

Abstract
Oncogenic Ras transforms immortal rodent cells to a tumorigenic state, in part, by constitutively transmitting mitogenic signals through the mitogen-activated protein kinase (MAPK) cascade. In primary cells, Ras is initially mitogenic but eventually induces premature senescence involving the p53 and p16INK4a tumor suppressors. Constitutive activation of MEK (a component of the MAPK cascade) induces both p53 and p16, and is required for Ras-induced senescence of normal human fibroblasts. Furthermore, activated MEK permanently arrests primary murine fibroblasts but forces uncontrolled mitogenesis and transformation in cells lacking either p53 or INK4a. The precisely opposite response of normal and immortalized cells to constitutive activation of the MAPK cascade implies that premature senescence acts as a fail-safe mechanism to limit the transforming potential of excessive Ras mitogenic signaling. Consequently, constitutive MAPK signaling activates p53 and p16 as tumor suppressors.
Keywords: MAPK signaling, premature senescence, MEK, tumor suppression, p53, p16
Cancer arises through the accumulation of genetic changes that each enhances the growth or survival of developing tumor cells (Fearon and Vogelstein 1990). Perhaps the simplest experimental model of this multistep process involves transformation of primary cultures by ras oncogenes (Weinberg 1989; Ruley 1990). Oncogenic Ras transforms most immortal rodent cells to a tumorigenic state, whereas transformation of primary cells requires either a cooperating oncogene (e.g., E1A) or the inactivation of tumor suppressors such p53 or INK4a (Gallimore et al. 1986; Tanaka et al. 1994; Serrano et al. 1996). Importantly, these transforming interactions have been validated in animal models and, in several instances, the genetic changes that cooperate with ras in primary cells are comutated with ras in spontaneous tumors (Fearon and Vogelstein 1990; Burns et al. 1991; Kemp et al. 1993; Linardopoulos et al. 1995; Chin et al. 1997).
Despite the widespread use of oncogene cooperation assays to model genetic interactions during tumorigenesis, a biological explanation for this phenomenon is only now emerging. Primary rodent and human cells undergo a limited number of cell doublings in culture and then undergo a process of cellular senescence (Hayflick 1965). When expressed alone, most oncogenes that cooperate with ras in transformation extend cellular life span in human cells, and facilitate the establishment of primary rodent cells into immortal cell lines (Ruley 1990). This observation suggested that oncogenic Ras requires ‘immortalizing’ changes to promote oncogenic transformation; these could occur spontaneously in immortal cell lines or be provided by a cooperating oncogene (for review, see Weinberg 1997). At the same time, other studies suggested that normal cells actively resist transformation by Ras (Franza et al. 1986; Hirakawa and Ruley 1988), implying that cooperating oncogenes interfere with natural anti-oncogenic defenses.
We demonstrated recently that prolonged expression of oncogenic Ras induces a permanent cell-cycle arrest in primary human and rodent fibroblasts that is phenotypically indistinguishable from cellular senescence (Serrano et al. 1997). Consequently, these studies explain the requirement for immortalizing changes in ras transformation assays, and identify a mechanism whereby normal cells actively counter ras’ transforming potential. For example, because immortal rodent cell lines have already lost aspects of the senescence program, they are readily transformed by oncogenic Ras alone. Similarly, immortalizing oncogenes disrupt the senescence program retained in primary cells; hence, such changes interfere with the ability of oncogenic Ras to provoke senescence. In either case, cells escape normal growth controls that actively limit transformation by Ras. Consequently, premature senescence may function as a bona fide mechanism of tumor suppression (Serrano et al. 1997; for review, see Weinberg 1997).
Consistent with this view, p53 and p16INK4a appear critical for premature senescence induced by oncogenic Ras (Serrano et al. 1997). Both p53 and INK4a are mutated at high frequency in many tumor types, implying that their action is central to tumor development. Nevertheless, the precise circumstances in which either is engaged to function as tumor suppressors remain poorly understood. p53 can promote cell-cycle arrest or apoptosis in response to a variety of cellular stresses, including DNA damage and hypoxia (Kastan 1993; Graeber et al. 1996). In both settings, cells acquiring p53 mutations have a selective advantage over their p53-normal counterparts. p16 increases gradually as cells proceed towards senescence (Alcorta et al. 1996; Zindy et al. 1997). Consequently, loss of p16 might uncouple the cell-cycle machinery from a cell-doubling clock, allowing developing tumor cells to proliferate beyond their normal life span. The fact that oncogenic Ras activates p53 and p16 provides an additional explanation for their action as tumor suppressors: p53 and p16 act in a compensatory mechanism that suppresses Ras-induced transformation. Hence, cells acquiring p53 and INK4a mutations would tolerate ras mutations occurring early in tumor development (Serrano et al. 1996).
In immortal rodent lines, transformation by oncogenic Ras involves its ability to bind and activate a series of effector proteins, including Raf-1, phosphoinositide 3-OH kinase [PI(3)K], and Ral.GDS (Van Aelst et al. 1993; Rodriguez-Viciana et al. 1994; Spaargaren and Bischoff 1994). Each of these proteins, in turn, activates distinct downstream targets, thereby producing different aspects of the transformed phenotype (for review, see Katz and McCormick 1997). For example, the Ras–Raf interaction initiates the mitogen-activated protein kinase (MAPK) cascade, which involves the sequential activation of a series of protein kinases that transmit mitogenic signals to nuclear transcription factors (Chen et al. 1992; Gille et al. 1995). These kinases include Raf-1, the MEKs (MEK1 and MEK2), and the MAPKs (ERK1 and ERK2). In contrast, the ability of Ras to activate PI(3)K promotes membrane ruffling (Joneson et al. 1996; Rodriguez-Viciana et al. 1997), perhaps through Rac and Rho (for review, see Van Aelst and D’Souza-Schorey 1997), and may also suppress apoptosis through activation of Akt/PKB (for review, see Downward 1998). Finally, the Ral.GDS proteins act as exchange factors that can activate the Ral family of small GTPases (Spaargaren and Bischoff 1994) which, in turn, can regulate phospholipase D (Jiang et al. 1995). Although each of these effector pathways contributes to the transforming activity of Ras in immortal rodent fibroblasts (Khosravi-Far et al. 1995, 1996; White et al. 1995; Urano et al. 1996; Rodriguez-Viciana et al. 1997), activation of the MAPK cascade is clearly sufficient (Cowley et al. 1994; Mansour et al. 1994; Bottorff et al. 1995; Stang et al. 1997).
Almost all of the studies examining Ras signaling in mammalian systems have utilized immortal or tumor-derived lines harboring unknown genetic alterations. Because premature senescence induced by Ras is restricted largely to nonimmortal cells (Serrano et al. 1997), Ras might signal premature senescence through an unidentified pathway lost during the immortalization process. Alternatively, Ras could promote premature senescence through a known effector pathway or by activating a combination of effector pathways. In the current study, we identified the effector pathway responsible for Ras-induced senescence. We find that Ras induces senescence in normal cells through the primary mechanism whereby it forces uncontrolled proliferation and transformation in immortal cell lines. These results have important implications for our understanding of oncogene cooperation, tumor suppression, and the regulation of cellular senescence.
Results
Oncogenic Ras is initially mitogenic in normal diploid human fibroblasts
Microinjection of oncogenic ras into human diploid fibroblasts induces S-phase entry (Lumpkin et al. 1986). On the other hand, retroviral transduction of oncogenic ras into human fibroblasts provokes permanent cell-cycle arrest (Serrano et al. 1997). These observations raise the possibility that the initial effect of Ras is forced proliferation and, only later, cell-cycle arrest. Therefore, the immediate and long-term consequences of oncogenic Ras expression were examined in the same population of normal diploid human IMR90 fibroblasts. In this and subsequent experiments, oncogenic ras (H-RasV12) was introduced into whole-cell populations using high-titer retroviral vectors coexpressing a selectable marker. After a brief selection to eliminate uninfected cells, cells were plated for all assays described below (designated day −1). Using this protocol, the percentage of transduced cells ranged between 70% and 90% as estimated in parallel infection with viruses expressing a lacZ reporter (data not shown).
To examine the proliferation properties of Ras-expressing populations, [3H]thymidine and BrdU incorporation were measured shortly after ras transduction (48–72 hr postinfection) in the presence and absence of serum. In normal growth conditions (10% serum), vector and Ras-expressing cells incorporated similar amounts of [3H]thymidine (Fig. 1A). As expected, transfer of vector-containing cells to low serum for 24 hr resulted in a marked reduction in [3H]thymidine incorporation. By contrast, Ras-expressing cells continued to incorporate high levels of [3H]thymidine even after serum depletion. Similarly, Ras-expressing cells placed in low serum incorporatated twice as much BrdU as cells containing an empty vector (Fig. 1B). Initially, proliferation in the presence of oncogenic Ras was accompanied by morphological changes characteristic of constitutive Ras activity in immortal cells, becoming small and refractile with thin cytoplasmic projections (Fig. 1C). Only later do Ras-expressing cells acquire the characteristic senescent morphology (Fig. 1C; see also Serrano et al. 1997). Therefore, the immediate consequences of oncogenic Ras expression are forced proliferation, implying that cell-cycle arrest is an antiproliferative cellular response.
Figure 1.

Oncogenic Ras is initially mitogenic in primary fibroblasts. (A) [3H]thymidine incorporation assay using IMR90 cell populations containing empty vector (V) or H–RasV12 (R). Twenty-four hours postinfection, 2 × 14 of the indicated cells were plated and grown in medium containing 10% FBS or 0.5% FBS for 24 hr followed by a 3H]thymidine pulse. Values were normalized to those obtained from control populations in 10% serum; the average and standard deviation of three measures are shown. (B) Cell-cycle analysis of IMR90 cell populations containing empty vector (V) or H–RasV12 (R) in low-serum conditions. Twenty-four hours postinfection, cells were transferred to 0.5% FBS-containing medium, 24 hr later, the cells werepulsed with BrdU for 4 hr; and analyzed for BrdU incorporation and DNA content by two-color flow cytometry (see Materials and Methods). The box represents cells incorporating BrdU (S-phase); the percentage of BrdU positive cells are indicated. (C) Representative photomicrographs of IMR90 cells transduced with empty vector or a H–RasV12-expressing construct. Cells were transduced with retroviruses containing either empty vector or H-RasV12; the photomicrographs were taken at day 2 postselection (prearrest) and day 4 postselection (postarrest) according to the time scheme described in Materials and Methods.
Ras effectors and cell-cycle arrest
As a first step in identifying the Ras-signaling pathway(s) required for premature senescence, we exploited a series of Ras ‘effector loop’ mutants that interact preferentially with specific Ras effector proteins (White et al. 1995; Joneson et al. 1996). For example, H–RasV12/S35 binds preferentially to Raf-1 and activates MAPK but has no effect on membrane ruffling (White et al. 1995). In contrast, H-RasV12/C40 associates with PI(3)K and promotes membrane ruffling and cell survival but does not bind Raf-1 or induce mitogenesis (Joneson et al. 1996; Rodriguez-Viciana et al. 1997). H-RasV12/G37 binds Ral.GDS but fails to bind Raf-1 or PI(3)K (White et al. 1995). IMR90 human fibroblasts were infected with retroviruses expressing individual Ras mutants, and cell proliferation was determined by assessing relative cell accumulation at various times postplating (Serrano et al. 1997).
Consistent with previous results, constitutive expression of oncogenic Ras induced cell-cycle arrest at subconfluent densities and in the presence of serum, whereas cells harboring the empty vector grew exponentially to confluence (Fig. 2A). Cell populations expressing the H–RasV12/G37 or H-RasV12/C40 mutants failed to arrest, suggesting that neither the Ras–PI(3)K interaction nor the Ras–Ral.GDS interaction is sufficient to induce cell-cycle arrest. In contrast, cells expressing H–RasV12/S35 grew slowly, at rates similar to cells expressing H–RasV12 (Fig. 2A). Of note, each Ras mutant was expressed at levels equal to (or greater than) H–RasV12, ranging from a 5- to 10-fold increase over endogenous Ras expression (data not shown). Therefore, the differential effects of each mutant were not caused by variations in Ras expression but, more likely, by their ability to interact with distinct effector proteins. These results correlate the Ras–Raf interaction with Ras-induced cell-cycle arrest in normal diploid human fibroblasts.
Figure 2.
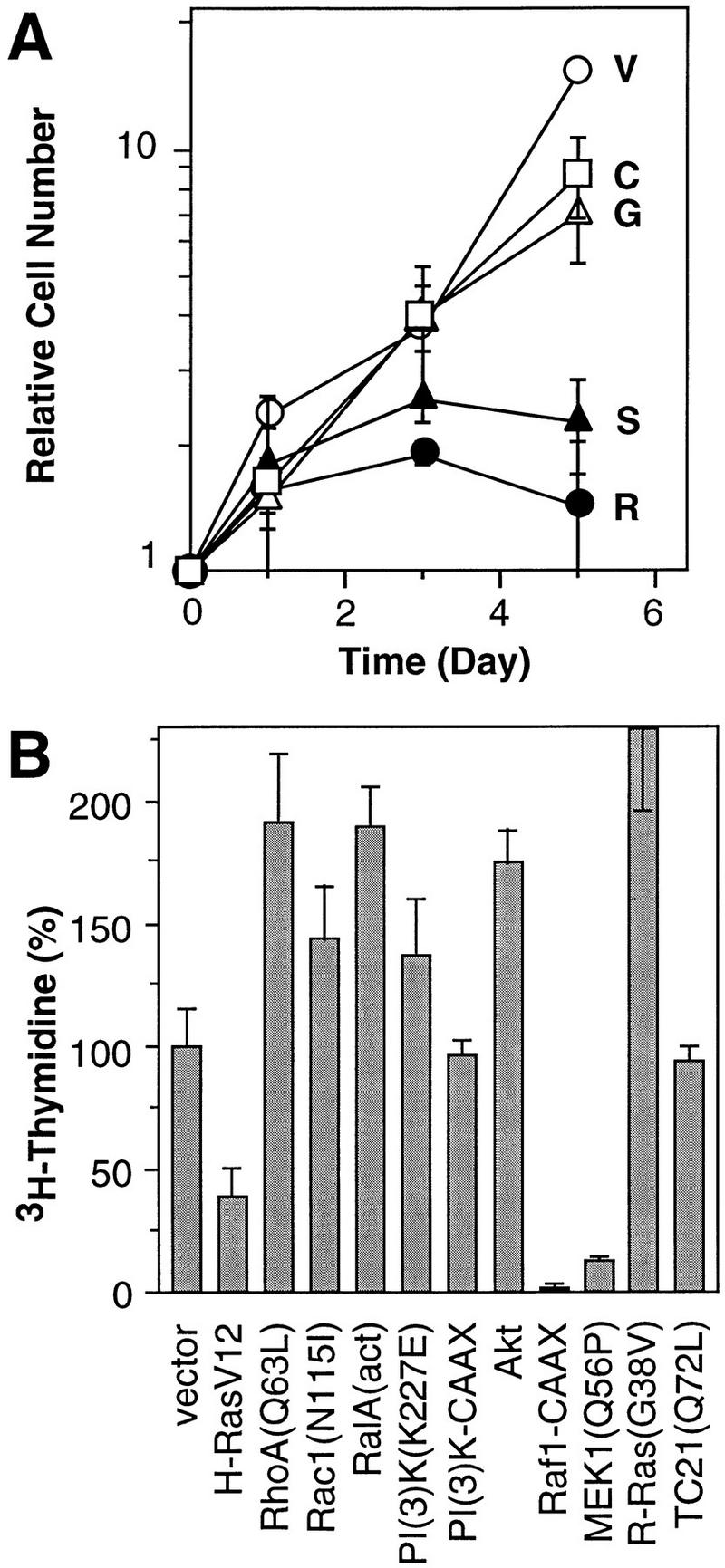
Effect of Ras-dependent signaling pathways on proliferation. (A) Representative growth curves corresponding to the indicated IMR90 cell populations containing empty vector (V, ○), H-RasV12 (R, •) H-RasV12/S35 (S, ▴), H-RasV12/G37 (G, ▵), H-RasV12/C40 (C, □). Each value was determined in triplicate and normalized to the cell number at day 0. (B) Representative [3H]thymidine incorporation assay of IMR90 cell populations expressing various Ras-related proteins, Ras effectors or Ras downstream components (see Materials and Methods). The values were normalized to those obtained from cells containing a control vector; the average and standard deviation of three measures are shown.
Because the Ras effector loop mutants were characterized in immortal cell lines, it is formally possible that they possess different activities in primary cells. As an alternative approach, selected Ras effectors (Raf-1, PI(3)K), downstream components (Rac, Akt, RalA, RhoA, MEK1) (see Introduction), or Ras-related proteins (R-Ras, TC21) were tested for their ability to cause growth arrest by retroviral transduction into IMR90 human fibroblasts. Of note, R-Ras is highly homologous to H-Ras, and interacts with PI(3)K and activates the PI(3)K/Akt pathway but not the MAPK pathway in vivo (Marte et al. 1997). TC21 is more closely related to H-Ras than any other known member of the Ras superfamily but does not activate Raf (Graham et al. 1996). Consequently, H-Ras, R-Ras, and TC21 stimulate cell growth and transformation via distinct signaling pathways. After selection for virus-infected populations, cells were plated at subconfluent densities and pulsed with [3H]thymidine in the presence of 10% serum. Among the Ras effectors and downstream components examined, some enhanced proliferation, whereas others had little effect (Fig. 2B). Only constitutively active Raf-1 and MEK—components of the MAPK cascade—inhibited proliferation (Fig. 2B; see also Zhu et al. 1998). Neither R-Ras nor TC21 produced cell-cycle arrest. Therefore, both the Ras mutant and effector analysis are consistent: Ras-induced arrest in normal diploid human fibroblasts involves activation of the MAPK cascade.
Activation of the MEK/MAPK cascade induces p53, p16, and premature senescence
The MEKs are dual specificity protein kinases that, upon activation, phosphorylate the threonine and tyrosine regulatory sites of MAPKs (for review, see Marshall 1994). MEK1Q56P is a MEK mutant identified by virtue of its ability to enhance the transforming activity of otherwise transforming-defective Ras mutants. Substitution of glutamine to proline at codon 56 results in a gain-of-function mutant that has increased kinase activity (Bottorff et al. 1995). To better characterize MEK-induced arrest, the proliferation properties of IMR90 cell populations containing a control vector, oncogenic Ras, or MEK1Q56P were measured by growth curves, or by BrdU-labeling and flow cytometry (Serrano et al. 1997). Activated MEK causes arrest even more rapidly than oncogenic Ras, with cells displaying a dramatic reduction in BrdU incorporation [compare 0.6% (MEK) and 0.3% (Ras) to 24.5% (vector) (Fig. 3A,C)]. Likewise, MEK-expressing cells arrest primarily with a G1 DNA content, although there is a clear G2 component (Fig. 3C). Importantly, both oncogenic Ras and MEK1Q56P activate MAPK in human fibroblasts (Fig. 3B).
Figure 3.
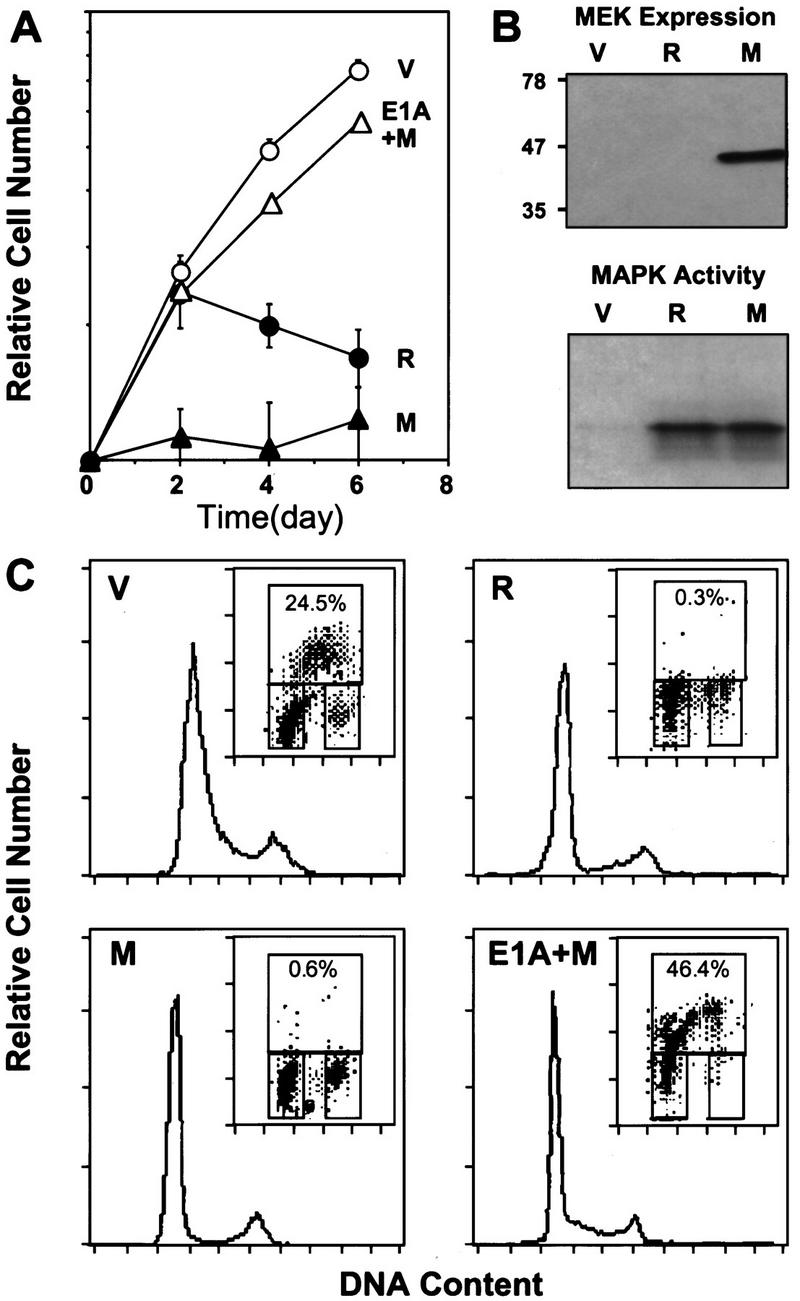
Activated MEK induces cell-cycle arrest in human diploid fibroblasts. (A) Representative growth curves corresponding to IMR90 cell populations transduced with empty vector (V, ○), H–RasV12 (R, •), MEK1Q56P (M, ▴) or E1A+MEK1Q56P (E1A+M, ▵). Each value was determined in triplicate and normalized to the cell number at day 0. (B) Expression of ectopic MEK1Q56P in retrovirally infected IMR90 cells was verified by immunoblot analysis at day 4 postselection. MAP kinase activity in cell populations at day 2 postselection was measured by immunoprecipitation of ERK2, followed by a kinase assay using [γ-32P]ATP and myelin basic protein as substrate (see Materials and Methods). (C) Cell-cycle analysis of the indicated cell populations (day 6 postselection) as determined by BrdU incorporation and DNA-content analysis (see Materials and Methods). The histogram displays the DNA profile of the indicated cell population as measured by propidium iodide. (Insets) The upper box indicates cells incorporating BrdU (S phase); the lower-left box displays G0/G1 cell population; the lower-right box indicates cells in G2/M.
The adenovirus E1A oncogene cooperates with ras to transform primary rodent fibroblasts (Ruley 1990) and abrogates Ras-induced senescence (Serrano et al. 1997). To determine the effect of E1A on MEK-induced arrest, IMR90 cells were infected sequentially with retroviruses expressing E1A, and then either MEK1Q56P or a control vector. Cell populations coexpressing E1A and MEK1Q56P accumulated at rates comparable to vector-containing cells and incorporated high levels of BrdU, showing no signs of cell-cycle arrest (Fig. 3A,C). Consequently, as in the case of Ras-expressing cells, E1A effectively counters MEK-induced arrest and allows uncontrolled proliferation.
Cellular senescence is accompanied by a series of changes that, together, distinguish senescence from quiescence or differentiation. These changes include altered expression of cell-cycle proteins, upregulation of p53, p16, and p21, and the accumulation of senescence-associated β-galactosidase (SA-β-gal) (Noda et al. 1994; Atadja et al. 1995; Dimri et al. 1995; Alcorta et al. 1996; Reznikoff et al. 1996). Like cells expressing oncogenic Ras, MEK1Q56P-expressing cells harbored underphosphorylated Rb and lost cyclin A expression (Fig 4). Also, MEK induced p53 (about threefold induction), p21 (about fivefold induction), and p16 (about sixfold induction) (Fig. 4). Finally, MEK1Q56P-expressing cells exhibited high levels of SA-β-gal activity, with a percentage of positive cells similar to that observed in Ras-expressing or late passage IMR90 cells (Fig. 5A,B). Of note, these effects require constitutive MEK activation, because cells overexpressing a wild-type MEK allele grow normally, display normal levels of p53, p21, p16, and are negative for SA-β-gal activity (data not shown).
Figure 4.
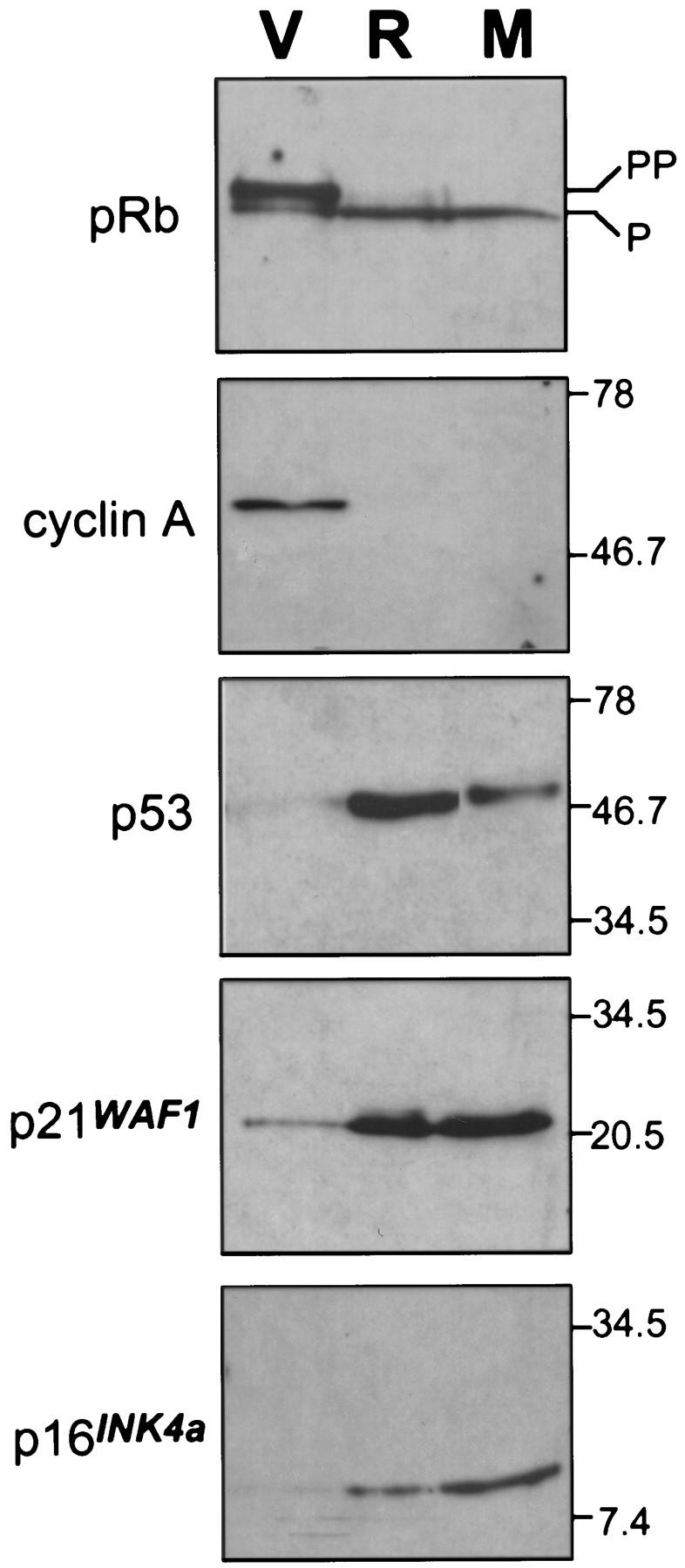
Activated MEK induces accumulation of p53 and p16. Immunoblots of cell-cycle regulatory proteins in lysates from cells containing empty vector (V), H–RasV12 (R), or MEKQ56P (M) at day 4 postselection.
Figure 5.
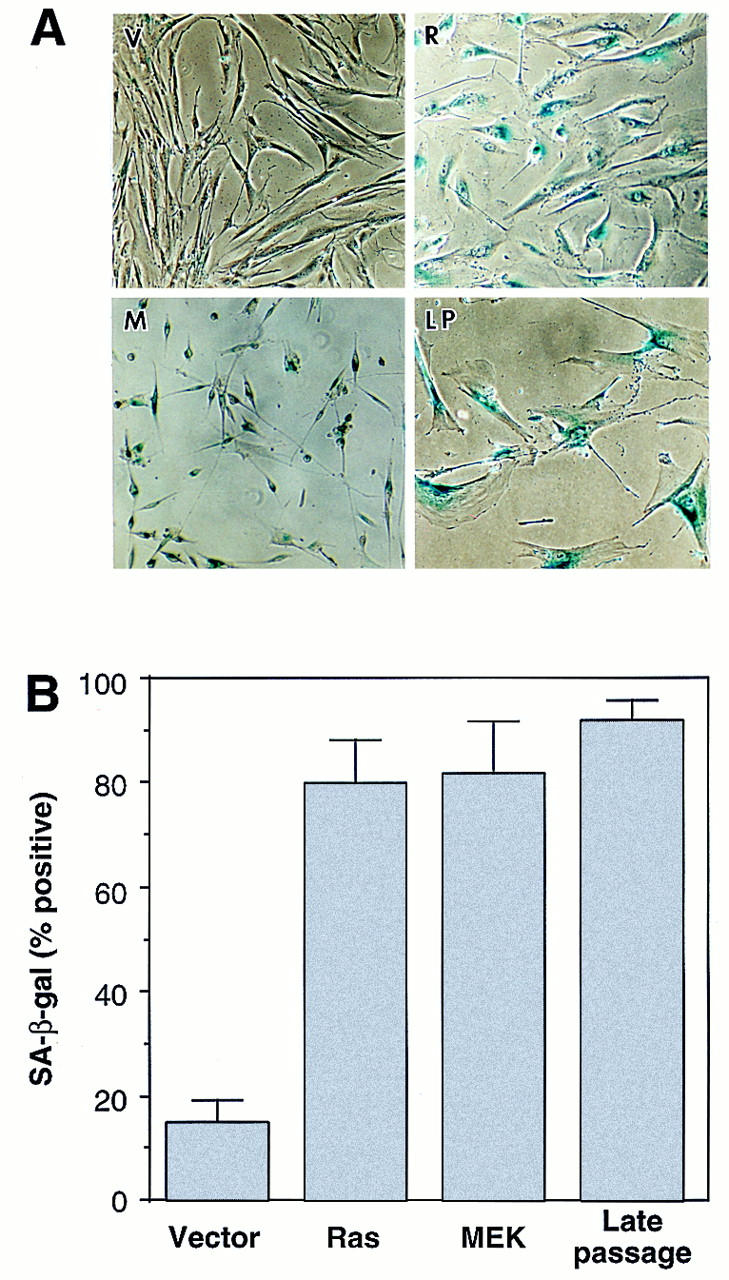
MEK activates premature senescence. (A) Photomicrographs of IMR90 cell populations containing an empty vector (V), H–RasV12 (R), or MEK1Q56P (M) stained for SA-β-galactosidase activity (pH 6.0) at day 6 postselection. Late-passage IMR90 cells (LP) having undergone replicative senescence are shown for comparison. (B) Percentage of cells positive for SA-β-galactosidase 6 days after drug selection. Cells (∼ 200) from each cell population were scored; the average and standard deviation of data from at least three separate experiments are shown.
These data indicate that the cell-cycle arrest produced by oncogenic Ras and activated MEK is virtually identical and is characteristic of cellular senescence. Indeed, the only difference we observed between these populations is their morphology: Whereas Ras-arrested cells became large and flat, MEK-arrested cells were small and refractile (Fig. 5A). Nevertheless, MEK-expressing cells remain arrested in a metabolically active state for as long as we have observed them (>2 weeks). Because Rho family members mediate changes in cell morphology in other settings (for review, see Van Aelst and D’Souza-Schorey 1997), they may contribute to the morphological changes observed in Ras-arrested cells. Nevertheless, cell morphology has been dissociated from senescence in other settings (Wistrom and Villeponteau 1990). Thus, activation of the MAPK cascade is sufficient to promote premature senescence.
MEK activation is required for premature senescence
To determine whether MEK activity was required for senescence, we took advantage of PD98059, a highly specific noncompetitive MEK inhibitor that prevents its activation by Raf (Alessi et al. 1995; Dudley et al. 1995). IMR90 cells were infected with control or oncogenic Ras-expressing retroviruses, and treated daily with 50 μm PD98059 prior to the onset of cell-cycle arrest and the appearance of senescence-related markers (3 days postinfection). Notably, this drug concentration has been found previously to be compatible with the proliferation of fibroblasts albeit at slower rates (Pumiglia and Decker 1997). All cultures were monitored for proliferation and senescence as described above.
Remarkably, PD98059 prevented the onset of Ras-induced arrest but had no effect on the accumulation of vector-containing cells (Fig. 6A). Although the accumulation rate of Ras-expressing cells treated with PD98059 did not achieve that of untreated cells harboring an empty vector, they displayed an approximately sixfold increase in BrdU incorporation compared to Ras-expressing cells treated with solvent alone (Fig. 6B). Similarly, treatment with PD98059 prevented the appearance of the senescence-like morphology and the upregulation of SA-β-galactosidase in Ras-expressing cells (Fig. 6C). Moreover, PD98059 had no effect on the detection of SA-β-gal activity in late passage (senescent) cells, demonstrating that PD98059 does not directly inhibit this enzyme. Thus, these data imply that MEK—and hence the MAPK cascade—is required for premature senescence induced by oncogenic Ras.
Figure 6.
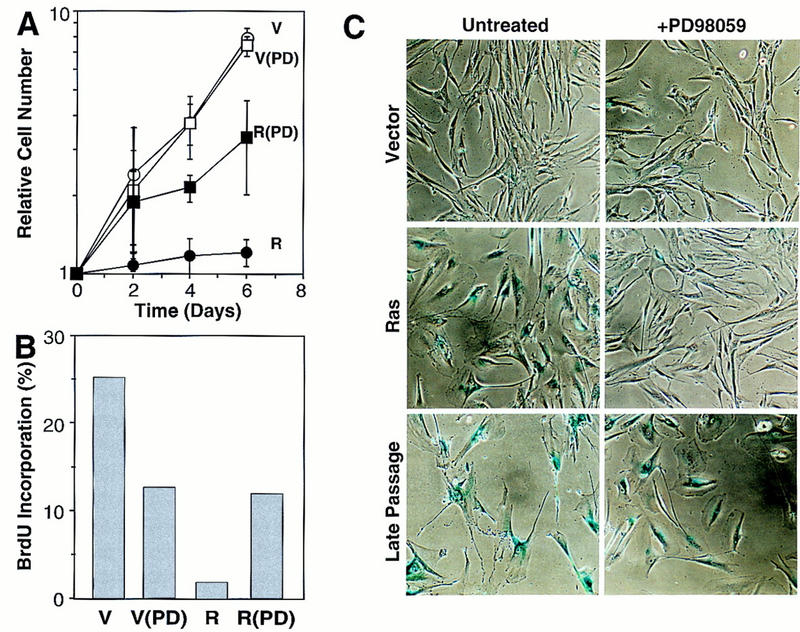
MEK is required for Ras-induced cell-cycle arrest and premature senescence. (A) Representative growth curves documenting the effects of PD98059 on Ras-induced growth arrest. IMR90 cells were infected with an empty vector- or an H-RasV12-expressing retrovirus. Immediately after selection, cell populations were treated daily with medium containing 50 μm PD98059 or 0.25% DMSO (carrier). Each value was determined in triplicate and normalized to the cell number at day 0. (B) Cell populations containing an empty vector (V) or expressing H-RasV12 (R) were treated daily with PD98059 (PD). On day 6 postselection, cells were pulsed with BrdU and analyzed for BrdU incorporation by flow cytometry. The calculated percentage of BrdU-positive cells is shown. (C) Photomicrographs of control and PD98059-treated cell populations stained for SA-β-gal activity on day 6 postselection. The percentage of positive cells in each population was 9% (vector); 12.5% (vector + PD98059); 80% (Ras); 15.4% (Ras + PD98059); 90% (late passage); 91% (late passage + PD98059).
MEK arrests primary murine fibroblasts but transforms fibroblasts lacking p53 or p16INK4a
Premature senescence may act as a fail-safe mechanism that suppresses transformation by Ras (Serrano et al. 1997). Thus, whereas oncogenic Ras arrests primary rodent fibroblasts, it readily transforms primary fibroblasts lacking p53 or INK4a (Tanaka et al. 1994; Serrano et al. 1996). Because MEK is both necessary and sufficient for Ras-induced senescence in human fibroblasts, we asked whether inactivation of p53 or INK4a would overcome MEK-induced arrest in primary rodent cells. Therefore, wild-type, p53-null, and INK4a-null primary mouse embryo fibroblasts (MEFs) were infected with control vector-, Ras-, and MEK1Q56P-expressing retroviruses, and the rate of cell accumulation was examined in the resulting populations. Of note, our INK4a-null cells have deletions in INK4a exon 2 that may also inactivate p19ARF (Haber 1997, see discussion); hence, these cells are referred to as INK4aex2−/− for clarity.
As in human fibroblasts, MEK promoted cell-cycle arrest in wild-type MEFs (Fig. 7A). However, unlike in human cells, MEK-arrested MEFs acquired the enlarged and flat senescent morphology characteristic of Ras-expressing MEFs (data not shown). Remarkably, when MEK1Q56P was expressed at comparable levels in primary p53−/− and INK4aex2−/− MEFs (Fig. 7B), the resulting populations grew at rates similar to parallel cultures expressing oncogenic Ras, with no indication of a growth delay or arrest. Moreover, MEK-expressing p53−/− and INK4aex2−/− MEFs appeared transformed morphologically (data not shown) and continued to proliferate in low serum (Fig. 7C). Therefore, deletion of either p53 or INK4aex2 was sufficient to alter the cellular response to MEK from permanent arrest to uncontrolled mitogenesis.
Figure 7.


Inactivation of either p53 or INK4aex2 reveals the mitogenic and transforming activity of activated MEK. (A) Representative growth curves corresponding to wild-type (WT), p53-null (p53−/−) or p16-null (INK4aex2−/−) MEFs transduced with empty vector (V), H-RasV12 (R), or MEK1Q56P (M) expressing retroviruses. Each value was determined in triplicate and normalized to the cell number at day 0. (B) MEK1 expression in the indicated cell populations was determined by immunoblotting on day 4 postselection. (C) [3H]thymidine incorporation in the indicated cell populations containing empty vector (gray bars) or MEK1Q56P (black bars). On day 6 postselection, cells were transferred to 0.5% FBS-containing medium for 36 hr, followed by a 24-hr pulse with [3H]thymidine. Each value was determined in triplicate and normalized to the value obtained for wild-type cells containing the empty vector. (D) Analysis of anchorage-independent growth of the indicated MEF populations. Immediately following selection for virus-transduced populations, ∼2 × 14 cells were plated in soft agar (see Materials and Methods). Photomicrographs were taken 3 weeks after plating.
The transformation properties of virus-infected populations were assessed by their ability to grow in suspension and form tumors in immunocompromised mice. In soft-agar assays, MEK-expressing populations grown minimally in culture derived from p53−/− and INK4aex2−/− cells formed colonies at high frequency (Fig. 7D). Similarly, MEK-expressing populations derived from p53−/− and INK4aex2−/− MEFs formed tumors at high frequency when injected subcutaneously into athymic nude mice (Table 1). Importantly, the transformation properties of MEK-expressing cells required cooperation between MEK and loss of either p53 (see also Fukasawa and Vande Woude 1997) or INK4aex2, because p53−/− and INK4aex2−/− MEFs harboring an inert vector neither grew in soft agar nor formed tumors in nude mice. However, MEK1Q56P was not as potent as oncogenic Ras in transforming p53−/− and INK4aex2−/− MEFs, because both soft-agar colonies and tumors expressing MEK were typically smaller than those expressing oncogenic Ras (Fig. 7D; Table 1). The inability of activated MEK to recapitulate the potency of oncogenic Ras in transformation assays is consistent with previous studies showing that multiple effector pathways contribute to Ras-mediated transformation (White et al. 1995).
Table 1.
Tumorigenicity assays
| MEF genotypea
|
Tumor frequencyb
|
Tumor volumec (cm3)
|
|---|---|---|
| WT (V) | 0/4 | N.A. |
| WT (R) | 0/10 | N.A. |
| WT (M) | 0/4 | N.A. |
| p53−/− (V) | 0/8 | N.A. |
| p53−/− (R) | 8/8 | 1.40 ± 0.66 |
| p53−/−(M) | 7/8 | 0.11 ± 0.087 |
| INK4aex2−/−(V) | 0/8 | N.A. |
| INK4aex2−/−(R) | 8/8 | 1.34 ± 1.29 |
| INK4aex2−/− (M) | 7/8 | 0.21 ± 0.11 |
Primary MEFs were isolated from wild-type (WT), p53-deficient (p53−/−), and p16-deficient (INK4aex2−/−) mice and infected with control vector (V), H-RasV12 (R), or MEK1Q56P (M).
Number of tumors arising per number of sites injected.
Tumor volume measured at 2 weeks [V = (L × W2)/2].
These data demonstrate that activated MEK efficiently transforms primary MEFs lacking p53 or INK4aex2, cells otherwise predisposed to immortalization. Because these MEK-expressing cell populations were derived from primary fibroblasts grown minimally in culture, it seems highly unlikely that additional factors contribute to the transformed phenotype of these cells. Similarly, we show that human fibroblasts expressing an immortalizing oncogene (i.e., E1A) overcome MEK-induced arrest. Together, these data demonstrate that constitutive activation of the MEK/MAPK cascade can produce either premature senescence or forced mitogenesis depending on the integrity of a senescence program controlled by p53 and p16. The precisely opposite response of primary and immortal cells to MEK activation implies that premature senescence is a cellular fail-safe mechanism that suppresses the transforming potential of aberrant Ras mitogenic signaling.
Discussion
Oncogenic Ras transforms immortal rodent fibroblasts, in part, by constitutively transmitting mitogenic signals through the MAPK cascade. By marked contrast, oncogenic Ras acutely activates senescence in primary fibroblasts through the p53 and p16 tumor suppressors (Serrano et al. 1997). Here we demonstrate that Ras signals premature senescence through activation of the MAPK cascade. In normal diploid human fibroblasts, Ras effector loop mutants that retain their ability to bind Raf-1 promoted premature senescence and, among a series of Ras downstream components examined, only activated MEK was capable of inducing p53, p16, and features of senescence. Moreover, a MEK inhibitor prevented Ras-induced cell-cycle arrest and senescence. In primary murine fibroblasts, activated MEK arrested wild-type MEFs but forced uncontrolled mitogenesis and transformation when expressed at comparable levels in p53−/− or INK4aex2−/− MEFs. Thus, oncogenic Ras arrests normal cells through the primary mechanism whereby it transforms immortal cells or primary fibroblasts defective in p53 or p16 function.
Role of the MAPK cascade in cell-cycle arrest
Signaling through the MAPK cascade is relevant to several physiological processes, including mitogenesis, differentiation, and even apoptosis, depending on the cellular context (for review, see Marshall 1998). In some settings, the difference between proliferation and arrest is related to the intensity or duration of the signal (Sewing et al. 1997; Woods et al. 1997; Kerkhoff and Rapp 1998). We show that oncogenic Ras initially forces uncontrolled proliferation, and only later do cells arrest. Importantly, the cell-cycle arrest induced by expression of Ras or MEK is abolished in cells expressing E1A (IMR90), or lacking p53 or p16 (MEFs). In contrast to our observations, others have found that high levels of Raf activity can produce an immediate cell-cycle arrest that occurs independently of p53 or p16 (Sewing et al. 1997; Woods et al. 1997). Perhaps the difference between these studies and ours reflects the fact that other Ras effector pathways can attenuate Raf-induced arrest and impinge upon its ability to activate components of the arrest machinery (Olson et al. 1998); alternatively, they may arise from different levels and kinetics of activation of the Raf/MAPK cascade. Nevertheless, the results presented here demonstrate clearly that Ras-mediated MAPK activation can produce two precisely opposite outcomes—cell-cycle arrest or forced mitogenesis—depending on the integrity of the senescence program controlled by p53 and p16.
Other mitogenic oncogenes can display antiproliferative potential. For example, the c-myc and E1A oncogenes induce proliferation, but both activate p53 to promote apoptosis (for review, see Weinberg 1997). The Myc functions involved in both proliferation and apoptosis are related (Evan et al. 1992; Amati et al. 1993) and, at least in primary cells, the E1A domains required for p53 accumulation, apoptosis, and oncogenic transformation are inseparable (Samuelson and Lowe 1997). The fact that oncogenic Ras uses the same signal-transduction pathway to promote both arrest and forced mitogenesis reinforces the view that normal cells counter malignant transformation by actively responding to hyperproliferative signals; in this case, by sensing excessive MEK/MAPK activity and activating senescence. The necessity to overcome these fail-safe mechanisms can explain the phenomenon of oncogene cooperation and may be important during multistep carcinogenesis.
The MAPK cascade and senescence
The cell-cycle arrest induced by deregulated MEK activity in primary fibroblasts reflects bona fide cellular senescence and not an unusual form of quiescence or differentiation. First, unlike quiescence, both oncogenic Ras and MEK arrest cells at subconfluent densities in the presence of mitogenic growth factors. This arrest is permanent: whereas the MEK inhibitor PD98059 prevents Ras-induced arrest when added prior to the appearance of senescence markers, it cannot reverse the arrest at later times (data not shown). Second, although p53 and p16 play fundamental roles in Ras-induced arrest, neither have essential roles in differentiation. Indeed, most p53- and INK4aex2-deficient mice develop normally (Donehower et al. 1992; Serrano et al. 1996). Cell-cycle arrest induced by Ras and MEK is accompanied by accumulation of p53, p21, p16, and SA-β-gal activity. Importantly, these markers together are associated with cellular senescence in both fibroblasts and epithelial cells but rarely, if ever, with quiescence or differentiation (Atadja et al. 1995; Dimri et al. 1995; Palmero et al. 1997). Finally, cDNA array analysis of ras-arrested fibroblasts reveals global changes in gene expression consistent with that reported for senescent cells (G. Ferbeyre and S.W. Lowe, unpubl.).
The characterization of Ras-induced arrest in primary fibroblasts as cellular senescence, and the involvement of the MEK/MAPK cascade in this process, have important implications for our understanding of senescence biology. Senescence was defined originally by the observation that primary cells have a genetically determined limit to their proliferative potential in cell culture, after which they arrest permanently with characteristic features (Hayflick 1965). Owing to the ‘end-replication problem’, telomeres shorten during each cell division unless telomerase is expressed, and it has been proposed that some aspect of excessive telomere shortening activates cell-cycle arrest and other characteristics of senescence (for review, see Greider 1998). However, a number of exogenous stimuli can also produce a phenotype suggestive of senescence, including DNA damage, DNA-demethylating drugs, ceramide, or inhibitors of histone deacetylation (Holliday 1986; Venable et al. 1995; Ogryzko et al. 1996; Linke et al. 1997), raising the possibility that telomere shortening is not the only signal capable of activating the senescence program.
Our data demonstrate that senescence can be activated in response to deregulated MAPK activation through MEK. Consistent with these results, forced Raf-1 expression also activates senescence in normal human fibroblasts (see Fig. 2B; Zhu et al. 1998). Whereas it remains possible that telomere shortening or some other cell-doubling-sensitive mechanism activates senescence through Ras/MAPK signaling, our hypothesis is that senescence can be activated by diverse stimuli leading to engagement of a common cell-cycle arrest program. In this view, the process of senescence is related conceptually to apoptosis, a genetically controlled program carried out by a common cell-death machinery. Diverse stimuli and signaling pathways feed into this machinery, allowing regulation of cell death to be highly versatile. Although less understood, the possibility that multiple signaling pathways induce senescence is consistent with cell fusion studies (for review, see Vojta and Barrett 1995). Consequently, the biological roles of cellular senescence may go well beyond the control of cellular or organismal aging, acting as a global anti-proliferative response to a variety of cellular stresses.
The role of p16 and p53 in premature senescence
The precise role of p16 in tumor suppression is confounded by the complexity of the INK4a locus, which encodes a second tumor suppressor translated in an alternative reading frame designated p19ARF (Quelle et al. 1995). Mutations at the INK4a locus often disrupt both p16 and p19ARF (for review, see Haber 1997) and mice lacking p19ARF develop tumors (Kamijo et al. 1997). p19ARF also promotes cell-cycle arrest and primary cells derived from ARF-deficient mice can be transformed by oncogenic Ras alone (Kamijo et al. 1997). Because the mutation in our INK4aex2−/− MEFs may disrupt p19ARF function, it is possible that p19ARF is the sole product of the INK4a/ARF locus responsible for senescence in response to oncogenic Ras. Oncogenes can signal p53 through p19ARF (Zindy et al. 1998; de Stanchina et al. 1998); moreover, oncogenic Ras and MEK induce ARF message and protein and ARF-null MEFs are defective in Ras-induced arrest (Palmero et al. 1998; G. Ferbeyre, A.W. Lin, F. Zindy, M. Roussel, C.J. Sherr, and S.W. Lowe, unpubl.). Nevertheless, p16 contributes to Ras-induced senescence. For example, p16 accumulates in response to oncogenic Ras and MEK, and expression of a p16-insensitive Cdk4 mutant (R24C) (Wolfel et al. 1995) counters Ras-induced growth arrest (Serrano et al. 1997). Furthermore, in human cells, p16 can directly induce senescence (Uhrbom et al. 1997; McConnell et al. 1998) and can be inactivated spontaneously during the immortalization process (Reznikoff et al. 1996; Foster et al. 1998). Regardless of the relative contribution of p16 and p19ARF to this process, the INK4aex2-deficient cells used accurately recapitulate mutations in human tumors.
In rodent cells, the cell-cycle arrest induced by activated MEK is abrogated by inactivation of either p53 or p16 (Fig. 7; see also Serrano et al. 1997). Nevertheless, p53 and p16 do not function in a simple linear pathway. p16 levels increase in response to Ras in p53−/− MEFs and, conversely, p53 levels increase in INK4aex2−/− MEFs (data not shown). Introduction of oncogenic Ras or MEK into INK4aex2−/−;p53−/− double-mutant MEFs produces more transformed foci than in either single mutant (A.W. Lin and S.W. Lowe, unpubl.). Thus, p53 and p16 (and perhaps p19ARF) act cooperatively to promote Ras-induced arrest, and disruption of either p53 or p16 prevents arrest and is sufficient for transformation. In human cells, p53 and p16 also appear to cooperate to promote arrest, although inactivation of both pathways appears necessary to escape senescence (see discussion in Serrano et al. 1997). Consistent with this view, ectopic expression of p21 (a p53 target) and p16 can induce senescence in human cells (McConnell et al. 1998; Vogt et al. 1998).
The activation of p53 and p16 in response to deregulated MEK/MAPK signaling provides a relevant setting in which these tumor suppressors act to limit malignant transformation. In this view, mutations that deregulate the MAPK cascade provide an initial proliferative advantage but also accelerate senescence. However, cells acquiring mutations in p53 or at the INK4a/ARF locus escape senescence, thereby revealing the full mitogenic potential of the MAPK cascade. Consistent with this scenario, ras mutations often precede mutations in p53 or INK4a during tumor development (Fearon and Vogelstein 1990; Burns et al. 1991; Linardopoulos et al. 1995).
Premature senescence and tumor suppression
In summary, our data demonstrate that oncogenic Ras promotes premature senescence through activation of the MAPK cascade. Remarkably, this is the same pathway whereby Ras induces mitogenesis in immortal cells. Escape from Ras-induced senescence allows inappropriate proliferation and, in rodent cells, oncogenic transformation. Consequently, the biological outcome of constitutive MAPK activation—cell-cycle arrest or forced mitogenesis—is largely context-dependent and determined by the integrity of the senescence machinery. These data strongly support the notion that normal cells possess fail-safe mechanisms that limit the consequences of Ras mitogenic signaling. Because these safeguards involve p53 and p16, the activation of premature senescence in response to forced mitogenic signaling may be an important mechanism of tumor suppression.
Materials and methods
Cell culture
Normal diploid human IMR90 fibroblasts (early–mid passages) expressed the murine ecotropic receptor (Serrano et al. 1997), and primary mouse embryo fibroblasts derived from wild-type, p53−/− and INK4aex2−/− day 13.5 embryos were prepared as described previously (Jacks et al. 1994; Serrano et al. 1997). All cultures were maintained in Dulbecco’s modified Eagle medium (DMEM; GIBCO) supplemented with 10% fetal bovine serum (FBS; Sigma) and 1% penicillin G/streptomycin sulfate (Sigma).
Retroviral vectors and gene transfer
Oncogenic Ras (H–RasV12) (Serrano et al. 1997) and Ras effector loop mutants (White et al. 1995; Joneson et al. 1996) were expressed using the pBabe–Puro vector (Morgenstern and Land 1990). cDNA encoding Rac1 (N115I), RalA, RhoA (Q63L), R–Ras (G38V), TC21 (Q72L), PI(3)Kp110α (K227E), PI(3)Kp110α–CAAX, wild-type Akt, or E1A12S, respectively, were cloned into a pLPC retroviral vector. Raf-1–CAAX was cloned into a Babe–Puro retroviral vector. MEK1Q56P (Bottorff et al. 1995) was expressed using Babe–Puro- or WZL–Hygro-based retroviral vectors (Ariad Pharmaceuticals, unpubl.). Retroviral gene transfer was performed as described (Serrano et al. 1997) using high-titer retroviral stocks generated by transient transfection of the Phoenix ecotropic packaging line (G. Nolan, Stanford University, CA). Infected cell populations were selected by culture in puromycin (2.5 μg/ml, 3 days) or in hygromycin (200 μg/ml, 5 days) to eliminate uninfected cells. Day 4 postinfection was designated as day 0 (Serrano et al. 1997). In all experiments, cells infected with an empty vector were used as control.
Analysis of cell proliferation
Cell proliferation was assessed by three criteria: growth curves, [3H]thymidine incorporation and flow cytometry of BrdU-labeled cells. For growth curves, cells were plated at a density of 2.5 × 104 per well in 12-well-plates following drug selection. Cells were fixed in 10% formalin at the indicated times according to a time scheme and method described previously (Serrano et al. 1997). Crystal violet (0.1%; Sigma) was used to stain cells. Cell-associated dye was extracted in 2 ml of 10% acetic acid and the optical density was measured at 590 nm with each value normalized to day 0. Each point was done in triplicate, each growth curve was performed at least twice. For experiments using PD98059 (Calbiochem), treatment was initiated 2 days postinfection. Cells were fed daily with fresh medium containing 50 μm PD98059 containing 0.25% DMSO. Solvent-containing medium was used to treat control cultures in parallel.
For [3H]thymidine incorporation assay, 2 × 104 cells per well in 12-well-plates were plated in triplicate. Cells were pulsed for 20–24 hr or 4 hr with 5 μCi/ml [methyl-3H]thymidine (Amersham, 2 Ci/mmoles). After washing with PBS, cells were trypsinized and transferred to glass fiber filters (Filtermat, Wallac) followed by scintillation counting. For BrdU incorporation and DNA-content analysis, subconfluent cultures were labeled with 10 μm BrdU (Amersham) for 4 hr. Cells were trypsinized and fixed in 75% ethanol for 30 min. Cells were washed in PBS and were treated subsequently with 2 m HCl for 20 min at room temperature. After washing in washing buffer (0.1% BSA/PBS), cells were neutralized in 0.1 m sodium borate for 2 min followed by washing in washing buffer. Cells were incubated subsequently with FITC-conjugated anti-BrdU antibody (Pharmingen) for 1 hr at room temperature or overnight at 4°C. Finally, cells were washed in washing buffer three times followed by treatment with propidium iodide (10 μg/ml) and RNase A (100 μg/ml) at 37°C for 30 min. Samples were analyzed by two-dimensional flow cytometry to detect both fluorescein and propidium iodide.
Protein expression and activity
Western blot analysis was carried out as described previously with minor modifications (Serrano et al. 1997). Whole-cell lysates were derived by lysing cell pellets in Laemmli SDS sample buffer. Samples corresponding to 30 μg of protein were resolved on SDS-PAGE gels and transferred to immobilone-P membranes (Millipore). The following antibodies were used: CM1 (Novocastra) at 1:1000 dilution for the detection of p53, anti-WAF1(p21) antibody (C-19; Santa Cruz) at 1:500 dilution, anti-p16 antibody (DCS-50; Novocastra) at 1:200 dilution, anti-Rb (G3-245; Pharmingen) at 0.5 μg/ml together with C-36 and XZ-55 hybridoma supernatant at 1:100 dilution each. Anti-cyclin A (Santa Cruz) at 1:500 dilution. Anti-Ras antibody (Santa Cruz) at 1:500 dilution. Western blot analysis was accomplished according to standard procedures using ECL detection (Amersham).
For MAPK assays, cells were lysed in lysis buffer (20 mm Tris-HCl at pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerolphosphate, 1 mm Na3VO4, 1 μg/ml leupeptin, 1 mm PMSF) accompanied by brief sonication on ice. Protein lysate (100 μg) was incubated with a polyclonal antibody against ERK2 (1:50 dilution; Santa Cruz) for 3 hr at 4°C. Afterwards, 25 μl of protein A–Sepharose beads (50%) were added to the mixture and were incubated for another 3 hr at 4°C. Samples were washed subsequently and resuspended in 20 μl of kinase buffer (25 mm Tris-HCl at pH 7.5, 10 mm MgCl2, 5 mm β-glycerolphosphate, 2 mm DTT, 0.1 mm Na3VO4) supplemented with 10 μCi [γ-32P]ATP (3000 Ci/mmoles) and 5 μg of myelin basic protein (MBP; Sigma) and the reaction mixtures were incubated at 30°C for 30 min. The samples were resolved on 12% SDS–polyacrylamide gels and exposed to Kodak X-OMAT AR.
SA-β-galactosidase activity was detected as described previously (Dimri et al. 1995) with minor modifications. Cells were washed with PBS, fixed in 0.5% glutaraldehyde, and were incubated with staining solution [1 mg/ml 5-bromo-4-chloro-3-indolyl β-galactoside (X-gal), 5 mm potassium ferrocyanide, 5 mm potassium ferricyanide, 1 mm MgCl2 in PBS at pH 6.0] for ∼12 hr at 37°C.
Transformation assays
For soft-agar assays, cells were resuspended in 0.3% Noble agar (in DMEM supplemented with 10% FBS) at a density of 2 × 104 cells per well (in six-well plates), and were plated onto solidifed 0.5% Noble agar-containing bottom layer medium. Cultures were fed weekly and photomicrographs of colonies were taken 3 weeks postplating. Experiments were repeated twice, and MEFs from two different embryo preparations were used in each assay.
For in vivo tumorigenicity assays, 106 cells/0.25 ml of PBS were injected subcutaneously into male NSW athymic nude mice (Tatonic Farms). Each animal received two injections, one on each rear flank. Virus-infected MEFs from two different embryo preparations were used for each genotype, and at least two animals were examined for each cell population (i.e., four tumors). Tumor formation was monitored three times per week by palpation at the sites of injection. The length (L) and width (W) of tumors were measured using a caliper and the volume of tumor formed was determined using the formula V = (L × W2)/2.
Acknowledgments
We are very grateful to G. Clark, P. Rodriguez-Viciana, J. Downward, and I. Mérida for generously providing reagents and to M. McMahon for discussing unpublished work. We thank D. Beach, B. Stillman, W. Herr, and G. Hannon for support and advice, and the Cold Spring Harbor Laboratory Flow Cytometry Facility and Graphic Arts Department for their patience and help. We also thank our colleagues for helpful comments and criticisms. S.W.L. and L.V.A. are Kimmel Scholars, M.S. is supported by grant PM95-0014 from Direccion general de investigacion y tecnica, Spain, and by a core grant from Pharmacia and Upjohn and from the Spanish Research Council, L.V.A. and S.W.L. are supported by grants CA72982 and CA13106, respectively, from the National Cancer Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lowe@cshl.org; FAX (516) 367-8454.
References
- Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci. 1996;93:13742–13747. doi: 10.1073/pnas.93.24.13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Amati B, Brooks MW, Levy N, Littlewood TD, Evan GI, Land H. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell. 1993;72:233–245. doi: 10.1016/0092-8674(93)90663-b. [DOI] [PubMed] [Google Scholar]
- Atadja P, Wong H, Garkavtsev I, Veillette C, Riabowol K. Increased activity of p53 in senescing fibroblasts. Proc Natl Acad Sci. 1995;92:8348–8352. doi: 10.1073/pnas.92.18.8348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottorff D, Stang S, Agellon S, Stone JC. RAS signalling is abnormal in a c-raf1 MEK1 double mutant. Mol Cell Biol. 1995;15:5113–5122. doi: 10.1128/mcb.15.9.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns PA, Kemp CJ, Gannon JV, Lane DP, Bremner R, Balmain A. Loss of heterozygosity and mutational alterations of the p53 gene in skin tumours of interspecific hybrid mice. Oncogene. 1991;6:2363–2369. [PubMed] [Google Scholar]
- Chen RH, Sarnecki C, Blenis J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol Cell Biol. 1992;12:915–927. doi: 10.1128/mcb.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Pomerantz J, Polsky D, Jacobson M, Cohen C, Cordon-Cardo C, Horner JWN, DePinho RA. Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes & Dev. 1997;11:2822–2834. doi: 10.1101/gad.11.21.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- de Stanchina E, McCurrach ME, Zindy F, Shieh S, Ferbeyre G, Samuelson AV, Prives C, Roussel MF, Sherr CJ, Lowe SW. E1A signaling to p53 involves the p19ARF tumor suppressor. Genes & Dev. 1998;12:2434–2442. doi: 10.1101/gad.12.15.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Downward J. Ras signalling and apoptosis. Curr Opin Genet Dev. 1998;8:49–54. doi: 10.1016/s0959-437x(98)80061-0. [DOI] [PubMed] [Google Scholar]
- Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Foster SA, Wong DJ, Barrett MT, Galloway DA. Inactivation of p16 in human mammary epithelial cells by CpG island methylation. Mol Cell Biol. 1998;18:1793–1801. doi: 10.1128/mcb.18.4.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franza BR, Jr, Maruyama K, Garrels JI, Ruley HE. In vitro establishment is not a sufficient prerequisite for transformation by activated ras oncogenes. Cell. 1986;44:409–418. doi: 10.1016/0092-8674(86)90462-9. [DOI] [PubMed] [Google Scholar]
- Fukasawa K, Vande Woude GF. Synergy between the Mos/mitogen-activated protein kinase pathway and loss of p53 function in transformation and chromosome instability. Mol Cell Biol. 1997;17:506–518. doi: 10.1128/mcb.17.1.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallimore PH, Grand RJ, Byrd PJ. Transformation of human embryo retinoblasts with simian virus 40, adenovirus and ras oncogenes. Anticancer Res. 1986;6:499–508. [PubMed] [Google Scholar]
- Gille H, Kortenjann M, Thomae O, Moomaw C, Slaughter C, Cobb MH, Shaw PE. ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation. EMBO J. 1995;14:951–962. doi: 10.1002/j.1460-2075.1995.tb07076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379:88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- Graham SM, Vojtek AB, Huff SY, Cox AD, Clark GJ, Cooper JA, Der CJ. TC21 causes transformation by Raf-independent signaling pathways. Mol Cell Biol. 1996;16:6132–6140. doi: 10.1128/mcb.16.11.6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greider CW. Telomeres and senescence: the history, the experiment, the future. Curr Biol. 1998;8:R178–R181. doi: 10.1016/s0960-9822(98)70105-8. [DOI] [PubMed] [Google Scholar]
- Haber DA. Splicing into senescence: the curious case of p16 and p19ARF. Cell. 1997;91:555–558. doi: 10.1016/s0092-8674(00)80441-9. [DOI] [PubMed] [Google Scholar]
- Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- Hirakawa T, Ruley HE. Rescue of cells from ras oncogene-induced growth arrest by a second, complementing, oncogene. Proc Natl Acad Sci. 1988;85:1519–1523. doi: 10.1073/pnas.85.5.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday R. Strong effects of 5-azacytidine on the in vitro lifespan of human diploid fibroblasts. Exp Cell Res. 1986;166:543–552. doi: 10.1016/0014-4827(86)90499-4. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Jiang H, Luo JQ, Urano T, Frankel P, Lu Z, Foster DA, Feig LA. Involvement of Ral GTPase in v-Src-induced phospholipase D activation. Nature. 1995;378:409–412. doi: 10.1038/378409a0. [DOI] [PubMed] [Google Scholar]
- Joneson T, White MA, Wigler MH, Bar-Sagi D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of RAS. Science. 1996;271:810–812. doi: 10.1126/science.271.5250.810. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- Kastan MB. P53: A determinant of the cell cycle response to DNA damage. Adv Exp Med Biol. 1993;339:291–293. doi: 10.1007/978-1-4615-2488-5_28. [DOI] [PubMed] [Google Scholar]
- Katz ME, McCormick F. Signal transduction from multiple Ras effectors. Curr Opin Genet Dev. 1997;7:75–79. doi: 10.1016/s0959-437x(97)80112-8. [DOI] [PubMed] [Google Scholar]
- Kemp CJ, Donehower LA, Bradley A, Balmain A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell. 1993;74:813–822. doi: 10.1016/0092-8674(93)90461-x. [DOI] [PubMed] [Google Scholar]
- Kerkhoff E, Rapp UR. High-intensity Raf signals convert mitotic cell cycling into cellular growth. Cancer Res. 1998;58:1636–1640. [PubMed] [Google Scholar]
- Khosravi-Far R, Solski PA, Clark GJ, Kinch MS, Der CJ. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol Cell Biol. 1995;15:6443–6453. doi: 10.1128/mcb.15.11.6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linardopoulos S, Street AJ, Quelle DE, Parry D, Peters G, Sherr CJ, Balmain A. Deletion and altered regulation of p16INK4a and p15INK4b in undifferentiated mouse skin tumors. Cancer Res. 1995;55:5168–5172. [PubMed] [Google Scholar]
- Linke SP, Clarkin KC, Wahl GM. p53 mediates permanent arrest over multiple cell cycles in response to gamma-irradiation. Cancer Res. 1997;57:1171–1179. [PubMed] [Google Scholar]
- Lumpkin CK, Knepper JE, Butel JS, Smith JR, Pereira-Smith OM. Mitogenic effects of the proto-oncogene and oncogene forms of c-H-ras DNA in human diploid fibroblasts. Mol Cell Biol. 1986;6:2990–2993. doi: 10.1128/mcb.6.8.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande Woude GF, Ahn NG. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- Marshall CJ. MAP kinase kinase kinase, MAP kinase kinase and MAP kinase. Curr Opin Genet Dev. 1994;4:82–89. doi: 10.1016/0959-437x(94)90095-7. [DOI] [PubMed] [Google Scholar]
- ————— Signal transduction. Taking the Rap. Nature. 1998;392:553–554. doi: 10.1038/33293. [DOI] [PubMed] [Google Scholar]
- Marte BM, Rodriguez-Viciana P, Wennstrom S, Warne PH, Downward J. R-Ras can activate the phosphoinositide 3-kinase but not the MAP kinase arm of the Ras effector pathways. Curr Biol. 1997;7:63–70. doi: 10.1016/s0960-9822(06)00028-5. [DOI] [PubMed] [Google Scholar]
- McConnell BB, Starborg M, Brookes S, Peters G. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr Biol. 1998;8:351–354. doi: 10.1016/s0960-9822(98)70137-x. [DOI] [PubMed] [Google Scholar]
- Morgenstern JP, Land H. Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR. Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res. 1994;211:90–98. doi: 10.1006/excr.1994.1063. [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Hirai TH, Russanova VR, Barbie DA, Howard BH. Human fibroblast commitment to a senescence-like state in response to histone deacetylase inhibitors is cell cycle dependent. Mol Cell Biol. 1996;16:5210–5218. doi: 10.1128/mcb.16.9.5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson M F, Paterson H F, Marshall C J. Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature. 1998;394:295–299. doi: 10.1038/28425. [DOI] [PubMed] [Google Scholar]
- Palmero I, McConnell B, Parry D, Brookes S, Hara E, Bates S, Jat P, Peters G. Accumulation of p16INK4a in mouse fibroblasts as a function of replicative senescence and not of retinoblastoma gene status. Oncogene. 1997;15:495–503. doi: 10.1038/sj.onc.1201212. [DOI] [PubMed] [Google Scholar]
- Palmero I, Pantoja C, Serrano M. p19ARF links the tumour-suppressor p53 to ras. Nature. 1998;395:125–126. doi: 10.1038/25870. [DOI] [PubMed] [Google Scholar]
- Pumiglia KM, Decker SJ. Cell cycle arrest mediated by the MEK/mitogen-activated protein kinase pathway. Proc Natl Acad Sci. 1997;94:448–452. doi: 10.1073/pnas.94.2.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Reznikoff CA, Yeager TR, Belair CD, Savelieva E, Puthenveettil JA, Stadler WM. Elevated p16 at senescence and loss of p16 at immortalization in human papillomavirus 16 E6, but not E7, transformed human uroepithelial cells. Cancer Res. 1996;56:2886–2890. [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- Ruley HE. Transforming collaborations between ras and nuclear oncogenes. Cancer Cells. 1990;2:258–268. [PubMed] [Google Scholar]
- Samuelson AV, Lowe SW. Selective induction of p53 and chemosensitivity in RB-deficient cells by E1A mutants unable to bind the RB-related proteins. Proc Natl Acad Sci. 1997;94:12094–12099. doi: 10.1073/pnas.94.22.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Sewing A, Wiseman B, Lloyd AC, Land H. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol Cell Biol. 1997;17:5588–5597. doi: 10.1128/mcb.17.9.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaargaren M, Bischoff JR. Identification of the guanine nucleotide dissociation stimulator for Ral as a putative effector molecule of R-ras, H-Ras, K-ras, and Rap. Proc Natl Acad Sci. 1994;91:12609–12613. doi: 10.1073/pnas.91.26.12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stang S, Bottorff D, Stone JC. Interaction of activated Ras with Raf-1 alone may be sufficient for transformation of rat2 cells. Mol Cell Biol. 1997;17:3047–3055. doi: 10.1128/mcb.17.6.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Ishihara M, Kitagawa M, Harada H, Kimura T, Matsuyama T, Lamphier MS, Aizawa S, Mak TW, Taniguchi T. Cellular commitment to oncogene-induced transformation or apoptosis is dependent on the transcription factor IRF-1. Cell. 1994;77:829–839. doi: 10.1016/0092-8674(94)90132-5. [DOI] [PubMed] [Google Scholar]
- Uhrbom L, Nister M, Westermark B. Induction of senescence in human malignant glioma cells by p16INK4a. Oncogene. 1997;15:505–514. doi: 10.1038/sj.onc.1201227. [DOI] [PubMed] [Google Scholar]
- Urano T, Emkey R, Feig LA. Ral-GTPases mediate a distinct downstream signaling pathway from Ras that facilitates cellular transformation. EMBO J. 1996;15:810–816. [PMC free article] [PubMed] [Google Scholar]
- Van Aelst L, D’Souza-Schorey C. Rho GTPases and signaling networks. Genes & Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- Van Aelst L, Barr M, Marcus S, Polverino A, Wigler M. Complex formation between RAS and RAF and other protein kinases. Proc Natl Acad Sci. 1993;90:6213–6217. doi: 10.1073/pnas.90.13.6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venable ME, Lee JY, Smyth MJ, Bielawska A, Obeid LM. Role of ceramide in cellular senescence. J Biol Chem. 1995;270:30701–30708. doi: 10.1074/jbc.270.51.30701. [DOI] [PubMed] [Google Scholar]
- Vogt M, Haggblom C, Yeargin J, Christiansen-Weber T, Haas M. Independent induction of senescence by p16INK4a and p21CIP1 in spontaneously immortalized human fibroblasts. Cell Growth Differ. 1998;9:139–146. [PubMed] [Google Scholar]
- Vojta PJ, Barrett JC. Genetic analysis of cellular senescence. Biochim Biophys Acta. 1995;1242:29–41. doi: 10.1016/0304-419x(95)00002-w. [DOI] [PubMed] [Google Scholar]
- Weinberg RA. Oncogenes, antioncogenes, and the molecular bases of multistep carcinogenesis. Cancer Res. 1989;49:3713–3721. [PubMed] [Google Scholar]
- ————— The cat and mouse games that genes, viruses, and cells play. Cell. 1997;88:573–575. doi: 10.1016/s0092-8674(00)81897-8. [DOI] [PubMed] [Google Scholar]
- White MA, Nicolette C, Minden A, Polverino A, Van Aelst L, Karin M, Wigler MH. Multiple Ras functions can contribute to mammalian cell transformation. Cell. 1995;80:533–541. doi: 10.1016/0092-8674(95)90507-3. [DOI] [PubMed] [Google Scholar]
- Wistrom C, Villeponteau B. Long-term growth of diploid human fibroblasts in low serum media. Exp Gerontol. 1990;25:97–105. doi: 10.1016/0531-5565(90)90040-9. [DOI] [PubMed] [Google Scholar]
- Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, De Plaen E, Hankeln T, Meyer zum Buschenfelde KH, Beach D. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269:1281–1284. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol. 1997;17:5598–5611. doi: 10.1128/mcb.17.9.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, J., D. Woods, M. McMahon, and J.M. Bishop. 1998. Senescence of human fibroblasts induced by oncogenic Raf. Genes & Dev. (this issue). [DOI] [PMC free article] [PubMed]
- Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4a tumor suppressor verses other INK4 family members during mouse development and aging. Oncogene. 1997;15:203–211. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes & Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]