

Abstract
Complexins facilitate and inhibit neurotransmitter release through distinct domains, and their function was proposed to be coupled to the Ca2+ sensor Synaptotagmin-1. However, the mechanisms underlying Complexin function remain unclear. We now uncover an interaction between the Complexin N terminus and the SNARE complex C terminus, and show that disrupting this interaction abolishes the facilitatory function of Complexins in mouse neurons. Analyses of hypertonically induced exocytosis demonstrate that Complexins enhance synaptic vesicle fusogenicity. Genetic experiments crossing Complexin and Synaptotagmin-1 null mice indicate a functional interaction between these proteins, but also show that Complexins can promote Ca2+-triggered release in the absence of Synaptotagmin-1. We propose that the Complexin N terminus stabilizes the SNARE complex C terminus and/or helps release the inhibitory function of Complexins, thereby activating the fusion machinery in a manner that may cooperate with Synaptotagmin-1, but does not require Synaptotagmin-1.
Introduction
Neurotransmitter release is a key event in neuronal communication. Central components of the release apparatus are the SNARE proteins Syntaxin-1, Synaptobrevin-2, and SNAP-25, which form a stable four-helix bundle called the SNARE complex through their SNARE motifs1. Formation of the SNARE complex is key for membrane fusion2. In addition to the SNAREs, the exquisitely precise timing and high speed of neurotransmitter release require a diversity of specialized proteins including Complexins among others3,4.
Complexins constitute a family of neuronal SNARE complex-binding proteins5-8 that have no tertiary structure in solution9, but contain distinct functional domains (Fig. 1a). They bind to the tertiary SNARE complex with high affinity10-12 through a central α-helix13,14. This central α-helix is preceded by an accessory α-helix that does not contact the SNARE complex13. The precise role of Complexins in exocytosis has been controversial15. Genetic deletion of Complexin in Drosophila and knockdown of Complexins by RNA interference in mouse cortical neurons lead to a decrease in evoked release and an increase in spontaneous release16,17. In contrast, knockout of Complexins in mice causes a reduction in evoked and spontaneous release at multiple central synapses18-20, and a decrease in Ca2+-triggered exocytosis in adrenal chromaffin cells21. Moreover, in vitro lipid mixing and cell-cell fusion assays have suggested that Complexins inhibit22,23 or stimulate24,25 membrane fusion. These data suggest that Complexins may have both facilitatory and inhibitory functions.
Figure 1.

The N terminus of CplxI facilitates Ca2+-triggered neurotransmitter release. (a) Schematic diagram of CplxI domains. Numbers above the diagram indicate the boundary residues. (b,c,f) Representative traces of Cplx-KO neurons and Cplx-KO neurons rescued by CplxI WT, CplxI 16–134, or CplxI 8–134. The arrows represent stimulations. Artifacts and action potentials are blanked. (b) Basal evoked EPSCs. (c) Five consecutive EPSCs evoked at 20 ms inter-stimulus interval (ISI). (f) Evoked EPSCs in standard external solutions (4 mM Ca2+, 4 mM Mg2+) and in solutions with high Ca2+ concentrations (12 mM Ca2+, 1 mM Mg2+) or low Ca2+ concentrations (1 mM Ca2+, 1 mM Mg2+). (d,e,g,h) Bar graphs show the summary data of paired-pulse ratio (d), Pvr (e), EPSC amplitude potentiation by elevating Ca2+ concentration (g), and EPSC amplitude depression by lowering Ca2+ concentration (h). Data are normalized to the mean values of the corresponding CplxI WT rescue (black dashed lines and error bars). Data are expressed as mean ± SEM. **, P < 0.001; ***, P < 0.0001 compared to the corresponding CplxI WT rescue. The numbers of neurons analyzed are indicated on the bars.
Indeed, in vivo structure-function analyses and cross-species rescue experiments show that both murine and Drosophila Complexins contain similar functional domains that can facilitate or inhibit exocytosis26,27. Binding of the central α-helix to the SNARE complex is necessary but not sufficient for Complexin function21,26,27. The N terminus of Complexins facilitate release, whereas the accessory α-helix inhibits release26,27. Chimeric analyses reveal that the strength of facilitation or inhibition associated with each domain is different between murine and Drosophila Complexins, leading to apparently different phenotypes in mice and flies lacking Complexins27.
The emerging picture is that Complexins play dual roles in neurotransmitter release by activating and inhibiting vesicle fusion via distinct domains26. However, the molecular mechanism underlying the crucial facilitatory function of the Complexin N terminus is unclear. The N terminus of Complexins has been speculated to interact with lipid membranes17,26, but no such biochemical interaction has been reported. Furthermore, a widespread model proposes that Complexins clamp vesicles at the primed state and the Ca2+ sensor Synaptotagmin-1 releases the inhibition upon Ca2+ influx22,23,28,29. However, the nature of the interplay between Complexins and Synaptotagmin-1 in neurotransmitter release is still debated4,30, and it is unclear whether Complexin function is strictly coupled to Synaptotagmin-124,26.
To address these questions, we performed physiological and biophysical analyses of Complexin I (CplxI), a major paralog in the mouse brain. We find that the N terminus of CplxI binds to the C terminus of the SNARE complex, and mutations that disrupt this interaction specifically abolish the facilitatory function of CplxI in hippocampal neurons. We show that this interaction increases synaptic vesicle fusogenicity at a step that precedes Ca2+ triggering. Moreover, genetic experiments crossing Complexin and Synaptotagmin-1 null mice demonstrate that Complexins facilitate neurotransmitter release even in the absence of Synaptotagmin-1. Thus, Complexins may cooperate with Synaptotagmin-1 but can function independently of this Ca2+ sensor.
Results
To identify key functional residues in the CplxI N terminus, we employed a knockout-rescue approach26. We used cultured autaptic hippocampal neurons from CplxI/II double knockout (CplxI/II-DKO) or CplxI/II/III triple knockout (CplxI/II/III-TKO) mice because CplxI and CplxII are the major Complexin paralogs in the mouse brain and CplxI/II-DKO and CplxI/II/III-TKO hippocampal neurons show indistinguishable phenotypes19. CplxI/II-DKO and CplxI/II/III-TKO are referred together as Cplx-KO unless indicated otherwise.
Overexpression of wild-type (WT) CplxI completely rescues the transmitter release defects of CplxI/II-DKO neurons and does not interfere with release in WT neurons26. We therefore rescued Cplx-KO neurons by different CplxI variants and compared them to those rescued by WT CplxI (control). Western blotting showed that the expression levels of different CplxI variants are 4–7 fold of the endogenous CplxI level (Supplementary Fig. 1). Immunostaining revealed that all of the CplxI variants are diffusely distributed throughout both axonal and dendritic processes, and are present at presynaptic termini (Supplementary Fig. 2). To allow for comparisons of different CplxI variants across different rescue experiments, we normalized the data to the mean values of the corresponding control neurons (black dashed lines and error bars in Figures 1 and 2). We often included Cplx-KO neurons only expressing enhanced green fluorescent protein (EGFP) as negative controls. All the data from Cplx-KO neurons are pooled together and shown in Figures 1 and 2, as the normalized data of Cplx-KO neurons are not significantly different across experiments (P > 0.05 for all groups).
Figure 2.

Identification of Met5 and Lys6 as crucial residues for CplxI N-terminal function. (a) Helical wheel model of rat CplxI N terminus. Residues are numbered and labeled as orange (hydrophobic), red (negatively charged), and blue (positively charged). (b) N terminus amino acid sequence alignment of Complexin paralogs and orthologs. Identical residues are marked as red and highly conserved residues are marked as blue. Hs, Homo sapiens; Rn, Rattus norvegicus; Mm, Mus musculus; Lp, Loligo pealeii; Dm, Drosophila melanogaster; Ce, Caenorhabditis elegans. Asterisks mark residues Met5 and Lys6 in RnCplxI. (c–f) Summary data of evoked release from Cplx-KO neurons and Cplx-KO neurons rescued by WT or mutant CplxI. Bar graphs show paired-pulse ratio (c), Pvr (d), EPSC amplitude potentiation by elevating Ca2+ concentration (e), and EPSC amplitude depression by lowering Ca2+ concentration (f). Data are normalized to the mean values of the corresponding CplxI WT rescue (black dashed lines and error bars). Data are expressed as mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.0001 compared to the corresponding CplxI WT rescue. The numbers of neurons analyzed are indicated on the bars.
Facilitatory role of the CplxI N terminus
Deletion of the first 26 residues of CplxI (denoted as CplxI 27–134) abolishes its facilitatory function in CplxI/II-DKO neurons26. To narrow down the critical regions, we generated two additional N-terminal deletion mutants, CplxI 16–134 and CplxI 8–134. In response to an action potential, the excitatory postsynaptic current (EPSC) amplitude of Cplx-KO neurons is reduced about 50% when compared to WT neurons18,19. This defect is fully rescued by the overexpression of WT CplxI (ref. 26; Fig. 1b; Cplx-KO: 4.2 ± 0.3 nA, n = 147, WT CplxI rescue: 8.7 ± 0.6 nA, n = 142, P < 0.0001). In contrast, both CplxI 16–134 and CplxI 8–134 fail to rescue the decreased evoked release of Cplx-KO neurons (Fig. 1b; CplxI 16–134 rescue: 3.1 ± 0.4 nA, n = 25, corresponding WT CplxI rescue: 5.8 ± 1.0 nA, n = 18, P < 0.01; CplxI 8–134 rescue: 2.8 ± 0.3 nA, n = 37, corresponding WT CplxI rescue: 5.5 ± 0.6 nA, n = 30, P < 0.001).
In response to a train of high-frequency repetitive stimulations, the synaptic responses of WT neurons exhibit depression, whereas those of Cplx-KO neurons show initial facilitation (refs.18,19, Fig. 1c,d), indicating that the release probability is decreased in Cplx-KO neurons. Overexpression of WT CplxI converts synaptic facilitation to synaptic depression, whereas both CplxI 16–134 and CplxI 8–134 fail to rescue Cplx-KO neurons (Fig. 1c,d).
To directly assess the Ca2+-triggered release efficacy, we determined vesicular release probability (Pvr), the fraction of readily releasable pool (RRP) released by one action potential. We measured RRP sizes by hypertonic sucrose solution31. RRP size is not altered in Cplx-KO neurons18,19, and neither CplxI 16–134 nor CplxI 8–134 significantly changes RRP size (data not shown, P > 0.4 for both mutants when compared to WT CplxI). Consequently, overexpression of WT CplxI rescues the decreased Pvr of Cplx-KO neurons, whereas CplxI 16–134 and CplxI 8–134 do not (Fig. 1e).
To examine the apparent Ca2+ sensitivity of release, we changed the external standard Ca2+ concentrations (4 mM Ca2+, 4 mM Mg2+) to high Ca2+ concentrations (12 mM Ca2+, 1 mM Mg2+) or low Ca2+ concentrations (1 mM Ca2+, 1 mM Mg2+), and measured EPSC amplitude potentiation or depression, respectively (Fig. 1f). Cplx-KO neurons show greater potentiation or depression upon elevation or lowering of Ca2+ concentrations, respectively, than WT neurons, indicating a lower apparent sensitivity to Ca2+ 19,26. The reduced Ca2+ sensitivity of Cplx-KO neurons is rescued by WT CplxI, but not by either CplxI 16–134 or CplxI 8–134 (Fig. 1g,h). Pvr, paired-pulsed ratio, and apparent Ca2+ sensitivity of Cplx-KO neurons rescued by CplxI 16–134 or CplxI 8–134 do not significantly differ from those of Cplx-KO neurons (Fig. 1d,e,g,h, P > 0.05 for all parameters). Collectively, these results indicate that the N terminus is essential for CplxI to facilitate Ca2+-triggered neurotransmitter release, and the facilitatory function of CplxI involves a stretch of 7 residues at its very N terminus.
N-terminal point mutations impair CplxI facilitatory function
We next screened a series of point mutations in the very N terminus of CplxI, guided in part by the realization that the N terminus of CplxI can form an amphipathic α-helix (Fig. 2a). The sequence in this region is relatively conserved across paralogs and orthologs, and the potential to form an amphipathic α-helix is conserved in all of them (Fig. 2b).
Mutation of residues 3–6 (F3A V4A M5A K6Q) abolishes the rescue of Cplx-KO neurons in any of the measured parameters (Fig. 2c–f). Surprisingly, mutating the most conserved residue, Phe3 (ref. 8), to alanine (F3A) does not affect the ability of CplxI to rescue Cplx-KO neurons (Fig. 2c–f). However, mutating Met5 and Lys6, which are expected to contribute to the amphipathicity of the putative N-terminal α-helix, to glutamate (M5E K6E) completely abolishes rescue activity (Fig. 2c–f). A milder, single point mutation in the highly conserved residue Lys6 (K6Q) partially impairs the rescue (Fig. 2c–f). Hence, both Met5 and Lys6 are important for the CplxI facilitatory function, and this function is abrogated when both residues are mutated.
We also examined the effect of N-terminal mutations on spontaneous neurotransmitter release. Cplx-KO neurons show about 30% reduction in mEPSC frequency as compared to WT neurons19 and this defect can be rescued by overexpression of WT CplxI (Fig. 3). In contrast, the mEPSC frequencies of CplxI F3A V4A M5A K6Q- and CplxI M5E K6E-rescued Cplx-KO neurons are only about 15-25% of WT CplxI-rescued Cplx-KO neurons (Fig. 3a,b), showing that the N-terminal mutations further suppress mEPSC frequency in Cplx-KO neurons. Consistent with these results, a CplxI deletion mutant lacking the 26 N-terminal residues also strongly suppresses mEPSC frequency in Cplx-KO neurons (CplxI WT rescue: 10.4 ± 1.2 Hz, n = 33; CplxI 27-134 rescue: 3.0 ± 0.6 Hz, n = 37, P < 0.0001). The mEPSC amplitudes are slightly smaller in Cplx-KO neurons and mutant CplxI-rescued Cplx-KO neurons compared to WT CplxI-rescued Cplx-KO neurons, but only the CplxI F3A V4A M5A K6Q mutant reaches statistical significance (Fig. 3c). These data show that the N terminus of CplxI also facilitates spontaneous release and suggest that the N-terminal mutations do not affect the inhibitory function of Complexins.
Figure 3.

The CplxI N terminus facilitates spontaneous release. (a) Representative traces of mEPSCs from Cplx-KO neurons and Cplx-KO neurons rescued by WT or mutant CplxI. (b,c) Bar graphs show mEPSC frequency (b) and amplitude (c). Data are expressed as mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared to CplxI WT-rescued Cplx-KO neurons. The numbers of neurons analyzed are indicated on the bars.
The CplxI N terminus binds to the SNARE complex C terminus
The anti-parallel binding of the CplxI central α-helix to the SNARE complex13,14 positions the N terminus in close proximity to the site of membrane fusion. This observation and the potential of the CplxI N terminus to form a positively charged amphipathic α-helix (Fig. 2a) led us to hypothesize that, upon CplxI binding to the SNARE complex through the central α-helix, the CplxI N terminus may interact with the membrane(s). To test this hypothesis, we used nuclear magnetic resonance (NMR) spectroscopy.
Analysis of 15N-labeled CplxI binding to unlabeled SNARE complex-containing proteoliposomes by 1H-15N heteronuclear single quantum correlation (HSQC) spectra is challenging because SNARE complexes aggregate on membranes unless they are reconstituted at protein-to-lipid molar ratios of 1:1000 or lower30, resulting in difficulties to obtain high concentrations of reconstituted SNARE complexes. Moreover, severe overlap in the CplxI 1H-15N HSQC spectrum (Fig. 4) limits the number of cross-peaks that can be monitored, and often there are time-dependent changes in a few C-terminal cross-peaks that can be attributed to partial proteolysis. Despite these difficulties, 1H-15N HSQC experiments using 2 μM 15N-CplxI clearly show that the CplxI N terminus binds to SNARE complex-containing proteoliposomes, since addition of the proteoliposomes leads to broadening beyond detection of cross-peaks not only from the central α-helix but also from the N terminus (Fig. 4a,g,h). Cross-peaks that are still observable correspond to residues that remain flexible upon proteoliposome binding, which include the sequence connecting the N terminus of CplxI to the accessory α-helix.
Figure 4.

The CplxI N terminus binds to the SNARE complex and the M5E K6E mutation disrupts this interaction. (a,d,e) Expansions of 1H-15N HSQC spectra of WT CplxI alone (a,d,e, black contours) or in the presence of SNARE complex-containing proteoliposomes (a, red contours), plain liposomes (d, green contours), or soluble SNARE complex (e, red contours). Selected cross-peaks are labeled. Cross-peaks from residues arising from the expression vector are indicated with asterisks. (b) Expansions of 1H-15N HSQC spectra of WT CplxI alone (black contours) and CplxI M5E K6E alone (blue contours). New peaks from CplxI M5E K6E are labeled with “N”. (c,f) Expansions of 1H-15N HSQC spectra of CplxI M5E K6E alone (c,f, blue contours) or in the presence of SNARE complex-containing proteoliposomes (c, orange contours) or soluble SNARE complex (f, orange contours). (g–j) Bar diagrams showing the relative cross-peak intensities of 1H-15N HSQC spectra of CplxI alone (g) and after adding SNARE complex-containing proteoliposomes (h), plain liposomes (i), or soluble SNARE complex (j). The corresponding spectra are shown in panels (a,d,e). The relative intensities for each spectrum are obtained by dividing absolute intensities by the average of all cross-peak intensities. Only well-resolved cross-peaks are quantified. Cross-peaks that disappeared below the noise level are assigned zero intensity. Note that, because of limited sensitivity under the conditions of our experiments, the individual cross-peak intensities exhibit substantial variability in repeated experiments, but the overall relative intensity patterns conclusively illustrate the conclusions drawn in the text.
We next studied whether the M5E K6E mutation affects proteoliposome binding. Comparison of 1H-15N HSQC spectra of WT and M5E K6E mutant CplxI (Fig. 4b) shows that most cross-peaks are unaltered by the mutation. Several cross-peaks from the N terminus of WT CplxI are not observed for the M5E K6E mutant, while new cross-peaks from the mutant N terminus appear (labeled by “N” in Fig. 4b). Therefore, the mutation causes natural perturbations in the mutated and neighboring residues but does not alter most of the protein, as expected from the lack of tertiary structure in isolated CplxI9. Importantly, the N-terminal cross-peaks of the M5E K6E mutant are not perturbed by addition of SNARE complex-containing proteoliposomes (Fig. 4c), whereas cross-peaks from the central α-helix still broaden beyond detection. Hence, the M5E K6E mutation preserves the binding of the CplxI central α-helix to the SNARE complex, but disrupts the interaction of the N terminus with the proteoliposomes.
To determine whether binding of the CplxI N terminus to the proteoliposomes involves the SNARE complex or the phospholipids, we acquired a 1H-15N HSQC spectrum of WT CplxI in the presence of plain liposomes, but the spectrum remains largely unaltered (Fig. 4d,g,i). Hence, CplxI does not bind to membranes with the lipid composition used in our experiments, which includes a fraction of negatively charged phospholipids (15%) that is close to physiological. Note that CplxI was recently reported to bind to membranes32, but the liposomes used in that study contain a much higher fraction of negatively charged phospholipids (35%). Importantly, 1H-15N HSQC spectra of WT CplxI in the presence of soluble SNARE complex reveal broadening beyond detection of the cross-peaks from the central α-helix and the N terminus of CplxI (Fig. 4e,g,j), showing that the N terminus interacts with the SNARE complex. Moreover, the N-terminal cross-peaks of the CplxI M5E K6E mutant are unperturbed upon addition of the soluble SNARE complex (Fig. 4f), showing that the mutation disrupts binding of the CplxI N terminus to the SNARE complex.
To test our conclusions by a different method, we used fluorescence spectroscopy with a CplxI mutant labeled at residue 12 with 7-nitrobenz-2-oxa-1,3-diazole (NBD), a probe that is highly sensitive to the environment33. The fluorescence spectrum of NBD-labeled CplxI mutant exhibits a substantial increase in intensity upon addition of the SNARE complex-containing proteoliposomes (Supplementary Fig. 3), which is much less dramatic than that observed upon insertion of NBD into a phospholipid bilayer33 and is compatible with an alteration in probe environment due to SNARE complex binding. Indeed, the soluble SNARE complex induces the same increase in NBD fluorescence intensity as the proteoliposomes, whereas plain phospholipids do not induce any significant change (Supplementary Fig. 3), confirming that the CplxI N terminus binds to the SNARE complex.
To map the approximate binding site of the CplxI N terminus on the SNARE complex, we used 1H-15N transverse-relaxation optimized spectroscopy (TROSY)-enhanced HSQC spectra of four separate SNARE complex samples where each SNARE motif was individually 2H,15N-labeled. We measured the paramagnetic broadening effects (PBEs) induced on the SNARE complex by the binding of a CplxI fragment (residues 2–82) bearing an A12C mutation and labeled at residue 12 with the paramagnetic probe (1-oxyl-2,2,5,5-tetramethyl-3-pyrroline-3-methyl)methanethiosulfonate (MTSL)34 (Fig. 5). For this purpose, we compared spectra acquired before and after reduction of MTSL with dithionite, which eliminates the broadening effects. Fig. 5a illustrates the data obtained for the SNARE complex with 2H,15N-labeled SNAP-25 C-terminal SNARE motif. Mapping of the PBEs onto the crystal structure of the CplxI 26–83/SNARE complex (Fig. 5b) indicates that residue 12 of CplxI is located at the very C terminus of the SNARE complex, a natural location based on the position of the accessory α-helix and the fact that this helix is flexibly linked to the N terminus.
Figure 5.
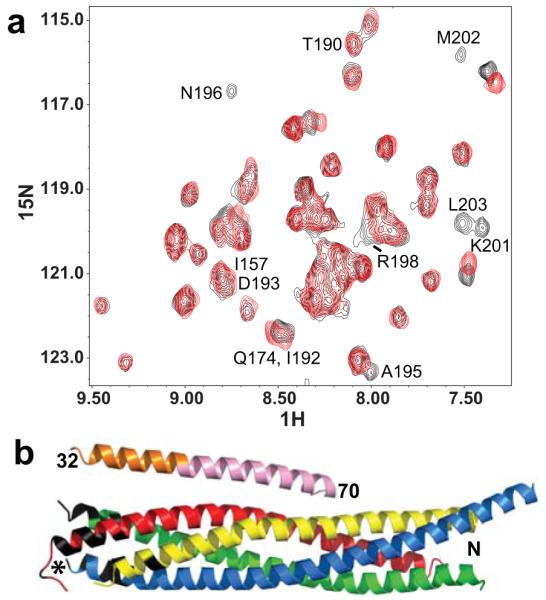
The CplxI N terminus binds to the C terminus of the SNARE complex. (a) Expansions of 1H-15N TROSY-HSQC spectra of SNARE complex with the SNAP-25 C-terminal SNARE motif 2H,15N-labeled, in the presence of CplxI 2–82 A12C-MTSL before (red contours) and after (black contours) reduction with dithionite. Selected cross-peaks are labeled with the residue number. (b) Ribbon diagram of the crystal structure of the CplxI 26–83/SNARE complex13 illustrating the PBEs caused by CplxI 2–82 A12C-MTSL on the 1H-15N TROSY-HSQC cross-peaks of the SNARE complex. CplxI is colored pink (central α-helix) and orange (accessory α-helix), Synaptobrevin-2 red, Syntaxin-1 yellow, and SNAP-25 blue and green. Residues in black correspond to well-resolved cross-peaks whose intensities decrease by more than 50% due to the PBEs induced by MTSL (residues 78, 81, 82, 86, 87 and 90 of Synaptobrevin-2, residues 242 and 247 of Syntaxin-1, and residues 73-75, 78, 80, 195, 196, 198–204 of SNAP-25). All these residues are clustered at the C terminus of the SNARE complex, and the cross-peaks from other residues in this region are not observable or overlapped. We estimate that the amide protons of these residues are approximately within 18 Å or less from the probe34. The asterisk indicates the estimated position of the MTSL probe in the bound CplxI 2–82 A12C-MTSL.
Cplx regulates the fusogenicity of synaptic vesicles
Our findings suggest a simple model whereby binding of the CplxI N terminus provides critical stabilization to the SNARE complex C terminus, helping the SNAREs to exert mechanical force on the membranes to induce fusion. Binding of the CplxI N terminus may also help remove the inhibition caused by the CplxI accessory α-helix. Both models are compatible and lead to important functional predictions. For instance, the stabilization model suggests that Complexins may increase the propensity of primed synaptic vesicles to fuse, a notion that is already supported by our spontaneous release data (Fig. 3). Moreover, both models predict that Complexin function may not strictly depend on Synaptotagmin-1 and Ca2+.
To test these hypotheses, we analyzed hypertonically-induced synaptic vesicle fusion, a Ca2+-independent process. The magnitude and kinetics of the release reflect the fusogenicity of vesicles31,35-37. While the standard saturating 500 mM sucrose solution depletes all fusion-competent vesicles in WT and Cplx-KO neurons18,19, we noted a slight slowing of the release kinetics and a slight reduction of peak amplitudes in Cplx-KO neurons when we averaged all responses and overlaid the traces from the different groups (Fig. 6a). This observation is consistent with our hypothesis that the fusogenicity of synaptic vesicles is reduced in Cplx-KO neurons.
Figure 6.

Complexins regulate synaptic vesicle fusogenicity. (a) Average traces of synaptic responses induced by sucrose solutions from control (500 mM, n = 82; 250 mM, n = 27), CplxI/II-DKO (500 mM, n = 78; 250 mM, n = 25), and CplxI/II/III-TKO (500 mM, n = 78; 250 mM, n = 28) neurons. (b,c) Summary data of 250 mM sucrose solution-induced response onset latency (b), peak release rate and fraction of RRP released (c) from control, CplxI/II-DKO, and CplxI/II/III-TKO neurons. Data are expressed as mean ± SEM; *, P < 0.05; ***, P < 0.001 compared to control. Control neurons are either CplxII−/− or CplxII−/− CplxIII+/−. (d,e) Summary data of 250 mM sucrose solution-induced response onset latency (d), peak release rate and fraction of RRP released (e) from Cplx-KO neurons and Cplx-KO neurons rescued by WT or mutant CplxI variants. Data are expressed as mean ± SEM; **, P < 0.01; ***, P < 0.001 compared to CplxI WT-rescued Cplx-KO neurons. The numbers of neurons analyzed are indicated in the figures.
As a more sensitive assay, we measured the response onset latency, peak release rate, and the fraction of RRP released (see Methods) by an intermediate sucrose concentration (250 mM), which causes smaller response amplitudes with slower kinetics (refs. 36,37; Fig. 6a). We found that, compared to control neurons, the response onset is delayed about 150 ms (Fig. 6b), and the peak release rate and the fraction of RRP released are reduced by 50% (Fig. 6c) in Cplx-KO neurons. Similar results are also observed in autaptic striatal GABAergic neurons (Supplementary Fig. 4) and mass-cultured hippocampal neurons (Supplementary Fig. 5). These data indicate that, in the absence of Complexins, the fusogenicity of vesicle decreases, i.e. the energy required for vesicle fusion increases in mouse neurons. Thus, murine Complexins exert a facilitatory action independent of Ca2+ triggering.
In contrast to the facilitating function of murine Complexins, Drosophila Complexin (dmCplx) practically abolishes both evoked and spontaneous release in Cplx-KO neurons27, indicating that dmCplx decreases vesicle fusogenicity. Indeed, we found that the onset of the synaptic response induced by 500 mM sucrose solution is delayed about 290 ms (Supplementary Fig. 6a), and the peak release rate is reduced by 40% (Supplementary Fig. 6b) in Cplx-KO neurons expressing dmCplx, compared to Cplx-KO neurons expressing EGFP. Furthermore, 250 mM sucrose solution is barely able to induce release in Cplx-KO neurons expressing dmCplx (Supplementary Fig. 6a,c,d), consistent with a drastic increase in energy required for vesicle fusion. Altogether, these experiments indicate that Complexins regulate synaptic vesicle fusogenicity.
We next studied the role of the interactions of the CplxI central α-helix and N terminus with the SNARE complex in regulating vesicle fusogenicity. The delayed onset, the decreased peak release rate, and the reduced fraction of RRP released in Cplx-KO neurons are rescued by overexpression of WT CplxI (Fig. 6d,e). In contrast, a mutant CplxI carrying two mutations (K69A Y70A) in the central α-helix, which neither binds to the SNARE complex nor rescues the evoked release of Cplx-KO neurons26, is unable to rescue Cplx-KO neurons in response onset latency, peak release rate, and the fraction of RRP released by 250 mM sucrose solution (Fig. 6d,e). Hence, the ability of CplxI to boost vesicle fusogenicity requires binding to the SNARE complex. Finally, a CplxI bearing the M5E K6E mutation, which disrupts the binding of the CplxI N terminus to the SNARE complex, also fails to rescue Cplx-KO neurons (Fig. 6d,e and Supplementary Fig. 5). Thus, the CplxI N-terminal interaction with the SNARE complex plays an essential role in augmenting synaptic vesicle fusogenicity.
Cplx functions independently and cooperatively with Syt1
A key prediction of both the stabilization and the disinhibition models is that Complexins may function independently of Synaptotagmin-1 during Ca2+-triggered release, even if their function is normally coupled to this Ca2+ sensor. To test this hypothesis, we compared the Ca2+-evoked release efficacy of Synaptotagmin-1-deficient neurons38 to that of neurons lacking both Synaptotagmin-1 (Syt1) and Complexins.
We generated Syt1/CplxI/CplxII-TKO mice, and they died shortly after birth, sooner than Syt1-KO and CplxI/II-DKO mice, suggesting a more severe phenotype than Syt1-KO and CplxI/II-DKO mice. The synaptic characteristics of CplxII-KO neurons are indistinguishable from WT neurons18. We thus used CplxII-KO neurons as controls and compared them to CplxI/CplxII-DKO, Syt1/CplxII-DKO, and Syt1/CplxI/CplxII-TKO neurons. The fast, synchronous evoked EPSCs are reduced in CplxI/CplxII-DKO neurons, and are diminished in Syt1/CplxII-DKO and Syt1/CplxI/CplxII-TKO neurons compared to the control neurons (Fig. 7a,b). The RRP sizes are not significantly different among all groups (P > 0.05, data not shown). The Pvrs of CplxI/CplxII-DKO and Syt1/CplxII-DKO neurons are about 58% and 33% of the control value, respectively (Fig. 7c). Importantly, the Pvr of Syt1/CplxI/CplxII-TKO is further reduced to 18% of the control value and 56% of the Syt1/CplxII-DKO value (P < 0.01 for Syt1/CplxI/CplxII-TKO versus Syt1/CplxII-DKO). These results indicate that both Complexins and Synaptotagmin-1 regulate the efficacy of Ca2+-triggered release, and that Complexins can facilitate Ca2+-evoked release independently from Synaptotagmin-124,26, although this does not imply that Complexins bind Ca2+.
Figure 7.

Complexins facilitate Ca2+-evoked neurotransmitter release independently and cooperatively with Synaptotagmin-1. (a) Representative evoked EPSC traces of neurons from various genotypes. The arrows represent stimulations. Artifacts and action potentials are blanked. (b,c) Bar graphs show EPSC amplitude (b) and Pvr of evoked release (c). CplxII-KO neurons serve as control, as they are indistinguishable from WT neurons. Data are normalized to the mean values of control neurons. Genotypes are indicated below the bars. (d) Pvr of evoked release from WT and Syt1 heterozygous neurons. (e) Pvr of evoked release from CplxI/II-DKO and Syt1-Het/CplxI-KO/CplxII-KO neurons. Data are expressed as mean ± SEM; *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared to control neurons (b,c) or to CplxI/II-DKO neurons (e). The numbers of neurons analyzed are indicated above the bars.
However, we do find evidence indicating a genetic interaction between Complexins and Synaptotagmin-1. Loss of one functional Syt1 allele has no effect on evoked release, as the Pvr of Syt1 heterozygous neurons is similar to that of WT neurons (Fig. 7d, P = 0.9). However, Syt1 heterozygosity reduces the Pvr of evoked release in the absence of Complexins because the Pvr of Syt1-Het/CplxI-KO/CplxII-KO neurons decreases about 36% compared to CplxI/CplxII-DKO neurons (Fig. 7e). Conversely, CplxI heterozygosity does not significantly reduce Pvr in Syt1/CplxII-DKO neurons (Pvr: Syt1-KO/CplxI-Het/CplxII-KO, 3.8 ± 0.5% verse Syt1-KO/CplxII-KO, 4.2 ± 0.3%, P = 0.5). These data show that half the amount of Synaptotagmin-1 is normally sufficient to maintain WT-like evoked release but becomes insufficient in the absence of Complexins, and indicate that Complexins and Synaptotagmin-1 may functionally interact in Ca2+-triggered release.
Discussion
Complexin function was initially associated with two hallmarks: a specific active role in the Ca2+ triggering step of release18 and the binding of its central α-helix to the SNARE complex13,14. It later became clear that Complexin function involves both facilitatory and inhibitory functions. A popular model predicts that Complexins inhibit release before Ca2+ influx and Synaptotagmin-1 releases this inhibition22,23,28, but it is difficult to account for the facilitatory function of Complexins based solely on this model. The finding that the Complexin N terminus is essential for its function showed that a crucial interaction for Complexins remained to be uncovered26. The Complexin N terminus was recently proposed to assist the SNAREs in exerting force on the membranes17, but no biochemical data supporting this notion was described. Our data now reveal three novel aspects fundamental for understanding Complexin function: the interaction of the Complexin N terminus with the SNARE complex C terminus, the identification of a role for Complexins in a priming step before Ca2+ influx, and the observation that Complexin function can be at least partially separated from the action of Synaptotagmin-1. Our hypertonic-sucrose data indicate that Complexins facilitate release by lowering the energy required for fusion, thus enhancing vesicle fusogenicity. This provides a natural explanation for why Complexins facilitate both evoked and spontaneous release in mouse hippocampal neurons19. Moreover, we identify two residues in the CplxI N terminus that are essential for binding to the SNARE complex C terminus and for the active function of Complexins. This correlation suggests that Complexins enhance vesicle fusogenicity by binding to the SNARE complex not only through the central α-helix but also through the N terminus.
Since binding of Complexins to the SNARE complex through the central α-helix places Complexins right at the site of fusion, the SNAREs and the membranes are natural targets for the Complexin N terminus to perform its function. Indeed, the stimulatory effects of Complexins in some reconstitution experiments24,25 indicate that their active function can be at least partially recapitulated in these in vitro systems containing only lipids and SNAREs. Our results strongly favor the notion that binding of the Complexin N terminus to the SNARE complex C terminus underlies in part its active role in membrane fusion and neurotransmitter release. However, we cannot rule out the possibility that, in the context of trans-SNARE complexes assembled between two membranes, the Complexin N terminus may also interact with the lipids.
The finding that the M5E K6E mutation abrogates the Complexin facilitatory function in each of the electrophysiological parameters that we measured (Figs. 2, 3, and 6) highlights the functional importance of the Complexin N terminus. We propose that the interaction between the Complexin N terminus and the SNARE complex C terminus plays two roles (Fig. 8). First, since the C terminus of the SNARE complex is not very stable13, and its assembly is expected to help overcome the energy barrier for membrane fusion, our findings naturally lead to the notion that the Complexin N terminus may promote membrane fusion by stabilizing the SNARE complex C terminus. Second, the Complexin N terminus may help release the inhibition caused by the accessory α-helix, switching the release machinery from an inhibited state (Fig. 8b) to an activated state (Fig. 8c,d). This model can explain the stimulatory24,25 and inhibitory22,23 effects of Complexins in reconstitution assays, and is supported by multiple physiological observations.
Figure 8.

Proposed model for the key facilitatory function of the Complexin N terminus. (a) The SNARE complex (Synaptobrevin-2, red; Syntaxin-1, yellow; and SNAP-25, green) assembles partially during priming. “N” and “C” indicate the N and C termini, respectively. (b–d) Complexins bind to the SNARE complex in at least two different modes that correspond to an inhibited (b) and an activated state (c,d). (b) The Complexin central α-helix binds to the partially assembled SNARE complex and helps to assemble the SNARE complex further. At the same time, the Complexin accessory α-helix replaces the C terminus of the Synaptobrevin-2 SNARE motif in the four-helix bundle, preventing C-terminal assembly26,41. (c) The Complexin accessory α-helix is released from the SNARE complex by the Complexin N terminus, Synaptotagmin-1/Ca2+, or both, allowing full assembly of the SNARE complex C terminus. (d) The SNARE complex is fully assembled. Binding of the Complexin N terminus to the SNARE complex C terminus is proposed to help releasing the inhibition of the accessory α-helix and/or to stabilize the C terminus of the SNARE complex to assist in exerting force on the membranes. Note that the location of the Complexin N terminus is only tentative in (c) and (d). The Complexin C terminus is not shown for simplicity.
The finding that removal of the N terminus abolishes the rescue activity of CplxI, but deletion of the N terminus and the accessory α-helix leads to partial rescue26, together with examination of the crystal structure of CplxI 26–83 bound to the SNARE complex (Fig. 5b), suggested that the accessory α-helix may inhibit release by replacing the C terminus of the Synaptobrevin-2 SNARE motif in the four-helix bundle, thus hindering C-terminal assembly of the SNARE complex (Fig. 8b; ref. 26). This proposal has recently been supported by the finding that Complexins bind to Syntaxin-1/SNAP-25 complexes24,39,40 and by cell-cell fusion data41. Our model postulates that the Complexin N terminus helps release the inhibition by the accessory helix, which is supported by the recent EPR data42 and could arise because binding of the Complexin N terminus to the SNARE complex may require dissociation of the accessory α-helix. However, this disinhibition activity alone cannot explain that removal of the CplxI N terminus and accessory α-helix leads to only partial rescue26. Since the Complexin central α-helix provides some stabilization for the SNARE complex13 but this effect is not sufficient for the Complexin facilitatory function26, our model proposes that the activating function of the Complexin N terminus involves stabilization of the SNARE complex C terminus. Note that in our model binding of the Complexin central α-helix to the SNARE complex also plays a key role by strategically positioning the accessory α-helix and the N terminus near their binding sites on the SNAREs26.
This model also provides a clear explanation for the notion that Complexin function does not depend strictly on Synaptotagmin-1/Ca2+, as shown by the findings that lack of Complexins leads to impaired spontaneous release (Fig. 3; ref. 19), decreased sensitivity of release to 250 mM sucrose (Fig. 6), and exacerbation of the impairment of release observed in the absence of Synaptotagmin-1 (Fig. 7a–c). Several studies indicated a tight link between the functions of both proteins22,23,28, and biophysical data28 indicated that Complexin is displaced from the SNARE complex by Synaptotagmin-1, but this competition may only be partial and both proteins may be able to bind simultaneously to the SNARE complex30. Moreover, the key role of the N terminus for Complexin function argues strongly against the notion that Complexins are released from the SNARE complex26. In our model, the stabilizing effect of the Complexin N terminus on the SNARE complex C terminus is Ca2+-independent and the release of the inhibition of the accessory α-helix can be promoted by the Complexin N terminus without an absolute requirement for Synaptotagmin-1 and Ca2+. In fact, although most current models assume that the transition from state B to state C in Fig. 8 occurs upon Ca2+ influx, an alternative possibility is that these two states are in equilibrium in primed vesicles before Ca2+ influx. In our model, vesicles in all states can undergo spontaneous, hypertonically-induced, or Ca2+-triggered fusion, and Complexins increase fusogenicity of all states. This proposal can explain the observation that Complexins and Synaptotagmin-1 interact genetically and yet their functions are not strictly interdependent. Nevertheless, Synaptotagmin-1 could still help promoting the transition from state B to state C upon Ca2+ influx, cooperating with the Complexin N terminus in releasing the accessory α-helix from the SNARE complex.
Clearly, further research will be required to fully elucidate how Complexins function, but it now seems clear that Complexins orchestrate synaptic vesicle fusion through built-in facilitatory and inhibitory mechanisms, and that the facilitatory function depends on their N-terminal interaction with the C terminus of the SNARE complex.
Methods
Viral constructs and production
WT rat CplxI was used to generate all mutant CplxI variants26. Myc-tagged CplxI variants were generated by fusing a linker (SGGSGGTGG) followed by a c-Myc (EQKLISEEDL) to the C terminus of CplxI variants. The cDNAs were subsequently cloned into a Semliki-Forest virus vector described previously26. A lentiviral vector expressing WT Drosophila Cplx was described previously27,43. The corresponding vectors without CplxI or dmCplx served as control constructs. Virus production and infection were performed as described27,44 with modification. The procedures are described in detail in Supplementary Methods online.
Mice, neuronal culture, Western blotting, and immunocytochemistry
CplxI/II-DKO and CplxI/II/III-TKO mice were obtained as described18,19. Syt1/CplxI/CplxII-TKO mice were obtained by interbreeding of mice homozygous for the CplxII mutations, and heterozygous for both the Syt1 mutation38 and the CplxI mutation. All procedures to maintain and use these mice were approved by the Institutional Animal Care and Use Committee for Baylor College of Medicine and Affiliates. Primary neuronal culture, Western blotting, immunocytochemistry, and confocal microscopy were performed as described26.
Electrophysiology of cultured neurons
Whole-cell voltage clamp recordings were performed at room temperature (23–24°C) on cultured neurons during 9–14 day in vitro as described19. SFV rescue experiments were performed between 9 hours and 12 hours after neurons were infected with SFV. For hippocampal glutamatergic neurons, the patch pipette solution contained (mM): K-Gluconate, 146; HEPES, 17.8; EGTA, 1; MgCl2, 0.6; ATP-Mg, 4; GTP-Na, 0.3; Phosphocreatine, 12; Phosphocreatine kinase, 50U ml−1; 300 mOsm; pH 7.4. For striatal GABAergic neurons, the patch pipette solution contained (mM): KCl, 136; HEPES, 17.8; EGTA, 1; MgCl2, 0.6; ATP-Mg, 4; GTP-Na, 0.3; Phosphocreatine, 12; Phosphocreatine kinase, 50U ml−1; 300 mOsm; pH 7.4. The standard extracellular solution contained (mM): NaCl, 140; KCl, 2.4; HEPES, 10; glucose, 10; CaCl2, 4; MgCl2, 4; 300 mOsm; pH 7.4. For Ca2+-sensitivity experiments, Mg2+ concentration in extracellular solution was kept at 1 mM and Ca2+ concentration was 12 or 1 mM. Hypertonic sucrose solutions were made by adding 500 mM or 250 mM sucrose to the standard extracellular solution.
Basal evoked EPSC, RRP, paired-pulse ratio, apparent Ca2+-sensitivity of release, and mEPSC were measured as described19,26 with minor modifications. The details are described in Supplementary Methods.
To measure the synaptic vesicle fusogenicity, 250 mM sucrose solution were directly applied onto the neuron for 10 s (hippocampal glutamatergic neurons) or 12 s (striatal GABAergic neurons). The charge transfer of the transient synaptic current was measured and divided by the RRP size from the same neuron to obtain the fraction of RRP released by 250 mM sucrose solution. The onset of sucrose response was visually identified. The time from the onset of the sucrose solution application to the onset of the response was measured (T1). An “open tip” response was used to determine the time for sucrose solution to reach the neuron (T2). The sucrose response onset latency was defined as the interval between the time when sucrose solution reaches the neuron and the onset time of sucrose response (T1-T2). To determine the peak release rate, synaptic current traces were first digitally filtered at 10 Hz. The peak amplitude of sucrose response was then divided by the RRP size from the same neuron to obtain the peak release rate. For the experiments in hippocampal mass-cultured neurons, 0.5 μM tetrodotoxin and 15 μM gabazine were included in the sucrose solutions. Data were analyzed offline using AxoGraph X (AxoGraph Scientific). Statistic significances were tested using Student’s t-test, the nonparametric Mann-Whitney test, or one-way analysis of variance.
Recombinant protein expression and purification
Full-length rat Syntaxin-1 and rat SNAP-25 used to generate the membrane-bound SNARE complex were coexpressed and purified as described previously45,46. A fragment of rat Synaptobrevin-2 (residues 1–96) was used for membrane-bound SNARE complex. For soluble SNARE complexes, we used fragments spanning rat Syntaxin-1A (residues 191–253), rat Synaptobrevin-2 (residues 29–93), human SNAP-25 (residues 11–82), and human SNAP-25 (residues 141–203). All fragments were expressed and purified as described13,30. Full-length rat CplxI and its mutants were expressed and purified as described9. SNARE complexes were assembled as described12. Labeling of CplxI 2–82 A12C with MTSL was performed as described34.
Liposome reconstitution
Liposome reconstitution was performed using the direct method46 with a lipid mixture of 85% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine and 15% 1,2-dioleoyl-sn-glycero-3-[phospho-L-serine] (w/w) (Avanti Polar Lipids). The protein: lipid molar ratio was 1:1000.
NMR spectroscopy
All NMR spectra were performed on Varian INOVA 600 MHz spectrometers. Samples for experiments with proteoliposomes and control contained 2 μM 15N CplxI without or with proteoliposomes (3 μM SNARE complex, 3 mM lipid), in buffer containing 20 mM HEPES, 100 mM KCl, 1 mM EDTA and 1 mM TCEP. Acquisition time for each 1H-15N HSQC spectrum was approximately 12 hours on a cold probe. Additional 1H-15N spectra were acquired with samples containing 5 μM 15N CplxI and 3 mM lipids, or 80 μM 15N-CpxI and 120 μM SNARE complex. 1H-15N TROSY-HSQC spectra of soluble SNARE complexes where each SNARE motif was individually 2H,15N-labeled (ca. 80 μM) were acquired as described13 in the presence 100 μM CplxI 2–82 A12C-MTSL before and after addition of 1 mM dithionite.
Fluorescence spectroscopy
Fluorescently labeled CpxI A12C C105S was obtained by reacting CplxI A12C C105S with the cysteine-specific reagent N,N’-dimethyl-N-(iodoacetyl)-N’-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) ethylenediamine (NBD) as described33. Fluorescence spectra were acquired on a PTI spectrofluorimeter with excitation at 465 nm using 50 nM NBD-labeled CpxI A12C C105S alone or in the presence of 2 μM soluble SNARE complex, SNARE complex proteoliposomes (3 μM SNAREs, 3 mM lipids), or plain liposomes (3 mM lipids).
Supplementary Material
Acknowledgements
We thank Hui Deng, Hongmei Chen, and Xuan Zhu for technical assistances, Kerstin Reim and Nils Brose (Max Planck Institute of Experimental Medicine, Germany) for providing CplxI-KO, CplxII-KO and CplxIII-KO mice, and Thomas Sudhof (Stanford University, USA) for providing Syt1-KO mice. This work was supported by the US National Institute of Health (F31MH078678 to H.T.C., NS037200 to J.R., and NS050655 to C.R.), the Welch Foundation (grant I-1304 to J.R.), Baylor Research Advocates for Student Scientists and a McNair Fellowship (both to H.T.C.), and Baylor College of Medicine Mental Retardation and Developmental Disabilities Research Center.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
References
- 1.Wickner W, Schekman R. Membrane fusion. Nat Struct Mol Biol. 2008;15:658–64. doi: 10.1038/nsmb.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jahn R, Scheller RH. SNAREs - engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–43. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- 3.Wojcik SM, Brose N. Regulation of membrane fusion in synaptic excitation-secretion coupling: speed and accuracy matter. Neuron. 2007;55:11–24. doi: 10.1016/j.neuron.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 4.Rizo J, Rosenmund C. Synaptic vesicle fusion. Nat Struct Mol Biol. 2008;15:665–74. doi: 10.1038/nsmb.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McMahon HT, Missler M, Li C, Sudhof TC. Complexins: cytosolic proteins that regulate SNAP receptor function. Cell. 1995;83:111–9. doi: 10.1016/0092-8674(95)90239-2. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi S, et al. Identification of two highly homologous presynaptic proteins distinctly localized at the dendritic and somatic synapses. FEBS Lett. 1995;368:455–60. doi: 10.1016/0014-5793(95)00713-j. [DOI] [PubMed] [Google Scholar]
- 7.Ishizuka T, Saisu H, Odani S, Abe T. Synaphin: a protein associated with the docking/fusion complex in presynaptic terminals. Biochem Biophys Res Commun. 1995;213:1107–14. doi: 10.1006/bbrc.1995.2241. [DOI] [PubMed] [Google Scholar]
- 8.Reim K, et al. Structurally and functionally unique complexins at retinal ribbon synapses. J Cell Biol. 2005;169:669–80. doi: 10.1083/jcb.200502115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pabst S, et al. Selective interaction of complexin with the neuronal SNARE complex. Determination of the binding regions. J Biol Chem. 2000;275:19808–18. doi: 10.1074/jbc.M002571200. [DOI] [PubMed] [Google Scholar]
- 10.Pabst S, et al. Rapid and selective binding to the synaptic SNARE complex suggests a modulatory role of complexins in neuroexocytosis. J Biol Chem. 2002;277:7838–48. doi: 10.1074/jbc.M109507200. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Augustine GJ, Weninger K. Kinetics of complexin binding to the SNARE complex: correcting single molecule FRET measurements for hidden events. Biophys J. 2007;93:2178–87. doi: 10.1529/biophysj.106.101220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bowen ME, Weninger K, Ernst J, Chu S, Brunger AT. Single-molecule studies of synaptotagmin and complexin binding to the SNARE complex. Biophys J. 2005;89:690–702. doi: 10.1529/biophysj.104.054064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, et al. Three-dimensional structure of the complexin/SNARE complex. Neuron. 2002;33:397–409. doi: 10.1016/s0896-6273(02)00583-4. [DOI] [PubMed] [Google Scholar]
- 14.Bracher A, Kadlec J, Betz H, Weissenhorn W. X-ray structure of a neuronal complexin-SNARE complex from squid. J Biol Chem. 2002;277:26517–23. doi: 10.1074/jbc.M203460200. [DOI] [PubMed] [Google Scholar]
- 15.Brose N. For better or for worse: complexins regulate SNARE function and vesicle fusion. Traffic. 2008;9:1403–13. doi: 10.1111/j.1600-0854.2008.00758.x. [DOI] [PubMed] [Google Scholar]
- 16.Huntwork S, Littleton JT. A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat Neurosci. 2007;10:1235–7. doi: 10.1038/nn1980. [DOI] [PubMed] [Google Scholar]
- 17.Maximov A, Tang J, Yang X, Pang ZP, Sudhof TC. Complexin Controls the Force Transfer from SNARE Complexes to Membranes in Fusion. Science. 2009;323:516–521. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reim K, et al. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. 2001;104:71–81. doi: 10.1016/s0092-8674(01)00192-1. [DOI] [PubMed] [Google Scholar]
- 19.Xue M, et al. Complexins facilitate neurotransmitter release at excitatory and inhibitory synapses in mammalian central nervous system. Proc Natl Acad Sci U S A. 2008;105:7875–80. doi: 10.1073/pnas.0803012105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strenzke N, et al. Complexin-I is required for high-fidelity transmission at the endbulb of held auditory synapse. J Neurosci. 2009;29:7991–8004. doi: 10.1523/JNEUROSCI.0632-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai H, et al. Complexin II plays a positive role in Ca2+-triggered exocytosis by facilitating vesicle priming. Proc Natl Acad Sci U S A. 2008;105:19538–43. doi: 10.1073/pnas.0810232105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giraudo CG, Eng WS, Melia TJ, Rothman JE. A clamping mechanism involved in SNARE-dependent exocytosis. Science. 2006;313:676–80. doi: 10.1126/science.1129450. [DOI] [PubMed] [Google Scholar]
- 23.Schaub JR, Lu X, Doneske B, Shin YK, McNew JA. Hemifusion arrest by complexin is relieved by Ca(2+)-synaptotagmin I. Nat Struct Mol Biol. 2006;13:748–50. doi: 10.1038/nsmb1124. [DOI] [PubMed] [Google Scholar]
- 24.Yoon TY, et al. Complexin and Ca2+ stimulate SNARE-mediated membrane fusion. Nat Struct Mol Biol. 2008;15:707–13. doi: 10.1038/nsmb.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malsam J, et al. The carboxy-terminal domain of complexin I stimulates liposome fusion. Proc Natl Acad Sci U S A. 2009;106:2001–6. doi: 10.1073/pnas.0812813106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xue M, et al. Distinct domains of complexin I differentially regulate neurotransmitter release. Nat Struct Mol Biol. 2007;14:949–58. doi: 10.1038/nsmb1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xue M, et al. Tilting the Balance between Facilitatory and Inhibitory Functions of Mammalian and Drosophila Complexins Orchestrates Synaptic Vesicle Exocytosis. Neuron. 2009;64:367–80. doi: 10.1016/j.neuron.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang J, et al. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 2006;126:1175–87. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 29.Roggero CM, et al. Complexin/synaptotagmin interplay controls acrosomal exocytosis. J Biol Chem. 2007;282:26335–43. doi: 10.1074/jbc.M700854200. [DOI] [PubMed] [Google Scholar]
- 30.Dai H, Shen N, Arac D, Rizo J. A quaternary SNARE-synaptotagmin-Ca2+-phospholipid complex in neurotransmitter release. J Mol Biol. 2007;367:848–63. doi: 10.1016/j.jmb.2007.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- 32.Seiler F, Malsam J, Krause JM, Sollner TH. A role of complexin-lipid interactions in membrane fusion. FEBS Lett. 2009;583:2343–8. doi: 10.1016/j.febslet.2009.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arac D, et al. Close membrane-membrane proximity induced by Ca(2+)-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat Struct Mol Biol. 2006;13:209–17. doi: 10.1038/nsmb1056. [DOI] [PubMed] [Google Scholar]
- 34.Battiste JL, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–65. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 35.Stevens CF, Wesseling JF. Augmentation is a potentiation of the exocytotic process. Neuron. 1999;22:139–46. doi: 10.1016/s0896-6273(00)80685-6. [DOI] [PubMed] [Google Scholar]
- 36.Basu J, Betz A, Brose N, Rosenmund C. Munc13-1 C1 domain activation lowers the energy barrier for synaptic vesicle fusion. J Neurosci. 2007;27:1200–10. doi: 10.1523/JNEUROSCI.4908-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerber SH, et al. Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science. 2008;321:1507–10. doi: 10.1126/science.1163174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geppert M, et al. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- 39.Guan R, Dai H, Rizo J. Binding of the Munc13-1 MUN domain to membrane-anchored SNARE complexes. Biochemistry. 2008;47:1474–81. doi: 10.1021/bi702345m. [DOI] [PubMed] [Google Scholar]
- 40.Weninger K, Bowen ME, Choi UB, Chu S, Brunger AT. Accessory Proteins Stabilize the Acceptor Complex for Synaptobrevin, the 1:1 Syntaxin/SNAP-25 Complex. Structure. 2008;16:308–20. doi: 10.1016/j.str.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Giraudo CG, et al. Alternative Zippering as an On-Off Switch for SNARE-Mediated Fusion. Science. 2009;323:512–516. doi: 10.1126/science.1166500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu B, Song S, Shin YK. Accessory alpha-Helix of Complexin I Can Displace VAMP2 Locally in the Complexin-SNARE Quaternary Complex. J Mol Biol. 2010;396:602–609. doi: 10.1016/j.jmb.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xue M, Ma C, Craig TK, Rosenmund C, Rizo J. The Janus-faced nature of the C(2)B domain is fundamental for synaptotagmin-1 function. Nat Struct Mol Biol. 2008;15:1160–8. doi: 10.1038/nsmb.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ashery U, Betz A, Xu T, Brose N, Rettig J. An efficient method for infection of adrenal chromaffin cells using the Semliki Forest virus gene expression system. Eur. J. Cell Biol. 1999;78:525–532. doi: 10.1016/s0171-9335(99)80017-x. [DOI] [PubMed] [Google Scholar]
- 45.Weber T, et al. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–72. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 46.Chen X, et al. SNARE-mediated lipid mixing depends on the physical state of the vesicles. Biophys J. 2006;90:2062–74. doi: 10.1529/biophysj.105.071415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.