

Abstract
Background
Human heart failure (HF) is associated with decreased cardiac voltage-gated Na+ channel current (encoded by SCN5A), and the changes have been implicated in the increased risk of sudden death in HF. Nevertheless, the mechanism of SCN5A downregulation is unclear. A number of human diseases are associated with alternative mRNA splicing, which has received comparatively little attention in the study of cardiac disease. Splicing factor expression profiles during human HF and a specific splicing pathway for SCN5A regulation were explored in this paper.
Methods and Results
Gene array comparisons between normal human and heart failure tissues demonstrated that 17 splicing factors, associated with all major spliceosome components, were upregulated. Two of these splicing factors, RBM25 and LUC7L3, were elevated in human heart failure tissue and mediated truncation of SCN5A mRNA in both Jurkat cells and human embryonic stem cell-derived cardiomyocytes (hESC-CMs). RBM25/LUC7L3-mediated abnormal SCN5A mRNA splicing reduced Na+ channel current 91.1 ± 9.3% to a range known to cause sudden death. Overexpression of either splicing factor resulted in an increase in truncated mRNA and a concomitant decrease in the full-length SCN5A transcript.
Conclusions
Of the 17 mRNA splicing factors upregulated in HF, RBM25 and LUC7L3 were sufficient to explain the increase in truncated forms and the reduction in full length Na+ channel transcript. Since the reduction in channels was in the range known to be associated with sudden death, interruption of this abnormal mRNA processing may reduce arrhythmic risk in heart failure.
Keywords: heart failure, SCN5A, splicing regulation, RBM25, LUC7L3
Introduction
Up to 95% of human genes have multi-exon alternative spliced forms, suggesting that alternative splicing is one of the most significant components of the functional complexity of the human genome.1,2 Although our understanding of the role of alternative mRNA splicing is elemental, a growing list of human diseases, such as cancer,3 neurodegenerative disorders,4 and autoimmune diseases5 are associated with alternative splicing. Alternative splicing events have been identified in cancer allowing for ‘splicing signatures’ associated with different tumor subgroups.6 Important goals of future studies of alternative splicing regulation include understanding how it occurs, its implications for disease, and its stimuli.7
Alternative splicing regulation has received comparatively little attention in the study of cardiac diseases. Splicing variants associated with the cardiac sodium,8 calcium,9 and HERG potassium channels10 have been reported. SCN5A is the gene encoding the cardiac sodium channel (Nav1.5), a protein responsible for generating the main current for excitation propagation in cardiomyocytes.11 Previously, we have shown that cardiac Na+ channel mRNA is alternatively spliced in human heart failure (HF) and that this splicing contributes to a reduction in current of a magnitude likely to contribute to the arrhythmic risk in this condition.8 Here, we identified upstream stimuli, hypoxia and angiotensin II (Ang II), and mediators, RBM25 and LUC7L3, of this abnormal splicing event.
RBM25 belongs to a family of RNA-binding proteins whose members share an arginine-glutamic acid (RE)/arginine-aspartic acid (RD) rich central region and a C-terminal proline-tryptophan-isoleucine (PWI) motif.12 RBM25 localizes to the nuclear speckles and associates with multiple splicing components such as splicing cofactors SRm160/300, U1 small nuclear (sn) RNAs, assembled splicing complexes, and spliced mRNAs. Characterization of RBM25 strongly suggests that it functions in pre-mRNA processing and that this regulation is gene specific.12 LUC7L3, a human homolog of yeast U1 snRNP-associated factor, is also a nuclear protein with a role in pre-mRNA splicing. LUC7L3 has two zinc finger motifs.12,13 The first cross-links the pre-mRNA and is required for LUC7L3 splicing activity. LUC7L3 acts as a bridge between the pre-mRNA and the U1 snRNP through its second zinc finger. According to a recent report, RBM25 associates selectively with the LUC7L3 and activates proapoptotic Bcl-xS 5′ splicing via its interaction with the exonic splicing enhancer cis-element, CGGGCA.12,13 Here, we tested whether the RBM25/LUC7L3 complex mediates pathological SCN5A mRNA splicing in HF.
Methods
Microarray Assay
(see Supplemental Material)
Cell Culture
Jurkat T cell clones E6.1 (ATCC, Manassas, VA) were cultured in RPMI 1640 medium supplemented with 10% heat inactivated fetal calf serum, 4 mM glutamine, 75 units/mL streptomycin and 100 units/mL penicillin.
Human embryonic stem (ES) cells were maintained on mouse embryonic fibroblasts (MEFs) as previously described.14 Cardiomyocytes were differentiated from WA09 (H9) ES cells using a directed differentiation approach in defined media for efficient cardiogenesis. After 30 days of differentiation, the human embryonic stem cell-derived cardiomyocytes (hESC-CMs) were used in this study.
Real-Time PCR Quantification
Total RNA was isolated from cultured cells and human ventricular tissue using the RNeasy Mini Kit and RNeasy Lipid Tissue Mini Kit respectively (Qiagen, Valencia, CA). Human heart tissue was obtained from a tissue bank maintained at Advocate Christ Cardiac Surgery Clinical Research Center. For primers for the target genes, see the Supplemental Material.
Transfection and Infection Assays
Fugene 6 reagents from Roche (Madison, WI) were used for transfection assays by following the manufacturer’s instructions. Small inhibitory RNAs (siRNAs) for LUC7L3 and RBM25 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Human pGIPZ lentiviral short hairpin RNAmir particles were purchased from Open Biosystems (Huntsville, AL). Human cardiomyocytes were placed in a 24-well plate at the density of 200,000/well. RBM25 short hairpin RNA (shRNA; 5 µL for each well based on pre-titer results) was pre-incubated with polybrene (Sigma, Milwaukee, WI) at final concentration 8 µg/mL for 1 h and aliquoted to each well. The scrambled shRNA group followed the same protocol. The media was replaced by regular culture media after 5 h. The infection rates and RBM25 knockdown rates were evaluated by confocal microscope (Carl Zeiss GmbH, Oberkochen, Germany) and qPCR respectively on day 2 and day 3. Green fluorescent protein (GFP)-tagged open reading frame clones of Homo sapiens LUC73 and RBM25 were purchased from ORIGENE (Rockville, MD). The transfection assays followed the manufacturer’s instructions.12
Electrophysiology
hESC-CMs were trypsinized (2.5%, Invitrogen) for 10 min and plated on 35 mm glass bottomed culture dish (MatTek, Ashland, MA) at cell density 40,000 cells/dish on the day before the experiments. Na+ channel currents were measured by using the whole-cell patch-clamp technique in the voltage-clamp configuration at room temperature. To measure Na+ channel currents, pipettes (3 to 4 MΩ) were filled with a pipette solution containing (in mmol/L): CsCl 80, cesium aspartate 80, EGTA 11, MgCl2 1, CaCl2 1, HEPES 10, and Na2ATP 5 (adjusted to pH 7.4 with CsOH). The bath solution consisted of (in mmol/L): NaCl 130, CsCl 5, CaCl2 2, MgCl2 1.2, HEPES 10, and glucose 5 (adjusted to pH 7.4 with CsOH).15 The holding potential was −100 mV. A voltage step protocol ranging from −80 to +70 mV with steps of 10 mV was applied to establish the presence of Na+ channel currents. The peak current density was used to plot current-voltage (I–V) curves. Nifedipine (10 µM, Sigma) was added in the bath solution to block L-type Ca+ channel currents.
Gel Mobility Shift Assays
RNA gel mobility shift assays were performed by using the LightShift Chemiluminescent RNA electrophoretic mobility shift assay (EMSA) Kit (Pierce, Appleton, WI). In brief, biotinylated wild-type (CAGCAGGCGGGCAGCGGCCU) and mutant (CAGCAGGUUAGAGGCGGCCU) RNA substrates were synthesized by Invitrogen. Binding of biotinylated RNA to RBM25 was achieved by incubating 0.2 nmol/L of RNA and variable amounts of protein for 30 min at 4°C in 20 µL of binding buffer. For the competition assays, a molar excess of unlabeled competitor RNAs at various fold levels was added to the pre-incubated reaction mixture. Samples were fractionated in a native 5% polyacrylamide gel and transferred to Hybond-N+nylon membranes (Pierce, Appleton, WI). The biotin-labeled RNA was detected using the streptavidin horseradish peroxidase conjugate and a chemiluminescent substrate.12
Western Blots Assays
The Mini-PROTEAN® Tetra Electrophoresis System from BioRad (Hercules, CA) was used for Western blots analysis. Anti-RBM25 antibodies were provided by Dr Shu-Ching Huang (Dana-Farber Cancer Institute). Anti-LUC7L3 antibodies were purchased from Millipore (Billerica, MA). Anti-GFP was purchased from ORIGENE (Rockville, MD).
Statistics
Data are presented as means ± standard error of the mean (SEM). Means were compared using unpaired Student's t test or one-way analysis of variance (ANOVA). A probability value P <0.05 was considered statistically significant.
Results
Altered mRNA Profiles of Splicing Factors in Human HF Tissue
A mRNA microarray analysis was used to identify and compare splicing factors in both normal and human HF tissues. Of the 181 known human splicing factors analyzed, 17 were upregulated in HF. These splicing factors were grouped according to known pathogenic regulators, such as hypoxia,16 inflammation,17 wall tension,18 or hormonal factors,19 involved in HF (Table 1, and data in the Supplemental Material).
Table 1.
Comprehensive list for human HF-related splicing factors
| Pathogenic regulation involved | Gene symbol |
|---|---|
| Hypoxia | RBM25; LUC7L3; TARDBP; HNRPH3; PPIG |
| Inflammation | BAT1; RBM39; IVNS1ABP |
| Hormone level changing | BAT1 |
| Overloading pressure | RBM39 |
| Others | YTHDC1; SFPQ; QKI; HNRNPA1; TRA2A |
Upregulation of RBM25 and LUC7L3 in Human HF Tissue
The cis-element, CGGGCA, of splicing factor RBM25 was found to be near the splicing sites of SCN5A variants E28C and E28D. RBM25 requires LUC7L3 to be active in splicing regulation,12 so both RBM25 and LUC7L3 were evaluated further for a role in SCN5A mRNA splicing regulation. Of the other 45 splicing factors upregulated, based on known cis-element sequence, none is known to bind to or has canonical binding sequences that are present in SCN5A.
The upregulation of splicing factors RBM25 and LUC7L3 was confirmed in human HF tissue by qPCR. Compared to the normal human heart tissue, the results indicated that the relative abundances of RBM25 and LUC7L3 were increased by 1.1-fold and 0.6-fold in HF tissue respectively (P < 0.05, Figure 1A). Full length SCN5A mRNA was reduced by 0.6-fold in HF tissue (P < 0.05, Figure 1A), a result similar to our previous report.8 mRNA findings were correlated with protein expression by Western blots. The representative Western blots are shown in Figure 1B. Compared to the control group (mixture of 4 normal human heart tissue samples), Western blot quantification showed that RBM25 was increased by 0.5- to 0.6-fold in HF tissue samples 1–4, respectively (P < 0.05, three replications for each sample). The gel density of LUC7L3 was increased by 0.6- to 0.7-fold in the same HF tissue samples (P < 0.05, three replications for each sample). The clinical information on these HF tissue samples is shown in Table 3 of the Supplemental Material.
Figure 1.
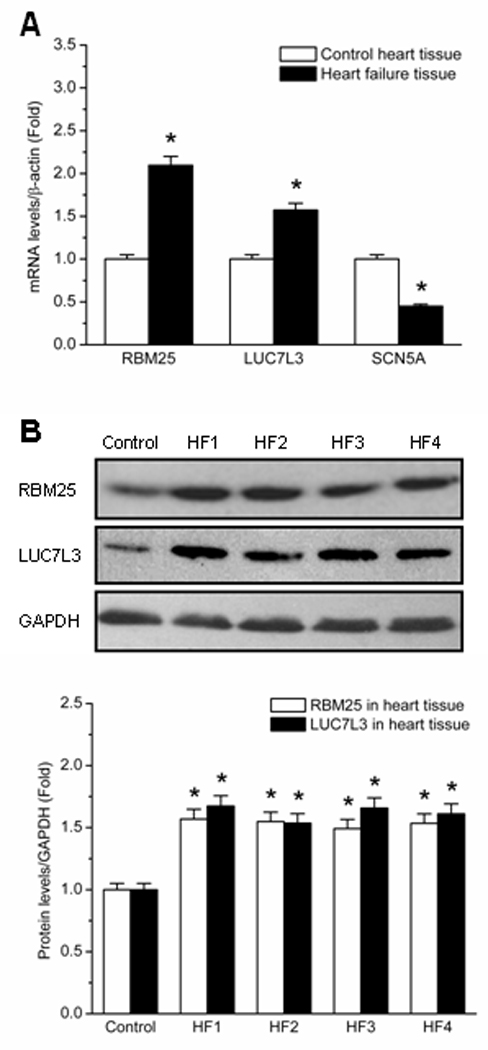
Expressions of RBM25 and LUC7L3 in human HF tissue. (A) qPCR demonstrates the upregulation of RBM25 and LUC7L3 in human HF tissue. The relative expression changes of RBM25 and LUC7L3 in both normal control (white bars) and failing heart tissue (black bars) are shown. All mRNA abundances are normalized by β-actin. * P < 0.05 when compared with control. (B) Western blots quantification confirms the upregulation of RBM25 and LUC7L3 in human HF tissue. Control represents the mixture of 4 normal human heart tissue samples. HF1, HF2, HF3 and HF4 represent the HF tissue sample 1, sample 2, sample 3 and sample 4, respectively. Quantification is based on three replications for each sample. All protein levels are normalized by GAPDH. * P < 0.05 when compared with control.
RBM25 Associates with SCN5A and Interacts with CGGGCA
Gel mobility shift assays showed that RMB25 was bound to the canonical sequence, CGGGCA, in SCN5A exon 28 (Figure 2). Scanning the entire SCN5A RNA sequence revealed only a single binding site for RBM25 at the place where SCN5A splicing variants were detected (Figure 2A).8 Binding of biotinylated wild-type (CAGCAGGCGGGCAGCGGCCU) RNA to RBM25 was observed in Figures 2B–D. The results illustrated that RBM25 binding was specific to the sequence CGGGCA. RBM25 was bound to the wild-type SCN5A sequence in a concentration-dependent manner (Figure 2B). For the competition assays, a molar excess of unlabeled competitor RNAs at various fold levels was added to the pre-incubated reaction mixture (Figure 2D). Specificity was confirmed by showing a lack of this binding to a mutated canonical binding sequence (Figure 2C) and the inability of unlabeled probe to compete with labeled probe for RBM25 binding (Figure 2D).
Figure 2.
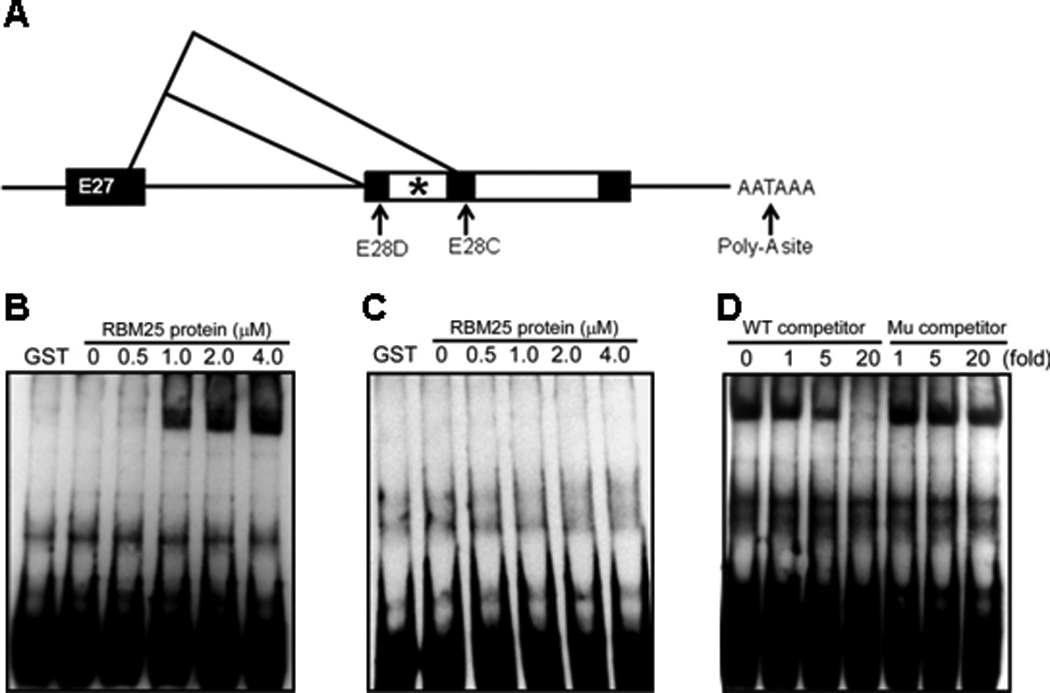
Illustration of the C-terminal structure of SCN5A and the variants E28C and E28D. (A) * indicates the RBM25 binding site CGGGCA in exon 28 (982bp–987bp). Gel mobility shift assays are performed using the biotinylated wild type (WT) probe (B) or the mutant (Mu) probe (C) and purified RBM25 protein. For loading samples from left to right, the amount of RBM25 in each binding reaction is increased by a fold. For the competition assay (D), 0, 1-, 5-, or 20-fold molar excesses of unlabeled WT or Mu probes are added in each binding reaction.
Ang II and Hypoxia Regulated RBM25, LUC7L3 Expression as well as SCN5A mRNA Splicing
Ang II and hypoxia are common pathogenic factors in HF and were identified in the microarray analysis as possible upstream stimuli responsible for the changes in mRNA splicing factors (Table 1).20 SCN5A mRNA is known to be transcribed in skeletal muscle and leukocytes. We have reported that leukocytes have a similar mRNA splicing pattern to that in heart.8 Moreover, circulating leukocytes from HF patients showed a four-fold increase in the Ang II type 1 receptor (data not shown). Therefore, Jurkat cells, an immortalized line of T lymphocyte cells, which prominently express SCN5A,21 were chosen to be as an initial model to study the SCN5A regulation mechanism.
Jurkat cells were divided into three experiment groups: untreated control, hypoxia-treated (1% O2), and Ang II-treated (200 nmol/L). The cells were harvested from each experiment group at four time points (30 min, 24 h, 48 h, and 72 h), and total mRNA was extracted. The expressions of RBM25 and LUC7L3 were examined by qPCR, and the results at 48 h are shown in Figure 3A. HIF-1α was used as the indicator of cellular hypoxic stress. Under the hypoxia-treated condition, the expressions of RBM25 and LUC7L3 in Jurkat cells were increased by 2.4-fold and 4.9-fold, respectively (P < 0.05). Under the Ang II-treated condition, the expressions of RBM25 and LUC7L3 in Jurkat cells were increased by 2.1-fold and 1.9-fold respectively (P < 0.05). The expressions of RBM25 and LUC7L3 were analyzed by Western blots at three time points (12 h, 24 h, and 48 h) for the hypoxia-treated group and at four time points (24 h, 48 h, 72 h, and 96 h) for the Ang II-treated group. Western blot quantification showed that RBM25 was increased by 1.9-, 2.0- and 1.5-fold in the hypoxia-treated group at time points 12 h, 24 h and 48 h, respectively and was increased by 2.1-, 2.1-, 1.9- and 2.0-fold in the Ang II-treated group at 24 h, 48 h, 72 h and 96 h, respectively (P < 0.05). The gel density of LUC7L3 was increased by 2.4-, 2.4- and 2.7-fold in the hypoxia-treated group at time points 12 h, 24 h and 48 h, respectively and was increased by 2.6-, 2.5-, 2.8- and 2.8-fold in the Ang II-treated group at time points 24 h, 48 h, 72 h and 96 h, respectively (P < 0.05). The representative Western blots and quantification (based on three replications for each group) are shown in Figure 3B.
Figure 3.
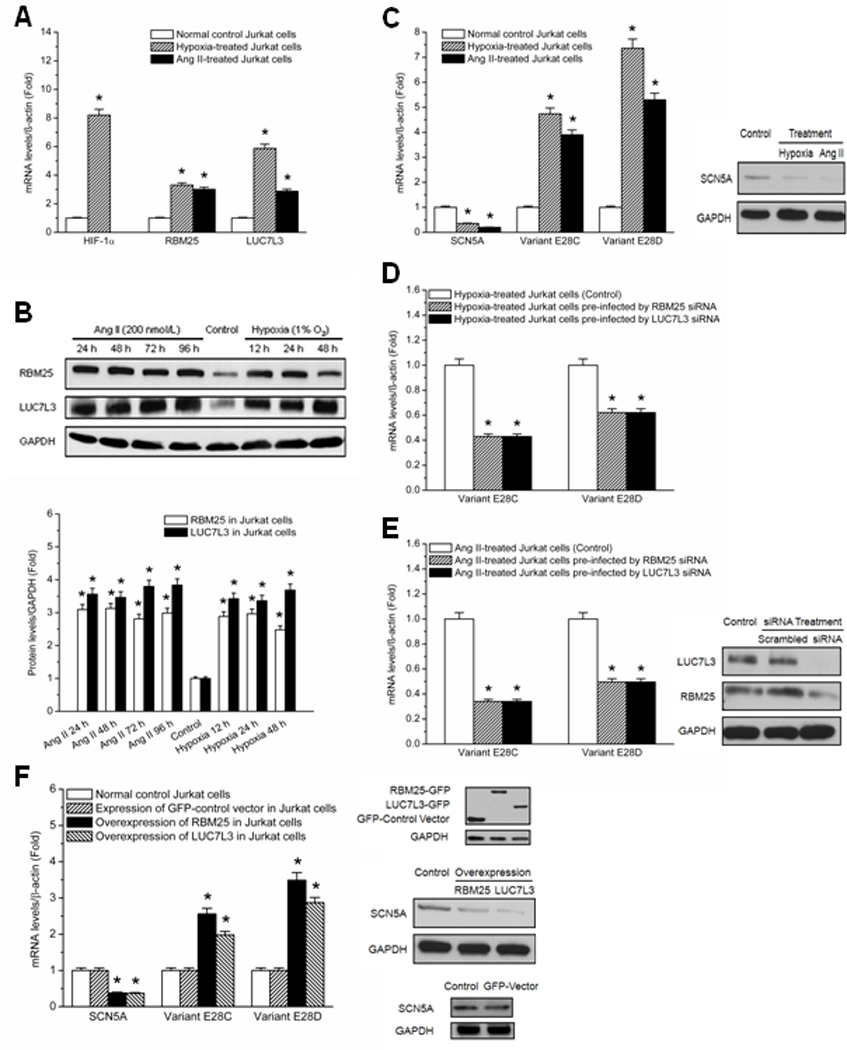
RBM25 and LUC7L3 are involved in SCN5A regulation in Jurkat cells. (A) qPCR demonstrates the upregulation of RBM25 and LUC7L3. The expression changes of RBM25 and LUC7L3 in hypoxia-treated (shaded bars) and Ang II-treated (black bars) vs. normal control Jurkat cells (white bars) are shown at 48 h. HIF-1α is an indicator of hypoxia. mRNA abundances are normalized by β-actin. * P < 0.05 when compared with control (N=6). (B) Western blots quantification confirms the upregulation of RBM25 and LUC7L3 in Jurkat cells. Expressions of RBM25 and LUC7L3 are analyzed by time course. * P < 0.05 when compared with control (N=6). (C) The expression changes of SCN5A and the variants E28C and E28D in hypoxia-treated (shaded bars) and Ang II-treated (black bars) vs. untreated Jurkat cells (white bars) are shown at 48 h. * P < 0.05 when compared with control (N=6). Western blots indicate the downregulation of SCN5A in Jurkat cells with Ang II or hypoxia. (D) qPCR demonstrates that RBM25 and LUC7L3 siRNAs could block the induction of hypoxia on variants E28C and E28D. The representative results at 24h are shown. * P < 0.05 when compared with control (N=6). (E) qPCR demonstrates that RBM25 and LUC7L3 siRNAs could block the induction of variants E28C and E28D by Ang II. Representative results at 48 h are shown. * P < 0.05 when compared with control (N=6). Scrambled siRNA had no effect on the induction by Ang II (data not shown). The knockdown efficiency of RBM25 and LUC7L3 siRNAs are evaluated by Western blots and compared to control and scrambled RNA. (F) qPCR demonstrates that RBM25 and LUC7L3 overexpressions decrease the full length SCN5A transcript and increase the variants E28C and E28D at 48h. * P < 0.05 when compared with control (N=6). Exogenously expressed RBM25-GFP and LUC7L3-GFP are detected by Western blot analysis with anti-GFP at 48 h after transfection in Jurkat cells. The representative Western blots show the downregulation of the full-length SCN5A transcript in Jurkat cells with exogenously expressed RBM25 and LUC7L3 as compared to the control group. Full-length SCN5A RNA is unchanged with GFP expression alone.
The effect of hypoxia and Ang II on the SCN5A variants E28C and E28D in Jurkat cells was studied also to correlate SCN5A variants with RBM25 and LUC7L3 abundances. The expressions of the full length SCN5A transcript and SCN5A variants E28C and E28D at 48 h are shown in Figure 3C. With hypoxia, the expressions of SCN5A variants E28C and E28D were increased by 3.7-fold and 6.4-fold, respectively (P < 0.05), while the expression of the full length SCN5A transcript was decreased by 0.7-fold (P < 0.05). With Ang II, the expressions of SCN5A variants E28C and E28D were increased by 2.9-fold and 4.3-fold, respectively (P < 0.05), while the expression of the full length SCN5A transcipt was decreased by 0.8-fold (P < 0.05).
siRNAs for these two splicing factors were found to block partially the increases in the hypoxia or Ang II-induced SCN5A variants E28C and E28D at 48 h (Figure 3D–E). The partial effect on reducing abnormal splicing may be due to the siRNA knockdown efficiencies were 60 ± 5% and 70 ± 5% estimated by qPCR and 63 ± 7% and 69 ± 6%. by Western blots for RBM25 and LUC7L3, respectively.
While downregulation of the two splicing factors in Jurkat cells reduced the SCN5A variants E28C and E28D, overexpression of RBM25 and LUC7L3 increased E28C and E28D and decreased the full length SCN5A mRNA abundances. The expressions of the full length SCN5A transcript and SCN5A variants E28C and E28D at 48 h are shown in Figure 3F. With overexpression of RBM25, the expressions of SCN5A variants E28C and E28D were increased by 1.6-fold and 2.5-fold, respectively (P < 0.05), while the expression of the full length SCN5A transcript was decreased by 0.6-fold (P < 0.05). With overexpression of LUC7L3, the expressions of SCN5A variants E28C and E28D were increased by 1.1-fold and 1.9-fold, respectively (P < 0.05), while the expression of the full length SCN5A transcript was decreased by 0.7-fold (P < 0.05).
The Effect of Ang II on Na+ Channels in hESC-CMs
The effect of Ang II on the cardiac Na+ channel was investigated in hESC-CMs. hESC-CMs were plated on a 24-well culture plate on day 30 of differentiation. The cells were divided into three experiment groups: Ang II-treated (200 nmol/L), Ang II-treated (200 nmol/L) and pre-infected by pGIPZ lentiviral RBM25 shRNAmir, and Ang II-treated (200 nmol/L) and pre-infected by scrambled shRNA. Ang II (200 nmol/L) treatment was given to all the experiment groups on infection day 3. When cells were pre-infected by RBM25 shRNA, the expression of the full length SCN5A transcript was increased by 0.4-fold, while the expressions of SCN5A variants E28C and E28D were decreased by 0.4-fold and 0.5-fold, respectively (P < 0.05). qPCR measurements were performed in each experiment group at 24 h after Ang II treatment and normalized by β-actin. No changes were observed when cells were pre-infected by scrambled shRNA, however. The results indicated that Ang II-mediated SCN5A downregulation was dependent on the splicing factor RBM25 (Figure 4A). The infection rate was 90 ± 6%, evaluated by the ratio of GFP positive cells (pGIPZ lentiviral infected cells) to total cells. RBM25 knockdown efficiency was 70 ± 5%, evaluated by both qPCR and Western blots (Figure 4B).
Figure 4.
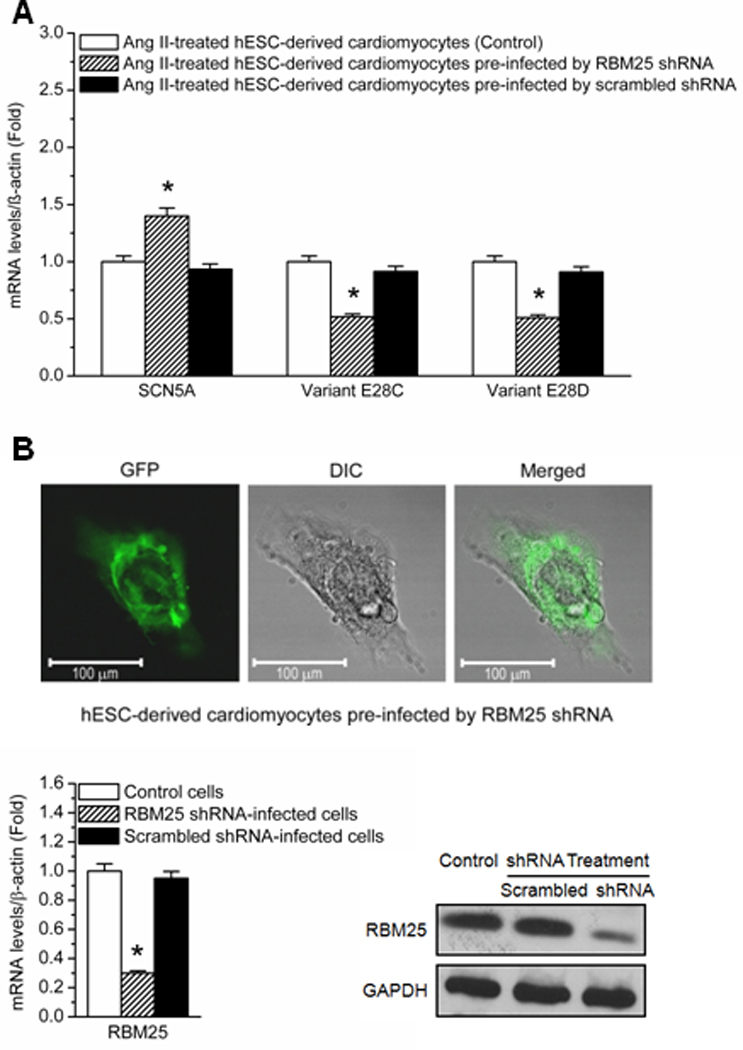
The effect of RBM25 shRNA on the expressions of SCN5A and the variants E28C and E28D in Ang II-treated hESC-CMs. (A) Ang II (200 nmol/L) treatment is given to all the experiment groups on infection day 3. RT-PCR measurements are done at 24 h after Ang II treatment and normalized by β-actin. The expression changes of SCN5A and the variants E28C and E28D in Ang II-treated cardiomyocytes pre-infected by RBM25 pLKO.1 shRNA (shaded bars) and pre-infected by scrambled shRNA (black bars) vs. normal Ang II-treated cardiomyocytes (white bars) are shown at 24 h. * P < 0.05 compared with normal Ang II-treated cardiomyocytes (N=6). (B) Confocal microscopy shows a hESC-CM. The GFP fluorescence indicates the infection by pGIPZ lentiviral shRNAmir. RBM25 knockdown efficiency is evaluated by qPCR and Western blot (N=6). Scrambled shRNA has no effect on RBM25.
Abnormal Na+ Channel mRNA Processing Altered Na+ Channel Current
The implications of Na+ channel mRNA processing changes were tested by measuring Na+ current in hESC-CMs by the whole-cell voltage-clamp technique. hESC-CMs were used to most accurately mimic clinical conditions and because a suitable animal model has not been validated. The cells were divided into four experiment groups: Control, Ang II-treated (200 nmol/L), Ang II-treated (200 nmol/L) and pre-infected by RBM25 shRNA, and Ang II-treated (200 nmol/L) and pre-infected by scrambled shRNA. Given that RBM25 regulates pre-mRNA alternative splicing by recruiting LUC7L3,12 loss-of-function of RBM25 with lentiviral shRNA was used exclusively to suppress abnormal channel splicing. The macroscopic Na+ channel currents in each experiment group were measured. The results from the first three experiment groups at 24 h after Ang II treatment are shown in Figure 5. There was a significant difference in peak currents between control cells and Ang II-treated cells at membrane potentials ranging from −40 to +30 mV (P < 0.05). The effect of Ang II on peak current was not observed when the cells were pre-infected by RBM25 shRNA before Ang II treatment. Non-specific effects of the lentivirus were excluded by comparing with the cells pre-infected by scrambled shRNA, which had no effect on the I–V relationship compared to Ang II alone (data not shown). The results indicated that Ang II could downregulate Na+ channel currents in hESC-CMs and that this downregulation was dependent on the splicing factor RBM25.
Figure 5.
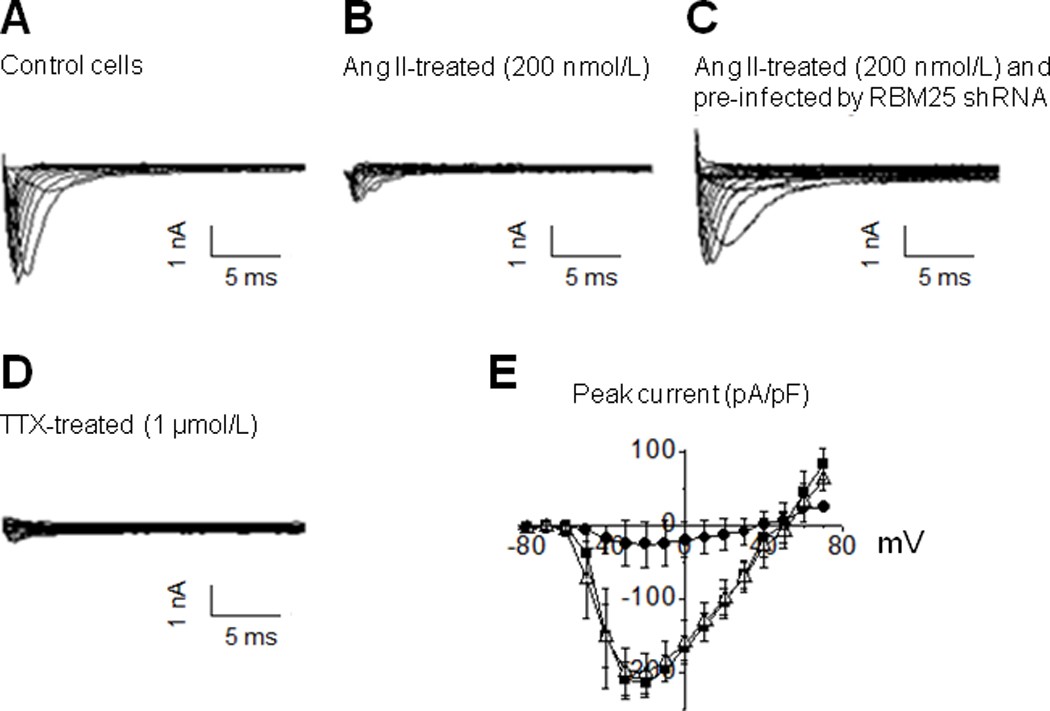
The effect of Ang II (200 nmol/L) on Na+ currents in hESC-CMs at 24 h after treatment. The representative current traces are shown in the control (A), Ang II-treated (B), Ang II-treated with pre-infection of RBM25 shRNA (C), and TTX -treated groups (D). The peak current statistics of the three experiment groups are shown in I–V curves (E), where the control group (n=3) is represented by filled squares, the Ang II-treated group (n=3) by filled circles, and the Ang II-treated group with pre-infection of RBM25 shRNA (n=3) by open triangles. The data are represented as mean ± SEM. The peak current is significantly reduced from −40 to +30 mV in the Ang II-treated group compared to the control group (P < 0.05), the maximum reduction was 91.1 ± 9.3 % at −30 mV. No difference is observed between the Ang II-treated group with pre-infection of RBM25 shRNA and the control group (E). The specificity of sodium channel current is tested with TTX (a specific sodium channel blocker) in Group D. Scrambled shRNA had no effect on the I-V relationship compared to Ang II alone (data not shown).
Discussion
HF remains a substantial clinical problem affecting millions of Americans, and HF-associated arrhythmia remains a cause of the high morbidity and mortality.22 The role of abnormal mRNA splicing in this condition is unknown. Gene array comparisons of splicing factor transcription between normal and HF tissues demonstrated that 17 splicing factors were upregulated during HF. The spliceosome assembles onto the pre-mRNA is in a stepwise manner. U1 snRNP binds to the 5’ splice site, SF1 binds to the branch site, and the U2 auxiliary factor (U2AF) binds to the 3’ splice site to form the E complex. Next, U2 snRNP replaces SF1 at branch site to form the A complex. Subsequently, the B complex is formed when the U4, U5, and U6 tri-snRNP enters the spliceosome. Finally, a rearrangement occurs to form the catalytically active C complex.23 Splicing factors in all major spliceosome components were altered in human HF. Upregulated splicing factors were located in U1 related components (RBM25, LUC7L3, PRPF40A), U2 (SF3B), U4/U6 (PRPF31), U4/U6 related components (SNRPD3), Prp19 complex (CDC5), Prp19 related complex (RBM22), EJC/TREX (BAT1, RBM8A), and common components (SRSF11, NCBP 2, HNRNPC). This suggested that alternative mRNA splicing may be a major pathological feature of and contribute to the arrhythmic risk in HF.
We evaluated the effects of abnormal mRNA splicing in HF on the cardiac Na+ channel (encoded by SCN5A), the main ion channel generating current for conduction in the heart. Previously, we have demonstrated that HF results in an increase in two SCN5A variants, designated as E28C (39 bp) and E28D (114 bp).8 SCN5A mRNA variants result from splicing at cryptic splice sequences in the terminal exon of SCN5A (exon 28). In comparison with the full-length Na+ channel, SCN5A variants are shorter and encode prematurely truncated, nonfunctional Na+ channel proteins missing the segments from domain IV, S3 or S4 to the C-terminus.8 A mouse model in which one allele of the SCN5A gene is substituted by a model truncation variant showed a >80% reduction in cardiac Na+ current and a significant reduction in electrical conduction velocity. 8,24
Based on the presence of a cis-element binding site in exon 28 of SCN5A, the role of the upregulation of U1 related components RBM25 and LUC7L3 in abnormal SCN5A mRNA splicing was investigated. RBM25 was bound to the canonical CGGGCA sequence in exon 28 near the splicing sites of SCN5A variants E28C and E28D. The binding specificity of RMB25 in exon 28 was confirmed by gel mobility shift assays. The upstream signal pathway of RBM25/LUC7L3-mediated splicing is not clear. Nevertheless, we showed that two common features present in HF, Ang II and hypoxia,25 were able to induce these splicing factors. These results are consistent with clinical data suggesting that renin-angiotensin system inhibition and revascularization have antiarrhythmic effects.26 Both hypoxia and Ang II increase oxidative stress and HIF-1α.27 LUC7L3 is known to be regulated by hypoxia, acidosis, and HIF-1α.28 It is possible that HIF-1α is an intermediate mediator of RBM25/LUC7L3-dependent splicing regulation of SCN5A. These signals are unlikely to be the only upstream regulators of abnormal Na+ channel splicing. Other factors, such as inflammation29 and stretch,30 may be important in mediated abnormal Na+ channel splicing in HF. Finally, while experimental manipulations of RBM25 and LUC7L3 were sufficient to cause or prevent SCN5A mRNA dysregulation, it remains possible that some of the other splicing factors increased in HF contribute to abnormal SCN5A splicing.
Abnormal SCN5A splicing variants appear confined to humans for unclear reasons.8 The mechanism of splicing does not appear to be tissue restricted, however, since a similar pattern of SCN5A splicing variants has been found in both lymphoblast and skeletal muscle.8 Moreover, Jurkat cells showed similar responses to cardiomyocytes.
Because there are no animal models of human abnormal Na+ channel splicing, it was impossible to determine directly the physiological significance of the downregulation of Na+ channel current mediated by abnormal splicing. Nevertheless, Ang II-treatment of hESC-derived cardiomyocytes showed current changes in the range known to contribute to arrhythmic risk.31
In summary, we showed that human HF is associated with dysregulation of mRNA splicing factors and that two of these splicing factors contribute to abnormal SCN5A regulation. SCN5A regulation was similar in Jurkat cells and hESC-derived cardiomyocytes. LUC7L3 and RBM25 were elevated in human HF tissue and mediated abnormal SCN5A splicing regulation in the two cell types tested. The resultant current reductions were on an order large enough to contribute to arrhythmic risk, especially in patients challenged by Na+ channel blockers. While Ang II and hypoxia were sufficient to signal SCN5A abnormal splicing, there could be other heart failure-associated factors that could contribute to SCN5A downregulation in vivo. Therapies directed at reducing the activation of RBM25 and LUC7L3 or their upstream inducing stimuli may prevent the reduction in cardiac Na+ channels seen in HF and reduce arrhythmic risk in this condition.
Clinical Summary.
Previously, we have shown that human heart failure is associated with abnormal mRNA splicing of the cardiac sodium channel. This abnormal mRNA splicing results in truncated sodium channels that are nonfunctional and a reduction in sodium channel current to levels known to cause sudden death. In this manuscript, we explore the mechanisms by which this abnormal splicing occurs. Using microarray comparisons of disease and normal human hearts, we identified two splicing factors, RBM25 and LUC7L3, that were necessary and sufficient to cause the abnormal sodium channel splicing. These factors were upregulated by hypoxia and elevated angiotensin II, conditions known to be present in heart failure. Moreover, we showed that the responses of white cells and heart to these two inciting stimuli were equivalent. The potential clinical implications of these findings include a possible mechanism whereby hypoxia is arrhythmogenic and blockade of the renin-angiotensin system is antiarrhythmic. Moreover, if white cell sodium channel splicing in vivo correlates with that in the myocardium, it may be possible to develop a blood test to assess sodium channel availability and arrhythmic proclivity. Finally, this work defines potential therapeutic targets to address arrhythmic risk in human heart failure.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by grants from the National Heart, Lung, and Blood Institute (R01 HL085558, R01 HL073753, P01 HL058000), and a Veterans Affairs MERIT grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
Dr. Dudley holds patents related to this work, Human Sodium Channel Isoforms (11, 707882) and Potential Drug Targets to Prevent Arrhythmia in Heart Disease.
References
- 1.Modrek B, Lee C. A genomic view of alternative splicing. Nat Genet. 2002;30:13–19. doi: 10.1038/ng0102-13. [DOI] [PubMed] [Google Scholar]
- 2.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 3.Ryan BM, Robles AI, Harris CC. Genetic variation in microRNA networks: the implications for cancer research. Nat Rev Cancer. 2010;10:389–402. doi: 10.1038/nrc2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, Hall MP, Shiue L, Swanson MS, Thornton CA, Ares M., Jr Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol. 2010;17:187–193. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Novak AJ, Slager SL, Fredericksen ZS, Wang AH, Manske MM, Ziesmer S, Liebow M, Macon WR, Dillon SR, Witzig TE, Cerhan JR, Ansell SM. Genetic variation in B-cell-activating factor is associated with an increased risk of developing B-cell non-Hodgkin lymphoma. Cancer Res. 2009;69:4217–4224. doi: 10.1158/0008-5472.CAN-08-4915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reyal F, van Vliet MH, Armstrong NJ, Horlings HM, de Visser KE, Kok M, Teschendorff AE, Mook S, van ' V, Caldas C, Salmon RJ, van de Vijver MJ, Wessels LF. A comprehensive analysis of prognostic signatures reveals the high predictive capacity of the proliferation, immune response and RNA splicing modules in breast cancer. Breast Cancer Res. 2008;10:R93. doi: 10.1186/bcr2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Licatalosi DD, Darnell RB. RNA processing and its regulation: global insights into biological networks. Nat Rev Genet. 2010;11:75–87. doi: 10.1038/nrg2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shang LL, Pfahnl AE, Sanyal S, Jiao Z, Allen J, Banach K, Fahrenbach J, Weiss D, Taylor WR, Zafari AM, Dudley SC., Jr Human heart failure is associated with abnormal C-terminal splicing variants in the cardiac sodium channel. Circ Res. 2007;101:1146–1154. doi: 10.1161/CIRCRESAHA.107.152918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartoszewski R, Rab A, Jurkuvenaite A, Mazur M, Wakefield J, Collawn JF, Bebok Z. Activation of the unfolded protein response by δF508 CFTR. Am J Respir Cell Mol Biol. 2008;39:448–457. doi: 10.1165/rcmb.2008-0065OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keller SH, Platoshyn O, Yuan JX, Long QT. syndrome-associated I593R mutation in HERG potassium channel activates ER stress pathways. Cell Biochem Biophys. 2005;43:365–377. doi: 10.1385/CBB:43:3:365. [DOI] [PubMed] [Google Scholar]
- 11.Shibata EF, Brown TL, Washburn ZW, Bai J, Revak TJ, Butters CA. Autonomic regulation of voltage-gated cardiac ion channels. J Cardiovasc Electrophysiol. 2006;17 Suppl 1:S34–S42. doi: 10.1111/j.1540-8167.2006.00387.x. [DOI] [PubMed] [Google Scholar]
- 12.Zhou A, Ou AC, Cho A, Benz EJ, Jr, Huang SC. Novel splicing factor RBM25 modulates Bcl-x pre-mRNA 5' splice site selection. Mol Cell Biol. 2008;28:5924–5936. doi: 10.1128/MCB.00560-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Puig O, Bragado-Nilsson E, Koski T, Seraphin B. The U1 snRNP-associated factor Luc7p affects 5' splice site selection in yeast and human. Nucleic Acids Res. 2007;35:5874–5885. doi: 10.1093/nar/gkm505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Wilson GF, Soerens AG, Koonce CH, Yu J, Palecek SP, Thomson JA, Kamp TJ. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009;104:e30–e41. doi: 10.1161/CIRCRESAHA.108.192237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu M, Sanyal S, Gao G, Gurung IS, Zhu X, Gaconnet G, Kerchner LJ, Shang LL, Huang CL, Grace A, London B, Dudley SC., Jr Cardiac Na+ current regulation by pyridine nucleotides. Circ Res. 2009;105:737–745. doi: 10.1161/CIRCRESAHA.109.197277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stearman RS, Dwyer-Nield L, Zerbe L, Blaine SA, Chan Z, Bunn PA, Jr, Johnson GL, Hirsch FR, Merrick DT, Franklin WA, Baron AE, Keith RL, Nemenoff RA, Malkinson AM, Geraci MW. Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen-induced murine model. Am J Pathol. 2005;167:1763–1775. doi: 10.1016/S0002-9440(10)61257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ricco R, Kanduc D. Hepatitis B virus and Homo sapiens proteome-wide analysis: A profusion of viral peptide overlaps in neuron-specific human proteins. Biologics. 2010;4:75–81. doi: 10.2147/btt.s8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Inuzuka Y, Okuda J, Kawashima T, Kato T, Niizuma S, Tamaki Y, Iwanaga Y, Yoshida Y, Kosugi R, Watanabe-Maeda K, Machida Y, Tsuji S, Aburatani H, Izumi T, Kita T, Shioi T. Suppression of phosphoinositide 3-kinase prevents cardiac aging in mice. Circulation. 2009;120:1695–1703. doi: 10.1161/CIRCULATIONAHA.109.871137. [DOI] [PubMed] [Google Scholar]
- 19.Claus EB, Park PJ, Carroll R, Chan J, Black PM. Specific genes expressed in association with progesterone receptors in meningioma. Cancer Res. 2008;68:314–322. doi: 10.1158/0008-5472.CAN-07-1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li YL, Xia XH, Zheng H, Gao L, Li YF, Liu D, Patel KP, Wang W, Schultz HD. Angiotensin II enhances carotid body chemoreflex control of sympathetic outflow in chronic heart failure rabbits. Cardiovasc Res. 2006;71:129–138. doi: 10.1016/j.cardiores.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 21.Fraser SP, Diss JK, Lloyd LJ, Pani F, Chioni AM, George AJ, Djamgoz MB. T-lymphocyte invasiveness: control by voltage-gated Na+ channel activity. FEBS Lett. 2004;569:191–194. doi: 10.1016/j.febslet.2004.05.063. [DOI] [PubMed] [Google Scholar]
- 22.Bardy GH, Lee KL, Mark DB, Poole JE, Packer DL, Boineau R, Domanski M, Troutman C, Anderson J, Johnson G, McNulty SE, Clapp-Channing N, Davidson-Ray LD, Fraulo ES, Fishbein DP, Luceri RM, Ip JH. Amiodarone or an implantable cardioverter-defibrillator for congestive heart failure. N Engl J Med. 2005;352:225–237. doi: 10.1056/NEJMoa043399. [DOI] [PubMed] [Google Scholar]
- 23.Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 24.Shang LL, Dudley SC., Jr Tandem promoters and developmentally regulated 5'- and 3'-mRNA untranslated regions of the mouse Scn5a cardiac sodium channel. J Biol Chem. 2005;280:933–940. doi: 10.1074/jbc.M409977200. [DOI] [PubMed] [Google Scholar]
- 25.Choudhary G, Dudley SC., Jr Heart failure, oxidative stress, and ion channel modulation. Congest Heart Fail. 2002;8:148–155. doi: 10.1111/j.1527-5299.2002.00716.x. [DOI] [PubMed] [Google Scholar]
- 26.Moro C, Hernandez-Madrid A, Matia R. Non-antiarrhythmic drugs to prevent atrial fibrillation. Am J Cardiovasc Drugs. 2010;10:165–173. doi: 10.2165/11537270-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 27.Gao G, Dudley SC., Jr Redox regulation, NF-κB, and atrial fibrillation. Antioxid Redox Signal. 2009;11:2265–2277. doi: 10.1089/ars.2009.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishii Y, Morishima M, Kakehi Y, Umehara K, Kioka N, Terano Y, Amachi T, Ueda K. CROP/Luc7A, a novel serine/arginine-rich nuclear protein, isolated from cisplatin-resistant cell line. FEBS Lett. 2000;465:153–156. doi: 10.1016/s0014-5793(99)01744-5. [DOI] [PubMed] [Google Scholar]
- 29.De CR, Ruigomez A, Rodriguez LA. Long-term Use of Anti-inflammatory Drugs and Risk of Atrial Fibrillation. Arch Intern Med. 2010;170:1450–1455. doi: 10.1001/archinternmed.2010.305. [DOI] [PubMed] [Google Scholar]
- 30.Hutchinson KR, Stewart JA, Jr, Lucchesi PA. Extracellular matrix remodeling during the progression of volume overload-induced heart failure. J Mol Cell Cardiol. 2010;48:564–569. doi: 10.1016/j.yjmcc.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burashnikov A, Antzelevitch C. Atrial-selective sodium channel block for the treatment of atrial fibrillation. Expert Opin Emerg Drugs. 2009;14:233–249. doi: 10.1517/14728210902997939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.