

Abstract
The oncogenes RAS and RAF came to view as agents of neoplastic transformation. However, in normal cells, these genes can have effects that run counter to oncogenic transformation, such as arrest of the cell division cycle, induction of cell differentiation, and apoptosis. Recent work has demonstrated that RAS elicits proliferative arrest and senescence in normal mouse and human fibroblasts. Because the Raf/MEK/MAP kinase signaling cascade is a key effector of signaling from Ras proteins, we examined the ability of conditionally active forms of Raf-1 to elicit cell cycle arrest and senescence in human cells. Activation of Raf-1 in nonimmortalized human lung fibroblasts (IMR-90) led to the prompt and irreversible arrest of cellular proliferation and the premature onset of senescence. Concomitant with the onset of cell cycle arrest, we observed the induction of the cyclin-dependent kinase (CDK) inhibitors p21Cip1 and p16Ink4a. Ablation of p53 and p21Cip1 expression by use of the E6 oncoprotein of HPV16 demonstrated that expression of these proteins was not required for Raf-induced cell cycle arrest or senescence. Furthermore, cell cycle arrest and senescence were elicited in IMR-90 cells by the ectopic expression of p16Ink4a alone. Pharmacological inhibition of the Raf/MEK/MAP kinase cascade prevented Raf from inducing p16Ink4a and also prevented Raf-induced senescence. We conclude that the kinase cascade initiated by Raf can regulate the expression of p16Ink4a and the proliferative arrest and senescence that follows. Induction of senescence may provide a defense against neoplastic transformation when the MAP kinase signaling cascade is inappropriately active.
Keywords: Raf, MAP kinase signaling, senescence, oncogenesis
Normal cells proliferate in vitro for a finite number of cell divisions after which they enter a state known as senescence (Hayflick and Moorhead 1961; Campisi 1996, 1997). Senescent cells withdraw irreversibly from the cell division cycle, but remain viable indefinitely, develop a distinctive morphology, and display characteristic phenotypic markers, such as the senescence-associated, acidic β-galactosidase activity (SA-β-gal; Dimri et al. 1995). Although the biochemical mediators of senescence in human cells remain uncertain, candidates include the p53 tumor suppressor protein, the cyclin-dependent kinase (CDK) inhibitors p21Cip1 and p16Ink4a, and regulators of telomere length and function, such as telomerase and TRF1 and TRF2. Increased expression of p53, p16Ink4a, and p21Cip1 is detected in a variety of senescent cells, and overexpression of telomerase leads to immortalization of human cells in culture (Kulju and Lehman 1995; Alcorta et al. 1996; Hara et al. 1996; Reznikoff et al. 1996; Shay and Bacchetti 1997; Bodnar et al. 1998; van Steensel et al. 1998). Furthermore primary human cells that are deficient in the expression of p21Cip1 have increased replicative capacity in vitro (Brown et al. 1997). p53, p16Ink4a, and p21Cip1 can arrest the cell division cycle: p21Cip1 and p16Ink4a do so by inhibiting CDKs required for progression through the cell cycle (Lees 1995), and p53 does so by inducing the expression of p21Cip1 (El-Deiry et al. 1993).
Cell lines derived from most tumors are capable of extended proliferation as if the capability to become senescent has been somehow repressed or lost. Accordingly, either p53, p16Ink4a, or both are frequently defective in tumor cells (Hall and Peters 1996; Hainaut et al. 1997), and restoration of p53 to deficient tumor cells elicits prompt senescence (Sugrue et al. 1997). Furthermore, tumor cells frequently express telomerase activity, which may contribute to the extended proliferation of these cells in culture (Meyerson et al. 1997). Clearly the extension of proliferative life span might make an important contribution to tumorigenesis by increasing the likelihood that mutant variants of cells can arise.
Oncoproteins such as Ras and Raf can transform established lines of cells to neoplastic growth (Stanton et al. 1989; Samuels et al. 1993). However, in cultures of normal cells, these two genes can have paradoxical effects, including arrest of the cell cycle, differentiation, or even apoptosis (O’Shea et al. 1996; Kauffmann-Zeh et al. 1997; Lloyd et al. 1997; Woods et al. 1997). In culture, Ras and Raf transform normal cells only with the cooperation of other gene products, such as E1A, T-antigen, or Myc, which apparently serve to extend the proliferative life span of the cells (Hunter 1991). Recent work has shown that the H-Ras oncoprotein can also elicit premature senescence in IMR-90 cells, a widely studied strain of human fibroblasts derived from lung tissue (Serrano et al. 1997). Induction of senescence by H-Ras is accompanied by increased expression of p53, p21Cip1, and p16Ink4a.
Ras proteins are membrane-associated GTPases that, in their GTP-bound state, transmit signals into the interior of the cell. The signal is transmitted by several means, but prominent among these is a cascade of protein kinases initiated by the Raf-1 protein serine/threonine kinase (Avruch et al. 1994; Marshall 1996). Binding of Raf to activated Ras predisposes it to activation by other means that likely include phosphorylation by Src-family protein tyrosine kinases at key regulatory tyrosine residues (Y340 and Y341 in Raf-1; Marais et al. 1995). Then, activated Raf phosphorylates to activate the second kinase in the cascade (MEK), and this, in turn, phosphorylates to activate the p42/p44 MAP kinases, also known as ERK2 and ERK1, respectively (Marshall 1994). Activated MAP kinases are pleiotropic modulators of cell function that phosphorylate cytosolic, membrane-bound, and nuclear substrates including several transcription factors (Treisman 1996). Activation of this signaling pathway has been implicated both in normal proliferation and differentiation and in the aberrant proliferation observed in cancer cells.
The signaling from Ras through Raf prompted us to ask whether Raf itself could elicit senescence. Using a conditionally active version of Raf that can be activated by estrogen and its analogs (Samuels et al. 1993), we demonstrated that activation of Raf suffices to both arrest the proliferation of IMR-90 cells and elicit premature senescence. Raf-induced senescence is irreversible and accompanied by a consistent elevation in the expression of p16Ink4a and appears to have no obvious requirement for either p53 or p21Cip1. Moreover, p16Ink4a can itself elicit senescence when expressed ectopically in IMR-90 cells. The signal from Raf that elicits senescence is apparently transmitted through MEK because pharmacological inhibition of this enzyme prevents Raf-induced senescence. Consequently, we propose that the kinase cascade initiated by Raf is likely the major signaling pathway that mediates senescence in response to Ras and that p16Ink4a is likely an important mediator of this response.
Results
Isolation of IMR-90 cells expressing a conditional version of Raf
To achieve conditional activation of the Raf/MEK/MAP kinase cascade in IMR-90 human lung fibroblasts, we employed a previously described chimera in which the catalytic domain of human Raf-1 was fused to the hormone-binding domain of the human estrogen receptor (Samuels et al. 1993). To provide a marker for cell sorting, an enhanced version of green fluorescent protein (EGFP) was fused to the amino terminus of the chimera as illustrated in Figure 1A and described previously (Woods et al. 1997). A variant was also created in which a pair of adjacent regulatory tyrosine residues (equivalent to Y340 and Y341 [YY] in full-length Raf-1) were mutated to aspartic acid [DD], leading to a 10-fold increase in the activity of Raf-1 as described previously (Woods et al. 1997). These chimeras are hereafter designated as GFPΔRaf-1[YY]:ER and GFPΔRaf-1[DD]:ER, respectively. GFPΔRaf-1:ER proteins have little or no kinase activity until activated with estrogen or its analogs such as 4-hydroxy-tamoxifen (4-HT), whereupon they rapidly activate downstream signaling pathways (Samuels et al. 1993). By use of a replication-deficient retroviral vector, GFPΔRaf-1:ER proteins were expressed in IMR-90 cells that had reached 25–35 doublings. IMR-90 cells normally become senescent after 50–60 population doublings (Hayflick and Moorhead 1961). Pooled populations of infected cells were selected with puromycin and then sorted by FACS to obtain cells expressing equivalent levels of GFPΔRaf-1:ER. As a control, a pooled population of IMR-90 cells infected with a retrovirus encoding only the puromycin-resistance gene was derived in parallel (Babe).
Figure 1.
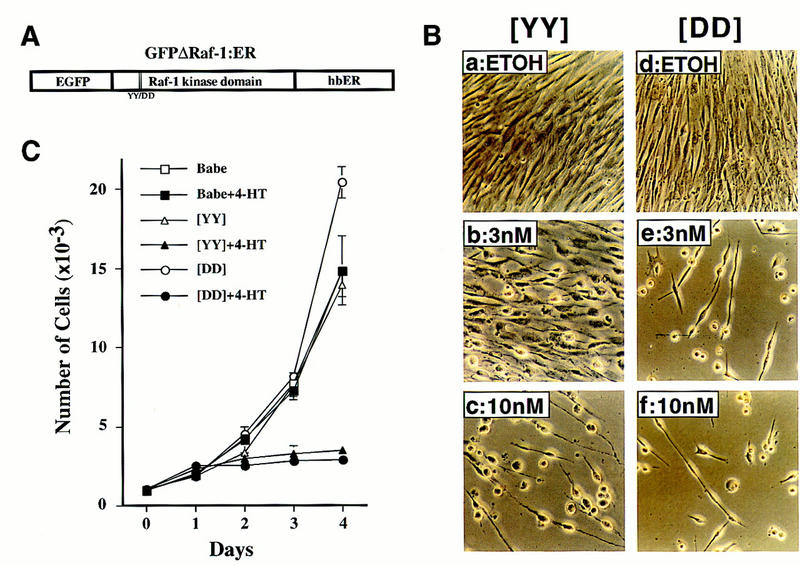
Activation of Raf kinase induces morphological alterations and arrests the proliferation of IMR-90 cells. (A) An inducible and tagged version of Raf. The illustrated construct was inserted into the replication-defective retrovirus described in Materials and Methods. (EGFP) Enhanced version of green fluorescent protein; (hbER) hormone-binding domain of the human estrogen receptor. The double vertical lines within the Raf-1 kinase domain and YY/DD below indicate the location of the tyrosine residues that were mutated to aspartic acid to generate the more active GFPΔRaf-1[DD]:ER as described in Materials and Methods and previously (Woods et al. 1997). (B) Morphological effects of Raf. IMR-90 cells expressing either the [YY] (a–c) or [DD] (d–e) forms of GFPΔRaf-1:ER were treated for 4 days with either ethanol as solvent control (ETOH) or different concentrations of 4-HT as indicated, then photographed with a phase-contrast microscope. (C) Proliferation of IMR-90 cells. Control (Babe) or GFPΔRaf-1:ER-expressing IMR-90 cells ([YY] or [DD]) were cultured in DMEM containing 15% vol/vol FCS in the absence or presence of 100 nm 4-HT as indicated. Cells were harvested at daily intervals and counted with a hemocytometer. Each data point represents the average of four independent measurements. A graph representative of multiple experiments is presented. (□) Babe; (█) Babe+4-HT; (▵) [YY]; (▴) [YY]+4-HT; (○) [DD]; (•) [DD]+4-HT.
Activation of Raf kinase arrests the proliferation of IMR-90 cells
Treatment of exponentially proliferating IMR-90 cells expressing GFPΔRaf-1:ER with 4-HT elicited pronounced morphological changes. Within 24 hr, the cells became more refractile and, after 2–4 days, most displayed an extended cell morphology with a spherical cell body. The cells remained adherent to the substratum and viable for at least 2 weeks after the addition of 4-HT (data not shown). The population density of the cells did not change, suggesting an arrest of proliferation, and no evidence of apoptosis was detected in these cultures. Both the [YY] and [DD] forms of GFPΔRaf-1:ER elicited the same response, although the latter required less 4-HT to elicit maximal effects than the former (Fig. 1B, cf. a–c with d–f); this difference presumably reflects the greater kinase activity of GFPΔRaf-1[DD]:ER once activated (Woods et al. 1997).
Activation of GFPΔRaf-1:ER arrested the proliferation of IMR-90 cells (Fig. 1C). IMR-90 cells infected with only the viral vector (Babe), and cells expressing the [YY] and [DD] forms of GFPΔRaf-1:ER all proliferated at the same rate in the absence of 4-HT. Upon activation of GFPΔRaf-1:ER with 4-HT, IMR-90 cells ceased proliferation within 2 days and failed to reach confluence. The arrest of proliferation was confirmed by BrdU labeling and FACscan analysis and by the incorporation of methyl-[3H]thymidine into DNA (Table 1; data not shown). Activation of GFPΔRaf-1[YY]:ER reduced the fraction of cells in S phase by one-third within 2 days and by about 10-fold within 4 days; similar results were obtained with GFPΔRaf-1[DD]:ER, except that the reduction in cell proliferation occurred more rapidly and amounted to >20-fold at maximum. Neither the proliferation nor the cell cycle profiles of control IMR-90 cells was affected by the addition of 4-HT (Fig. 1C; Table 1). We conclude that activation of Raf-1 arrests the cell cycle of normal human fibroblasts.
Table 1.
Activation of GFPΔRaf-1:ER induces cell cycle arrest
| Cells
|
Days
|
4-HT (nm)
|
G1 (%)
|
G2/M (%)
|
S (%)
|
|---|---|---|---|---|---|
| Babe | 2 | 0 | 41.0 | 13.9 | 45.1 |
| 2 | 100 | 42.0 | 12.9 | 45.1 | |
| 4 | 0 | 33.8 | 8.4 | 57.9 | |
| 4 | 100 | 33.5 | 8.4 | 58.0 | |
| GFPΔRaf-1[YY]:ER | 2 | 0 | 51.4 | 14.5 | 34.1 |
| 2 | 100 | 57.9 | 19.1 | 23.0 | |
| 4 | 0 | 34.8 | 11.9 | 53.3 | |
| 4 | 100 | 69.4 | 25.3 | 5.4 | |
| GFPΔRaf-1[DD]:ER | 2 | 0 | 54.4 | 13.9 | 31.6 |
| 2 | 100 | 62.6 | 25.7 | 11.6 | |
| 4 | 0 | 35.0 | 10.4 | 54.6 | |
| 4 | 100 | 67.8 | 30.2 | 2.0 |
Puromycin-resistant IMR-90 cells infected with control (Babe) and GFPΔRaf-1:ER-encoding retroviruses were treated with 100 nm 4-HT for 2 or 4 days and labeled with 15 μm BrdU for 3 hr. Ethanol-fixed cells were stained with FITC-anti-BrdU and with propidium iodide, and the percentage of cells in the different phases of the cell cycle was assessed by flow cytometry.
Senescence-associated, acidic β-galactosidase activity in IMR-90 cells following Raf-1 activation
Given the effects of H-Ras on IMR-90 cells (Serrano et al. 1997), it seemed possible that activation of Raf might elicit senescence in these cells. Therefore, we tested the cells arrested by activated Raf for the presence of a senescence-associated acidic β-galactosidase (SA–β-galactosidase) activity that has been associated with senescence in human cells (Dimri et al. 1995). Activation of GFPΔRaf-1[YY]:ER in IMR-90 cells elicited the expression of acidic β-galactosidase, which was apparent within 2–3 days and maximal by 5–6 days after addition of 4-HT (Fig. 2). We conclude that the arrest of proliferation induced by Raf is accompanied by senescence as the arrested cells remained viable for an extended period of time, assumed a characteristic morphology, and expressed an enzymatic marker that is strongly associated with cellular senescence.
Figure 2.

Activity of senescence-associated, acidic β-galactosidase in IMR-90 cells. Actively proliferating IMR-90 cells expressing GFPΔRaf-1[YY]:ER were treated with either ethanol (ETOH) or with a range of 4-HT concentrations from 1–100 nm as indicated for 6 days, then stained to detect the activity of the senescence-associated, acidic β-galactosidase.
Expression of cell cycle regulatory proteins in IMR-90 cells
Senescent cells typically contain elevated levels of the tumor suppressor protein p53 and the cell cycle inhibitors p16Ink4a and p21Cip1 (Noda et al. 1994; Kulju and Lehman 1995; Alcorta et al. 1996; Hara et al. 1996). It is presumed that the increase in p53, in turn, augments transcription from the gene encoding p21Cip1 (El-Deiry et al. 1993); in contrast, the precise mechanism responsible for the induction of p16Ink4a is not known but may involve transcriptional activation of the INK4A gene (Hara et al. 1996). Both p16Ink4a and p21Cip1 could contribute to the arrest of cellular proliferation displayed by senescent cells (McConnell et al. 1998). Therefore, we examined the effect of GFPΔRaf-1:ER activation on the expression of these proteins.
Activation of both the [YY] and [DD] forms of GFPΔRaf-1:ER in IMR-90 cells led to induced expression of the chimeric protein, consistent with the selective stabilization of ΔRaf:ER fusion proteins that we observed in a variety of other cell types (Fig. 3A, left and middle panels; Samuels et al. 1993). No proteins cross-reactive with the anti-hbER antisera were detected in extracts from control IMR-90 cells (Babe; Fig. 3A, right panel). As observed in NIH-3T3 cells, rapid activation of the p42/p44 MAP kinases was observed by both Western blotting with phospho-specific antisera and by immune-complex kinase assays within 4 hr following activation of GFPΔRaf-1:ER. MAP kinase activation was maintained for up to 6 days after the addition of 4-HT (Samuels et al. 1993; data not shown).
Figure 3.






Effects of Raf on cell cycle regulators. Control (Babe) and GFPΔRaf-1:ER ([YY]- and [DD])-expressing IMR-90 cells were rendered quiescent for 24 hr, at which time 4-HT was added to a final concentration of 1 μm. Cell extracts were prepared at different times (2–48 hr) after the addition of 4-HT, and the expression of GFPΔRaf-1:ER (A), p16Ink4a (B), p21Cip1 (C), cyclin D1 (D), p27Kip1 (E), and p42 MAP kinase (F) was assessed by Western blotting with the appropriate antisera as described in Materials and Methods. As a control, an equivalent volume of ethanol was added to the cells for 48 hr (48−).
Following activation of GFPΔRaf-1:ER, we observed induction of cyclin D1 expression as well as induced expression of the CDK inhibitors p16Ink4a and p21Cip1 (Fig. 3B–D, left and middle panels). The extent and kinetics of induction of these proteins was more rapid in cells expressing the more active [DD] form of GFPΔRaf-1:ER compared to cells expressing the [YY] form of the protein, such that maximal levels of p16Ink4a and p21Cip1 in particular were induced by the [DD] form after 8–16 hr whereas it required 32–48 hr of activity of the [YY] form to induce equivalent expression of these proteins.
In these experiments, Raf activation had little or no effect on the expression levels of either p53 (see Fig. 6A, below), p27Kip1, or p42 MAP kinase the last of which serves as a convenient loading control for the Western blots presented in Figure 3. In addition treatment of control IMR-90 cells (Babe) with 4-HT had no effect on the expression of cyclin D1, p16Ink4a, p27Kip1, or p21Cip1 (Fig. 3, right panels).
Figure 6.


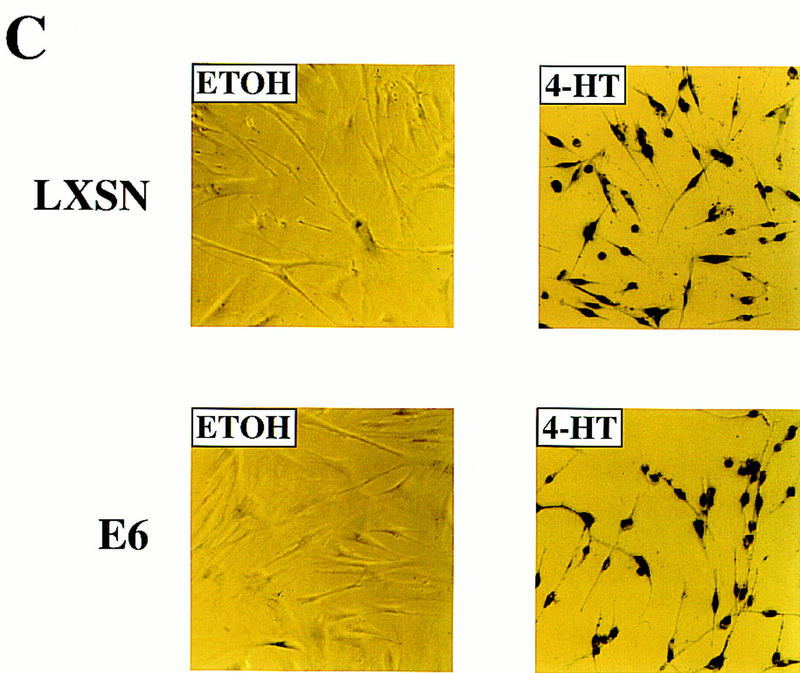
Raf-induced cell cycle arrest and senescence of IMR-90 cells in the absence of detectable p53 and p21Cip1. (A) Effect of the E6 oncoprotein on p53 and p21Cip1 expression. IMR-90 cells expressing GFPΔRaf-1[YY]:ER cells were infected with retroviruses encoding resistance to neomycin alone (LXSN) or expressing the E6 protein of HPV16 and resistance to neomycin (E6), and pooled populations of drug-resistant clones were established. Cells were then treated for 24 hr with either ethanol (lanes 1,3) or 1 μm 4-HT (lanes 2,4). Cell lysates were prepared, and the expression of p53, p21Cip1, and p42 MAP kinase was assessed by Western blotting with the appropriate antisera. (B) Raf-induced cell cycle arrest is unaffected by expression of HPV-16 E6 protein. IMR-90 cell populations described above expressing GFPΔRaf-1[YY]:ER in the absence (LXSN) or presence of the HPV16 E6 oncoprotein (E6) were treated with either ethanol or 4-HT for 4 days. Cells were labeled with BrdU for 3 hr prior to harvest, fixed, and stained with a FITC-conjugated anti-BrdU antibody and propidium iodide to detect S-phase cells. Cells were analyzed by use of a Becton-Dickinson FACscan to determine the percentage of cells in the G1, S, and G2/M phases of the cell cycle. (C) Raf-induced SA–β-galactosidase activity. IMR-90 cell populations described above expressing GFPΔRaf-1[YY]:ER in the absence (LXSN) or presence of the HPV16 E6 oncoprotein (E6) cells were treated with either ethanol or 4-HT for 6 days at which time the activity of SA–β-galactosidase was assessed as described in Materials and Methods.
Raf-induced cell cycle arrest is irreversible
Cell cycle arrest and senescence induced in response to prolonged passage of cells in culture is invariably an irreversible phenomenon (Campisi 1996, 1997). Because Raf-induced cell cycle arrest in NIH-3T3 cells is fully reversed following Raf inactivation (Samuels et al. 1993; Woods et al. 1997), we wished to determine whether the arrest and senescence phenotype observed in IMR-90 cells was irreversible. Control (Babe) and GFPΔRaf-1:ER ([YY] or [DD]) expressing IMR-90 cells were rendered quiescent in serum-free medium and then treated with either ethanol (solvent control) or 4-HT to activate GFPΔRaf-1:ER for 24 hr. At this time, cells were either untreated or washed extensively to remove 4-HT (to inactivate GFPΔRaf-1:ER) and then cultured in DMEM containing 10% FCS. The re-entry of cells into S phase was monitored over the following 6 days by the incorporation of [3H]thymidine into DNA.
Control (Babe) IMR-90 cells were unaffected by the addition of 4-HT to the culture media and proliferated as well as untreated cells (Fig. 4C). Cells expressing GFPΔRaf-1:ER failed to re-enter the cell cycle when maintained in the continuous presence of 4-HT (Fig. 4A,B) consistent with the ability of Raf to elicit cell cycle arrest in these cells (Fig. 1). Interestingly, cells in which the GFPΔRaf-1:ER was active for 24 hr and then inactivated also failed to proliferate upon culture in media containing 10% FCS for up to 6 days after the inactivation of GFPΔRaf-1:ER. These data indicate that Raf-induced cell cycle arrest in IMR-90 cells is irreversible.
Figure 4.
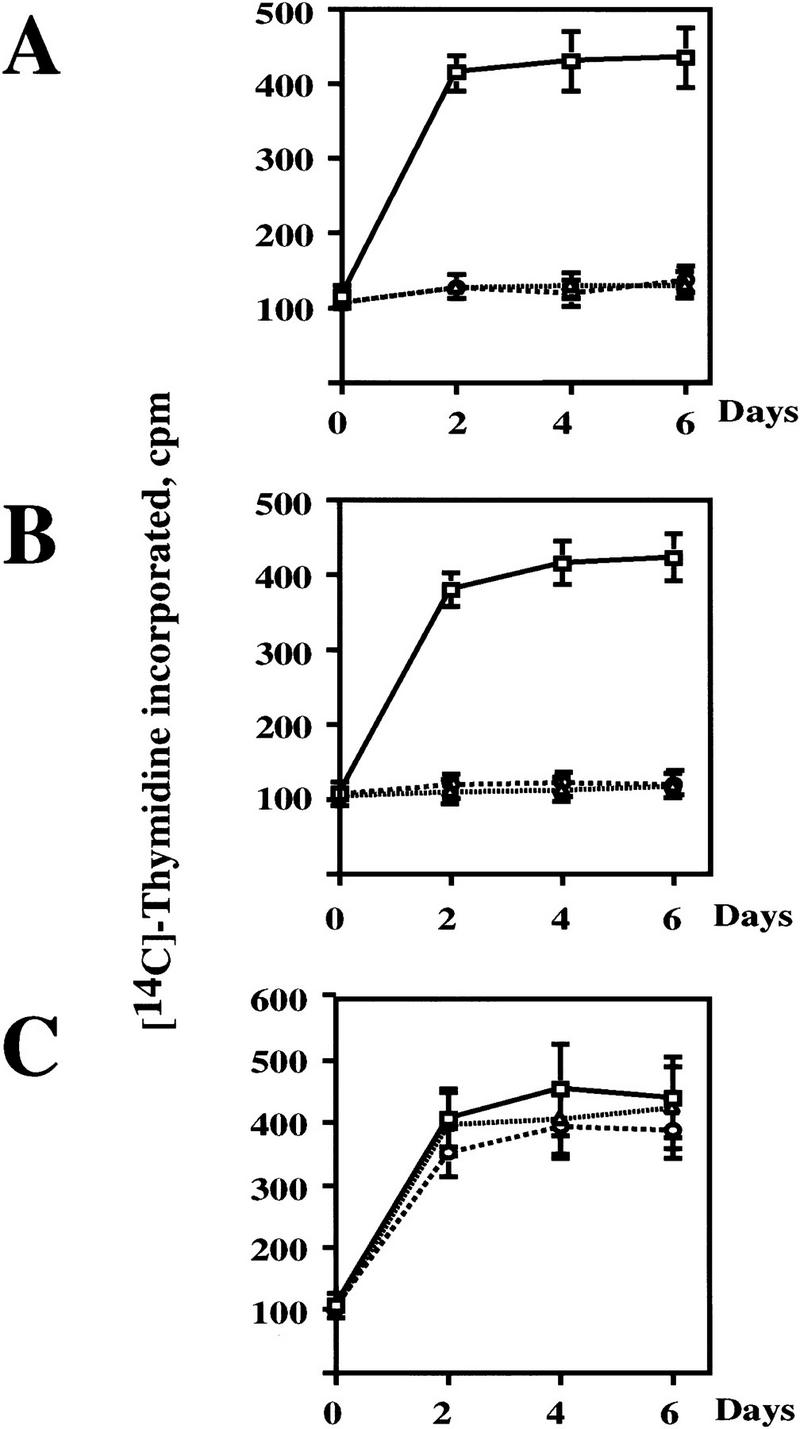
Raf-induced cell cycle arrest is irreversible. (A) GFPΔRaf-1[YY]:ER- and (B) GFPΔRaf-1[DD]:ER-expressing IMR-90 cells and (C) control cells (Babe) plated on Cytostar T plates were rendered quiescent in serum-free medium in the absence or presence of 1 μm 4-HT for 24 hr. At that time cells in the absence of 4-HT were stimulated with media containing 10% vol/vol FCS. Cells in the presence of 4-HT were either maintained in the continuous presence of 4-HT (4-HT continuous) or were washed extensively to remove the 4-HT (4-HT washed) prior to stimulation with media containing 10% vol/vol FCS. Cell proliferation was then measured over the following 6 days by the incorporation of methyl-[14C]thymidine into DNA. (□) 10% FCS; (▵) 4-HT continuous; (○) 4-HT washed.
To ensure that the inactivation of GFPΔRaf-1:ER did indeed lead to the inactivation of the MAP kinase pathway, we repeated the experiment described above in larger scale to analyze the effects of GFPΔRaf-1:ER inactivation on the activity of the MAP kinases and the expression of cyclin D1, p21Cip1, and p16Ink4a. Consistent with previous observations, withdrawal of 4-HT from IMR-90 cells expressing activated GFPΔRaf-1:ER led to the inactivation of the p42/p44 MAP kinases measured 24 or 48 hr after 4-HT withdrawal as assessed with phospho-specific antisera (P-MAPK, Fig. 5E). Consistent with the inactivation of the MAP kinases, the levels of p21Cip1 and cyclin D1 expression declined to basal levels following removal of 4-HT (Fig. 5B,C). However, the level of expression of p16Ink4a did not decrease after GFPΔRaf-1:ER inactivation, which may reflect the long half-life of both p16Ink4a mRNA and protein (Fig. 5A) (Hara et al. 1996). Equal loading of this Western blot was confirmed by reprobing with an antisera that recognizes p42 MAP kinase (Fig. 5D). These data indicate that continued GFPΔRaf-1:ER activity is required for maintained expression of cyclin D1 and p21Cip1 but is not required for the maintained expression of p16Ink4a.
Figure 5.
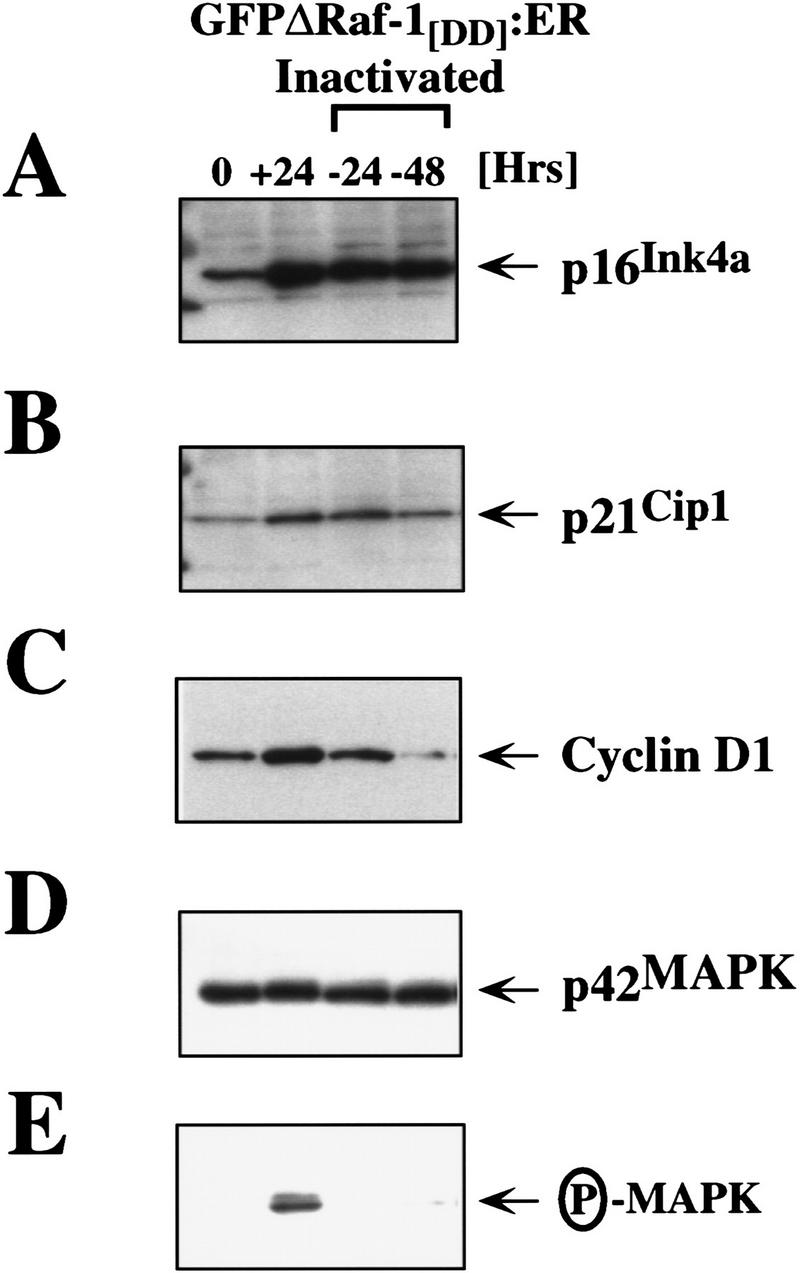
Sustained expression of p16Ink4a following inactivation of GFPΔRaf-1:ER. IMR-90 cells expressing GFPΔRaf-1[DD]:ER were either untreated (0) or treated with 1 μm 4-HT for 24 hr (all other samples). After 24 hr in 4-HT, (+24) cells were washed extensively and then cultured in the absence of 4-HT for a further 24 (−24) or 48 hr (−48). Cell extracts were prepared, and the expression of p16Ink4a (A), p21Cip1 (B), cyclin D1 (C), and p42 MAP kinase (D) and the activity of p42/p44 MAP kinases (E) was assessed by Western blotting with the appropriate antisera as described in Materials and Methods.
Neither p53 nor p21Cip1 is required for Raf-induced senescence in IMR-90 cells
Previous work has attributed the Raf-induced arrest of normal mouse embryo fibroblasts and primary rat Schwann cells to the induced expression of p21Cip1, which, under some circumstances, is mediated by p53 (Lloyd et al. 1997; Woods et al. 1997). To determine whether there is a role for p53 and p21Cip1 in Raf-induced cell cycle arrest and senescence, we used a second retrovirus vector to express the E6 oncoprotein of HPV16 in IMR-90 cells already expressing the [YY] form of GFPΔRaf-1:ER. Because E6 binds to p53 and facilitates its destruction by ubiquitin-mediated proteolysis, we anticipated that the expression of both p53 and p21Cip1 would be largely extinguished in these cells (Scheffner et al. 1990). As a control, we infected the same cells with a retrovirus encoding resistance to Neo alone (LXSN). Expression of HPV16 E6 in IMR-90 cells reduced both p53 and p21Cip1 to below detectable levels, and activation of GFPΔRaf-1:ER had no effect on the expression of either of these proteins (Fig. 6A, E6). The expression of p53 and p21Cip1 was readily detected in the control IMR-90 cell population (Fig. 6A, LXSN).
Activation of GFPΔRaf-1[YY]:ER in the absence of detectable p53 and p21Cip1 reduced the fraction of cells in S phase by 5- to 10-fold, similar to the response of cells that were not expressing E6 (Fig. 6B, LXSN and E6). Similar conclusions were reached by measurement of incorporation of methyl-[14C]thymidine into DNA (data not shown). Moreover, cells lacking p53 and p21Cip1 developed the SA–β-galactosidase marker of senescence and the characteristic cell morphology that is described above (Fig. 6C). We conclude that Raf-induced alterations in cell morphology, proliferative arrest, and senescence in IMR-90 cells do not require the action of either p53 or p21Cip1.
Overexpression of p16Ink4a elicits senescence
To explore the ability of p16Ink4a to induce senescence in cells, we infected parental IMR-90 cells with a retrovirus engineered to constitutively express p16Ink4a. Consistent with previous observations (McConnell et al. 1998), IMR-90 cells expressing p16Ink4a became positive for SA–β-galactosidase activity (Fig. 7A) whereas cells infected with a control retrovirus were negative. FACscan analysis revealed a threefold reduction in S-phase cells compared with the control infected populations (43% vs. 13.9%; Fig. 7B). We conclude that the sustained overexpression of p16Ink4a can arrest proliferation and elicit senescence in normal human fibroblasts and may be sufficient to explain the induction of senescence by Raf.
Figure 7.
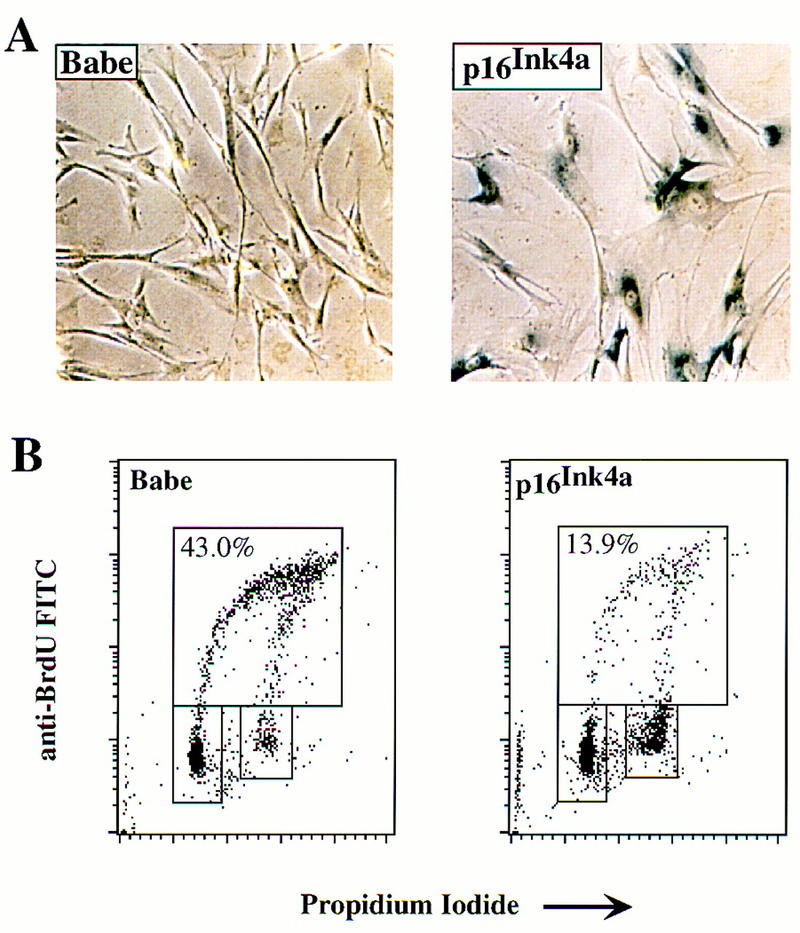
Cell cycle arrest and induction of senescence by p16Ink4a. IMR-90 cells were infected with a retroviral vector expressing either the selectable marker for puromycin resistance alone (Babe) or the marker plus human p16Ink4a as indicated. The infected cells were then subjected to selection for 6 days. Surviving cells were analyzed for SA–β-galactosidase activity (A) or labeled with BrdU for analysis by FACscan for relative content of DNA (B). (B) Upper boxes delineate cells in S phase; lower boxes delineate cells in G0/G1 (left) or G2/M (right). Numbers in upper boxes indicate percentage of cells in S phase.
Inhibition of the MAP kinase cascade blocks the induction of senescence by Raf
Signaling by Raf is executed, at least in part, through the MEK/MAP kinase pathway (Marshall 1994; Samuels et al. 1993). Therefore, we sought to implicate this enzyme cascade in Raf-induced cell cycle arrest and senescence by using PD098059 (PD), a specific and selective inhibitor of MEK1 (Alessi et al. 1995). As described above, activation of GFPΔRaf-1[YY]:ER led to the phosphorylation and activation of the p42/p44 MAP kinases (Fig. 8A). In the presence of PD, however, phosphorylation of the MAP kinases was reduced to levels comparable to those found in uninduced IMR-90 cells (Fig. 8A, cf. 4-HT with 4-HT+PD). Furthermore, the basal levels of MAP kinase phosphorylation in the absence of Raf activation were also reduced following treatment of cells with PD (Fig. 8A, cf. ETOH with PD). The levels of p16Ink4a mirrored the phosphorylation of the MAP kinases (Fig. 8A), as if one or more of the MAP kinases might control the expression of p16Ink4a.
Figure 8.
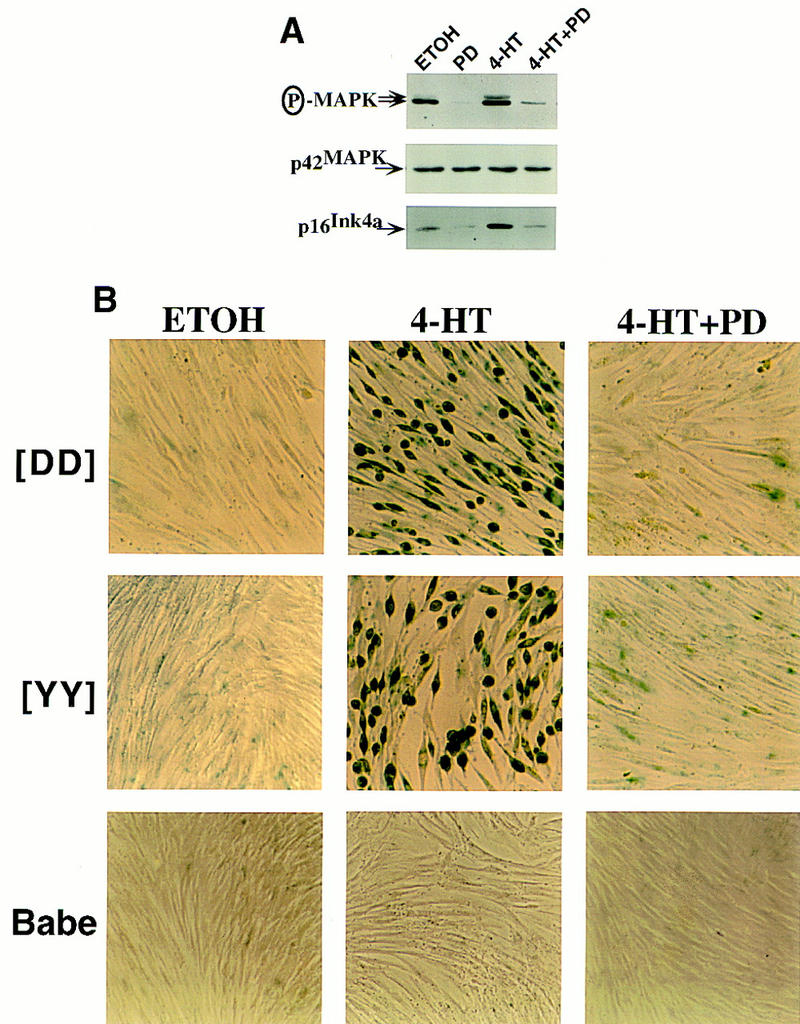
MEK activity is required for induction of p16Ink4a and senescence by Raf in IMR-90 cells. (A) Inhibition of MAP kinase activation and p16Ink4a expression by PD098059. Asynchronously growing IMR-90 cells expressing GFPΔRaf-1[YY]:ER were treated with either 10 nm 4-HT or an equivalent volume of ethanol (ETOH) for 4 days in the absence (DMSO) or presence of the MEK inhibitor PD098059 (PD, 25 μm) as indicated. The activity of p42/p44 MAP kinase and the expression of p42 MAP kinase and p16Ink4a were assessed by Western blotting with the appropriate antisera. (B) MEK activity is required for Raf-induced senescence in IMR-90 cells. Control (Babe) and GFPΔRaf-1:ER ([YY]- and [DD])-expressing IMR-90 cells were treated with various combinations of 4-HT (10 nm for [DD] and 3 nm for [YY]), PD (25 μm), or an equivalent volume of ethanol or DMSO for 6 days, and stained for SA–β-galactosidase activity.
Next we assessed the effect of PD on cellular proliferation. In the absence of activated Raf, inhibition of the MAP kinases reduced the fraction of cells in S phase by sixfold (Table 2). This result was in accord with the elimination of MAP kinase phosphorylation described above and with previous results (Alessi et al. 1995). When Raf was activated, however, the presence of PD largely prevented the inhibition of proliferation that would otherwise occur (Table 2). These results suggest that cell cycle arrest elicited by Raf is mediated by the MEK/MAP kinase cascade.
Table 2.
Prevention of Raf-induced arrest by inhibition of MEK
| Cells
|
4-HT (nm)
|
PD98059 (μm)
|
G1 (%)
|
G2/M (%)
|
S (%)
|
|---|---|---|---|---|---|
| Babe | 0 | 0 | 40.9 | 17.7 | 41.4 |
| 10 | 0 | 40.2 | 17.1 | 42.7 | |
| 10 | 25 | 67.1 | 26.0 | 6.9 | |
| GFPΔRaf-1[YY]:ER | 0 | 0 | 40.9 | 13.4 | 45.7 |
| 10 | 0 | 68.6 | 24.5 | 6.8 | |
| 10 | 25 | 50.4 | 16.1 | 33.5 |
Control (Babe) and GFPΔRaf-1[YY]:ER-expressing IMR-90 cells were treated with 10 nm 4-HT in the absence or presence of 25 μm of the MEK inhibitor PD098059 for 4 days and labeled with 15 μm BrdU for 3 hr. Ethanol-fixed cells were stained with FITC-anti-BrdU and with propidium iodide; and the percentage of cells in the different phases of the cell cycle was assessed by flow cytometry.
The presence of PD also prevented the appearance of SA–β-galactosidase activity and the anticipated morphological changes in response to the activation of GFPΔRaf-1:ER (Fig. 8B). The blockade was effective against both GFPΔRaf-1[YY]:ER and GFPΔRaf-1[DD]:ER induced with optimal concentrations of 4-HT. Neither 4-HT alone nor 4-HT in combination with PD had any morphological effect on the puromycin-resistant control IMR-90 cell population (Fig. 8B, Babe). Therefore, we conclude that activation of MEK is required for the arrest of proliferation and the induction of senescence triggered by Raf in IMR-90 cells. The results also reveal a pleiotropic response to MAP kinase activity that is dose dependent. As observed previously, there is an optima of activity that permits cellular proliferation (Woods et al. 1997). Either a deficit or a surfeit of the activity leads to cell cycle arrest, but only a surfeit elicits senescence.
Discussion
Activation of Raf elicits senescence in human fibroblasts
Previous work has indicated that Ras and Raf oncoproteins can elicit cell cycle arrest in both primary and immortalized cells (Lloyd et al. 1997; Woods et al. 1997). In the IMR-90 strain of human fibroblasts, Ras oncoproteins can elicit premature senescence (Serrano et al. 1997). Here we show that activation of Raf has a similar effect. Using a complementary approach with constitutively active forms of Ras and MEK, others have reached similar conclusions (Lin et al. 1998). The phenotypic response of IMR-90 cells to Raf displays many of the hallmarks of senescence: irreversible withdrawal from the cell division cycle; assumption of a characteristic morphology; retention of viability; and induction of an enzymatic marker for senescence, acidic β-galactosidase. However, a number of interesting differences were observed between the senescent phenotype elicited by Raf and that induced either by Ras or by prolonged passage in culture. Senescent cells are typically arrested in the G1 phase of the cell cycle (Hayflick and Moorhead 1961; Campisi 1996, 1997). In contrast, only a portion of IMR-90 cells rendered senescent by Raf are arrested in G1; the remainder appear to be in G2/M. In addition, Ras-induced senescence is characterized by a distinctive flattened cell phenotype that is in marked contrast to the rounded, refractile morphology of IMR-90 cells rendered senescent by Raf. This distinction may reflect the ability of Ras to influence pathways in addition to those activated by Raf such as PI3′ kinase, Ral–GDS (guanine-nucleotide dissociation stimulator), and members of the Rho family of GTPases that are known to play an integral role in the control of cell shape. Such differences between the effects of Ras and Raf on cell morphology have been observed in primary rat cardiac myocytes and bear further examination (Thorburn et al. 1994).
Senescent cells typically express high levels of p53, p21Cip1, and p16Ink4a (Zindy et al. 1997; Reznikoff et al. 1996; McConnell et al. 1998). Previous work has indicated that, in rodent cells, p21Cip1 is a major mediator of cell cycle arrest in response to either H-Ras or Raf (Lloyd et al. 1997; Woods et al. 1997). For example, Raf cannot arrest proliferation in mouse fibroblasts that are deficient in p21Cip1, whereas it arrests both primary and immortalized cells that lack p16Ink4a (D. Woods and M. McMahon, unpubl.). Furthermore, T antigen, a dominant-negative allele of p53, and expression of antisense p21Cip1 RNA (all of which impede the expression of p21Cip1) prevented Raf induced cell cycle arrest in primary rat Schwann cells (Lloyd et al. 1997). In contrast, p21Cip1 appears to be much less prominent in the response of IMR-90 cells to either Ras or Raf. First, Raf consistently induced high levels of p16Ink4a in IMR-90 cells, but did not induce high levels of either p53 or p21Cip1. Indeed there was no detectable induction of p53, and the induction of p21Cip1 showed significant experimental variation. Second, a dominant-negative allele of p53 failed to block the senescent response to H-Ras (data not shown). Third, Raf efficiently elicited cell cycle arrest and senescence in IMR-90 cells in which the levels of p53 and p21Cip1 were greatly reduced by the action of the E6 oncoprotein of HPV16.
Previous evidence suggested a role for p16Ink4a in the genesis of cellular senescence: The protein accumulates to high levels as cells approach senescence, and in many lines of cells that have escaped senescence, the INK4A gene is either defective, deleted, or inactivated by hypermethylation (Merlo et al. 1995; Reznikoff et al. 1996; Serrano et al. 1996; Foulkes et al. 1997; Loughran et al. 1997). The results described here add to that evidence by implicating p16Ink4a in the senescence induced by Raf in IMR-90 cells. First, induction by Raf produces a substantial rise in p16Ink4a that is coincident with the onset of cell cycle arrest and precedes the onset of senescence. Moreover, the irreversible nature of Raf-induced senescence is reflected by the fact that p16Ink4a levels remain elevated even after the inactivation of the signal from Raf. Finally, the ectopic overexpression of p16Ink4a is itself sufficient to elicit proliferative arrest and senescence in IMR-90 cells.
The locus that encodes p16Ink4a also encodes a second protein known as p19ARF. These two proteins share a common second exon but are entirely distinct in their amino acid sequences (Quelle et al. 1995). The belated discovery of the tumor suppressor properties of p19ARF have, at least temporarily, obscured the role of p16Ink4a in both normal cellular processes and tumorigenesis (Kamijo et al. 1997; Pomerantz et al. 1998; Zhang et al. 1998). Our results demonstrate that p16Ink4a itself can elicit senescence in human fibroblasts, but do not speak to the biological activities of p19ARF. Nor do our results address a possible role for other members of the INK4 family of CDK inhibitors. It is clear however that p16Ink4a is not unique amongst the CDK inhibitors in its ability to induce senescence in cells. Ectopic expression of p15Ink4b, p21Cip1, and p27Kip1 elicited proliferative arrest, senescence, and SA-β-gal activity in TIG-3 primary human fibroblasts (McConnell et al. 1998). It seems likely that the inhibition of cyclin–CDK complexes by such inhibitors triggers a senescence program of which the mechanism remains poorly defined.
The phenotypic response to Raf is determined by signal strength
Previous reports have indicated that the same intracellular signaling pathway can elicit different phenotypic outcomes, determined by the relative strength of that signal (Marshall 1995; Woods et al. 1997). The results presented here reveal a pleiotropic response to Raf activation that is similarly dose dependent. In normal proliferating cells Raf/MEK/MAP kinase activity is presumably maintained by the presence of external growth factors (Fig. 8A). Interruption of the signal from MEK to MAP kinase (with PD) arrests cellular proliferation; thus, MAP kinase activity is required for initiation and maintenance of the cell division cycle, as reported previously (Alessi et al. 1995). In contrast, activation of oncogenic Raf gave rise to a sustained elevation of MAP kinase activity, resulting in both the arrest of cell division and senescence. In this latter setting, reduction of MAP kinase activity to moderate levels with PD allowed the cells to continue proliferating (Fig. 8A; Table 2).
The dose-dependent response to Raf/MEK/MAP kinase activation may be explained, at least in part, by the differential sensitivity of cyclin D1 and p16Ink4a expression to activation of the MAP kinase pathway. Both in this study (Fig. 4D) and in a previously published report (Woods et al. 1997), cyclin D1 accumulation, which serves to promote cell cycle progression, appears to be activated more rapidly and in response to lower levels of signaling through the MAP kinase pathway compared with the CDK inhibitors p21Cip1 and p16Ink4a, which appear to require a stronger signal sustained over a prolonged period of time. However, once this latter threshold is breached, it is likely that the induced p16Ink4a inhibits cdk4 even in the presence of elevated cyclin D1 (Fig. 3B,D). It is not immediately apparent why p16Ink4a induction is accompanied by other manifestations of senescence, although it is possible that inhibition of E2F transcription factors, as a consequence of Rb hypophosphorylation, may be involved in modulating aspects of the senescence phenotype. It will ultimately be of considerable interest to dissect the biochemical pathways that lead from the activation of Raf to the induced expression of p16Ink4a and to determine whether these signaling pathways play a more general role in the induction of senescence in other contexts such as in response to prolonged passage in culture or to alterations in the properties of telomeres (van Steensel et al. 1998).
It is now well established that both qualitative and quantitative aspects of Raf/MEK/MAP kinase activation play a crucial role in determining the biological outcome of signaling through this pathway. In both human and mouse cells, depending on the level of activation, the MAP kinase pathway can be employed to elicit strikingly different biological outcomes, namely cell proliferation, cell cycle arrest, terminal differentiation, and senescence (Lloyd et al. 1997; Woods et al. 1997). The biochemical mechanisms underlying these disparate observations require further elucidation. Whatever its mechanism, induction of senescence could provide a defense against neoplastic transformation when Ras, Raf, or the downstream MAP kinase cascade is inappropriately active. In this regard, senescence may be an alternative to apoptosis as a defense against tumorigenesis (Campisi 1996, 1997; Serrano et al. 1997).
Materials and methods
Cell culture and virus infection
IMR-90 cells were obtained from the American Tissue Culture Collection and propagated either in phenol red-free MEM or DMEM as described previously (Samuels et al. 1993). Retrovirus vectors for the expression of GFPΔRaf-1:ER and resistance to puromycin have been described previously (Morgenstern and Land 1990; Woods et al. 1997). A cDNA encoding human p16Ink4a was subcloned into the pBabe retroviral vector that carries puromycin resistance as a selectable marker (Morgenstern and Land 1990). The E6 oncogene of HPV16 was subcloned into the LXSN retroviral vector, which also encodes resistance to neomycin as a selectable marker (Miller and Rosman 1989).
By use of a calcium phosphate protocol, retroviral vectors were packaged in the Phoenix A cell line from G. Nolan (Stanford University, CA) producing nonreplicating forms of amphotrophic virus (Pear et al. 1993; Kinsella and Nolan 1996). IMR-90 cells were infected with viral supernatants as described previously (Samuels et al. 1993). Pooled populations of drug-resistant cells expressing equivalent levels of GFPΔRaf-1:ER were isolated by FACs (Woods et al. 1997).
The proliferative capacity of cells was monitored by seeding of ∼1000 cells in 12-mm diameter wells in MEM containing 15% vol/vol fetal calf serum (FCS). 4-HT in ethanol or an equivalent volume of ethanol was added the next day. Cells were harvested by trypsination at intervals of 24 hr, stained with 0.1% trypan blue, and viable cells enumerated by use of a hemocytometer. Four wells of cells were counted separately and the results averaged. Cells were stained for the activity of the SA–β-galactosidase as described previously (Dimri et al. 1995).
DNA synthesis in IMR-90 cells was measured by plating of cells in Cytostar-T 96-well scintillating microplates and labeling with methyl-[14C]thymidine according to the manufacturer’s instructions (Amersham Life Science). Incorporation of methyl-[14C]thymidine was estimated by use of a Packard top-count scintillation machine as described previously (Woods et al. 1997).
Analysis of cell extracts by Western blotting
Cell extracts were prepared in ELB lysis buffer (50 mm HEPES at pH 7.0, 150 mm NaCl, and 0.1% (vol/vol, NP-40) containing a standard cocktail of protease and phosphatase inhibitors (Samuels et al. 1993). Aliquots (50 μg) of cell extract, separated by polyacrylamide gel electrophoresis, were analyzed by probing of Western blots with the appropriate antisera. Mouse monoclonal and rabbit polyclonal antibodies reactive against human proteins were as follows: monoclonal anti-p21Cip1 (Ab-1, Calbiochem); monoclonal anti-p16Ink4a (Ab-1 and Ab-2, NeoMarkers); monoclonal anti-p53 (Mixture of DO.1, Santa Cruz Biotechnology and Ab-1, Calbiochem); monoclonal anti-p27Kip1 (Clone 57, Transduction Labs); polyclonal anti-Erk2 (C14, Santa Cruz Biotechnology); polyclonal anti-hbER (HC-20, Santa Cruz Biotechnology); polyclonal anti-cyclin D1 (gift of Dave Parry and Emma Lees, DNAX), and polyclonal phosphospecific anti-p42/p44 MAP kinase (Thr-202/Tyr-204, New England Biolabs). Horseradish peroxidase-conjugated anti-mouse and anti-rabbit immunoglobulins were from Amersham and used at a dilution of 1: 5000. Western blots were developed with the SuperSignal ULTRA chemiluminescence detection kit (Pierce).
Analysis of IMR-90 cell cycle phase by BrdU labeling
Cells were replated below confluence 1 day prior to analysis for optimal cell cycle distribution and were labeled with BrdU (15 μm) 3 hr prior to harvest. Single nuclei suspensions derived from ethanol-fixed cells were washed and costained with an anti-BrdU-FITC conjugated antibody (Boehringer Mannheim) and 20 μg/ml propidium iodide prior to analysis with a Becton-Dickinson FACscan as described previously (Takagi et al. 1993).
Acknowledgments
We thank the members of the McMahon and Bishop laboratories for useful comments and suggestions. We are particularly grateful to Emma Lees and Dave Parry for the provision of antisera and for stimulating discussion. We thank Scott Lowe for communicating results prior to publication, Liang Zhu for the provision of the p16Ink4a cDNA, and Ken Aldape for assistance with FACscan analysis. We are grateful for the support of Gwen Jewell, Gary Burget, and Maribel Andonian in the preparation of the manuscript and figures. Work here was supported by the National Institutes of Health (CA 44338-12) and by funds from the G.W. Hooper Foundation. DNAX Research Institute is supported by Schering Plough Corporation.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL mcmahon@cc.ucsf.edu; FAX (415) 502-3179.
References
- Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16 (Ink4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci. 1996;93:13742–13747. doi: 10.1073/pnas.93.24.13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Avruch J, Zhang XF, Kyriakis JM. Raf meets Ras: Completing the framework of a signal transduction pathway. Trends Biochem Sci. 1994;19:279–283. doi: 10.1016/0968-0004(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- Brown JP, Wei W, Sedivy JM. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science. 1997;277:831–834. doi: 10.1126/science.277.5327.831. [DOI] [PubMed] [Google Scholar]
- Campisi J. Replicative senescence: An old lives’ tale? Cell. 1996;84:497–500. doi: 10.1016/s0092-8674(00)81023-5. [DOI] [PubMed] [Google Scholar]
- ————— The biology of replicative senescence. Eur J Cancer. 1997;33:703–709. doi: 10.1016/S0959-8049(96)00058-5. [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Foulkes WD, Flanders TY, Pollock PM, Hayward NK. The CDKN2A (p16) gene and human cancer. Mol Med. 1997;3:5–20. [PMC free article] [PubMed] [Google Scholar]
- Hainaut P, Soussi T, Shomer B, Hollstein M, Greenblatt M, Hovig E, Harris CC, Montesano R. Database of p53 gene somatic mutations in human tumors and cell lines: Updated compilation and future prospects. Nucleic Acids Res. 1997;25:151–157. doi: 10.1093/nar/25.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M, Peters G. Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv Cancer Res. 1996;68:67–108. doi: 10.1016/s0065-230x(08)60352-8. [DOI] [PubMed] [Google Scholar]
- Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regulation of p16CDKN2 expression and its implications for cell immortalization and senescence. Mol Cell Biol. 1996;16:859–867. doi: 10.1128/mcb.16.3.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- Hunter T. Cooperation between oncogenes. Cell. 1991;64:249–270. doi: 10.1016/0092-8674(91)90637-e. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- Kinsella TM, Nolan GP. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum Gene Ther. 1996;7:1405–1413. doi: 10.1089/hum.1996.7.12-1405. [DOI] [PubMed] [Google Scholar]
- Kulju KS, Lehman JM. Increased p53 protein associated with aging in human diploid fibroblasts. Exp Cell Res. 1995;217:336–345. doi: 10.1006/excr.1995.1095. [DOI] [PubMed] [Google Scholar]
- Lees E. Cyclin dependent kinase regulation. Curr Biol. 1995;7:773–780. doi: 10.1016/0955-0674(95)80060-3. [DOI] [PubMed] [Google Scholar]
- Lin, A.W., M. Baradas, J.C. Stone, L. van Aelst, M. Serrano, and S.W. Lowe. 1998. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK signaling. Genes & Dev. (this issue). [DOI] [PMC free article] [PubMed]
- Lloyd AC, Obermuller F, Staddon S, Barth CF, McMahon M, Land H. Cooperating oncogenes converge to regulate cyclin/cdk complexes. Genes & Dev. 1997;11:663–677. doi: 10.1101/gad.11.5.663. [DOI] [PubMed] [Google Scholar]
- Loughran O, Clark LJ, Bond J, Baker A, Berry IJ, Edington KG, Ly IS, Simmons R, Haw R, Black DM, Newbold RF, Parkinson EK. Evidence for the inactivation of multiple replicative lifespan genes in immortal human squamous cell carcinoma keratinocytes. Oncogene. 1997;14:1955–1964. doi: 10.1038/sj.onc.1201028. [DOI] [PubMed] [Google Scholar]
- Marais R, Light Y, Paterson H, Marshall CJ. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 1995;14:101–110. doi: 10.1002/j.1460-2075.1995.tb07316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CJ. MAP kinase kinase kinase, MAP kinase kinase and MAP kinase. Curr Opin Genet Dev. 1994;4:82–89. doi: 10.1016/0959-437x(94)90095-7. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- ————— Ras effectors. Curr Opin Cell Biol. 1996;8:197–204. doi: 10.1016/s0955-0674(96)80066-4. [DOI] [PubMed] [Google Scholar]
- McConnell BB, Starborg M, Brookes S, Peters G. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr Biol. 1998;8:351–354. doi: 10.1016/s0960-9822(98)70137-x. [DOI] [PubMed] [Google Scholar]
- Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, Ziaugra L, Beijersbergen RL, Davidoff MJ, Liu Q, Bacchetti S, Haber DA, Weinberg RA. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- Miller AD, Rosman GJ. Improved retroviral vectors for gene transfer and expression. BioTechniques. 1989;7:980–990. [PMC free article] [PubMed] [Google Scholar]
- Morgenstern JP, Land H. Advanced mammalian gene transfer: High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res. 1994;211:90–98. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- O’Shea CC, Crompton T, Rosewell IR, Hayday AC, Owen MJ. Raf regulates positive selection. Eur J Immunol. 1996;26:2350–2355. doi: 10.1002/eji.1830261012. [DOI] [PubMed] [Google Scholar]
- Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomerantz J, Schreiber-Agus N, Liégeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee HW, Cordon-Cardo C, DePinho RA. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 1998;92:713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Reznikoff CA, Yeager TR, Belair CD, Savelieva E, Puthenveettil JA, Stadler WM. Elevated p16 at senescence and loss of p16 at immortalization in human papillomavirus 16 E6, but not E7, transformed human uroepithelial cells. Cancer Res. 1996;56:2886–2890. [PubMed] [Google Scholar]
- Samuels ML, Weber MJ, Bishop JM, McMahon M. Conditional transformation of cells and rapid activation of the mitogen-activated protein kinase cascade by an estradiol-dependent human raf-1 protein kinase. Mol Cell Biol. 1993;13:6241–6252. doi: 10.1128/mcb.13.10.6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- Stanton VJ, Nichols DW, Laudano AP, Cooper GM. Definition of the human raf amino-terminal regulatory region by deletion mutagenesis. Mol Cell Biol. 1989;9:639–647. doi: 10.1128/mcb.9.2.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugrue MM, Shin DY, Lee SW, Aaronson SA. Wild-type p53 triggers a rapid senescence program in human tumor cells lacking functional p53. Proc Natl Acad Sci. 1997;94:9648–9653. doi: 10.1073/pnas.94.18.9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi S, McFadden ML, Humphreys RE, Woda BA, Sairenji T. Detection of 5-bromo-2-deoxyuridine (BrdUrd) incorporation with monoclonal anti-BrdUrd antibody after deoxyribonuclease treatment. Cytometry. 1993;14:640–648. doi: 10.1002/cyto.990140608. [DOI] [PubMed] [Google Scholar]
- Thorburn J, McMahon M, Thorburn A. Raf-1 kinase activity is necessary and sufficient for gene expression changes but not sufficient for cellular morphology changes associated with cardiac myocyte hypertrophy. J Biol Chem. 1994;269:30580–30586. [PubMed] [Google Scholar]
- Treisman R. Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol. 1996;8:205–215. doi: 10.1016/s0955-0674(96)80067-6. [DOI] [PubMed] [Google Scholar]
- van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92:401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol. 1997;17:5598–5611. doi: 10.1128/mcb.17.9.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15:203–211. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]