

Abstract
Functional states of mitochondria are often reflected in characteristic mitochondrial morphology. One of the most fundamental stress conditions, hypoxia–reoxygenation has been known to cause impaired mitochondrial function accompanied by structural abnormalities, but the underlying mechanisms need further investigation. Here, we monitored bioenergetics and mitochondrial fusion–fission in real time to determine how changes in mitochondrial dynamics contribute to structural abnormalities during hypoxia–reoxygenation. Hypoxia–reoxygenation resulted in the appearance of shorter mitochondria and a decrease in fusion activity. This fusion inhibition was a result of impaired ATP synthesis rather than Opa1 cleavage. A striking feature that appeared during hypoxia in glucose-free and during reoxygenation in glucose-containing medium was the formation of donut-shaped (toroidal) mitochondria. Donut formation was triggered by opening of the permeability transition pore or K+ channels, which in turn caused mitochondrial swelling and partial detachment from the cytoskeleton. This then favored anomalous fusion events (autofusion and fusion at several sites among 2–3 mitochondria) to produce the characteristic donuts. Donuts effectively tolerate matrix volume increases and give rise to offspring that can regain ΔΨm. Thus, the metabolic stress during hypoxia–reoxygenation alters mitochondrial morphology by inducing distinct patterns of mitochondrial dynamics, which includes processes that could aid mitochondrial adaptation and functional recovery.
Keywords: mitochondrial dynamics, microtubules, permeability transition, ROS, K+ channel
Physiological and pathophysiological conditions of mitochondrial metabolism are thought to evoke changes in the mechanisms of mitochondrial dynamics to control mitochondrial structure. Among the pathophysiological conditions, cellular hypoxia and reoxygenation are of particular interest as these represent two essential elements of ischemia–reperfusion injury. The hypoxia–reoxygenation-induced abnormalities in mitochondrial metabolic function and the ensuing changes in mitochondrial structure are widely believed to be important pathogenic factors that underlie ischemic cell injury in various tissues, including the brain, heart, kidney and liver.1, 2, 3, 4 Furthermore, mitochondrial function serves as a key effector in the pathways that mediate the protective effect that short periods of hypoxia–reoxygenation and some drugs provide against tissue injury caused by subsequent prolonged hypoxia–reoxygenation (preconditioning).1, 5, 6, 7 Thus, it is important to determine the hypoxia–reoxygenation-induced changes in mitochondrial dynamics and the underlying mechanisms.
Mitochondria use O2 as a substrate, so their respiration is inhibited during hypoxia. During reoxygenation, rapid restoration of respiration results in increased mitochondrial reactive oxygen species (ROS) production.8, 9 On the basis of ultrastructural studies, the mitochondrial morphology changes include some swelling in dog heart,10 no detectable changes in rabbit heart during ischemia11 and both shrinkage and swelling in rabbit kidney tubules during hypoxia.12 However, mitochondrial swelling is apparent in all these tissues during reoxygenation/reperfusion.11, 12, 13 Mitochondrial swelling during reoxygenation results, at least in part, from opening of the permeability transition pore (PTP), a high conductance channel in the inner mitochondrial membrane (IMM). Increased ROS production and calcium dysregulation are likely to contribute to PTP opening.1 Increased K+ conductance of the IMM is an alternative mechanism for matrix volume increase. Several studies have claimed that ATP-activated K+ channels (KATP) and the Ca2+-activated K+ channels (KCa) are present in the IMM14 and display changes in activity during hypoxia–reoxygenation.7, 15, 16 Furthermore, both PTP and KATP have been implicated as major factors in the mechanisms of protection against cell death.5, 6
Repetitive cycles of fusion and fission are central to mitochondrial dynamics. Fusion is supported by mitofusins (Mfn1 and 2) and optic atrophy 1 (Opa1) in the outer mitochondrial membrane (OMM) and IMM, respectively. Mitochondrial fusion is an effector of several signaling pathways and might play a role in disease mechanisms.17 Recent studies have indicated that ischemic cardiomyopathy and treatment with mitochondrial inhibitors lead to a decrease in Opa1 and in mitochondrial fusion activity,18, 19, 20 raising the possibility that mitochondrial fusion is altered during hypoxia–reoxygenation and might also contribute to the change in mitochondrial structure. In addition, inhibition of mitochondrial fission was reported to protect hearts against ischemia/reperfusion injury.21 Here, we monitored mitochondrial fusion–fission dynamics in real time simultaneously with the measurements of ΔΨm, ATP and Opa1 levels during hypoxia–reoxygenation. Our study provides evidence that hypoxia causes progressive loss of mitochondrial fusion activity and the appearance of novel processes of mitochondrial dynamics, including donut formation. The results also shed some light on the mechanisms of these changes and on their possible role in the mitochondrial adaptation to hypoxia–reoxygenation-induced cellular stress.
Results and Discussion
Effect of hypoxia–reoxygenation, FCCP on ΔΨm and mitochondrial morphology
H9c2 cells were exposed to hypoxia and reoxygenation in glucose-containing (+G) medium or glucose-free, methyl succinate-containing (−G) medium. In −G medium, the cells are forced to rely predominantly on oxidative phosphorylation for ATP synthesis, given the shortage of carbon source to feed the glycolytic pathway. Thus, −G medium might enhance the hypoxia-induced ATP deficit.
In +G medium, ΔΨm gradually decreased during 60 min of hypoxia (n=35, P<0.001), completely recovered in the first 30 min reoxygenation and decreased with longer reoxygenation (Figures 1a and b). As for the morphology, mitochondrial shortening and perinuclear redistribution were observed during hypoxia (Figures 1a and b). However, the most striking mitochondrial morphological change was the appearance of donut-shaped mitochondria with ΔΨm dissipation (Figure 1a). Donuts were observed during 60 min hypoxia in the majority of the cells (Figure 1b) and during 30–60 min of reoxygenation in 100% of cells. In −G medium, ΔΨm responses to hypoxia–reoxygenation were similar to that in +G medium, with more decrease (n=29, P<0.001) during hypoxia and less recovery during reoxygenation (Figure 1b). Mitochondrial redistribution and shortening were similar to that in +G medium (Figure 1b). However, donut-shaped mitochondria appeared during hypoxia and were maintained during reoxygenation (Figure 1b). Mitochondrial uncoupling by carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (5 μM, tested in +G medium), which induced collapse of ΔΨm, also elicited donut formation (Supplementary Figure 1B).22, 23 Notably, donut formation involved only a fraction of the mitochondria during both hypoxia–reoxygenation and FCCP exposure (10–30%). Thus, during hypoxia–reoxygenation, mitochondrial depolarization, a consequence of respiratory inhibition, is associated with mitochondrial redistribution, shortening and donut formation. Decreased O2 consumption is not required for donut formation, as uncoupling by FCCP also induced donuts. It is notable that donut formation is an early response to uncoupler, whereas mitochondrial fragmentation, a sign of enhanced fission or inhibition of fusion, occurs after 3–4 h uncoupling.18, 24 Hypoxia and reoxygenation might induce less donuts than FCCP because of the early mitochondrial shortening that does not favor donut formation (see below).
Figure 1.

Effect of hypoxia–reoxygenation on ΔΨm and mitochondrial morphology. (a) Images of two H9c2 cells transfected with mtYFP (green) and loaded with TMRE (red) acquired before (left) and during hypoxia (H) (60 min: left middle) and during reoxygenation (R) (30 min: right middle; 60 min: right). (b) Cumulative results for TMRE fluorescence in +G medium (upper left, n=35) and in −G medium (upper right, n=29) for mitochondrial morphology and distribution (middle left and right, +G medium: n=35, −G medium: n=56) and for the amount of donuts (lower left and right, +G medium: n=35, −G medium: n=29). *P<0.001
In addition to H9c2 cells, hypoxia- and/or FCCP-induced donut formation was observed in other cell types, including primary rat neonatal cardiomyocytes (Supplementary Figure 2), RBL-2H3 cells (Figure 6b), mouse embryonic fibroblasts (Supplementary Figure 5) and HeLa cells (data not shown). Thus, transition to donut-shaped mitochondria is not a cell-type-specific response and occurs in both primary cells and cell lines.
Effect of hypoxia–reoxygenation, FCCP and oligomycin on mitochondrial fusion
To visualize the exchange of matrix contents between individual mitochondria in real time, we have used mitochondrial matrix-targeted photoactivatable green fluorescence protein (mtPAGFP) in combination with mitochondrial matrix-targeted red fluorescent protein (mtDsRed) or TMRE. Using high-resolution confocal imaging with region of interest (ROI) scanning, we were able to achieve an irreversible photoactivation of mtPAGFP within subsets of the mitochondria. Monitoring cells for 7 min after photoactivation revealed interactions during which photoactivated PAGFP was transferred from one mitochondrion to another (Figure 2a). The interactions involved both complete fusion and transient fusion events.25, 26
Figure 2.

Effect of hypoxia–reoxygenation, FCCP and oligomycin on mitochondrial fusion and Opa1 cleavage. (a) Images showing the spread of photoactivated mtPAGFP in three subsets of mitochondria in naïve, hypoxia (H) in −G medium or FCCP-pretreated H9c2 cells. (b) In the graphs, the total number of mitochondrial fusion events per cell per 7 min is shown in each condition (n=4–6 cells, *P⩽0.01), whereas the immunoblots show the Opa1 levels. The higher molecular weight band is unaffected by H, reoxygenation (R), oligomycin, but gradually disappears during FCCP treatment
In +G medium, mitochondrial fusion activity decreased to 65% of basal during hypoxia and to 58% during reoxygenation. In −G medium, mitochondrial fusion activity decreased to 34% during hypoxia and to 10% during reoxygenation (Figure 2). Both complete and transient fusion events became less frequent. Recent studies have shown various experimental conditions of ΔΨm loss leading to the cleavage of Opa1 and fusion inhibition,26, 27, 28 and also heart failure and 10 h ischemia being associated with reduction of Opa1 level.19 However, Opa1 levels did not change during 1 h hypoxia–reoxygenation (Figure 2b), indicating fusion inhibition was not because of Opa1 depletion. On the other hand, the FCCP-induced mitochondrial fusion activity decrease (to 20% in 0–15 min and 1% in 15–30 min), closely followed the Opa1 cleavage (Figure 2b, see also ref. 26) Furthermore, overexpression of Opa1 attenuated the FCCP-induced inhibition of mitochondrial fusion (Figure 2b).
An alternative mechanism for the hypoxia–reoxygenation-induced fusion inhibition might be ATP depletion (though fusion was detected during significant ATP depletion in HeLa cells, ref. 18). ATP is required to support the production of GTP, which is needed for both outer and inner membrane fusion.20 Indeed, rapid ATP depletion evoked by an F1F0-ATPase inhibitor, oligomycin, induced a decrease in mitochondrial fusion activity to 21% in 15 min without Opa1 cleavage (Figure 2b). Table 1 shows a similar decrease in cellular ATP levels during 60 min hypoxia and 15 min treatment with oligomycin, suggesting that mitochondrial ATP synthesis inhibition can account for mitochondrial fusion inhibition during hypoxia. Notably, stress-induced mitochondrial hyperfusion has been observed in association with increased mitochondrial ATP levels,29, 30 indicating a direct relationship between mitochondrial ATP and fusion.
Table 1. Effect of hypoxia (H), FCCP and oligomycin on cellular ATP levels.
| Percent of control | H 60 min | FCCP 30 min | Oligomycin 5 min | Oligomycin 15 min |
|---|---|---|---|---|
| +G medium | 71±2* | 90±5 | 89±3* | 70±5* |
| −G medium | 53±4* | 13±4* |
*P<0.05
Donut formation is not induced by fusion inhibition or ATP depletion
To test the role of mitochondrial fusion inhibition and fission activity in donut formation, overexpression of Opa1 and a dominant-negative form of the mitochondrial fission protein Drp1, Drp1K38A were used, respectively. Opa1 overexpression did not induce donut formation and did not alter the hypoxia-induced donut formation (Supplementary Figure 3). FCCP-induced donut-shaped mitochondria also appeared in cells overexpressing Drp1K38A or Opa1 (Supplementary Figure 1B), which conditions have been shown to inhibit mitochondrial fission and to increase fusion, respectively.26 To discriminate between the respective roles of depolarization and loss of mitochondrial ATP production, oligomycin was used to inhibit ATP synthesis without causing depolarization. Oligomycin treatment (5 μg/ml) did not induce donut formation (data not shown). On the basis of the results with Drp1K38A- or Opa1-overexpressing cells, donut formation is not dependent on Drp1 activity or Opa1 depletion. Furthermore, as ATP depletion was not sufficient to trigger donut formation, distinct consequences of hypoxia and reoxygenation are relevant to inhibition of fusion versus the formation of donuts.
Dependence of donut formation on PTP or K+ channel opening and matrix swelling
Donut-shaped mitochondria were found in cells exposed to reoxygenation in +G medium, hypoxia in −G medium or FCCP (Figure 1 and Supplementary Figure 1). Reoxygenation in +G medium and FCCP have been linked to facilitation of PTP opening.3, 31 Indeed, donut formation during reoxygenation or FCCP treatment always occurred after dissipation of the ΔΨm. The depolarization events during reoxygenation were abrupt (Supplementary Figure 4A), which is often a sign of PTP opening. Indeed, depolarization during reoxygenation was suppressed in cells pretreated with cyclosporin A (CSA), an inhibitor of PTP (Supplementary Figure 4B). Further along this line, mastoparan, a potent facilitator of PTP,32 could also induce donut-shaped mitochondria with ΔΨm decrease, whereas Opa1 remained uncleaved during the 30 min treatment (Figure 3a, n=16). CSA alone did not cause a change in mitochondrial morphology but exerted protection against the FCCP-induced donut formation (Figure 3b). Similar to CSA, Me-Val-CSA, which binds mitochondrial cyclophilin but fails to inhibit calcineurin, could prevent the FCCP-induced donut formation, whereas FK506 that inhibits calcineurin without affecting the PTP did not exert protection (Figure 3b). Notably, CSA could not prevent FCCP-induced proteolytic processing of Opa1 (Figure 3b). Therefore, PTP opening seems to be involved in donut formation independent of uncoupler-induced Opa1 cleavage. CSA also prevented the appearance of donuts during reoxygenation, providing further evidence for the role of PTP opening (Figure 3c). However, CSA failed to affect donut formation during hypoxia in −G medium (Figures 3c and d).
Figure 3.

Role of PTP opening in donut formation. (a) Effect of mastoparan (Mas, 5 μM) on ΔΨm, mitochondrial morphology and Opa1 cleavage. (b) Effect of CSA, Me-Val-CSA (mCSA) and FK506 on the FCCP-induced donut formation and Opa1 cleavage. (c) Effect of CSA on the hypoxia–reoxygenation (H–R) induced donut formation (*P⩽0.001)
PTP opening commonly leads to mitochondrial swelling. Consistent with previous work,33, 34 valinomycin, a K+ ionophore, also induced mitochondrial swelling, leading to the appearance of ball-shaped mitochondria (Figure 4a). Swelling was paralleled by ΔΨm loss and Opa1 cleavage when 5–500 nM valinomycin was used. However, lower concentrations of the drug, 0.25–0.5 nM, induced only moderate swelling, while ΔΨm was maintained and Opa1 remained uncleaved (Figures 4b and c). Under these conditions, donut-shaped mitochondria were induced, indicating that donuts can be formed under conditions of moderate swelling (Figures 4d and e, n=9). Neither the large-scale swelling nor the donut formation evoked by valinomycin was sensitive to CSA (Figures 4a and d). Thus, mitochondrial swelling induced by two distinct mechanisms (PTP opening and increased K+ flux) can lead to donut formation.
Figure 4.

Role of K+ channel activity in donut formation. (a) Images of mtDsRed-transfected H9c2 cells show that a high concentration valinomycin (Val) causes mitochondria swelling (middle) that is insensitive to CSA (right). (b) Effect of various doses of valinomycin on Opa1 cleavage (b) and on ΔΨm (c). Hollow circles show the H9c2 cells treated with valinomycin, whereas the filled symbols show the corresponding time control. (d) Images of mtYFP-expressing (green) and TMRE-loaded H9c2 cells show donut formation by a low concentration valinomycin (0.5 nM) that did not evoke ΔΨm loss, Opa1 cleavage and massive matrix swelling. (e) Lack of protection by CSA against valinomycin-induced donut formation. (f and g) Dependence of donut formation during H–R on K+ channel opening. The concentrations of drugs were as follows: diazoxide (KATP agonist, 100 μM), NS1619 (KCa agonist, 30 μM), 5HD (KATP inhibitor, 500 μM), paxilline (Pax, KCa inhibitor, 10 μM). H9c2 cells were pretreated with drugs for 20 min before hypoxia (H) (*P⩽0.004)
We speculated that the CSA-insensitive donut formation evoked by hypoxia in −G medium might be mediated by an increase in K+ flux. Alteration of mitochondrial K+ flux has been reported in hypoxia and has been targeted by K+ channel drugs.7, 16, 35 These drugs were employed here to test the possible role of K+ flux in hypoxia-induced mitochondrial donut formation. None of the drugs caused an apparent change in mitochondrial morphology by itself (data not shown). However, donuts evoked by hypoxia in −G medium were prevented by either 5-hydroxydecanoate (5HD) or paxilline, inhibitors of the KATP and the KCa, respectively (Figure 4f). In addition, K+ channel agonists, diazoxide (for KATP) and NS1619 (for KCa), could facilitate donut formation during hypoxia in both +G and −G buffers (Figures 4f and g). Thus, the appearance of donut-shaped mitochondria, observed during hypoxia and reoxygenation, seems to be mediated through matrix swelling, which is dependent on either PTP opening during reoxygenation or K+ channel activity during hypoxia.
During hypoxia, increased activity of mitochondrial KCa has been reported16 and mitochondrial KATP was also suggested to be activated.36 The PTP remains closed during ischemia, but opens upon reperfusion.1, 37 These results complement our observations that donut formation depends on K+ channels and PTP opening in hypoxia and reoxygenation, respectively. In addition, electron microscopy studies have shown both mitochondrial matrix condensation and swelling in response to hypoxia (60 min) and more mitochondrial swelling during reoxygenation.12 Importantly, high concentrations of valinomycin (>1 nM) induced ball-shaped mitochondria reflecting a rapid and robust increase in matrix volume. By contrast, donuts appeared at low levels of valinomycin, when the swelling is moderate. K+ channel opening and the early period following PTP opening are also believed to cause moderate swelling.
Mechanism of mitochondrial donut formation
Time-lapse microscopy revealed that toroid formation resulted from end-to-end autofusion (Figures 5a and b), end-to-side autofusion (Figure 5c) or fusion between separate, elongated mitochondria (Figure 5d). During end-to-end (Figures 5a and b) or end-to-side (Figure 5c) autofusion, the mitochondrion first crooked at one end, eventually bending around to fuse with its side or opposite end, leading to the formation of a donut with or without a tail, respectively. When two different mitochondria came together to form a donut, the adjacent ends contacted and fused with the other mitochondrion's side, creating a hole (Figure 5d). Although many of the donuts maintained their shape, in several cases reopening of the ring occurred. These results would indicate that both complete and transient fusions are involved in donut formation. However, transient fusion could not be totally confirmed as it was difficult to determine whether reopening occurs at the exact site where the ends fused before. If donut formation involves mitochondrial fusion, it is expected to depend on the availability of the fusion proteins. FCCP induced donut formation in wild-type MEFs, but did not in Mfn1−/2− cells (Supplementary Figure 5). Furthermore, donut formation was confined to the initial minutes of FCCP treatment, before Opa1 cleavage took place (n=22). Thus, both OMM and IMM fusion proteins are required for donut formation. Focal photoactivation of mtPAGFP in a donut was followed by rapid spreading of the green fluorescence throughout its entirety, indicating continuity of the matrix (Figure 5e, upper donut). Matrix continuity also included the donuts' tails (Figure 5e). To assess whether the integrity of the OMM was maintained during donut formation cells were transfected with cytochrome c-GFP that resides in the mitochondrial intermembrane space. No release of GFP to the cytoplasm was observed (Supplementary Figure 6), indicating that the OMM barrier was sustained.
Figure 5.

Distinct mechanisms of donut formation. (a–d) Time-lapse images show formation of individual donuts in H9c2 cells exposed to hypoxia (H) (45–60 min in −G medium) (a) or to FCCP (0–15 min) (b–d). (e) Matrix continuity is indicated by rapid spreading of photoactivated mtPAGFP throughout two donuts formed during FCCP treatment (30–40 min). In each image sequence, the first image shown is labeled as 0 s
As we have shown previously that mitochondria were aligned with and attached to straight microtubules38 and donuts usually formed after mitochondrial bending, a prediction was made that mitochondrial detachment from the cytoskeleton is necessary for donut formation. Simultaneous labeling of the microtubules with tubulin-GFP and the mitochondria with mtDsRed allowed us to visualize detachment of one end of straight mitochondria before bending and autofusion during uncoupler treatment (n=15, Figure 6a). Detachment from the microtubules was observed in 89±1% of donut formation (n=18 cells). However, only partial appeared, parts of the donuts' surface remained closely associated with the microtubules (Figures 6a-c). Notably, dissociation of mitochondria induced by the microtubule disrupting agent, nocodazol, could not induce donut formation by itself. Three-dimensional (3D) reconstruction of the xyz image series also clarified that the rings observed in the two-dimensional images in fact correspond to toroidal mitochondria. In summary, partial detachment from the microtubules sets up the condition for mitochondrial bending and autofusion or inter-fusion at multiple sites. Interestingly, these fusion events could still occur despite the decrease in overall fusion activity during hypoxia–reoxygenation and uncoupler treatment. We propose that the force needed for mitochondrial detachment results from the mitochondrial volume change induced by PTP or K+ channel opening. As large-scale swelling per se precludes mitochondrial bending, donut formation can only be triggered by submaximal K+ fluxes (hypoxia, low-dose valinomycin) or during the initial phase of PTP opening (reoxygenation, FCCP, mastoparan). It is of significance that the mitochondrial donuts formed in different conditions have similar size. Their average diameters are as follows: 1.30±0.05 μm (reoxygenation in +G medium, n=63), 1.33±0.05 μm (hypoxia in −G medium, n=64), 1.32±0.04 μm (mastoparan, n=71), 1.33±0.04 μm (valinomycin, n=71) and 1.33±0.04 μm (FCCP, n=71).
Figure 6.

Spatial relationship between microtubules and mitochondria during donut formation. (a and b) Time-lapse images of RBL cells expressing tubulin-GFP (green) and mtDsRed (red) treated with FCCP (5 μM for 0–15 min). The first image shown is labeled as 0 s. (c) 3D reconstruction of a z-series
Fate and function of donut-shaped mitochondria
What happens to the mitochondrial donuts when the condition of their induction is discontinued? We examined the fate of the donuts after FCCP washout. Following FCCP washout, the donuts rapidly broke into pieces which subsequently recovered ΔΨm (Figures 7a and b). However, rotenone (25 min) and oligomycin (5 min) added before uncoupler washout to avoid the recovery of mitochondrial metabolism also prevented donut fragmentation during washout (Figure 7b, Supplementary Figure 7A). Most donut-shaped mitochondria grew several branches, forming complex, flower shapes (Figure 7b). Only rotenone or oligomycin added before uncoupler washout could slow the ΔΨm recovery and donut fragmentation (Figure 7c). Thus, at least partial restoration of energy metabolism is needed to allow donut fragmentation. To test the role of Drp1 in donut opening and fragmentation, FCCP treatment and washout were performed in Drp1K38A-overexpressing cells. Drp1K38A could not prevent disappearance of the ring or ΔΨm recovery, but abolished donut fragmentation (Figure 7c, Supplementary Figure 7B), indicating that donut fragmentation is dependent on Drp1-mediated fission. Thus, both donut formation and breakdown illuminate novel aspects of fusion–fission dynamics: mitochondria form donuts by auto- or inter-fusion, while partly detached from the cytoskeleton, and give rise to several offspring by Drp1-independent and, presumably, ATP-dependent break of the ring and Drp1-mediated fragmentation. The fate of the donuts during prolonged hypoxia is unclear. Mitophagy has been reported after 24–48 h hypoxia.39 It remains to be solved if donuts undergo mitophagy.
Figure 7.

Donut structure and possible roles in mitochondrial adaptation to metabolic stress. (a) Images show mitochondrial morphology (mtYFP, green) and TMRE uptake (red) during FCCP 30 min treatment and washout (1 and 8 min) in H9c2 cells. (b) Time-lapse images of individual donuts during FCCP washout incubated without (upper row) or with rotenone and oligomycin (middle row). In each image sequence, the first image shown is labeled as 0 s. (c) Cumulative data on donut number during FCCP washout in the absence or presence of rotenone, oligomycin (added 25 min before washout) and dominant-negative Drp1 (n=6, *P<0.02). (d and e) Volume increase-induced reshaping of tubular- and donut-shaped model mitochondria. Drawing shows the diameter and length changes evoked by 35% volume increase (d). Calculated volume–diameter relationships at two different initial length values (L0: 3 and 6 μm, e). (f and g) Images and bar charts show faster TMRE uptake by donuts descendents (n=95) than by linear mitochondria (n=44) during FCCP washout with rotenone (*P<0.001). (h) Summary of the cause–effect relationship between changes in mitochondrial composition, function and dynamics during hypoxia and reoxygenation (H–R)
As the donuts were formed during mitochondrial swelling, it is of interest to evaluate their tolerance to matrix expansion. The surface of a mitochondrion has been shown by 3D reconstruction of deconvolved confocal volumes to remain constant during swelling.33 We set up models to determine the surface–volume relationships for linear mitochondria and donut mitochondria of various diameters (Figure 7d). The calculation showed that a donut can accommodate a larger volume than linear mitochondria, and this difference is more obvious with the increase of d (Figure 7e). On the other hand, the smaller donut diameter at any given volume provides a favorable condition for the recruitment of and scission ring formation by Drp1.40 Fission has been suggested to govern mitochondrial segregation and elimination through generating uneven units.41
To evaluate the metabolic capacity of the donuts, TMRE uptake was measured during FCCP washout (Figures 7f and g). Descendents of the donuts showed very quick TMRE uptake similar to linear mitochondria. Furthermore, when rotenone was included during FCCP washout to delay the ΔΨm recovery, it was possible to visualize that the descendents of the donuts accumulated TMRE more quickly than the linear mitochondria (>4 μm) (Figures 7f and g). Thus, some components of the mitochondrial metabolic machinery were better preserved in the donuts than in the mitochondria that retained linear shape.
Conclusions
This study uncovered that metabolic inhibition induces a novel pattern of mitochondrial dynamics and delineated the underlying mechanisms (scheme in Figure7h). Hypoxia was shown to yield a pattern that includes (1) mitochondrial retention in the perinuclear area, (2) partial dissociation from the microtubules and anomalous fusion activity, which lead to the formation of toroidal mitochondria and (3) suppression of overall fusion activity leading to mitochondrial shortening. A key trigger for this response is suppression of mitochondrial ATP production and cellular ATP depletion. On the basis of the slower and lesser response in the presence of glycolytic substrates, cytoplasmic ATP production can partially compensate for the mitochondrial metabolic impairments. ATP is needed for the mechanisms of both mitochondrial movements and fusion. In addition, ATP depletion affects ion fluxes such as Ca2+ removal from the cytoplasm by plasma membrane and by sarco-endoplasmic reticulum Ca2+ ATPases and K+ flux mediated by KATP. Initiation of donut formation is likely to be determined by a chemico-mechanical switch that involves increased K+ and/or KCa activity or PTP opening to increase mitochondrial matrix volume. During hypoxia in −G medium, severe ATP depletion seems to be sufficient to enhance K+ fluxes and to cause some swelling. During hypoxia in +G medium, the lesser ATP depletion fails to activate this process, but PTP opening during the subsequent reoxygenation provides an alternative inducer for swelling. Notably, the relevance of mitochondrial KATP and KCa remain a subject of debate (e.g. ref. 14) However, the present results with KATP and KCa drugs and K+ ionophores are most consistent with the regulation of mitochondrial matrix volume by mitochondrial KATP and KCa. PTP induction during R and during FCCP treatment is likely to be facilitated by the combined effect of ΔΨm loss, ROS production and mitochondrial Ca2+ sequestration. Thus, different mechanisms (hypoxia in −G medium: K+ influx; reoxygenation in +G medium: solute influx) converge on water retention and ensuing matrix volume expansion to force partial dissociation of the mitochondria from the microtubules. Partial detachment permits bending of the mitochondria and anomalous fusion that produces donut-shaped mitochondria. As the overall mitochondrial fusion activity is decreased, the particular physical properties of the parent organelle are likely to promote fusion. These include the mobility of the free mitochondrial end and the increased association between IMM and OMM that might optimize the interaction between Mfns and Opa1. In addition to promoting anomalous fusion, partial detachment of the mitochondria from the microtubular tracks will also affect the movements of the mitochondria and might contribute to the retention of the organelles in the perinuclear region.
These results also show that donuts have some advantage over linear structures in tolerating a matrix volume increase. Donut formation eliminates the ends, which because of the high curvature and preferential Mfn localization are important for fusion and, in turn, for recycling of mitochondria by autophagy.41 Although donuts display loss of the ΔΨm during hypoxia and reoxygenation, they are competent to quickly regain ΔΨm. Importantly, donuts fall to several linear structures, when the metabolic stress condition is terminated. Thus, reshaping of mitochondria to donuts might be a component of a protective mechanism that helps to preserve the organelles under conditions of metabolic stress. As K+ channel activators support ischemic preconditioning in many tissues and promote donut formation during hypoxia, it is intriguing to speculate that donut formation might also have a role in preconditioning. Donuts have been documented in both primary cells and several cell lines, suggesting their formation in various tissues upon metabolic stress. However, in cardiac or skeletal muscle, a large fraction of mitochondria is located in the narrow interfibrillar space and so it remains to be seen whether these mitochondria can undergo donut formation like mitochondria of the brain, kidney or the subplasmalemmal space of the muscle.
Experimental Procedures
Cells
H9c2 cells and RBL-2H3 mucosal mast cells were cultured and plated for experiments as described previously.38, 42 For most measurements, the cells were incubated in an extracellular medium containing 0.25% BSA at 35 °C.38 For hypoxia and reoxygenation conditions, cells were mounted in an air-tight sealed perfusion chamber on the confocal stage and were perfused with +G medium (glucose 10 mM, n=35) or −G medium (methylsuccinate 1 mM n=29) solutions saturated with air or N2 (100%) (for hypoxia) at 35 °C.
Transient expression
Transfection was performed using electroporation (2–3 × 106 cells and 4–20 μg of each plasmid DNA per 250 μl). All mtFP vectors (GFP, YFP, DsRed, PAGFP) have the targeting sequence of cytochrome c oxidase subunit VIII to achieve mitochondrial matrix localization. Vectors encoding mtPAGFP, Opa1, Drp1K38A and a dominant-negative mutant of calcineurin devoid of the CaM binding and the autoinhibitory domains and harboring an inactivating His-151 to Gln point mutation (ΔCnAH151Q) were kindly provided by Jennifer Lippincott-Schwartz (NIH), Richard J Youle (NIH), Alex van der Bliek (UCLA) and Luca Scorrano (Venetian Institute of Molecular Medicine, Italy), respectively. Transfected cells were further incubated in the culture for 24–72 h before the imaging experiments. Neonatal cardiomyocytes were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Live cell microscopic imaging
To assess ΔΨm, H9c2 cells were loaded with TMRE (25 nM for 15 min). During the recording, TMRE (5 nM) was also added in buffer.
Most experiments were performed using a Bio-Rad Radiance system (Bio-Rad, Hercules, CA, USA) fitted to an Olympus IX70 microscope (Olympus, Tokyo, Japan) using a × 40 objective (Uapo340, NA 1.35; Olympus) recording 512 × 512 pixel image pairs or triplicates at 0.25–1 s. The HeCd laser (442 nm) was used for photoactivation of PAGFP. PAGFP was photoactivated using the ROI scanning option in the LaserSharp2000 software (Bio-Rad). Three 25 μm2 areas were chosen per cell and illuminated with maximum power at 442 nm excitation, respectively, for eight consecutive images to achieve irreversible photoactivation. Every run, each cell is monitored for 8 min. The dual-line Kr/Ar ion laser source was used for imaging of GFP and YFP at 488 nm excitation, and TMRE was used for DsRed at 568 nm. To create 3D-like reconstructions, a sequence of images was collected from below the adherent surface up in 0.25 μm steps (z-series) using the focus motor driven by the Lasersharp software.
Donuts were manually counted in each cell. We only considered structures that showed a central hole. Acquisition of z-series and 3D image reconstruction confirmed that these structures were real toroids (Figure 6). Although the pixel size was <140 nm, it is possible that we failed to detect some small donuts with this approach.
TMRE imaging was also performed using a CCD camera.26, 38 Image analysis was carried out using custom-designed imaging software. For quantitation of TMRE fluorescence, the whole-cell area was masked, excluding nuclei, and the background fluorescence obtained in the cell-free areas was subtracted. Complete depolarization evoked by FCCP caused approx. 90% decrease in TMRE fluorescence in confocal imaging and 60% decrease in CCD imaging.
3D rendering of the z image stacks was carried out using the Voxx software from Indiana University, Bloomington, IN, USA.
Western blot analysis
Equal amounts of cell lysates were run on 10% polyacrylamide gels and transferred to nitrocellulose filters. Filters were blocked overnight, followed by incubation with an anti-Opa1 antibody at dilution 1:500 (BD Biosciences, San Jose, CA, USA). After incubation with the primary antibody, bound antibodies were visualized using horseradish peroxidase-coupled secondary antibody and Dura Signal chemiluminescence-developing kit (Pierce, Rockford, IL, USA).
Drug treatment
FCCP (5 μM, 7 min, n=14; 15 min, n=19); mastoparan (5 μM, 30 min, n=16); valinomycin (0.05–500 nM, 15 min, n=9). These treatments were also performed in the presence of CSA (5 μM), Me-Val-CSA (10 μM), FK506 (10 μM), diazoxide (100 μM), NS1619 (30 μM), 5HD (500 μM) and paxilline (10 μM). For FCCP washout, cells (control or Drp1K38A-expressing cells) were treated with FCCP for 30 min, or together with rotenone (10 μM, 25 min) and oligomycin (5 μg/ml, 5 min), and then FCCP was washed out with 2% BSA once and 0.25% BSA twice (n=6).
Cellular ATP measurement
ATP levels were determined using the Bioluminescence Detection Kit for ATP (Promega, Madison, WI, USA). Briefly, H9c2 cells were extracted with 2.5% trichloroacetic acid, the sample was neutralized and diluted in 10 mM Tris-acetate (pH 7.75), and ATP levels were measured using the Luciferase/Luciferin reagent according to the manufacturer's protocol (n=4–12).
Model mitochondria
Linear mitochondria were drawn as l – length, d – diameter cylinders capped with d – diameter half-spheres at both ends. The following equations were used to describe the surface and volume of model mitochondria:
 |
Donut mitochondria were drawn as m – middle line length and d – diameter donut. The following equations are used to describe the surface and volume of model mitochondria:
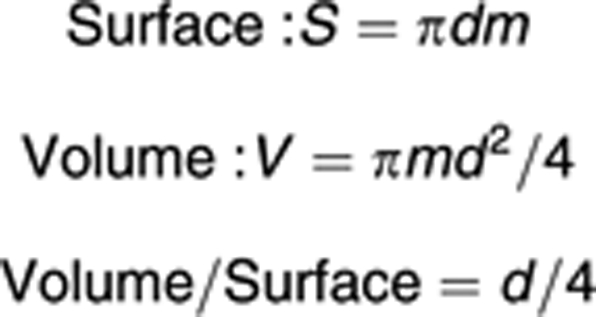 |
Surface of individual mitochondria has been shown to be constant during swelling,33 so S was kept constant during swelling here: S=S0
Linear mitochondria swelling:
 |
Range of d:
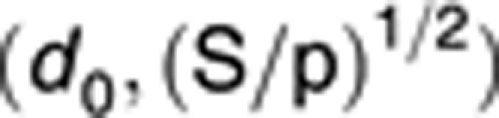 |
Donut-shaped mitochondria swelling:
 |
Range of d:
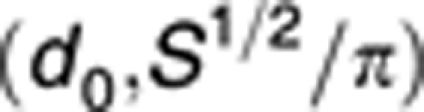 |
Statistics
The data are shown as mean±S.E. (n⩾3). Significance of differences from the relevant controls was calculated by Student's t-test.
Acknowledgments
We thank Miguel Aon, Tamas Balla, Paolo Bernardi, Andrew Halestrap, George Purkins, Orian Shirihai and David Weaver for discussions and for help in the preparation of the manuscript. This work was supported by NIH Grants to GH (AA017773).
Glossary
- FCCP
carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- +G medium
glucose-containing medium
- −G medium, glucose-free, methyl succinate-containing medium; IMM
inner mitochondrial membrane
- Kca
Ca2+-activated K+ channels
- KATP
ATP-activated K+ channels
- mtPAGFP
mitochondrial matrix-targeted photoactivatable green fluorescence protein
- mtDsRed
mitochondrial matrix-targeted red fluorescent protein
- OMM
outer mitochondrial membrane
- PTP
permeability transition pore
- R
reoxygenation
- ROS
reactive oxygen species
- CSA
cyclosporin A
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Edited by L Scorrano
Supplementary Material
References
- Di Lisa F, Canton M, Menabo R, Kaludercic N, Bernardi P. Mitochondria and cardioprotection. Heart Fail Rev. 2007;12:249–260. doi: 10.1007/s10741-007-9028-z. [DOI] [PubMed] [Google Scholar]
- Robertson CL, Scafidi S, McKenna MC, Fiskum G. Mitochondrial mechanisms of cell death and neuroprotection in pediatric ischemic and traumatic brain injury. Exp Neurol. 2009;218:371–380. doi: 10.1016/j.expneurol.2009.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP. Mitochondria and reperfusion injury of the heart – a holey death but not beyond salvation. J Bioenerg Biomembr. 2009;41:113–121. doi: 10.1007/s10863-009-9206-x. [DOI] [PubMed] [Google Scholar]
- Lemasters JJ. Modulation of mitochondrial membrane permeability in pathogenesis, autophagy and control of metabolism. J Gastroenterol Hepatol. 2007;22 (Suppl 1:S31–S37. doi: 10.1111/j.1440-1746.2006.04643.x. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Zorov DB, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Role of glycogen synthase kinase-3beta in cardioprotection. Circ Res. 2009;104:1240–1252. doi: 10.1161/CIRCRESAHA.109.197996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia–reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlid KD, Costa AD, Quinlan CL, Pierre SV, Dos Santos P. Cardioprotective signaling to mitochondria. J Mol Cell Cardiol. 2009;46:858–866. doi: 10.1016/j.yjmcc.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, et al. Superoxide flashes in single mitochondria. Cell. 2008;134:279–290. doi: 10.1016/j.cell.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korge P, Ping P, Weiss JN. Reactive oxygen species production in energized cardiac mitochondria during hypoxia/reoxygenation: modulation by nitric oxide. Circ Res. 2008;103:873–880. doi: 10.1161/CIRCRESAHA.108.180869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry CE, Richard VJ, Reimer KA, Jennings RB. Ischemic preconditioning slows energy metabolism and delays ultrastructural damage during a sustained ischemic episode. Circ Res. 1990;66:913–931. doi: 10.1161/01.res.66.4.913. [DOI] [PubMed] [Google Scholar]
- Hoppel CL, Tandler B, Fujioka H, Riva A. Dynamic organization of mitochondria in human heart and in myocardial disease. Int J Biochem Cell Biol. 2009;41:1949–1956. doi: 10.1016/j.biocel.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg JM, Venkatachalam MA, Roeser NF, Nissim I. Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc Natl Acad Sci USA. 2000;97:2826–2831. doi: 10.1073/pnas.97.6.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saris NE, Eriksson KO. Mitochondrial dysfunction in ischaemia–reperfusion. Acta Anaesthesiol Scand Suppl. 1995;107:171–176. doi: 10.1111/j.1399-6576.1995.tb04353.x. [DOI] [PubMed] [Google Scholar]
- Szewczyk A, Jarmuszkiewicz W, Kunz WS. Mitochondrial potassium channels. IUBMB Life. 2009;61:134–143. doi: 10.1002/iub.155. [DOI] [PubMed] [Google Scholar]
- Xu W, Liu Y, Wang S, McDonald T, Van Eyk JE, Sidor A, et al. Cytoprotective role of Ca2+-activated K+ channels in the cardiac inner mitochondrial membrane. Science. 2002;298:1029–1033. doi: 10.1126/science.1074360. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Gu XQ, Bednarczyk P, Wiedemann FR, Haddad GG, Siemen D. Hypoxia increases activity of the BK-channel in the inner mitochondrial membrane and reduces activity of the permeability transition pore. Cell Physiol Biochem. 2008;22:127–136. doi: 10.1159/000149790. [DOI] [PubMed] [Google Scholar]
- Wasilewski M, Scorrano L. The changing shape of mitochondrial apoptosis. Trends Endocrinol Metab. 2009;20:287–294. doi: 10.1016/j.tem.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Legros F, Lombes A, Frachon P, Rojo M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell. 2002;13:4343–4354. doi: 10.1091/mbc.E02-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res. 2009;84:91–99. doi: 10.1093/cvr/cvp181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeusen S, McCaffery JM, Nunnari J. Mitochondrial fusion intermediates revealed in vitro. Science. 2004;305:1747–1752. doi: 10.1126/science.1100612. [DOI] [PubMed] [Google Scholar]
- Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- De Vos KJ, Allan VJ, Grierson AJ, Sheetz MP. Mitochondrial function and actin regulate dynamin-related protein 1-dependent mitochondrial fission. Curr Biol. 2005;15:678–683. doi: 10.1016/j.cub.2005.02.064. [DOI] [PubMed] [Google Scholar]
- Plecita-Hlavata L, Lessard M, Santorova J, Bewersdorf J, Jezek P. Mitochondrial oxidative phosphorylation and energetic status are reflected by morphology of mitochondrial network in INS-1E and HEP-G2 cells viewed by 4Pi microscopy. Biochim Biophys Acta. 2008;1777:834–846. doi: 10.1016/j.bbabio.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Ishihara N, Jofuku A, Eura Y, Mihara K. Regulation of mitochondrial morphology by membrane potential, and DRP1-dependent division and FZO1-dependent fusion reaction in mammalian cells. Biochem Biophys Res Commun. 2003;301:891–898. doi: 10.1016/s0006-291x(03)00050-0. [DOI] [PubMed] [Google Scholar]
- Twig G, Liu X, Liesa M, Wikstrom JD, Molina AJ, Las G, et al. Biophysical properties of mitochondrial fusion events in pancreatic beta-cells and cardiac cells unravel potential control mechanisms of its selectivity. Am J Physiol Cell Physiol. 2010;299:C477–C487. doi: 10.1152/ajpcell.00427.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Weaver D, Shirihai O, Hajnoczky G. Mitochondrial ‘kiss-and-run': interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 2009;28:3074–3089. doi: 10.1038/emboj.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvezin-Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem. 2006;281:37972–37979. doi: 10.1074/jbc.M606059200. [DOI] [PubMed] [Google Scholar]
- Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009;28:1589–1600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1–S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA. 2009;106:11960–11965. doi: 10.1073/pnas.0904875106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaltowski AJ, Castilho RF, Vercesi AE. Opening of the mitochondrial permeability transition pore by uncoupling or inorganic phosphate in the presence of Ca2+ is dependent on mitochondrial-generated reactive oxygen species. FEBS Lett. 1996;378:150–152. doi: 10.1016/0014-5793(95)01449-7. [DOI] [PubMed] [Google Scholar]
- Pfeiffer DR, Gudz TI, Novgorodov SA, Erdahl WL. The peptide mastoparan is a potent facilitator of the mitochondrial permeability transition. J Biol Chem. 1995;270:4923–4932. doi: 10.1074/jbc.270.9.4923. [DOI] [PubMed] [Google Scholar]
- Safiulina D, Veksler V, Zharkovsky A, Kaasik A. Loss of mitochondrial membrane potential is associated with increase in mitochondrial volume: physiological role in neurones. J Cell Physiol. 2006;206:347–353. doi: 10.1002/jcp.20476. [DOI] [PubMed] [Google Scholar]
- Gerencser AA, Doczi J, Torocsik B, Bossy-Wetzel E, Adam-Vizi V. Mitochondrial swelling measurement in situ by optimized spatial filtering: astrocyte–neuron differences. Biophys J. 2008;95:2583–2598. doi: 10.1529/biophysj.107.118620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR. Preconditioning protects by inhibiting the mitochondrial permeability transition. Am J Physiol Heart Circ Physiol. 2004;287:H841–H849. doi: 10.1152/ajpheart.00678.2003. [DOI] [PubMed] [Google Scholar]
- Brayden JE. Functional roles of KATP channels in vascular smooth muscle. Clin Exp Pharmacol Physiol. 2002;29:312–316. doi: 10.1046/j.1440-1681.2002.03650.x. [DOI] [PubMed] [Google Scholar]
- Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307 (Part 1:93–98. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Weaver D, Hajnoczky G. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J Cell Biol. 2004;167:661–672. doi: 10.1083/jcb.200406038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, et al. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol. 2005;170:1021–1027. doi: 10.1083/jcb.200506078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Varnai P, Golenar T, Roy S, Purkins G, Schneider TG, et al. Imaging interorganelle contacts and local calcium dynamics at the ER–mitochondrial interface. Mol Cell. 2010;39:121–132. doi: 10.1016/j.molcel.2010.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.