

Abstract
Autocatalytic activation of initiator caspases is the link between pro-apoptotic signals and the destruction machinery of apoptosis. Activation of caspase-9, which mediates oncogene and drug-induced apoptosis, requires binding to the protein APAF-1. We found that the proteolytic activity of caspase-9 in a complex with APAF-1 is several orders of magnitude higher than that of the free enzyme. Thus, this complex functions as a holoenzyme in which caspase-9 is the catalytic subunit and APAF-1 its allosteric regulator. We argue that caspase-9 is activated by allosteric regulation and suggest that this mechanism is common for other initiator caspases.
Keywords: Apoptosis, cancer, caspases, capase-9, APAF-1
Caspases, a family of proteases, are a central part of the apoptotic machinery (Thornberry and Lazebnik 1998). Caspases are expressed as precursors that are activated in a cascade following a pro-apoptotic stimulus. This cascade begins with autocatalytic activation of initiator caspases, which process and activate the effector caspases that, in turn, disassemble a cell. Each initiator caspase mediates a subset of pro-apoptotic signals. Caspase-9 is required for normal brain development and mediates apoptosis induced by chemotherapeutic drugs and oncogenic transformation (Fearnhead et al. 1998; Hakem et al. 1998; Kuida et al. 1998). As a consequence, the loss of caspase-9 activity has been implicated in tumorigenesis (Soengas et al. 1999). Caspase-9 activation requires binding of the precursor to a complex of two proteins, APAF-1 and cytochrome c, and is dependent on hydrolysis of dATP or ATP (Li et al. 1997; Hu et al. 1999; Saleh et al. 1999; Zou et al. 1999). How formation of this complex results in caspase-9 activation is not clear. A current model (Muzio et al. 1998; Srinivasula et al. 1998; Yang et al. 1998a,b; Saleh et al. 1999) argues that the precursor of an initiator caspase has low activity that is sufficient for autocatalytic processing but only when two or more precursor molecules are brought into close proximity by adaptor proteins, such as APAF-1. The processed caspase becomes fully active and is free to cleave its substrates. Previously we postulated an alternative model in which proteins like APAF-1 facilitate autocatalysis of the caspase precursors by increasing their activity through a conformational change (Thornberry and Lazebnik 1998). The finding that the activity of recombinant caspase-9 increases in cell extracts suggested that caspase activity can be changed by cytosolic factors (Stennicke et al. 1999). Here we present evidence that caspase-9 activity increases several orders of magnitude when this caspase is in a complex with APAF-1. Hence, these two proteins function as a holoenzyme in which APAF-1 is an allosteric regulator of caspase-9.
Results and Discussion
Caspase-9 activation can be reproduced in a cell-free system by incubating extracts from 293 cells with either ATP or dATP (Liu et al. 1996; Fearnhead et al. 1998), resulting in an “active extract”. In active extract, as in cells, caspase-9 processes and activates caspase-3, a major effector caspase. We found that the caspase-3 activity increased linearly following addition of a nucleotide (Fig. 1A) until all caspase-3 was processed (Fig. 1B). The constant rate of caspase-3 activation meant that caspase-9 activity had to be constant throughout the incubation. Yet, the concentration of processed caspase-9, which is thought to be the active form of the enzyme, increased with time (Fig. 1C). This contradiction could be explained if not all processed caspase-9 is active. With this in mind, we noticed that the amount of APAF-1 bound to caspase-9 remained constant during the incubation (Fig. 1C,D). Moreover, this amount directly correlated with the rate of caspase-3 activation (Fig. 1, ATP vs. dATP; data not shown). Hence, the constant caspase-9 activity could be explained if caspase-9 is active only in a complex with APAF-1. A key condition for this hypothesis is that processed caspase-9 remains bound to APAF-1. To confirm that this complex is not an artifact of immunoprecipitation, we fractionated the inactive and active extracts by sedimentation (Fig. 1E,F). We found that a fraction of APAF-1 comigrated with processed caspase-9 as a putative complex that was detected only in the active extract. The size of the complex was similar to that reported for recombinant proteins (Saleh et al. 1999; Zou et al. 1999). All other caspases tested (3, 7, and 8) were not detectable in this complex by immunoblotting, in contrast to the observations of others (Cain et al. 1999; Hu et al. 1999).
Figure 1.
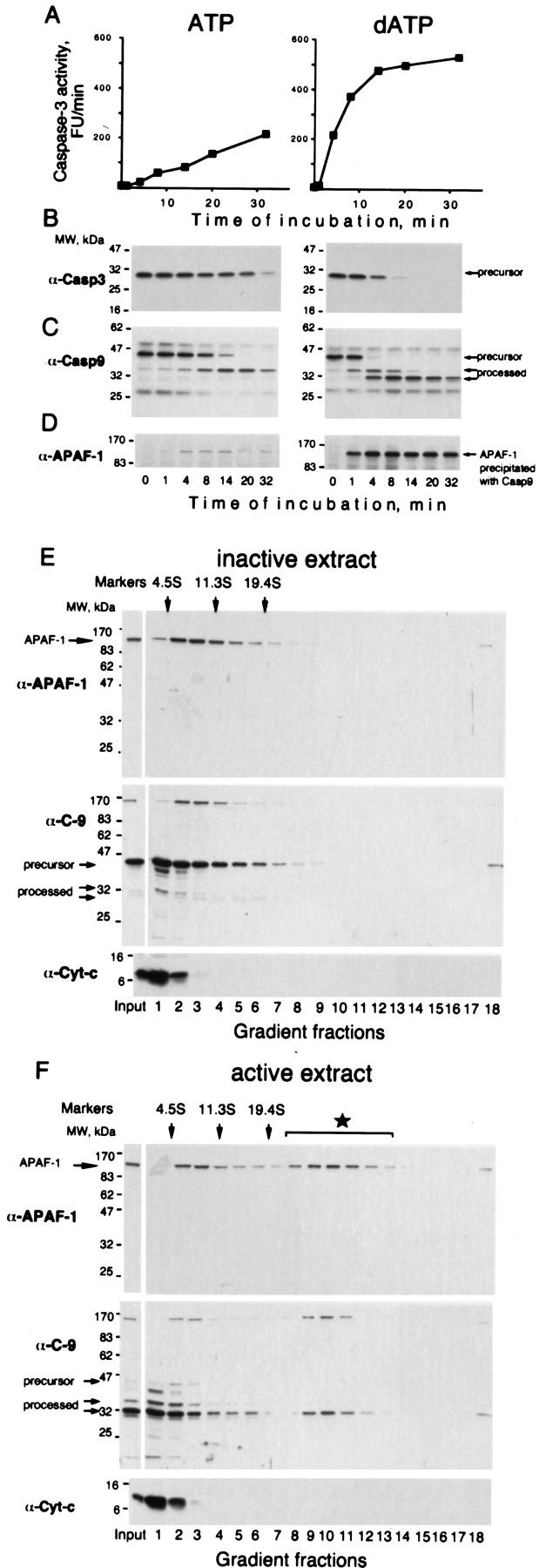
The rate of caspase-3 activation correlates with the amount of APAF-1 bound to caspase-9 (A,B) Caspases in 293 extract were activated by adding 5 mm ATP or dATP followed by incubation at 37°C. At the indicated times, caspase-3 activity using DEVD-AFC as a substrate (A) and caspase-3 processing, by immunoblotting with antibodies as indicated was assayed (B). (C,D) At the indicated times aliquots of the extracts were used to immunoprecipitate caspase-9. Caspase-9 (C) and APAF-1 (D) in the precipitate were detected by immunoblotting. (E,F) A fraction of processed caspase-9 is in a putative complex with APAF-1 (star in F). A total of 50 μl (1.5 mg) of 293 extract was supplemented with 5 mm dATP and incubated at 37°C for 8 min (active extract). A control aliquot was incubated with buffer (inactive extract). Both aliquots were fractionated on 5 ml of 5%–20% linear sucrose gradients and the fractions were analyzed by immunoblotting for APAF-1, caspase-9, and cytochrome c. Sedimentation markers have been described.
To test our hypothesis directly, we decided to separate free caspase-9 from the caspase-9–APAF-1 complex to compare their activities. We first established a caspase-9 activity assay using caspase-3 precursor or its catalytically inactive mutant as substrates. In this assay, caspase-9 immunoprecipitated from active extracts was active, whereas caspase-9 immunoprecipitated from inactive extracts or the mock precipitate from active extract was inactive (Fig. 2A,B). We then immunoprecipitated all caspase-9 from the active extract, eluted it with the epitope peptide, fractionated the eluate by sedimentation, and measured the activity in each fraction. Although the complex contained only a minor fraction of the total caspase-9 (Fig. 2C) it accounted for the majority of the caspase-9 activity (Fig. 2D). The difference in relative activity was at least 1000-fold as estimated by comparing the caspase-9 activity and concentration (determined by densitometry of the caspase-9 immunoblot) in the fractions. Similar results were obtained by doing this experiment by first fractionating an active extract and then measuring the activity of caspase-9 precipitated from the fractions (Fig. 3B). These results were consistent with the notion that caspase-9 is fully active only when it is bound to APAF-1.
Figure 2.
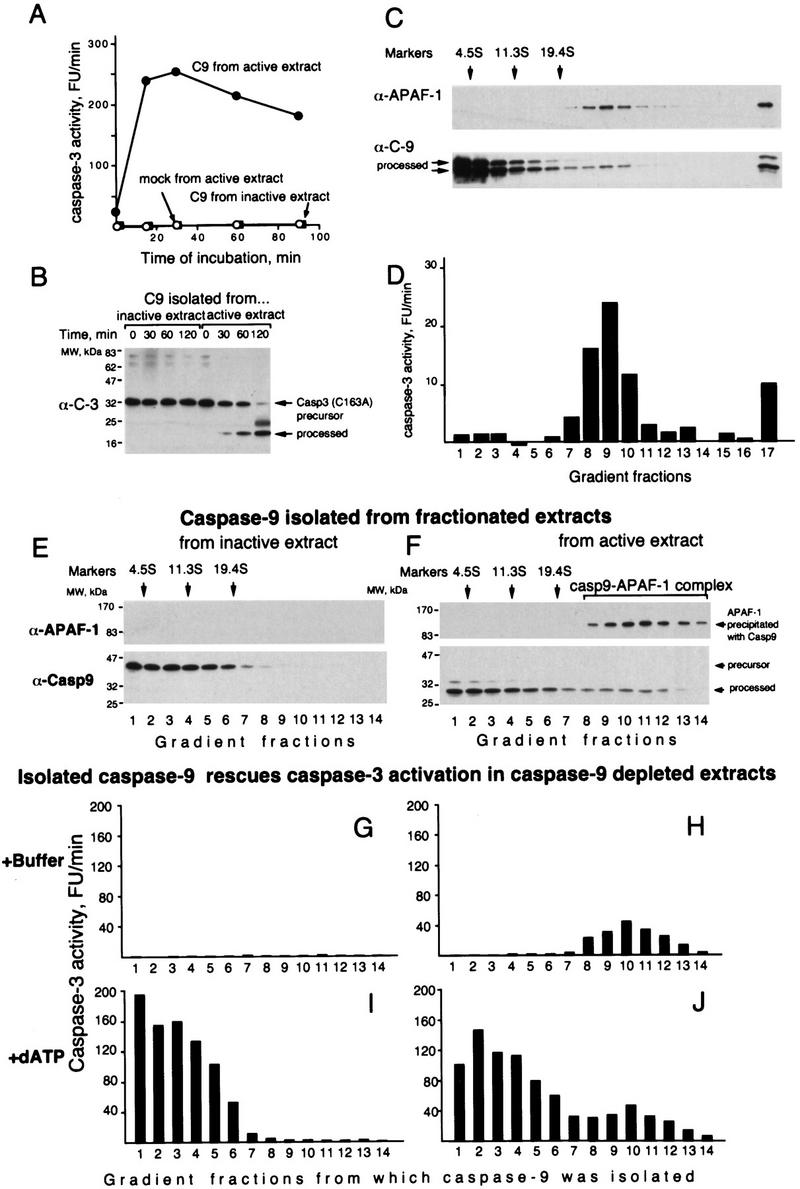
Caspase-9 is active only in a complex with APAF-1. (A) Caspase-9 depleted from active extracts can activate caspase-3. Caspase-9 was depleted from active or inactive extracts eluted with the epitope peptide, and its activity was analyzed using caspase-3 precursor as a substrate. Sepharose with no antibody bound was used for mock depletions. (B) Caspase-9 depleted from active extracts can process catalytically inactive caspase-3. The experiment was done as in A, but the caspase-3 used was made inactive by mutating the catalytic Cys-163 to serine. (C,D) Caspase-9 was depleted from 4 ml (∼135 mg) active extract as in A and fractionated, analyzed for APAF-1 and caspase-9 by immunoblotting (C) and for caspase-9 activity (D) using caspase-3 as in B (1-hr incubation). APAF-1 and caspase-9 in fraction 17 (the bottom of the gradient) are likely to be an aggregate formed by anti-caspase-9 antibodies that leached from Sepharose and were detected only in this fraction (not shown). (E,F) Both free processed caspase-9 and the caspase-9 holoenzyme can rescue caspase-3 activation in extracts depleted of caspase-9. A total of 240 μl (6.4 mg) of active or inactive extracts was fractionated by sedimentation in sucrose gradients and caspase-9 was immunoprecipitated from each fraction and eluted with the epitope peptide. The amounts of caspase-9 and APAF-1 that coprecipitated with caspase-9 from the active extract were estimated in the eluates by immunoblotting (E,F). The eluates were added to aliquots of inactive extract that was immunodepleted of caspase-9. The aliquots were then incubated for 15 min at 37°C either with buffer (G,H) or dATP (I,J) and caspase-3 activity was measured with DEVD–AFC as a substrate.
Figure 3.

(A–C) Binding to APAF-1 increases the catalytic activity of caspase-9. A total of 1.5 ml (60 mg) of active extract was fractionated by sedimentation. Each fraction was divided into two aliquots, one of which was immunoprecipitated with anti-caspase-9–Sepharose, and the other mock precipitated with Sepharose. Both precipitates were eluted with the epitope peptide. (A) The amount of caspase-9 and APAF-1 in the anti-caspase-9–Sepharose eluate as revealed by immunoblotting. The activity of the eluates [(solid bars) anti-caspase-9, (open bars) mock] was measured using either caspase-3 precursor (B) or a IETD–AFC peptide substrate (C). The open bars in B are not seen because the activity of the mock precipitates was low. (D) The main components of the holoenzyme are APAF-1 and caspase-9. Catalytically inactive caspase-9 containing the Flag epitope tag was stably expressed in 293 cells. A total of 500 μl (20 mg) of extract from these cells was incubated with 1 mm dATP at 37°C for 10 min, and caspase-9 was precipitated using anti-Flag antibodies linked to agarose (Sigma). An equal amount of extract from parental 293 cells was treated identically and used for a control precipitation. The precipitates from both extracts were fractionated on a 10% or 15% (not shown) SDS–polyacrylamide gel, which was then stained with Coomassie blue. Caspase-9 and APAF-1 were identified by immunoblotting and immunoprecipitation (data not shown).
However, an alternative explanation could be that the free caspase-9 is inactivated for reasons other than separation from APAF-1. If our hypothesis is correct, free caspase-9 should regain its activity if allowed to reform a complex with APAF-1. One way of testing this is to add free caspase-9 to an extract depleted of caspase-9. Caspase-3 activation in this extract is abolished but should be restored by adding functional caspase-9. Caspase-9 precursor isolated from inactive extract rescued caspase-3 activation (Fig. 2E,G,I). As expected, the activation was rescued only in the presence of dATP, which is required for APAF-1 oligomerization (Fig. 2, cf. G and I). Similarly, free processed caspase-9 rescued caspase-3 activation and did it only in the presence of dATP, indicating that the free caspase-9 is not irreversibly inhibited but requires the APAF-1 oligomer to restore its function (Fig. 2F,H,J). In contrast to free caspase-9, the caspase-9–APAF-1 complex activated caspase-3 even without dATP, consistent with the notion that dATP is required for caspase-9 activation but not for the activity of the complex. Thus, these results indicated that the processed caspase-9 is active only in a complex with APAF-1, that is, caspase-9 and APAF-1 form a holoenzyme in which caspase-9 is the catalytic and APAF-1 the regulatory subunit.
Considering how other proteases are regulated, APAF-1 could increase caspase-9 activity by two mechanisms. APAF-1 may provide an additional binding site for caspase-3 (form a bridge), increase caspase-9 activity through a conformational change (by allosteric regulation), or both mechanisms could be involved. To evaluate these possibilities we compared the activity of the caspase-9 holoenzyme on caspase-3 and the peptide substrate IETD-AFC (7amino-4-triflouromethylcoumarin) which is derived from the caspase-9 cleavage site in caspase-3. We reasoned that this peptide is too small to bind simultaneously to the catalytic site of caspase-9 and an additional site on APAF-1. Thus, if both free caspase-9 and the holoenzyme cleave the peptide at the same rate, but only holoenzyme cleaves caspase-3, then APAF-1 provides a bridge. If both the peptide and caspase-3 are cleaved preferentially by the holoenzyme, then APAF-1 allosterically regulates caspase-9. With both substrates, the activity of the holoenzyme was greater than that of free caspase-9, consistent with the model that APAF-1 increases the catalytic activity of caspase-9 by allosteric regulation. Because free caspase-9 did cleave IETD-AFC, although at a lower rate than the holoenzyme, but did not process caspase-3, APAF-1 may also have some bridging effect. An intrinsic caveat of this experiment is that in principle an activity other than that of caspase-9 could copurify with the holoenzyme and be responsible for cleavage of IETD-afc.
We isolated the caspase-9 holoenzyme from cells that overexpressed catalytically inactive caspase-9, we used this approach to obtain amounts of the complex large enough to obtain protein sequence of unknown components should there be any. However, the only proteins detected were APAF-1 and caspase-9 (Fig. 3D). This does not rule out the possibility that weakly associated or labile proteins regulate the caspase-9 holoenzyme. Moreover, factors other than polypeptides may be involved, as in blood clotting where not only cofactor Va but also calcium and phospholipids are cofactors for the protease factor Xa. Cytochrome c is required for caspase-9 activation and was a part of the recombinant caspase-9–APAF-1 complex (Zou et al. 1999). We failed to detect cytochrome c in the holoenzyme not only by Coomassie staining but also by immunoblotting (Fig. 1E,F). This can be explained either by inadequate detection techniques or by the lack of cytochrome c in the holoenzyme. The role of cytochrome c may be limited to APAF-1 oligomerization and once APAF-1 oligomer is formed, cytochrome c may not be required for caspase-9 binding and activation.
Having established that caspase-9 is active as a holoenzyme in a cell-free system, we investigated which form of caspase-9 is active in apoptotic cells. We used extracts from IMR90-E1A cells (human fibroblasts transformed with adenoviral oncogene E1A (Fearnhead et al. 1998) that were treated with the chemotherapeutic drug etoposide. Extracts from untreated cells were used as a control. Caspase-9 precipitated from extracts of apoptotic cells processed caspase-3, whereas caspase-9 precipitated from control extracts did not (Fig. 4A). We then fractionated extracts by sedimentation, immunoprecipitated caspase-9 and measured its activity. As in the cell-free system, caspase-9 activity was greater in the fractions corresponding to the holoenzyme (Fig. 4D). However, we did not detect a distinct caspase-9–APAF-1 complex by immunoblotting (Fig. 4C), perhaps due to the low synchrony of apoptosis in cell populations or the low solubility of the complex in cells.
Figure 4.

Caspase-9 is active in cells as a holoenzyme. Human fibroblasts transformed with the adenoviral E1A oncogene (IMR90-E1A) were either treated with 50 μm etoposide for 18 hr (sixteen 15-cm plates) or left untreated (8 plates) and used to prepare extracts (Liu et al. 1996). (A) Only caspase-9 from treated cells is active. Caspase-9 was precipitated from the extracts and assayed for activity using caspase-3. (B,C) A total of 600 μl (9 mg) of each extract was fractionated by sedimentation in sucrose gradients, and the amount of caspase-9 and APAF-1 in the fractions evaluated by immunoblotting. (D) Caspase-9 was immunoprecipitated from the fractions, eluted with the epitope peptide, and the activity was measured as in A (14 hr incubation). (E) A model for caspase-9 activation and activity.
The finding that APAF-1 increases caspase-9 activity, and the observations that autocatalytic cleavage of caspase-9 is intramolecular (Zou et al. 1999) and that a recombinant caspase-9 mutated at the processing sites has partial activity in a cell-free system (Stennicke et al. 1999), suggest the following model for caspase-9 activation (Fig. 4E). A pro-apoptotic agent induces release of cytochrome c. Binding of cytochrome c to APAF-1 results in the nucleotide-dependent formation of an APAF-1 oligomer (Zou et al. 1999). This oligomer, but not free APAF-1, binds to caspase-9 and increases its activity through allosteric interaction. This activity is sufficient to carry out intramolecular processing resulting in fully active caspase-9. The APAF-1-bound and free caspase-9 remain in a steady-state equilibrium in which only the bound form is active.
Regulation of protease activity is hardly a new concept. For example, tissue factor initiates the blood clotting cascade by binding and allosterically activating factors VII and VIIa (Jesty and Nemerson 1995). The adenoviral protease (AVP) is activated by the stepwise binding to two cofactors, the viral DNA and to a peptide excised from a viral protein by the AVP–DNA complex (Mangel et al. 1997). The increase in protease activity by cofactors can range from severalfold to 1 million (Mann et al. 1992). The mechanism of allosteric regulation is demonstrated most convincingly by solving the structure of the protein complex and the free components. The structure of the complex between the caspase-9 and APAF-1 caspase recruitment domains (CARDs) is an important step in this direction (Qin et al. 1999). Considering how other protease precursors are activated (Khan and James 1998), a reasonable hypothesis is that APAF-1 removes the caspase-9 prodomain from its catalytic site.
What are the implications of caspase-9 being active as a holoenzyme? A practical implication is that in vitro screens for caspase-9 substrates and inhibitors, including potential therapeutics, should use the caspase-9 holoenzyme rather than caspase-9 alone. Another is that caspase-9 activity may be regulated even after caspase-9 processing, for example, by sequestering caspase-9 from the holoenzyme. Also, the activity of caspase-9 in a cell can be limited by the amount of APAF-1 oligomer. As a consequence, a catalytically inactive caspase-9 should be a competitive inhibitor of caspase-9 activity even if all caspase-9 in the cell is processed. This is consistent with the otherwise difficult to explain high efficiency of caspase-9 dominant-negative mutant and caspase-9 “decoys” as inhibitors of apoptosis (Fearnhead et al. 1998; Srinivasula et al. 1998, 1999; Seol and Billiar 1999). The proposed mechanism would also mean that a mutant caspase-9 may inhibit apoptosis even if expressed only by one allele in a tumor cell. Although caspase-9 mutations in tumors have not yet been reported, a missense mutation in one allele of the caspase-10 gene can cause apoptotic defects underlying autoimmune lymphoproliferative syndrome (ALPS) type II (Wang et al. 1999).
Oncogenes sensitize cells to apoptosis by facilitating the activation of caspase-9, and this facilitation is not restricted to cytochrome c release (Fearnhead et al. 1998; Juin, et al. 1999). We suggest that oncogenic transformation regulates the subsequent steps of caspase-9 activation, such as the formation of the caspase-9 holoenzyme or its activity, perhaps by regulating the concentration of the APAF-1 oligomer.
Considering structural similarities among initiator caspases and among the adaptor proteins (e.g., CED-4, APAF-1, and putative APAF-1 homolog FLASH (Zou et al. 1997; Imai et al. 1999) we speculate that initiator caspases other than caspase-9 are also activated allosterically and function as holoenzymes. The mechanism of induced activity may also apply to the caspases that are involved in processes other than apoptosis.
Materials and methods
Antibodies
The monoclonal antibodies to caspase-9 (1-2), caspase-3 (4-1-18), and APAF-1 (1-19) were described previously (Fearnhead et al. 1998). Antibody to cytochrome c was provided by Dr. Jemmerson (University of Minnesota Medical School, Minneapolis).
Assay for caspase-9 activity
Caspase-3 as a substrate
An aliquot of caspase-9 was supplemented with recombinant caspase-3 precursor to the final concentration of 400 nm and incubated at 37°C for the times indicated in the legends to figures 1–4. Three-microliter aliquots of the reactions were then added to 200 μl of caspase-3 assay buffer (40 μm DEVD-AFC in 10 mm HEPES, 0.5 mm EGTA, 10% glycerol at pH 7.0), and the rate of DEVD cleavage was measured with a fluorescence plate reader (Cytofluor, Perseptive Biosystems) and presented as arbitrary fluorescence units per minute (FU/min); 1 FU is equivalent to 0.65 pmole released AFC.
IETD-AFC as a substrate
An aliquot of 440 μm IETD-AFC was added directly to the eluates to a final concentration of 40 μm and incubated for 9 hr at 37°C. The increase in fluorescence was measured with the fluorescence plate reader and presented in arbitrary fluorescence units.
Immunoprecipitation and sedimentation in sucrose gradients
Extracts or extract fractions were incubated for 2 hr at 4°C with CNBr–Sepharose beads cross-linked to caspase-9 mAb 1-2 (3 mg/ml) that was affinity purified on caspase-9-Sepharose. A total of 10 μl of beads was used per 1 ml (∼25 mg) of extract or per a 300-μl fraction. The beads were washed three times with NPM (50 mm PIPES, 50 mm NaCl, 5 mm EGTA, 1 mm MgCl, 5 mm DTT at pH 7.0) supplemented with 5% sucrose. Bound caspase-9 was eluted for 2 hr at 4°C with 200 μg/ml epitope peptide in NPM supplemented with 1 mg/ml BSA and 5% sucrose.
Cell extract (5 mg in 150 μl) or immunoprecipitate was loaded onto a 5-ml tube containing 5%–20% sucrose gradient in NMP and sedimented at 4°C for 5 hr (100,000g) at 36,000 rpm. Fractions (300-μl) were collected from the top. BSA (4.5S, 66 kD), catalase (11.3S, 240 kD) and thyroglobulin (19.4S, 669 kD) were used as sedimentation markers. Several tubes were used to fractionate larger amounts of extract, and the fractions from each tube were combined.
Expression and purification of caspase-3 precursor
Caspase-3 precursor and its catalytically inactive mutant, both containing the amino-terminal His6 tag, were expressed in Escherichia coli from the pQE30 vector (Qiagen) and purified by chromatography on Ni-agarose.
Cell lines
293 cells expressing catalytically inactive caspase-9 were obtained using the Marx IV retroviral expression system (kind gift of Dr. Greg Hannon, Cold Spring Harbor Laboratory).
Acknowledgments
We thank Ximena Opitz-Araya for excellent technical help; Ryuji Kobayashi and Nora Poppito for helping to map the antibody epitope; Jeanne Wiggins for massive cell culture; Jonathan Hoffman (Harborfields High School) for extract preparation; and Tatsuya Hirano, Arne Stenlund, and Adrian Krainer for helpful discussion. J.R. is a Howard Hughes Medical Institute predoctoral fellow. Y.L. is a Pew Scholar. This work was supported by National Institutes of Health grant CA 13106-25 to Y.L.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lazebnik@cshl.org; FAX (516) 367-8461.
References
- Cain K, Brown DG, Langlais C, Cohen GM. Caspase activation involves the formation of the aposome, a large (approximately 700 kDa) caspase-activating complex. J Biol Chem. 1999;274:22686–22692. doi: 10.1074/jbc.274.32.22686. [DOI] [PubMed] [Google Scholar]
- Fearnhead HO, Rodriguez J, Govek EE, Guo W, Kobayashi R, Hannon G, Lazebnik YA. Oncogene-dependent apoptosis is mediated by caspase-9. Proc Natl Acad Sci. 1998;95:13664–13669. doi: 10.1073/pnas.95.23.13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, et al. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339–352. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- Hu Y, Benedict M, Ding L, Nunez G. Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J. 1999;18:3586–3595. doi: 10.1093/emboj/18.13.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Kimura T, Murakami A, Yajima N, Sakamaki K, Yonehara S. The CED-4 homologous protein FLASH is involved in Fas-mediated activation of caspase-8 during apoptosis. Nature. 1999;398:777–785. doi: 10.1038/19709. [DOI] [PubMed] [Google Scholar]
- Jesty J, Nemerson Y. The pathway of blood coagulation. In: Beutker E, Lichtman MA, Coller BS, Kipps TJ, editors. Williams hematology. New York, NY: McGraw-Hill; 1995. pp. 1227–1238. [Google Scholar]
- Juin P, Hueber AO, Littlewood T, Evan G. c-Myc-induced sensitization to apoptosis is mediated through cytochrome c release. Genes & Dev. 1999;13:1367–1381. doi: 10.1101/gad.13.11.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, James M. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 1998;7:815–836. doi: 10.1002/pro.5560070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang XD. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Mangel WF, Toledo DL, Ding JZ, Sweet RM, McGrath WJ. Temporal and spatial control of the adenovirus proteinase by both a peptide and the viral DNA. Trends Biochem Sci. 1997;22:393–398. doi: 10.1016/s0968-0004(97)01123-7. [DOI] [PubMed] [Google Scholar]
- Mann K, Krishnaswamy S, Lawson J. Surface-dependent hemostasis. Semin Hematol. 1992;29:213–226. [PubMed] [Google Scholar]
- Muzio M, Stockwell BR, Stennicke HR, Salvesen GS, Dixit VM. An induced proximity model for caspase-8 activation. J Biol Chem. 1998;273:2926–2930. doi: 10.1074/jbc.273.5.2926. [DOI] [PubMed] [Google Scholar]
- Qin H, Srinivasula SM, Wu G, Fernandes-Alnemri T, Alnemri ES, Shi Y. Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature. 1999;399:549–557. doi: 10.1038/21124. [DOI] [PubMed] [Google Scholar]
- Saleh A, Srinivasula SM, Acharya S, Fishel R, Alnemri ES. Cytochrome c and dATP-mediated oligomerization of apaf-1 is a prerequisite for procaspase-9 activation. J Biol Chem. 1999;274:17941–17945. doi: 10.1074/jbc.274.25.17941. [DOI] [PubMed] [Google Scholar]
- Seol DW, Billiar TR. A caspase-9 variant missing the catalytic site is an endogenous inhibitor of apoptosis. J Biol Chem. 1999;274:2072–2076. doi: 10.1074/jbc.274.4.2072. [DOI] [PubMed] [Google Scholar]
- Soengas MS, Alarcon RM, Yoshida H, Giaccia AJ, Hakem R, Mak TW, Lowe SW. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science. 1999;284:156–159. doi: 10.1126/science.284.5411.156. [DOI] [PubMed] [Google Scholar]
- Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1:949–957. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]
- Srinivasula SM, Ahmad M, Guo Y, Zhan Y, Lazebnik Y, Fernandes-Alnemri T, Alnemri ES. Identification of an endogenous dominant-negative short isoform of caspase-9 that can regulate apoptosis. Cancer Res. 1999;59:999–1002. [PubMed] [Google Scholar]
- Stennicke HR, Deveraux QL, Humke EW, Reed JC, Dixit VM, Salvesen GS. Caspase-9 can be activated without proteolytic processing. J Biol Chem. 1999;274:8359–8362. doi: 10.1074/jbc.274.13.8359. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- Wang J, Zheng L, Lobito A, Chan F, Dale K, Sneller M, Yao X, Puck J, Straus S, Lenardo M. Inherited human caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell. 1999;98:47–58. doi: 10.1016/S0092-8674(00)80605-4. [DOI] [PubMed] [Google Scholar]
- Yang X, Chang HY, Baltimore D. Essential role of CED-4 oligomerization in CED-3 activation and apoptosis. Science. 1998a;281:1355–1357. doi: 10.1126/science.281.5381.1355. [DOI] [PubMed] [Google Scholar]
- ————— Autoproteolytic activation of pro-caspases by oligomerization. Mol Cell. 1998b;1:319–325. doi: 10.1016/s1097-2765(00)80032-5. [DOI] [PubMed] [Google Scholar]
- Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- Zou H, Li Y, Liu X, Wang X. An APAF-1.Cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]