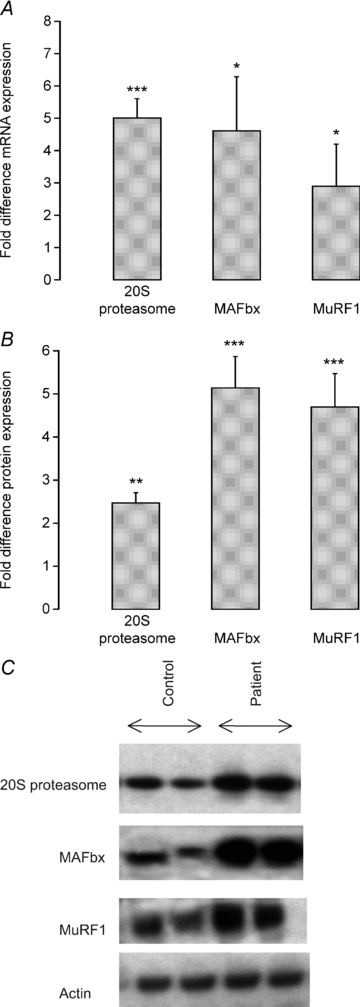
A, fold difference in muscle mRNA expression of genes thought to control muscle protein degradation (20S proteasome, MAFbx and MuRF1) in critically ill patients (n = 10). Values are expressed as 2−ΔΔCt normalized to endogenous HMBS. Muscle biopsies from healthy, age- and sex-matched controls (n = 10) were used as a calibrator with a value of 1. *, ***Significantly different from control (P < 0.05, P < 0.001). Data represent mean ± SEM. B, fold difference in the relative optical density of 20S proteasome, MAFbx and MuRF1 proteins in critically ill patients (n = 10) quantified by Western blotting. Biopsies from healthy, age- and sex-matched controls (n = 10) were used as a calibrator with a value of 1. **, ***Significantly different from control (P < 0.01, P < 0.001). Data represent mean ± SEM. C, typical Western blots for 20S proteasome, MAFbx and MurF1 proteins in muscle biopsies obtained from controls (n = 10) and critically ill patients (n = 10).
Figure 3.

A, fold difference in muscle mRNA expression of cathepsin-L, calpain-3 and myostatin in patients (n = 10). Values are expressed as 2−ΔΔCt normalized to endogenous HMBS. Muscle biopsies from healthy, age- and sex-matched controls (n = 10) were used as a calibrator with a value of 1. **Significantly different from healthy controls (P < 0.01). Data represent mean ± SEM. B, fold difference in relative optical density of calpain-3 and myostatin (42 kDa) proteins in critically ill patients (n = 10) quantified by Western blotting. Biopsies from healthy, age- and sex-matched controls (n = 10) were used as a calibrator with a value of 1. **, ***Significantly different from healthy controls (P < 0.01, P < 0.001). Data represent mean ± SEM. C, typical Western blots for calpain-3 and myostatin proteins in muscle biopsies obtained from controls (n = 10) and critically ill patients (n = 10).
Muscle myostatin mRNA and protein expression
Myostatin mRNA expression was significantly higher in patients (3-fold, P < 0.01, Fig. 3A), and was accompanied by a marked increase in myostatin protein expression over controls (8.5-fold, P < 0.001, Fig. 3B). A typical Western blot for myostatin from controls and patients is presented in Fig. 3C.
Signalling proteins regulating translation initiation of muscle protein synthesis
Gene expression of members of the PI3K/Akt signalling pathway involved in the control of translation initiation of muscle protein synthesis was found to be significantly higher in patients than in controls (Fig. 4A). Thus, Akt mRNA was 2-fold (P < 0.05), GSK3α 4-fold (P < 0.001), GSK3β 1-fold (P < 0.05), mTOR 2.3-fold (P < 0.01), 70 kDa ribosomal protein S6 kinase 1 (p70s6k) 1-fold (P < 0.05) and 4E-BP1 2.5-fold (P < 0.01) higher in patients compared with controls. On the other hand, the expression level of the corresponding proteins (represented as the ratio of phosphorylated protein to total protein level) was significantly lower in patients than in controls (Fig. 4B). Thus, PAkt/Akt was 3.0-fold (P < 0.01), PGSK3α/GSK3α 3.5-fold (P < 0.01), PGSK3β/GSK3β 5.4-fold (P < 0.01), PmTOR/mTOR 2.9-fold (P < 0.05), Pp70s6k/p70s6k 4.0-fold (P < 0.01) and P4E-BP1/4E-BP1 4.1-fold (P < 0.01) less in patients than in controls. Typical Western blots for total and phosphorylated proteins investigated in controls and patients are depicted in Fig. 4C.
Figure 4.
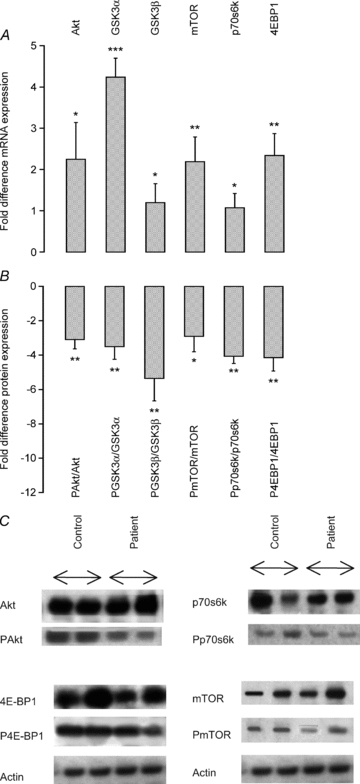
A, fold difference of muscle mRNA expression of genes involved in the phosphoinositide 3-kinase (PI3K)/Akt1/mTOR pathway in critically ill patients (n = 10). Values are expressed as 2−ΔΔCt normalized to endogenous HMBS. Muscle biopsies from healthy, age- and sex-matched controls (n = 10) were used as a calibrator with a value of 1. *, **, ***Significantly different from control (P < 0.05, P < 0.01, P < 0.001). Data represent mean ± SEM. B, fold difference of the ratio between the phosphorylated (P) AktSer473, (P) GSK3αSer21, (P) GSK3βSer9, (P) mTORSer2448, (P) p70S6KThr389 and (P) 4E-BP1Thr37/46 to the total proteins measured and quantified by Western blot in critically ill patients (n = 10). Biopsies from healthy, age- and sex-matched controls (n = 10) were used as a calibrator with a value of 1. *, **Significantly different from control (P < 0.05, P < 0.01). Data represent mean ± SEM. C, typical Western blots for phosphorylated (P) AktSer473, (P) mTORSer2448, (P) p70S6KThr389 and (P) 4E-BP1Thr37/46 proteins in muscle biopsies obtained from control (n = 10) and critically ill patients (n = 10).
Muscle PDK4 mRNA and protein expression
PDK4 mRNA and protein expression was ∼2- (P < 0.05) and 2.7-fold (P < 0.05) higher in patients than in controls, respectively (Fig. 5A and B). A typical Western blot for PDK4 protein in controls and patients is presented in Fig. 5B.
Figure 5.
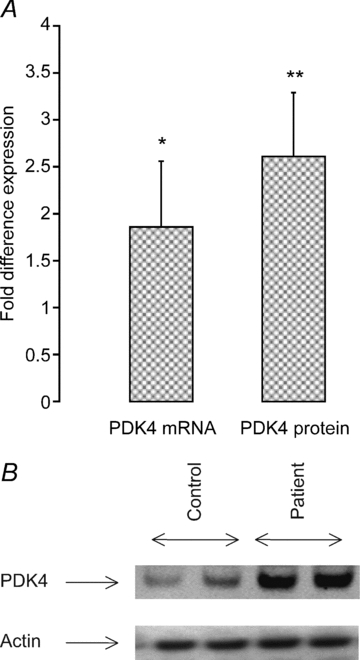
A, fold difference in muscle PDK4 mRNA and protein expression measured in critically ill patients (n = 10). Biopsies from healthy, age- and sex-matched controls (n = 10) were used as a calibrator with a value of 1. PDK4 mRNA is expressed as 2−ΔΔCt normalized to endogenous HMBS. *, **Significantly different from healthy controls (P < 0.05, P < 0.01). Data represent mean ± SEM. B, typical Western blots for PDK4 protein in muscle biopsies obtained from controls (n = 10) and critically ill patients (n = 10).
Discussion
Although based on a relatively small and heterogeneous population, this is the first study to comprehensively describe alterations in molecular events thought to control both muscle protein synthesis and protein degradation in critically ill patients. The major, novel findings from this study are as follows. (i) There was a wide-ranging decline in the magnitude of phosphorylation (activation) of signalling proteins thought to control translation initiation of muscle protein synthesis in patients compared to age- and sex-matched healthy control volunteers. (ii) This suppression of anabolic signalling was paralleled by upregulation of muscle protein breakdown pathways (ubiquitin proteasome complex and cathepsin L) at the level of both mRNA and protein expression. (iii) These catabolic events in patients were accompanied by clear evidence of a cellular programme of anabolic restoration by members of the Akt/mTOR signalling pathway, as reflected by extensive upregulation of mRNAs encoding the proteins in this pathway. (iv) Critical illness was also associated with the downregulation of muscle calpain-3 mRNA and protein. (v) For the first time, we show that a marked upregulation of myostatin mRNA and protein expression occurs in skeletal muscle from critically ill patients, which we believe to be functionally linked to the suppression of the Akt/mTOR signalling axis that we also observed. (vi) There was significant upregulation of muscle PDK4 mRNA and protein expression in patients, pointing to the inhibition of muscle CHO oxidation in these patients. Collectively, these data point to a simultaneous decrease in muscle protein synthesis and increase in muscle protein breakdown in patients, but these catabolic events are accompanied by initiation of a cellular programme of anabolic restoration at the transcriptional level. There is also molecular evidence for the development of muscle insulin resistance in this patient population. The present findings are consistent with increased protein turnover observed in acutely traumatized and ill patients (van Acker et al. 2000) (Shangraw & Turinsky, 1984), and in human and animal models of endotoxaemia (Crossland et al. 2008; Fredriksson et al. 2008). Immediately after or during trauma and major surgery, protein turnover is depressed, but after 48 h it is upregulated, such that fractional synthetic rates of muscle protein synthesis are not depressed or may even be increased (Emery & Ghusain-Choueiri, 1995). Conversely, in malnourished states (e.g. kwashiorkor in children) protein turnover is decreased (Golden et al. 1977).
The role of the ubiquitin–proteasome system in muscle in critical illness
Most, but not all, cellular proteins are degraded by the ATP-dependent ubiquitin–proteasome pathway (Jagoe & Goldberg, 2001). In line with this, the finding of increased concentrations of ubiquitinated proteins in muscle homogenates from myopathic septic patients (Rabuel et al. 2004), and our own observations of increased 20S proteasome mRNA and protein expression in patients in the present study (P < 0.001 and P < 0.05, respectively; Fig. 2A and B) confirms this contention. Furthermore, the two muscle-specific E3-ligases intimately associated with activation of ubiquitin–proteasome-mediated protein breakdown, i.e. atrogin–1/MAFbx and MuRF1, were also elevated at both mRNA (MAFbx; P < 0.05 and MuRF1; P < 0.05) and protein (MAFbx; P < 0.001 and MuRF1; P < 0.001) levels in patients (Fig. 2A and B). Although a comprehensive upregulation of the ubiquitin–proteasome system is widely considered to be a prerequisite during inflammation-mediated muscle atrophy (Wray et al. 2003; Crossland et al. 2008), it would appear from human- and animal-based research that this is not always the case in non-inflammatory states (Constantin et al. 2007; Greenhaff et al. 2008; Vary et al. 2008). In line with the presence of an inflammatory state, muscle TNF-α and IL-6 mRNA expression levels were increased 2.5-fold (P < 0.05) and 6.6-fold (P < 0.01), respectively) in patients in the present study. Such a response might reasonably be expected to reduce insulin receptor substrate-1 (IRS-1) binding (Rui et al. 2001), thereby dampening Akt signalling (Latres et al. 2005). Additionally, TNF-α is thought to directly inhibit Akt protein (Medina et al. 2005). The net result of both events would be the dephosphorylation (activation) of the forkhead (FOXO) family of transcription factors downstream, and the upregulation of FOXO target genes, which include MAFbx and MuRF1 (Sandri et al. 2004). In line with this, we were able to demonstrate downregulation of muscle Akt1 phosphorylation (Fig. 4B and C), and that this occurred concomitantly with increased expression of MAFbx and MuRF1 mRNA and protein (Fig. 2A and B).
The role of lysosomal-mediated muscle protein breakdown in critical illness
Animal-based research suggests that lysosomal-mediated protein degradation can account for almost one-third of muscle protein loss in cachexia (Baracos et al. 1995). Furthermore, human studies have demonstrated that muscle cathepsin-D mRNA expression increases in, for example, cancer cachexia (Lundholm et al. 1981) and that cathepsin-D enzyme activity increases in severe accident trauma (Guarnieri et al. 1988). For the first time, however, we show that upregulation of cathepsin-L mRNA (Fig. 3) occurs in patients, pointing to cathepsin-L being an active component of muscle wasting in this patient population.
Calpain-3 mRNA and protein expression is decreased in muscle in critical illness
Calpains are Ca2+-activated, non-lysosomal, cysteine proteases, of which μ- and m-isoforms (also known as calpain-1 and -2, respectively) are the most ubiquitously expressed, including in skeletal muscle. Calpain-1 and -2 are believed to degrade cellular ‘cement’ proteins, such as vinculin, talin, desmin, titin and troponin. Accordingly, increased expression and activity of calpain-1 and -2 is well documented in animal wasting models (Bartoli & Richard, 2005). Furthermore, since cytosolic Ca2+ concentrations are consistently elevated in sepsis, inflammation and ischaemia (Wagenmakers, 2001), it is not unreasonable to assume that calpain-1 and -2 activation will also be a feature of critical illness. The cellular role of calpain-3 (or p94), however, which is the only muscle-specific calpain, is less clear. Thus, calpain-3 expression appears to be either unaffected or decreased in cancer cachexia, muscle denervation (Smith et al. 2008) and immobilization (Jones et al. 2004). In line with these observations, a reduction in calpain-3 protein expression has also been associated with certain types of muscular dystrophies (Richard et al. 1995). Accordingly, calpain-3 mRNA and protein expression in the present study was found to be significantly reduced in patients (Fig. 3A and B). Furthermore, it has recently been reported that over-expression of the myostatin gene in rodent skeletal muscle is accompanied by a decrease in calpain-3 mRNA and protein expression and atrophy (Amirouche et al. 2009), pointing to a regulatory role of myostatin in the control of calpain-3 expression. In line with this contention, we observed that the upregulation of myostatin mRNA and protein in patients was accompanied by a reduction in the calpain-3 gene and protein expression (Fig. 3A and B).
Muscle myostatin mRNA and protein expression is increased in critical illness
Myostatin (GDF8), a member of the transforming growth factor (TGF)-β superfamily, functions as a negative regulator of satellite cell proliferation and differention, and muscle growth and development (Lee & McPherron, 2001). This is thought to occur by myostatin-mediated inhibition of the myogenic regulators MyoD, myogenin and Myf5 (Lee, 2004). Consistent with this role, increased myostatin mRNA and protein expression appears to be a regular feature of human muscle wasting conditions such as AIDS (Gonzalez-Cadavid et al. 1998) and chronic muscle disuse (Reardon et al. 2001) possibly by inhibiting protein synthesis via the Akt/mammalian target of rapamycin (mTOR)/p70S6K signalling pathway (Trendelenburg et al. 2009). Conversely, the expression of a non-functional allele of myostatin in humans results in extreme muscle growth (Schuelke et al. 2004). As far as we are aware, the present study is the first to report increased myostatin mRNA (P < 0.05) and protein (P < 0.01) expression in critically ill patients, which was quite marked (Fig. 3A and B). Furthermore, the present study supports the emerging contention that myostatin plays an important role in the regulation of human muscle mass because it has an involvement in the control of both muscle protein synthesis by inhibiting the anabolic signalling, i.e. translation initiation, through inhibiting Akt (Amirouche et al. 2009; Morissette et al. 2009) and muscle protein breakdown (McFarlane et al. 2006).
Translational control of muscle protein synthesis is inhibited in critical illness, but is accompanied by initiation of anabolic restoration at the level of gene expression
Changes in whole body or lower limb protein synthesis have been documented in critically ill patients (Essén et al. 1998; Tjader et al. 2004). However, our understanding of the molecular regulation of protein turnover in these patients is relatively limited and has been acquired mainly from cell-based investigation and animal models of muscle wasting. Such studies have provided evidence to suggest that the loss of muscle mass is the direct result of a decline in muscle protein synthesis arising from a reduction in translational efficiency, rather than a fall in ribosome number (Lang et al. 2007). Specifically, dephosphorylation (inactivation) of Akt1 decreases protein translation initiation by dampening the mTOR signalling cascade, thereby inhibiting ribosomal S6 kinase (p70s6k), and activating (i) 4E-BP1 (PHAS-1), a negative regulator of the translation initiation factor eIF-4E (Hara et al. 1997), and (ii) glycogen synthase kinase 3β (GSK3β), which is another negative regulator of protein synthesis via inhibition of eIF-2B (Hardt & Sadoshima, 2002). Furthermore, the reduction of Akt phosphorylation in catabolic conditions is purported to be involved in the upregulation of muscle protein breakdown through activation of MAFbx and MuRF1 (Sandri et al. 2004). Indeed, we did observe decreased Akt1, GSK3β, mTOR, p70s6k and 4E-BP1 protein phosphorylation in patients in the present study (Fig. 4), which was accompanied by widespread upregulation of molecular events controlling muscle protein breakdown. Moreover, these events were accompanied by the unexpected novel observation that the suppression of muscle anabolic signalling in patients was paralleled by a comprehensive increase in mRNA expression of these same signalling proteins (Fig. 4), pointing to the initiation of a cellular programme of anabolic restoration in patients. This finding may bear similarity to the observation that muscle total RNA in critically ill patients is significantly higher than in age- and sex-matched controls (Gamrin et al. 1996).
Muscle pyruvate dehydrogenase kinase 4 mRNA and protein expression is increased in critical illness – the link to muscle insulin resistance
Pyruvate dehydrogenase kinase 4 (PDK4) impairs the activation of mitochondrial pyruvate dehydrogenase complex (PDC) (Wu et al. 1999), the enzyme that controls the rate of CHO oxidation. The long-term impairment of muscle CHO oxidation by PDK4 has been implicated as a causative factor in muscle insulin resistance (Gorter et al. 2004). We have previously proposed a role for the Akt1/FOXO 1 (forkhead box, class O) signalling pathway in the simultaneous induction of muscle wasting and impairment of muscle CHO oxidation during endotoxaemia. Specifically, endotoxin-mediated elevation of muscle TNF-α and IL-6 inhibit Akt phosphorylation and initiate the transcription of the FOXO 1 target genes MAFbx, MuRF1 and PDK4 resulting in loss of muscle mass and impairment of PDC activation (Crossland et al. 2008). It may be therefore entirely reasonable to suggest that the present elevation of muscle pro-inflammatory cytokines (Fig. 1) and suppression of muscle Akt signalling (Fig. 4) in patients might have been responsible for the observed elevation of muscle PDK4 mRNA and protein expression (Fig. 5), and may have been in itself, or in combination with the upregulation of myostatin (also shown to inhibit muscle carbohydrate oxidation (McPherron & Lee, 2002), responsible for the reported induction of muscle insulin resistance typical of critical illness (Robinson & van Soeren, 2004). This is extremely important and relevant to the clinical setting as uncontrolled blood glucose concentrations in critically ill patients impacts on clinical outcome in terms of mortality, weaning time or ongoing neuromuscular complications (van den Berghe et al. 2001; Hermans et al. 2007).
Impaired muscle function has been reported to be a clinical trait of critical illness (Puthucheary et al. 2010). Furthermore, the reduction in muscle mass (and consequently the loss of muscle strength) is an independent predictor of survival during critical illness (De Jonghe et al. 2002; Ali et al. 2008; Sharshar et al. 2009). While no study has comprehensively investigated the temporal relationship between changes in the expression of genes and proteins thought to control muscle mass and the loss of muscle mass and strength during critical illness, the molecular events reported in the present study could be considered as potentially important factors underpinning muscle wasting and changes in muscle structure and function in critical illness. Although histopathological and muscle functional assessments were not performed in the present study, critically ill patients experience myosin filament loss (Helliwell et al. 1998) and/or necrosis (Helliwell et al. 1991). These two factors would contribute not only to muscle wasting per se, but also to the derangement of sarcomere structure, which could consequently impair energy metabolism and cross-bridge cycling.
In conclusion, this is the first study to describe comprehensive alterations in the molecular events thought to control muscle protein synthesis and breakdown in critically ill patients, as evidenced by the simultaneous downregulation of signalling proteins thought to increase muscle protein synthesis and activation of molecular pathways driving muscle protein breakdown. However, these catabolic changes were paralleled by initiation of a cellular programme of anabolic restoration at the transcriptional level. We also provide molecular evidence for an increase in muscle PDK4 protein expression, which may account for the reported induction of muscle insulin resistance in this patient population. These data point to a high plasticity of muscle in critically ill patients, and further efforts should be focused on delineating the temporal changes in gene and protein expression in critically ill patients, not least to determine the further impact of time-related events that will undoubtedly occur in this patient group. Furthermore, although challenging to perform, these studies should include concurrent measurements of muscle protein synthesis and breakdown. Finally, strategies aimed at preserving muscle mass and metabolic function in critical illness should focus on maintaining Akt phosphorylation and inhibiting myostatin expression.
Acknowledgments
This study was partly supported by the Medical Research Council (grant award G0501985) and by a Project Grant from the European Society of Anaesthesiology.
Glossary
Abbreviations
- Akt1
v-akt murine thymoma oncogene homologue 1
- CHO
carbohydrate
- 4E-BP1
eukaryotic translation initiation factor 4E-binding protein 1
- FOXO 1
forkhead box, class O
- GDF8
myostatin
- GSK3α,β
glycogen synthase kinase 3
- HMBS
hydroxymethylbilane synthase
- IL-6
interleukin 6
- IRS-1
insulin receptor substrate-1
- MAFbx
muscle atrophy F-box
- MuRF1
muscle RING-finger 1
- mTOR
mammalian target of rapamycin
- p70S6K
70 kDa ribosomal protein S6 kinase 1
- PDK4
pyruvate dehydrogenase kinase isozyme 4
- PI3K
phosphoinositide 3-kinase
- TNF-α
tumour necrosis factor α
- VF
ventricular fibrillation
Author contributions
Experiments in this study were conducted in the School of Biomedical Sciences, University of Nottingham and the Department of Anaesthesia and Intensive Care, Queen's Medical Centre, Nottingham University Hospital. All authors approved the final version of the manuscript to be published and all authors contributed to drafting the article and revising it critically for important intellectual content. All authors contributed to the conception and design, or analysis and interpretation of the data in the manuscript.
References
- Ali NA, O'Brien JM, Jr, Hoffmann SP, Phillips G, Garland A, Finley JC, Almoosa K, Hejal R, Wolf KM, Lemeshow S, Connors AF, Jr, Marsh CB. Acquired weakness, handgrip strength, and mortality in critically ill patients. Am J Respir Crit Care Med. 2008;178:261–268. doi: 10.1164/rccm.200712-1829OC. [DOI] [PubMed] [Google Scholar]
- Amirouche A, Durieux AC, Banzet S, Koulmann N, Bonnefoy R, Mouret C, Bigard X, Peinnequin A, Freyssenet D. Down-regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology. 2009;150:286–294. doi: 10.1210/en.2008-0959. [DOI] [PubMed] [Google Scholar]
- Baracos VE, DeVivo C, Hoyle DH, Goldberg AL. Activation of the ATP-ubiquitin-proteasome pathway in skeletal muscle of cachectic rats bearing a hepatoma. Am J Physiol Endocrinol Metab. 1995;268:E996–E1006. doi: 10.1152/ajpendo.1995.268.5.E996. [DOI] [PubMed] [Google Scholar]
- Bartoli M, Richard I. Calpains in muscle wasting. Int J Biochem Cell Biol. 2005;37:2115–2133. doi: 10.1016/j.biocel.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Bergstrom J, Hultman E. The effect of exercise on muscle glycogen and electrolytes in normals. Scand J Clin Lab Invest. 1966;18:16–20. doi: 10.3109/00365516609065602. [DOI] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- Constantin D, Constantin-Teodosiu D, Layfield R, Tsintzas K, Bennett AJ, Greenhaff PL. PPARδ agonism induces a change in fuel metabolism and activation of an atrophy programme, but does not impair mitochondrial function in rat skeletal muscle. J Physiol. 2007;583:381–390. doi: 10.1113/jphysiol.2007.135459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossland H, Constantin-Teodosiu D, Gardiner SM, Constantin D, Greenhaff PL. A potential role for Akt/FOXO signalling in both protein loss and the impairment of muscle carbohydrate oxidation during sepsis in rodent skeletal muscle. J Physiol. 2008;586:5589–5600. doi: 10.1113/jphysiol.2008.160150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jonghe B, Bastuji-Garin S, Durand MC, Malissin I, Rodrigues P, Cerf C, Outin H, Sharshar T. Respiratory weakness is associated with limb weakness and delayed weaning in critical illness. Crit Care Med. 2007;35:2007–2015. doi: 10.1097/01.ccm.0000281450.01881.d8. [DOI] [PubMed] [Google Scholar]
- De Jonghe B, Lacherade JC, Sharshar T, Outin H. Intensive care unit-acquired weakness: risk factors and prevention. Crit Care Med. 2009;37:S309–315. doi: 10.1097/CCM.0b013e3181b6e64c. [DOI] [PubMed] [Google Scholar]
- De Jonghe B, Sharshar T, Lefaucheur JP, Authier FJ, Durand-Zaleski I, Boussarsar M, Cerf C, Renaud E, Mesrati F, Carlet J, Raphael JC, Outin H, Bastuji-Garin S. Paresis acquired in the intensive care unit: a prospective multicenter study. JAMA. 2002;288:2859–2867. doi: 10.1001/jama.288.22.2859. [DOI] [PubMed] [Google Scholar]
- Deval C, Mordier S, Obled C, Bechet D, Combaret L, Attaix D, Ferrara M. Identification of cathepsin L as a differentially expressed message associated with skeletal muscle wasting. Biochem J. 2001;360:143–150. doi: 10.1042/0264-6021:3600143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery PW, Ghusain-Choueiri A. The local and systemic effects of minor injury on muscle protein synthesis in the rat. Int J Biochem Cell Biol. 1995;27:815–820. doi: 10.1016/1357-2725(95)00045-q. [DOI] [PubMed] [Google Scholar]
- Essén P, McNurlan MA, Gamrin L, Hunter K, Calder G, Garlick PJ, Wernerman J. Tissue protein synthesis rates in critically ill patients. Crit Care Med. 1998;26:92–100. doi: 10.1097/00003246-199801000-00022. [DOI] [PubMed] [Google Scholar]
- Fredriksson K, Tjader I, Keller P, Petrovic N, Ahlman B, Scheele C, Wernerman J, Timmons JA, Rooyackers O. Dysregulation of mitochondrial dynamics and the muscle transcriptome in ICU patients suffering from sepsis induced multiple organ failure. PLoS ONE. 2008;3:e3686. doi: 10.1371/journal.pone.0003686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamrin L, Andersson K, Hultman E, Nilsson E, Essen P, Wernerman J. Longitudinal changes of biochemical parameters in muscle during critical illness. Metabolism. 1997;46:756–762. doi: 10.1016/s0026-0495(97)90119-0. [DOI] [PubMed] [Google Scholar]
- Gamrin L, Essen P, Forsberg AM, Hultman E, Wernerman J. A descriptive study of skeletal muscle metabolism in critically ill patients: free amino acids, energy-rich phosphates, protein, nucleic acids, fat, water, and electrolytes. Crit Care Med. 1996;24:575–583. doi: 10.1097/00003246-199604000-00005. [DOI] [PubMed] [Google Scholar]
- Golden MH, Waterlow JC, Picou D. Protein turnover, synthesis and breakdown before and after recovery from protein-energy malnutrition. Clin Sci Mol Med. 1977;53:473–477. doi: 10.1042/cs0530473. [DOI] [PubMed] [Google Scholar]
- Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci U S A. 2001;98:14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, Sinha-Hikim I, Ma K, Ezzat S, Shen R, Lalani R, Asa S, Mamita M, Nair G, Arver S, Bhasin S. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc Natl Acad Sci U S A. 1998;95:14938–14943. doi: 10.1073/pnas.95.25.14938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter PM, Olijhoek JK, van der Graaf Y, Algra A, Rabelink TJ, Visseren FL. Prevalence of the metabolic syndrome in patients with coronary heart disease, cerebrovascular disease, peripheral arterial disease or abdominal aortic aneurysm. Atherosclerosis. 2004;173:363–369. doi: 10.1016/j.atherosclerosis.2003.12.033. [DOI] [PubMed] [Google Scholar]
- Greenhaff PL, Karagounis LG, Peirce N, Simpson EJ, Hazell M, Layfield R, Wackerhage H, Smith K, Atherton P, Selby A, Rennie MJ. Disassociation between the effects of amino acids and insulin on signaling, ubiquitin ligases, and protein turnover in human muscle. Am J Physiol Endocrinol Metab. 2008;295:E595–E604. doi: 10.1152/ajpendo.90411.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarnieri G, Toigo G, Situlin R, Del Bianco MA, Crapesi L. Cathepsin B and D activity in human skeletal muscle in disease states. Adv Exp Med Biol. 1988;240:243–256. doi: 10.1007/978-1-4613-1057-0_29. [DOI] [PubMed] [Google Scholar]
- Hara K, Yonezawa K, Kozlowski MT, Sugimoto T, Andrabi K, Weng QP, Kasuga M, Nishimoto I, Avruch J. Regulation of eIF-4E BP1 phosphorylation by mTOR. J Biol Chem. 1997;272:26457–26463. doi: 10.1074/jbc.272.42.26457. [DOI] [PubMed] [Google Scholar]
- Hardt SE, Sadoshima J. Glycogen synthase kinase-3β: a novel regulator of cardiac hypertrophy and development. Circ Res. 2002;90:1055–1063. doi: 10.1161/01.res.0000018952.70505.f1. [DOI] [PubMed] [Google Scholar]
- Helliwell TR, Coakley JH, Wagenmakers AJ, Griffiths RD, Campbell IT, Green CJ, McClelland P, Bone JM. Necrotizing myopathy in critically-ill patients. J Pathol. 1991;164:307–314. doi: 10.1002/path.1711640406. [DOI] [PubMed] [Google Scholar]
- Helliwell TR, Wilkinson A, Griffiths RD, McClelland P, Palmer TE, Bone JM. Muscle fibre atrophy in critically ill patients is associated with the loss of myosin filaments and the presence of lysosomal enzymes and ubiquitin. Neuropathol Appl Neurobiol. 1998;24:507–517. doi: 10.1046/j.1365-2990.1998.00144.x. [DOI] [PubMed] [Google Scholar]
- Hermans G, Wilmer A, Meersseman W, Milants I, Wouters PJ, Bobbaers H, Bruyninckx F, Van den Berghe G. Impact of intensive insulin therapy on neuromuscular complications and ventilator dependency in the medical intensive care unit. Am J Respir Crit Care Med. 2007;175:480–489. doi: 10.1164/rccm.200605-665OC. [DOI] [PubMed] [Google Scholar]
- Jagoe RT, Goldberg AL. What do we really know about the ubiquitin-proteasome pathway in muscle atrophy? Curr Opin Clin Nutr Metab Care. 2001;4:183–190. doi: 10.1097/00075197-200105000-00003. [DOI] [PubMed] [Google Scholar]
- Jones SW, Hill RJ, Krasney PA, O'Conner B, Peirce N, Greenhaff PL. Disuse atrophy and exercise rehabilitation in humans profoundly affects the expression of genes associated with the regulation of skeletal muscle mass. FASEB J. 2004;18:1025–1027. doi: 10.1096/fj.03-1228fje. [DOI] [PubMed] [Google Scholar]
- Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med. 1985;13:818–829. [PubMed] [Google Scholar]
- Lang CH, Frost RA, Vary TC. Regulation of muscle protein synthesis during sepsis and inflammation. Am J Physiol Endocrinol Metab. 2007;293:E453–E459. doi: 10.1152/ajpendo.00204.2007. [DOI] [PubMed] [Google Scholar]
- Latres E, Amini AR, Amini AA, Griffiths J, Martin FJ, Wei Y, Lin HC, Yancopoulos GD, Glass DJ. Insulin-like growth factor–1 (IGF–1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem. 2005;280:2737–2744. doi: 10.1074/jbc.M407517200. [DOI] [PubMed] [Google Scholar]
- Lee SJ. Regulation of muscle mass by myostatin. Annu Rev Cell Dev Biol. 2004;20:61–86. doi: 10.1146/annurev.cellbio.20.012103.135836. [DOI] [PubMed] [Google Scholar]
- Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci U S A. 2001;98:9306–9311. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundholm K, Edstrom S, Ekman L, Karlberg I, Schersten T. Metabolism in peripheral tissues in cancer patients. Cancer Treat Rep. 1981;65(Suppl. 5):79–83. [PubMed] [Google Scholar]
- McFarlane C, Plummer E, Thomas M, Hennebry A, Ashby M, Ling N, Smith H, Sharma M, Kambadur R. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-κB-independent, FoxO1-dependent mechanism. J Cell Physiol. 2006;209:501–514. doi: 10.1002/jcp.20757. [DOI] [PubMed] [Google Scholar]
- McPherron AC, Lee SJ. Suppression of body fat accumulation in myostatin-deficient mice. J Clin Invest. 2002;109:595–601. doi: 10.1172/JCI13562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina EA, Afsari RR, Ravid T, Castillo SS, Erickson KL, Goldkorn T. Tumor necrosis factor–α decreases Akt protein levels in 3T3-L1 adipocytes via the caspase-dependent ubiquitination of Akt. Endocrinology. 2005;146:2726–2735. doi: 10.1210/en.2004-1074. [DOI] [PubMed] [Google Scholar]
- Morissette MR, Cook SA, Buranasombati C, Rosenberg MA, Rosenzweig A. Myostatin inhibits IGF–I-induced myotube hypertrophy through Akt. Am J Physiol Cell Physiol. 2009;297:C1124–C1132. doi: 10.1152/ajpcell.00043.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthucheary Z, Montgomery H, Moxham J, Harridge S, Hart N. Structure to function: muscle failure in critically ill patients. J Physiol. 2010;588:4641–4648. doi: 10.1113/jphysiol.2010.197632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabuel C, Renaud E, Brealey D, Ratajczak P, Damy T, Alves A, Habib A, Singer M, Payen D, Mebazaa A. Human septic myopathy: induction of cyclooxygenase, heme oxygenase and activation of the ubiquitin proteolytic pathway. Anesthesiology. 2004;101:583–590. doi: 10.1097/00000542-200409000-00006. [DOI] [PubMed] [Google Scholar]
- Reardon KA, Davis J, Kapsa RM, Choong P, Byrne E. Myostatin, insulin-like growth factor-1, and leukemia inhibitory factor mRNAs are upregulated in chronic human disuse muscle atrophy. Muscle Nerve. 2001;24:893–899. doi: 10.1002/mus.1086. [DOI] [PubMed] [Google Scholar]
- Richard I, Broux O, Allamand V, Fougerousse F, Chiannilkulchai N, Bourg N, Brenguier L, Devaud C, Pasturaud P, Roudaut C, Hillaire D, Passos-Bueno M-R, Zatz M, Tischfield JA, Fardeau M, Jackson CE, Cohen D, Beckmann JS. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell. 1995;81:27–40. doi: 10.1016/0092-8674(95)90368-2. [DOI] [PubMed] [Google Scholar]
- Robinson LE, van Soeren MH. Insulin resistance and hyperglycemia in critical illness: role of insulin in glycemic control. AACN Clin Issues. 2004;15:45–62. doi: 10.1097/00044067-200401000-00004. [DOI] [PubMed] [Google Scholar]
- Rui L, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A, Dunaif A, White MF. Insulin/IGF-1 and TNF-α stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest. 2001;107:181–189. doi: 10.1172/JCI10934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin–1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- Shangraw RE, Turinsky J. Altered protein kinetics in vivo after single-limb burn injury. Biochem J. 1984;223:747–753. doi: 10.1042/bj2230747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharshar T, Bastuji-Garin S, Stevens RD, Durand MC, Malissin I, Rodriguez P, Cerf C, Outin H, De Jonghe B. Presence and severity of intensive care unit-acquired paresis at time of awakening are associated with increased intensive care unit and hospital mortality. Crit Care Med. 2009;37:3047–3053. doi: 10.1097/CCM.0b013e3181b027e9. [DOI] [PubMed] [Google Scholar]
- Smith IJ, Lecker SH, Hasselgren PO. Calpain activity and muscle wasting in sepsis. Am J Physiol Endocrinol Metab. 2008;295:E762–E771. doi: 10.1152/ajpendo.90226.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supinski GS, Wang W, Callahan LA. Caspase and calpain activation both contribute to sepsis-induced diaphragmatic weakness. J Appl Physiol. 2009;107:1389–1396. doi: 10.1152/japplphysiol.00341.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjader I, Essen P, Garlick PJ, McMnurlan MA, Rooyackers O, Wernerman J. Impact of surgical trauma on human skeletal muscle protein synthesis. Clin Sci (Lond) 2004;107:601–607. doi: 10.1042/CS20040192. [DOI] [PubMed] [Google Scholar]
- Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol. 2009;296:C1258–C1270. doi: 10.1152/ajpcell.00105.2009. [DOI] [PubMed] [Google Scholar]
- van Acker BA, Hulsewe KW, Wagenmakers AJ, Soeters PB, von Meyenfeldt MF. Glutamine appearance rate in plasma is not increased after gastrointestinal surgery in humans. J Nutr. 2000;130:1566–1571. doi: 10.1093/jn/130.6.1566. [DOI] [PubMed] [Google Scholar]
- van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R. Intensive insulin therapy in the critically ill patients. N Engl J Med. 2001;345:1359–1367. doi: 10.1056/NEJMoa011300. [DOI] [PubMed] [Google Scholar]
- Vary TC, Frost RA, Lang CH. Acute alcohol intoxication increases atrogin–1 and MuRF1 mRNA without increasing proteolysis in skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1777–R1789. doi: 10.1152/ajpregu.00056.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenmakers AJM. Muscle function in critically ill patients. Clin Nutr. 2001;20:451–454. doi: 10.1054/clnu.2001.0483. [DOI] [PubMed] [Google Scholar]
- Wray CJ, Mammen JM, Hershko DD, Hasselgren PO. Sepsis upregulates the gene expression of multiple ubiquitin ligases in skeletal muscle. Int J Biochem Cell Biol. 2003;35:698–705. doi: 10.1016/s1357-2725(02)00341-2. [DOI] [PubMed] [Google Scholar]
- Wu P, Inskeep K, Bowker-Kinley MM, Popov KM, Harris RA. Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes. 1999;48:1593–1599. doi: 10.2337/diabetes.48.8.1593. [DOI] [PubMed] [Google Scholar]