

Abstract
An important requirement for physiologic homeostasis is the detoxification and removal of endogenous hormones and xenobiotic compounds with biological activity. Much of the detoxification is performed by cytochrome P-450 enzymes, many of which have broad substrate specificity and are inducible by hundreds of different compounds, including steroids. The ingestion of dietary steroids and lipids induces the same enzymes; therefore, they would appear to be integrated into a coordinated metabolic pathway. Instead of possessing hundreds of receptors, one for each inducing compound, we propose the existence of a few broad specificity, low-affinity sensing receptors that would monitor aggregate levels of inducers to trigger production of metabolizing enzymes. In support of this model, we have isolated a novel nuclear receptor, termed the steroid and xenobiotic receptor (SXR), which activates transcription in response to a diversity of natural and synthetic compounds. SXR forms a heterodimer with RXR that can bind to and induce transcription from response elements present in steroid-inducible cytochrome P-450 genes and is expressed in tissues in which these catabolic enzymes are expressed. These results strongly support the steroid sensor hypothesis and suggest that broad specificity sensing receptors may represent a novel branch of the nuclear receptor superfamily.
Keywords: Steroid, xenobiotic receptor, nuclear receptor, transcriptional activity
Lipophilic hormones, such as steroids, retinoic acid, thyroid hormone, and vitamin D3, control broad aspects of animal growth, development, and adult organ physiology. The effects of these hormones are mediated by a large superfamily of intracellular receptors that function as ligand-dependent and sequence-specific transcription factors. The nonsteroidal nuclear receptors for thyroid hormone (TR), vitamin D3 (VDR), all-trans retinoic acid (RAR), and fatty acids and eicosanoids (PPAR) form heterodimers with the 9-cis retinoic acid receptor (RXR) that bind bipartite hormone-response elements (HREs) composed of directly repeated half sites related to the sequence AGGTCA (Mangelsdorf and Evans 1995). In contrast, the steroid receptors function as homodimers and bind to palindromic target sequences spaced by three nucleotides (Beato et al. 1995). In addition to the known receptors, a large group of structurally related ‘orphan’ nuclear receptors has been described; that these receptors possess obvious DNA and ligand-binding domains but lack identified ligands (Mangelsdorf et al. 1995). Each has the potential to regulate a distinct endocrine signaling pathway.
It is widely viewed that the hormone response is a consequence of the release from an endocrine gland of a ligand that circulates through the blood, and coordinately regulates responses in target tissues by acting through specific nuclear receptors. Hormone responsiveness is dependent on the ability to rapidly clear ligand from the blood and the body so that, in absence of a stimulus, target tissues return to a ground state. Hormonal homeostasis is thus achieved by the coordinated release and degradation of bioactive hormones. Steroid hormones and their many metabolites are primarily inactivated by reduction and oxidation in the liver. As there are >45 adrenal steroids identified (Norman and Litwack 1997), dozens of each of the sex steroids (androgens, estrogens, and progestins) (Norman and Litwack 1997), 25–35 vitamin D metabolites (Horst and Reinhardt 1997), and likely hundreds of fatty acids, eicosanoids, hydroxyfats, and related bioactive lipids, the problem of efficient ligand elimination is critical to physiologic homeostasis. In addition to a myriad of endogenous hormones, a similar diversity of ingested plant and animal steroids and bioactive xenobiotic compounds must also be degraded.
Selye (1971) first introduced the concept that exogenous steroids and pharmacologic substances may function to modulate the expression of enzymes that would protect against subsequent exposure to toxic xenobiotic substances. These compounds, which Selye called catatoxic steroids, are typified by the synthetic glucocorticoid antagonist pregnenolone-16-carbonitrile (PCN). PCN and a variety of xenobiotic steroids induce the proliferation of hepatic endoplasmic reticulum and the expression of cytochrome P-450 genes (Schuetz and Guzelian 1984; Gonzalez et al. 1986; Burger et al. 1992). One consequence of PCN treatment is the induction of nonspecific ‘protection’ against subsequent exposure to such diverse xenobiotics as digitoxin, indomethacin, barbiturates, and steroids (Selye 1971). Furthermore, it is known that a variety of such compounds can activate P-450 genes responsible for their detoxification or degradation (Denison and Whitlock 1995; Fernandez-Salguero and Gonzalez 1995; Hankinson 1995; Rendic and Di Carlo 1997).
Although it appears that catatoxic compounds must regulate the expression of cytochrome P-450s and other detoxifying enzymes, two lines of evidence argue that such regulation is independent of the classic steroid receptors. First, many of the most potent compounds (e.g., PCN, spironolactone, cyproterone acetate) are steroid receptor antagonists, whereas others (e.g., dexamethasone) are receptor agonists (Burger et al. 1992). Second, the nonspecific protective response remains after bilateral adrenalectomy (and presumably in the absence of adrenal steroids) but not after partial hepatectomy (Selye 1971). Therefore, hepatic orphan nuclear receptors regulated by these protective compounds would provide a novel pathway for the induction of xenobiotic metabolizing enzymes. Because such enzymes are induced by high (pharmacological) doses of xenobiotics, natural and synthetic steroids, and phytosteroids, we anticipate that the sensor would be a broad specificity, low-affinity receptor.
Here we describe the characterization of a novel human orphan nuclear receptor, termed the steroid and xenobiotic receptor (SXR), that responds to an enormous variety of natural and synthetic steroid hormones, including antagonists as well as xenobiotic drugs such as rifampicin and bioactive dietary compounds such as phytoestrogens. The ability of SXR to regulate expression of catabolic enzymes in response to this diversity of natural and pharmaceutical compounds is unprecedented for a nuclear receptor and provides a novel mechanism for direct regulation of metabolism.
Results
SXR is a novel human orphan nuclear receptor
SXR was isolated in a screen to identify potential human homologs of the Xenopus benzoate X receptor (BXR) (Blumberg et al. 1998). The cDNA encodes a predicted protein of 434 amino acids (Fig. 1A) that is 73% identical to BXR in the DNA-binding domain (DBD) and 43% identical in the ligand-binding domain (LBD) (Fig. 1B). SXR is most closely related to the recently described pregnane X receptor (PXR) (Kliewer et al. 1998) (95% identical in the DBD, 73% identical in the LBD). SXR is related more distantly to the vitamin D3 receptor and the orphan receptor CAR (constitutive androstane receptor) (Baes et al. 1994) (Fig. 1B). Other than these receptors, SXR shows no more similarity to other nuclear receptors than the different receptor subfamilies do to each other (Fig. 1B). Because true homologs among nuclear receptors typically share considerable similarity, especially in the DBD, we conclude that SXR and PXR comprise a new branch of the nuclear receptor superfamily.
Figure 1.

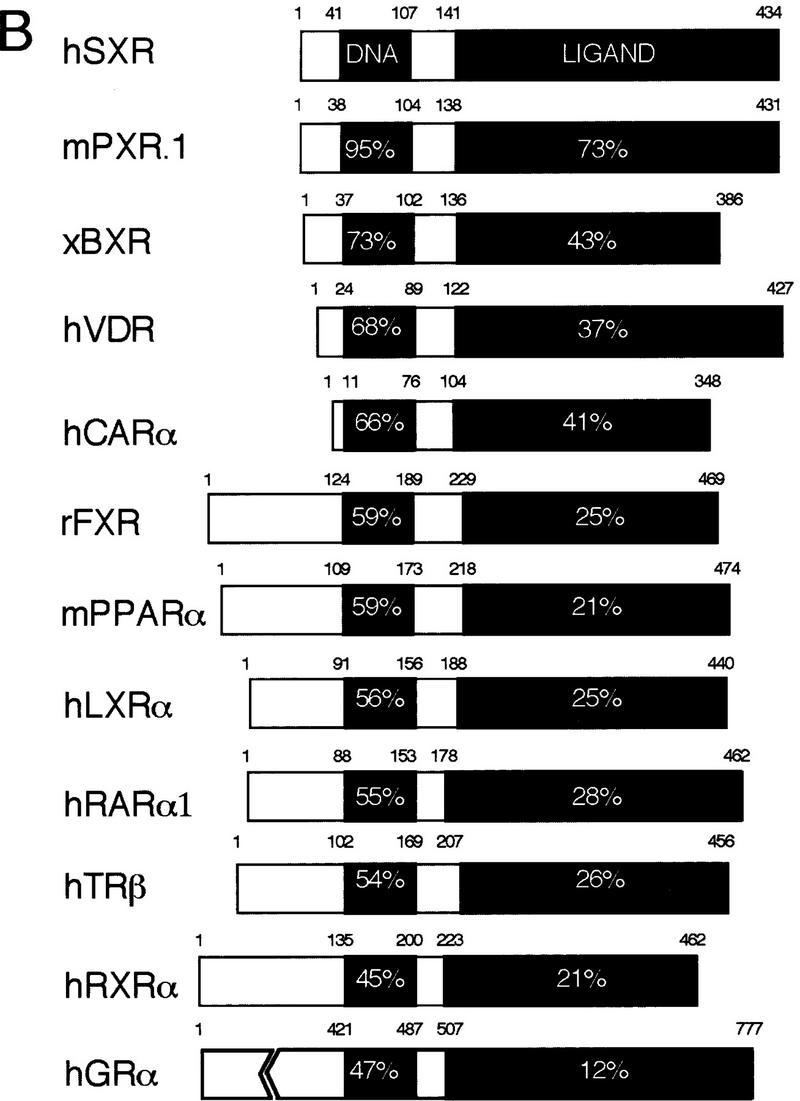

SXR is a novel orphan nuclear receptor. (A) Sequence of the longest SXR cDNA clone. The DBD is shown in boldface type; upstream termination codons in-frame with the putative initiator leucine are indicated by asterisks. Leu can function as an initiator, as demonstrated by SDS-PAGE analysis of labeled proteins produced from in vitro-transcribed, translated cDNAs. The unmodified cDNAs yielded a translation product indistinguishable in size from that produced when the leucine was changed to methionine, albeit not nearly as efficiently (data not shown). (B) Schematic comparisons among SXR and other RXR partners. Amino acid sequences were aligned using the program GAP (Devereaux et al. 1984). The similarity between SXR and other receptors is expressed as percent amino acid identity. LBD boundaries follow those for the canonical nuclear receptor LBD (Wurtz et al. 1996). (C) SXR mRNA expression. Full-length SXR cDNA was used to probe multiple-tissue Northern blots (Clontech). SXR mRNA is expressed abundantly in liver and strongly, but much less abundantly, in intestine. (Left) Exposed for 4 hr, (right) exposed for 24 hr. Longer exposures did not reveal hybridizing bands in any other tissues on these blots. The sizes of four of five mRNAs are shown; the fifth could not be sized accurately as it is much larger than the largest size marker.
Screening of a mouse liver cDNA library at reduced stringency resulted in the identification of 39 cDNAs, all of which encoded PXR.1 (data not shown). Because orthologous nuclear receptors typically share upward of 90% amino acid identity in the LBD when comparing rodent and human receptors [e.g., RARα, 98% human/mouse (h/m); PPARγ, 98% h/m; glucocorticoid receptor (GR), 95% h/m; TRβ, 98% h/rat; estrogen receptorα (ERα), 89% h/m], PXR and SXR may represent α and β subtypes of a new receptor family. Although this is supported by the distinct pharmacological properties of the receptors (see below) further screening of mouse and human liver cDNA libraries has failed to identify other family members. This suggests that PXR and SXR could represent unusually divergent orthologous genes. If correct, this divergence may reflect receptor adaptation to the different diets of rodents and primates and the requirement to detoxify appropriate food-borne compounds.
Northern blot analysis showed that SXR mRNA is expressed at high levels in liver and at more moderate levels in the intestine (Fig. 1C). Longer exposures did not reveal expression in any other tissues on these blots. Multiple mRNAs were detected, ranging from 3500 nucleotides to larger than 9000 nucleotides. Comparison of the four cDNAs obtained suggests that these differences may be attributable to alternative polyadenylation as they share the same protein coding and 5′-untranslated sequences, but each has a different 3′ end (data not shown).
SXR DNA-binding specificity
Electrophoretic mobility shift assays were used to determine the ability of SXR to heterodimerize with RXR and to analyze the selectivity and specificity of SXR DNA binding. Receptors that heterodimerize with RXR typically bind to direct repeats of AGGTCA or closely related sequences (Mangelsdorf and Evans 1995). We tested SXR alone and in combination with RXR on a series of ‘testor’ elements differing in the spacing between half sites from 0 to 15 nucleotides. No binding was seen on classic steroid response elements (data not shown). In contrast, strong binding was selective to a DR-4 motif with minimal binding to DR-3 and DR-5 and no binding to other spacings (Fig. 2A; data not shown). When the variant AGTTCA (βDR) half site was used, strong binding was seen on βDR-4 and βDR-5 and significant, but reduced, binding to βDR-3 (Fig. 2B). These results demonstrate that SXR binds DNA as a heterodimer with RXR rather than as a homodimer like the classic steroid receptors (Beato et al. 1995).
Figure 2.
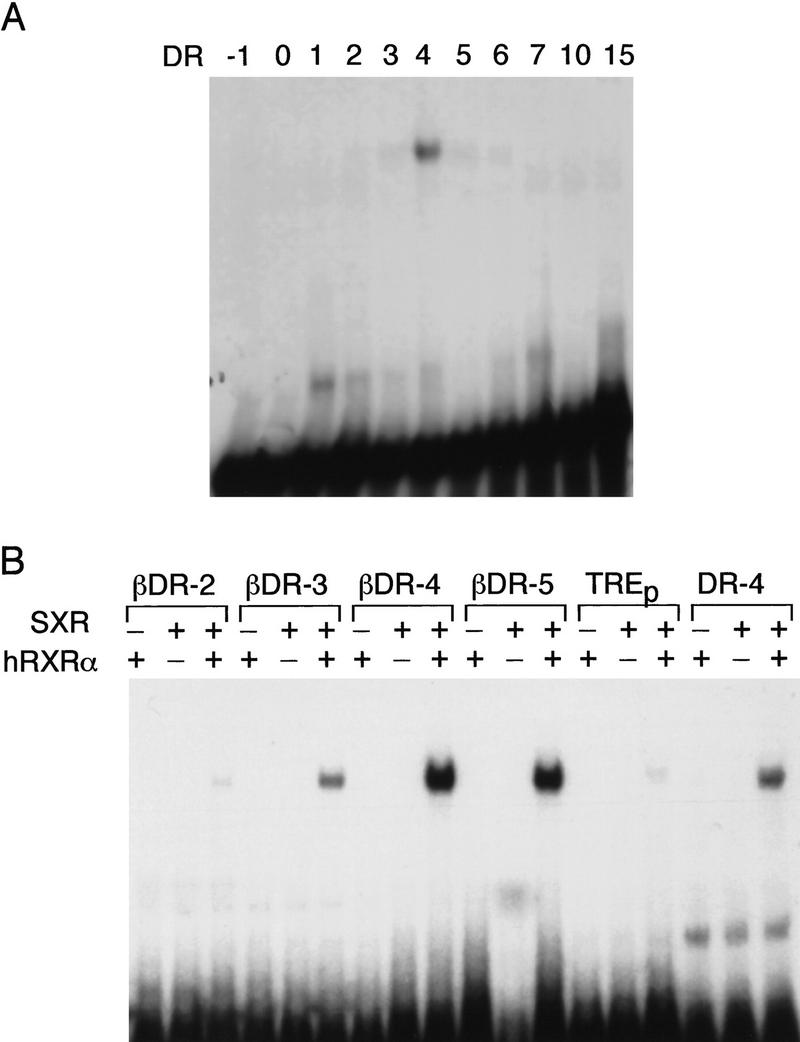
SXR DNA-binding specificity. (A) SXR:hRXRα heterodimers prefer DR-4 among a panel of AGGTCA-containing HREs. In vitro-transcribed and -translated SXR was incubated with 32P-labeled oligonucleotides and electrophoresed in native polyacrylamide gels. (B) AGTTCA is preferred to AGGTCA. SXR:hRXRα heterodimers were tested for their ability to bind half-sites of the sequence AGTTCA derived from the RARβ RA–responsive element (Sucov et al. 1990). We found that in addition to a spacing motif of 4 (βDR-4) they bind nearly as well to βDR-5 spacing and significantly to a βDR-3 motif. DR-4 and TREp are shown for reference.
SXR is activated by steroids
To determine whether the activity of SXR was ligand dependent, mixtures of natural and synthetic compounds were tested for their ability to activate SXR in transfection-based assays. A mixture containing dehydroepiandrosterone (DHEA) and pregnenolone was active, suggesting that SXR might be a new steroid receptor. To characterize its response properties, a large variety of steroids, including intermediate metabolites and major products of known steroid biosynthetic pathways were tested. Surprisingly, most of these compounds were active, although there were clear differences in potency (Fig. 3A). Of the >70 steroids tested most showed some activity at high doses (data not shown). Activation was dependent on the LBD of SXR, as both full-length receptors and GAL4-receptor LBD chimeras showed similar activity, whereas there was no activation of reporter gene expression in experiments with reporter alone or reporter plus GAL4 DNA-binding domain (Fig. 3A; data not shown). The most potent and efficacious activator of the numerous steroids tested is corticosterone (Fig. 3A). Estradiol and dihydrotestosterone are also remarkably effective activators, whereas aldosterone and 1,25-dihydroxy vitamin D3 are inactive, even at 50 μm (Fig. 3A; data not shown). Although ligands for the classic steroid receptors do show some overlap in receptor specificity, there is no example of a nuclear receptor that can be activated by so many different types of steroids. This broad ligand specificity of SXR parallels that of PPARα, which is activated by a very diverse group of dietary fatty acids at micromolar levels (Gottlicher et al. 1992; Forman et al. 1997; Kliewer et al. 1997).
Figure 3.
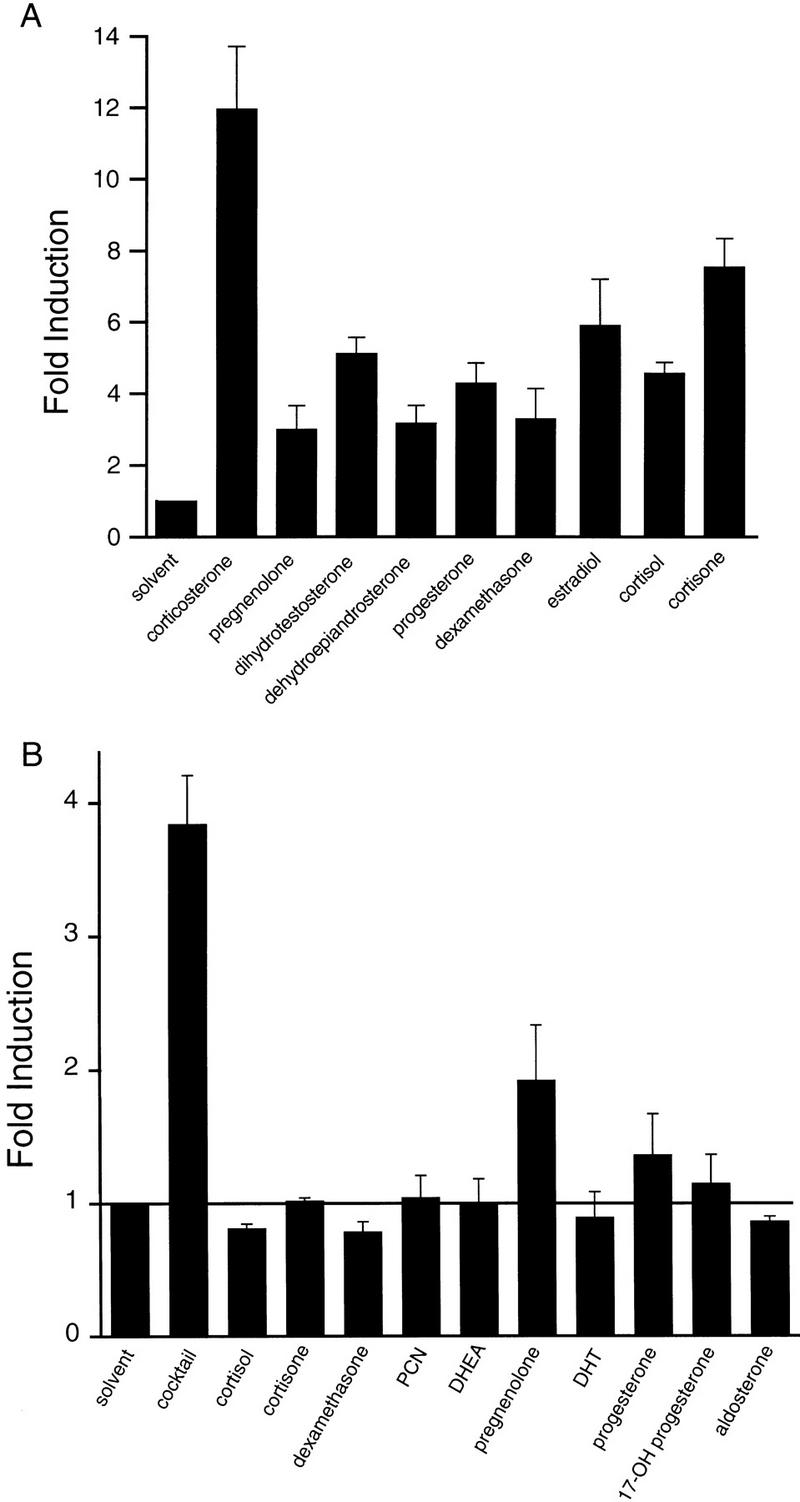
SXR is activated by many steroids. (A) Chimeric receptors composed of the GAL4 DNA-binding domain and the SXR-ligand binding domain were cotransfected into CV-1 cells with the reporter gene tk(MH100)4–luc (Forman et al. 1995). DHEA and pregnenolone activated this chimeric receptor; therefore, other steroids were tested for activation. Results are shown as fold induction over solvent (DMSO) control for 50 μm steroid and represent the averages and standard error from triplicate assays. Reporter alone or reporter plus GAL4–DBD was not activated by any of these compounds (data not shown). Similar results were obtained using full-length receptors and appropriate reporters (see below). (B) The ability of steroidal activators to act additively was tested using full-length SXR and the reporter tk(LXRE)3–luc (Willy et al. 1995). The cocktail contained 10 μm of each steroid for an overall concentration of 100 μm total steroid. The cocktail and its individual components were tested at 100, 10, and 1 μm; results are shown for 100 μm cocktail and 10 μm component steroids. Similar results were obtained using GAL–SXR (not shown).
The diversity of steroids showing activity on SXR led us to hypothesize that it might be able to sense cumulative, as well as individual steroid levels, predicting that combinations of activators might be more active than the individual components. As shown in Figure 3B, a cocktail containing 10 steroids each at 10 μm (for an overall concentration of 100 μm) was considerably more active than its individual components at 10 μm, a concentration at which most were inactive. These results support the proposal that SXR is a broad specificity, low-affinity, steroid-activated receptor.
SXR may regulate the activity of steroid-inducible P-450s
A search of the GenBank database for genes expressed in liver containing potential SXR response elements identified the steroid hydroxylases CYP2A1, CYP2A2, CYP2C1, CYP2C6, CYP3A1, CYP3A2, P-450 oxidoreductase, and UDP–glucuronosyltransferase as candidate target genes (Fig. 4A). The data shown in Figure 4B verify that SXR can activate DR-3, DR-4, and DR-5 elements that are present in these genes. In this series of transfections, corticosterone along with pregnenolone, progesterone, dihydrotestosterone (DHT), estradiol, and PCN are consistently among the best activators. Dexamethasone, cortisone, and DHEA are in the intermediate group with little response from either aldosterone or cortisol (Fig. 4B). Consistent with the DNA-binding data, maximal activities are achieved on βDR-3, βDR-4, and βDR-5 elements (Fig. 4B).
Figure 4.

SXR can activate responsive elements found in steroid and xenobiotic-inducible P-450 enzymes. (A) Putative DR-series response elements are found in inducible cytochrome P-450 enzymes. A database search for repeats of the sequence RGKTCA was performed, and some of the hits for enzymes involved in hepatic steroid hydroxylation are indicated. The standard nomenclature for P-450 enzymes has been used. P-450R is the single P-450 oxidoreductase required for hydroxylation of steroids. UGT1A6 is a rat UDP–glucuronosyltransferase that conjugates glucuronic acid to hydroxylated steroids. (B) SXR has a broad specificity for both response elements and steroidal activators. Full-length SXR was tested in cotransfection experiments for its ability to activate elements similar to those in A in response to a panel of steroids at 50 μm. DR-1, DR-2, and TREp were only very slightly activated; hence, results are shown only for corticosterone and PCN. The actual response elements and the number of copies are as follows, the base vector is tk–luc in all cases (Hollenberg et al. 1985): DR-1, tk(ApoAI)4 (Ladias and Karathanasis 1991); DR-2, tk(Hox-B1-RARE)2 (Ogura and Evans 1995); βDR-3, tk(CYP3A2)3 (Kliewer et al. 1998); DR-4, tk(MLV-TRE)2 (Umesono et al. 1991); βDR-4, tk(LXRE)3 (Willy et al. 1995); βDR-5, tk(βRARE)3 (Sucov et al. 1990); TREp, tk(TREp)2 (Umesono et al. 1991). The data shown are expressed as mean fold induction over solvent control ± s.e. from triplicate assays. (C) Conserved glucocorticoid-responsive elements found in human CYP3 genes. The region of human CYP3A4 shown to be necessary and sufficient for glucocorticoid and rifampicin induction of the full-length promoter is shown along with the corresponding regions of CYP3A5 and CYP3A7 (Barwick et al. 1996). (D) SXR:RXR heterodimers bind to IR-6 elements. The ability of SXR to bind to a panel of IR elements with spacers from 0 to 6 were tested along with βDR-4. SXR binds only as a homodimer with RXR to CYP3A4 IR-6 and βDR-4 elements. (E) SXR:RXR heterodimers bind to βDR-4 and IR-6 elements with similar affinity. Competition binding experiments were performed to estimate the relative affinity of SXR:RXR binding to CYP3A4 IR-6 element or βDR-4. The IR-M competitor has half-site mutations that prevent SXR:RXR binding (D). (F) SXR can activate through inducible, but not uninducible CYP3 promoter elements. The ability of SXR to activate tk–CYP3–luc response elements in response to various inducers was tested. (Open bars) Rifamipicin; (solid bars) corticosterone. Results are shown for 50 μm compound and represent the mean of triplicate determinations.
The inducibility of SXR by PCN and other steroids led us to consider whether P-450s known to be inducible by these compounds could be SXR targets. The primary human steroid-inducible P-450 is the CYP3A4 gene (Beaune et al. 1986; Molowa et al. 1986). Unlike the rat and mouse CYP3A genes, all of which contain a DR-3 element that SXR can activate (Fig. 4B), the human and rabbit promoters do not contain such an element. Steroid and xenobiotic inducibility of CYP3A4 has been localized to an 19-bp element that is functional in transient transfection assays (Barwick et al. 1996). This element contains an IR-6 motif (TGAACTcaaaggAGGTCA) and similar elements are also present in the human CYP3A5, CYP3A7, and the rabbit CYP3A6 genes (Fig. 4C; Barwick et al. 1996). We tested the ability of SXR to bind a series of inverted repeat elements with spacings from 0 to 6 nucleotides and found that only an IR-6 showed significant binding that, as with the direct repeats, was RXR dependent (Fig. 4D; data not shown). Competition binding experiments demonstrated little difference in the apparent affinity of SXR:RXR heterodimers for the βDR-4 or CYP3A4 IR-6 response elements (Fig. 4E). In accord with the known inducibility of the parent promoters, SXR could activate reporter constructs containing the CYP3A4, but not the CYP3A5 or CYP3A7 motifs (Fig. 4F).
We then asked whether compounds known to induce CYP3A4 could activate SXR, as would be predicted from our model. Compounds tested included drugs such as rifampicin and nifedipine, steroid antagonists such as tamoxifen, spironolactone, and PCN, natural and synthetic steroids such as dexamethasone (DEX), diethylstilbestrol (DES), estradiol, DHT, corticosterone, and cortisone, and phytoestrogens such as coumestrol, equol, and genistein. Of these compounds, rifampicin, nifedipine, corticosterone, estradiol, DES, and coumestrol were the most potent activators. We note that SXR response to PCN is variable between experiments, typically ranging from low to modest (cf. Figs. 4B and 5A). The CYP3A4 promoter responds to PCN with similar variability in cultured hepatocytes (Barwick et al. 1996). Remarkably, PXR responded poorly to these inducers, showing preferential activation by PCN, a weak activator of SXR (Fig. 5B). Interestingly, although PXR is reported to prefer pregnanes (C21 steroids such as DEX and pregnenolone; Kliewer et al. 1998) we find that it is similarly activated by C19 androstanes like testosterone, and C18 estranes like estradiol (Fig. 5B). Similar results were obtained with other natural steroids, including progesterone, pregnenolone, and DHEA (data not shown). To demonstrate that the activation of SXR and PXR by high steroid concentrations is not a general property of all steroid receptors, we tested the human estrogen receptor for its response to the same panel of compounds. Among steroids, only DHT and estradiol were activators of ER, along with the synthetic ER agonist, DES, and the phytoestrogens, including coumestrol (Fig. 5C).
Figure 5.
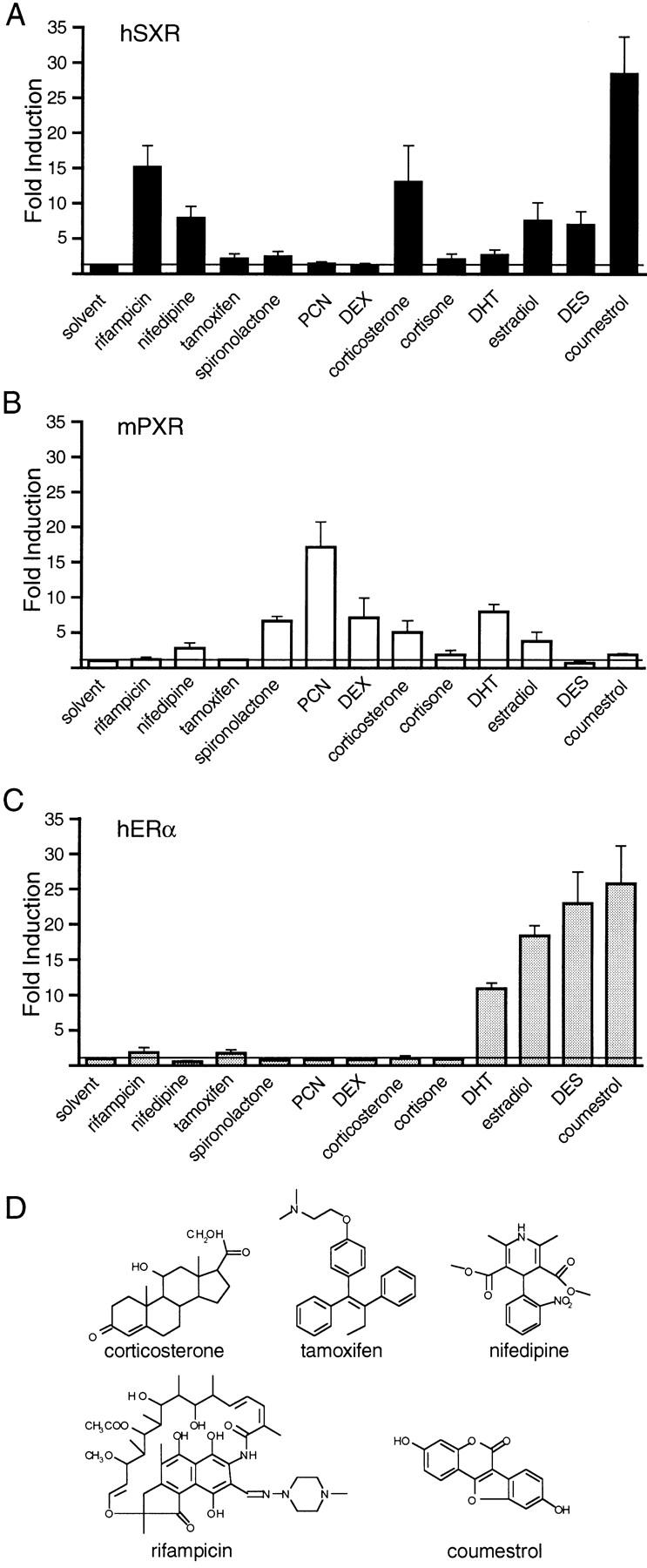
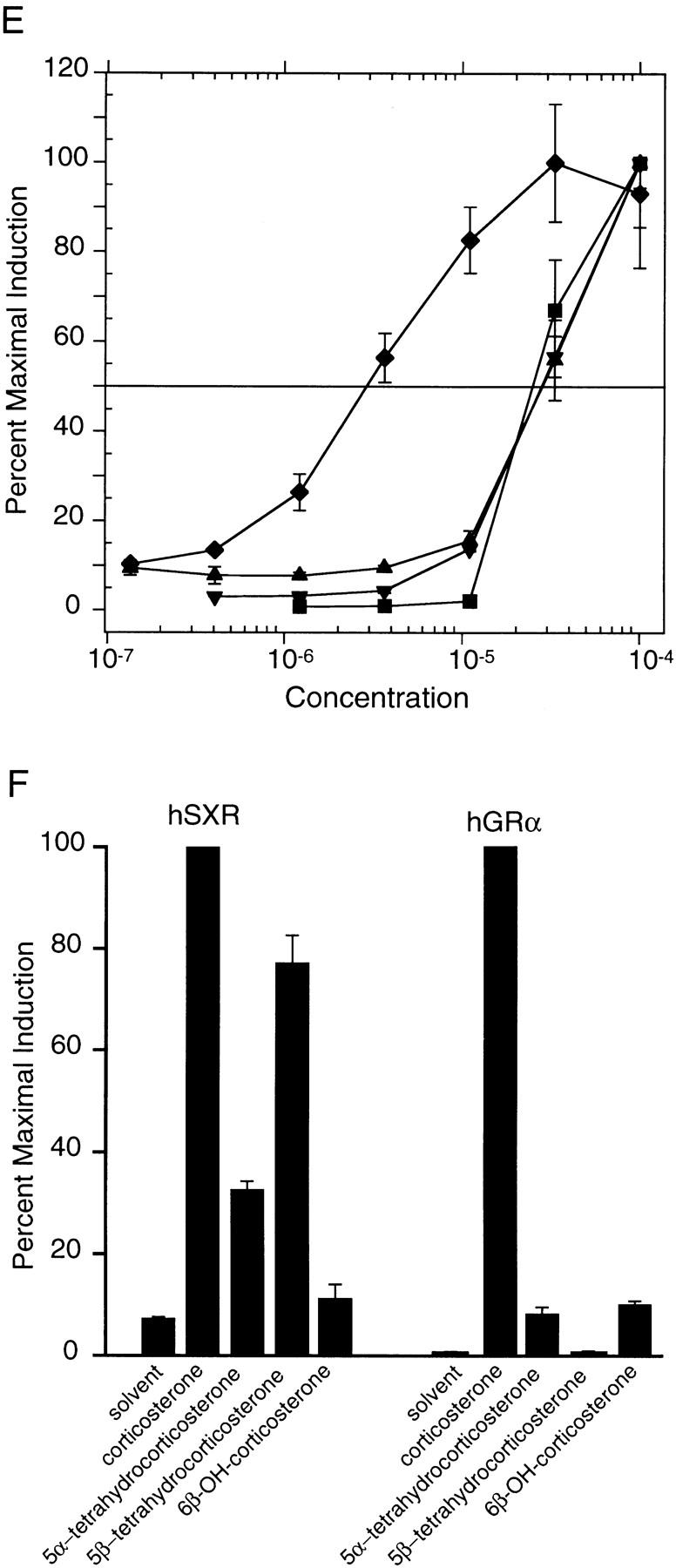
Pharmacology of SXR activation. (A–C) The ability of a panel of compounds to activate SXR, PXR.1, or ER was tested. Results are shown for 50 μm of compound with the following exceptions: 5 μm tamoxifen was used, DES concentration is 50 μm in A and B and 5 μm in C. (D) Chemical structures of some selected SXR activators from A–C. (E) Efficacy of SXR activation by selected compounds. The ability of a dilution series of compounds to activate full-length SXR was tested using several response elements as in Fig. 4. Results are shown for tk(LXRE)3–luc and represent the mean of triplicate determinations. Similar results were obtained for other response elements that SXR can activate. (♦) Rifamipicin; (▴) corticosterone; (▾) estradiol; (█) coumestrol. (F) Reduction of the 4–5 double bond does not inactivate corticosterone. 6β-hydroxylated, nonreduced, 5α and 5β reduced forms of corticosterone were tested for their ability to activate GAL–SXR on tk(MH100)4–luc and hGRα on MTV–luc at 50 μm. Similar results were obtained using full-length SXR.
To evaluate the efficacy of SXR activation by various compounds, we determined EC50 (50% effective concentration) values in dose-response experiments. The chemical structures of compounds are shown in Figure 5D and the dose responsiveness in Figure 5E. In contrast to the rank order of potency (coumestrol > rifampicin > corticosterone > nifedipine, estradiol, DES shown in Fig. 5A), the most efficacious activator of SXR was rifampicin (EC50 of 3 μm) and the order was rifampicin > corticosterone > estradiol > coumestrol.
Despite continued effort, we have been unable to demonstrate specific binding of any of these activators to baculovirus-expressed, full-length SXR:RXR heterodimers, using protease protection, corepressor dissociation, and coactivator association. Unfortunately, the most efficacious activator rifampicin is not available in radiolabeled form; we did test radiolabeled corticosterone without success. It is possible that all of our activators have too little affinity for SXR to demonstrate binding above background and this could be taken as evidence that a high-affinity, endogenous ligand remains to be identified as has been postulated for PXR (Kliewer et al. 1998). However, we believe that the number of SXR activators that are also CYP3A4 inducers is too large to be coincidental and conclude it is more likely that SXR is acting as a broad specificity, low-affinity sensor that regulates catabolism through CYP3A4 and other steroid and xenobiotic inducible P-450 enzymes.
Partially metabolized steroids activate SXR
The localization of apparent SXR-responsive elements in genes encoding steroid hydroxylases led us to consider whether products of steroid catabolism, such as reduced or hydroxylated corticosterone derivatives, could also activate SXR. Figure 5F shows that both 5α and 5β reduced forms of corticosterone are effective SXR activators, whereas 5α is slightly active and 5β is completely inactive on GR. Although a few 5α-reduced steroids remain active (e.g., DHT), 5β-reduced steroids fail to activate classic steroid receptors (Russell and Wilson 1994). Therefore, the activation of SXR by 5β-reduced steroids may reflect a previously undetected regulatory pathway for these compounds. Interestingly, 6β-hydroxy corticosterone is virtually inactive on SXR (Fig. 5F), suggesting that CYP3A4 catalyzed hydroxylation is a potential definitive regulatory step in steroid metabolism.
Discussion
We have proposed a novel model, termed the steroid sensor hypothesis, in which the induction of some xenobiotic-metabolizing enzymes by pharmacological levels of steroids, drugs, and xenobiotic compounds is regulated by a broad specificity sensor, rather than numerous specific receptors. In support of our hypothesis we show SXR is a novel member of the nuclear receptor superfamily that is activated by a diverse group of steroids and their metabolites. These include molecules that have high-affinity receptors such as progesterone, testosterone, estrogen, and corticosterone as well as their reduced catabolites that are, for the most part, inactive on the high-affinity receptors. In addition to the natural steroids, SXR is activated by synthetic steroids including PCN and DEX as well as xenobiotic drugs and phytosteroids. Direct regulation by a broad specificity sensor is biologically economical as much of the detoxification and catabolism of such compounds is mediated by cytochrome P-450 enzymes, particularly members of the CYP3A family, which both metabolize and are induced by a wide spectrum of diverse compounds, including steroids.
Our hypothesis leads to several predictions concerning the relationship among target genes, the sensor, and its activators. First, the sensor should be expressed in tissues that catabolize steroids and xenobiotics. SXR is highly expressed in liver, the major expression site of steroid and xenobiotic-metabolizing enzymes (Fig. 1C). Prominent expression of SXR mRNA is also found in the intestine (Fig. 1C). Although less is known about the role of this tissue in steroid metabolism, the gut is known to play an important role in first pass metabolism of dietary and orally administered compounds (Kolars et al. 1991; Holtbecker et al. 1996) and CYP3A4 is highly expressed in enterocytes (Kolars et al. 1992). Thus, SXR is expressed at high levels in two key tissues for steroid and xenobiotic catabolism. Second, catabolic enzymes expressed in these tissues should be induced by the sensor. Putative SXR response elements are found in the well-characterized, CYP3A4 promoter as well as those of P-450 oxidoreductase CYP2A, CYP2C, CYP2E, and glucuronosyl transferase, all known to be involved in steroid and xenobiotic catabolism (Fig. 4A; Gonzalez 1992). Third, compounds known to induce catabolic enzymes should activate the sensor. SXR is activated by a variety of xenobiotic compounds, including drugs such as rifampicin and nifedipine, steroid receptor agonists and antagonists such as estrogen and tamoxifen, and bioactive dietary compounds such as phytoestrogens (Figs. 4 and 5). In particular, CYP3A4 has been shown to be inducible by virtually all known SXR activators (Figs. 4 and 5; Rendic and Di Carlo 1997). Last, because some partially metabolized (reduced) steroids retain biological activity, it would be desirable that these continue to activate the sensor thereby ensuring their complete inactivation and elimination. As expected, products of earlier catabolic steps, such as reduced steroids, are activators of SXR but not classic steroid receptors (Fig. 5D; data not shown). Taken together, these observations provide strong support for the sensor hypothesis.
The observation that SXR can be activated by drugs and xenobiotic compounds suggests the possibility that these compounds could affect endogenous steroid metabolism indirecly. However, because steroid levels are tightly regulated, increased catabolism will be compensated by the pituitary (in healthy individuals) leading to adrenocorticotropin (ACTH) release, increased biosynthesis, and maintenance of plasma steroid levels. The increased catabolism will, however, be reflected by elevated urinary levels of steroid metabolites. Indeed, treatment with rifampicin, a strong SXR activator and CYP3A4 inducer, increases urinary metabolites such as 6β-hydroxycortisol (Ohnhaus et al. 1989; Watkins et al. 1989), and bile acid metabolites such as 6α-hydroxy hyocholic and 6α-hyodeoxycholic acids (Wietholtz et al. 1996), whereas the plasma levels of many circulating steroids increase slightly as a result of increased synthesis (Edwards et al. 1974; Lonning et al. 1989; Bammel et al. 1992). When synthetic steroids, such as prednisolone (McAllister et al. 1983; Lee et al. 1993) or 17α-ethynylestradiol (Guengerich 1990) are administered together with rifampicin, plasma levels are rapidly decreased due to enhanced urinary clearance. In some patients undergoing rifampicin therapy for tuberculosis, the increase in urinary steroid levels has led to misdiagnosis of Cushing’s syndrome (Kyriazopoulou and Vagenakis 1992; Terzolo et al. 1995; Zawawi et al. 1996). Steroid production and clearance normalized when rifampicin was withdrawn. In patients with Addison’s disease, who mostly lack the ability to synthesize adrenal steroids, rifampicin treatment leads to rapid depletion of endogenous and administered steroids, confirming that induction of CYP3A4 causes increased steroid catabolism as predicted by the model (Edwards et al. 1974; Kyriazopoulou et al. 1984).
The induction of CYP3A4 by SXR activators has implications for drug interactions. In principle, strong SXR activators should lead to higher levels of CYP3A4, which is involved in the clearance of 60% of clinically relevant drugs (Cholerton et al. 1992). For example, rifampicin leads to increased clearance of calcium channel blockers such as nifedipine (Holtbecker et al. 1996; Ndanusa et al. 1997) and verapamil (Barbarash et al. 1988), anti-arhythmics such as pirmenol (Stringer et al. 1988), and β-blockers such as propranolol (Herman et al. 1983), in addition to the steroid interactions mentioned above. It should be noted that, although most CYP3A4 inducers are SXR activators, a few such as cyclosporine A fail to activate SXR. This could be the result of the presence of additional pathways for CYP3A4 regulation. However, the ability of a particular compound to induce catabolic P-450s by activating SXR places it as a candidate for drug–drug interactions. Thus, screening against SXR provides a potential in vitro molecular test for such drug interactions.
Activation of SXR also provides a molecular explanation for the paradoxical induction of the CYP3A genes (a.k.a. P-450PCN) by both glucocorticoid receptor agonists and antagonists and the differential response of orthologous enzymes in different species. The inducible CYP3A genes harbor a SXR activatable response element in their promoters that has been shown to be responsible for PCN and glucocorticoid induction (see Fig. 4A,C) (Schuetz and Guzelian 1984; Gonzalez et al. 1986; Burger et al. 1992; Barwick et al. 1996; Kliewer et al. 1998). Despite their common role in steroid and xenobiotic catabolism, CYP3A genes from different species, and particularly the glucocorticoid-responsive promoter elements, show considerable differences in the pharmacology of their inducers (Barwick et al. 1996). For example, PCN is a strong inducer of rat CYP3A2 and CYP3A23, but a weak inducer of human CYP3A4 and rabbit CYP3A6, whereas rifampicin is a strong inducer of the human and rabbit but not the rat genes (Barwick et al. 1996). However, when these elements are tested by transient transfection into primary hepatocytes from rats or rabbits the responsiveness changes to that of the host cell type. Glucocorticoid-responsive elements from the rat CYP3A2 and CYP3A23 promoters were able to be induced by DEX in both rat and rabbit hepatocytes, by PCN only in rat hepatocytes, and by rifampicin only in rabbit hepatocytes (Barwick et al. 1996). Similarly, the glucocorticoid-responsive element from the human CYP3A4 promoter was inducible by DEX in both rat and rabbit hepatocytes, by PCN only in rat hepatocytes, and rifampicin only in rabbit hepatocytes (Barwick et al. 1996). The activation profiles in rat cells correspond to the responsiveness of PXR to the inducers (Fig. 5C), whereas the responsiveness in rabbit cells corresponds to that of SXR. Because the rabbit 3A6 promoter lacks the rodent DR-3 element but has the human IR-6 element (Barwick et al. 1996), we infer that rabbit liver will likely have a receptor more closely related to SXR than PXR. Thus, the pharmacology of SXR and PXR activation explains the different inducibility of the rat versus rabbit or human members of the cytochrome P-4503A family. This also suggests that rabbit hepatocytes behave more like their human counterparts and that rabbits are perhaps better suited to testing for human-like drug interaction than rodents.
The data presented strongly suggest the existence of a steroid and xenobiotic sensing mechanism and support our proposal for a broad specificity, low-affinity nuclear hormone receptor such as SXR. The origin of this sensing system may perhaps be illuminated by its expression in digestive tissues. Many plants produce compounds that have endocrine activities in animals as a protective strategy (for review, see Baker 1995), suggesting that the sensor evolved to defend against possible toxic nutrients and xenobiotic compounds. We also note that the aryl hydrocarbon receptor controls the transcriptional activity of P-450 genes in response to ingested xenobiotics (for review, see Denison and Whitlock 1995; Hankinson 1995) and that it represents a distinct catabolic regulator that is responsive to a discrete set of compounds. The correlation between the expression of SXR in liver and intestine and these organs as the major sites of absorption and processing for dietary compounds is particularly intriguing and suggests that P-450 enzyme systems may be dually regulated to enable broad responsiveness to the plethora of compounds to which we are exposed as well as providing regulated catabolism to ensure physiologic homeostasis.
Materials and methods
cDNA identification
SXR was identified from a human genomic library (Clontech) hybridized with a full-length cDNA encoding Xenopus BXR (Blumberg et al. 1998) under reduced stringency conditions [hybridization in 0.5 m NaPO4 (pH 7.0), 7% SDS, 5% dextran sulfate at 65°C overnight, washing three times for 20 min in 2× SSC, 0.1% SDS at 37°C]. Restriction mapping and Southern analysis showed that three exons were contained within the 9-kb EcoRI hybridizing fragment. This fragment was used to probe a human multiple tissue Northern blot (Clontech) at high stringency (hybridization as above, washing twice for 20 min in 0.1× SSC, 0.1% SDS at 50°C) and hybridization was detected in liver. A human liver cDNA library (Stratagene) was screened subsequently using the same conditions and four independent clones identified. Each of these was sequenced on both strands within the protein-coding region. DNA sequences were compiled and aligned using the programs of Staden (1986), University of Wisconsin Genetics Computer Group (Devereaux et al. 1984). Database searching was performed using the BLAST network server at the National Center for Biotechnology Information (Altschul et al. 1990). PXR was isolated from a mouse liver cDNA library (Stratagene) by screening with the SXR protein-coding region at reduced stringency (5× SSC, 43% formamide, 5× Denhardt’s, 0.1% SDS, 0.1 mg/ml denatured, sonicated salmon sperm DNA at 37°C). Three, 20-min washes were performed in 0.5× SSC, 0.1% SDS at 50°C.
DNA-binding analysis
Electrophoretic mobility shift assays were performed using in vitro transcribed, translated proteins (TNT, Promega). Proteins (1 μl each) were incubated for 20 min at room temperature with 100,000 cpm of Klenow-labeled probes in 10 mm Tris (pH 8), 100 mm KCl, 6% glycerol, 0.05% NP-40, 1 mm DTT, 100 ng/μl poly[d(I-C)] (Pharmacia) and then electrophoresed through a 5% polyacrylamide gel in 0.5× TBE (45 mm Tris-base, 45 mm boric acid, 1 mm EDTA) at room temperature. For competition binding, protein plus unlabeled oligonucleotides at 5 or 50-fold molar excess were preinucbated for 10 min on ice, labeled probes added, and incubated for 20 min at room temperature. Electrophoresis was as above. βDR-series oligonucleotides were described previously (Perlmann et al. 1993). DR0-15 oligonucleotides had the following sequences (DR-0, catagtcAGGTCAAGGTCAgatcaac; DR-1, catagtcAGGTCAtAGGTCAgatcaac; DR-2, catagtcAGGTCAatAGGTCAgatcaac; DR-3, catagtcAGGTCAtatAGGTCAgatcaac; DR-4, catagtcAGGTCAtataAGGTCAgatcaac; catagtcAGGTCAtatatAGGTCAgatcaac; DR-6, catagtcAGGTCAtatataAGGTCAagatcaac; DR-7, catagtcAGGTCAtatatatAGGTCAgatcaac; DR-10, catagtcAGGTCAtatatatataAGGTCAgatcaac; DR-15, catagtcAGGTCAtagtagtagtagtagAGGTCAgatcaac). IR series oligonucleotides had the following sequences (IR-0, agcttAGGTCATGACCTa; IR-1, agcttAGGTCAgTGACCTa; IR-2, agcttAGGTCAcgTGACCTa; IR-3, agcttAGGTCAcagTGACCTa, IR-4, agcttAGGTCAcatgTGACCTa; IR-5, agcttAGGTCAcactgTGACCTa; IR-M, agcttACGTCATGACGTa). CYP3A oligonucleotides were the following (CYP3A4, tagaataTGAACTcaaaggAGGTCAgtgagtgg; CYP3A5, tagaataTGAACTcaaaggAGGTAAgcaaaggg; CYP3A7, tagaataTTAACTcaatggAGGCAgtgagtgg).
Plasmid construction and transfection
The protein-coding region of SXR was PCR amplified and subcloned into NcoI and BamH1 sites of the vector pCDG1 (Blumberg et al. 1998) using ExoIII-mediated ligation independent cloning (Li and Evans 1997). During this process the putative initiator Leu was converted to Met with a Kozak consensus sequence CCATGG. GAL4–SXR was constructed by subcloning amino acids 107–434 into pCMX–GAL4 (Perlmann et al. 1993). Similarly, the PXR.1 protein-coding region was PCR amplified and subcloned into NcoI–BamHI cut pCDG1, whereas amino acids 104–431 were subcloned into CMX–GAL4. Reporter plasmids were constructed by synthesizing three-copy response elements and subcloning into HindIII–BamHI cut pTk-luc (Hollenberg et al. 1987).
CV-1 cells were maintained in DMEM containing 10% resin charcoal stripped calf bovine serum. Liposome-mediated transient transfections were performed using DOTAP reagent (Boehringer Mannheim) at a concentration of 5 μg/ml in DMEM containing 10% resin charcoal stripped fetal bovine serum in 96-well format using a Beckman Biomek 1000 laboratory workstation as described (Blumberg et al. 1996). Ligands were added the next day in DMEM containing 10% delipidated FBS. After 18–24 hr incubation, the cells were lysed and luciferase reporter gene assays and β-galactosidase transfection control assays performed as described (Blumberg et al. 1996). Reporter gene expression was normalized to the β-galactosidase transfection control and expressed as relative light units per OD per minute of β-galactosidase activity or fold induction over solvent control. Each data point represents the average of triplicate experiments ± s.e. and was replicated in independent experiments.
Acknowledgments
We thank Tanya A. Moreno for assistance in the early stages of this work, Drs. Debu Chakravarti, Frank Gonzalez, Valentine Lance, Enrique Saez, and Robert Tukey for critical reading of the manuscript and Barry Forman for IR-series oligonucleotides and various reporter plasmids. Supported by National Institutes of Health grant GM-26444 and the G. Harold and Leila Y. Mathers Charitable Foundation (to R.M.E.), and the American Cancer Society (DB-36 to B.B.). Ronald M. Evans is an investigator of the Howard Hughes Medical Institute at the Salk Institute for Biological Studies.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL blumberg@salk.edu; FAX (619) 455-1349.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Baes M, Gulick T, Choi HS, Martinoli MG, Simha D, Moore DD. A new orphan member of the nuclear hormone receptor superfamily that interacts with a subset of retinoic acid response elements. Mol Cell Biol. 1994;14:1544–1551. doi: 10.1128/mcb.14.3.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker ME. Endocrine activity of plant-derived compounds: An evolutionary perspective. Proc Soc Exp Biol Med. 1995;208:131–138. doi: 10.3181/00379727-208-43845. [DOI] [PubMed] [Google Scholar]
- Bammel A, van der Mee K, Ohnhaus EE, Kirch W. Divergent effects of different enzyme-inducing agents on endogenous and exogenous testosterone. Eur J Clin Pharmacol. 1992;42:641–644. doi: 10.1007/BF00265929. [DOI] [PubMed] [Google Scholar]
- Barbarash RA, Bauman JL, Fischer JH, Kondos GT, Batenhorst RL. Near-total reduction in verapamil bioavailability by rifampin. Electrocardiographic correlates. Chest. 1988;94:954–959. doi: 10.1378/chest.94.5.954. [DOI] [PubMed] [Google Scholar]
- Barwick JL, Quattrochi LC, Mills AS, Potenza C, Tukey RH, Guzelian PS. Trans-species gene transfer for analysis of glucocorticoid-inducible transcriptional activation of transiently expressed human CYP3A4 and rabbit CYP3A6 in primary cultures of adult rat and rabbit hepatocytes. Mol Pharmacol. 1996;50:10–16. [PubMed] [Google Scholar]
- Beato M, Herrlich P, Schutz G. Steroid hormone receptors: Many actors in search of a plot. Cell. 1995;83:851–857. doi: 10.1016/0092-8674(95)90201-5. [DOI] [PubMed] [Google Scholar]
- Beaune PH, Umbenhauer DR, Bork RW, Lloyd RS, Guengerich FP. Isolation and sequence determination of a cDNA clone related to human cytochrome P-450 nifedipine oxidase. Proc Natl Acad Sci. 1986;83:8064–8068. doi: 10.1073/pnas.83.21.8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg B, Bolado J, Jr, Derguini F, Craig AG, Moreno TA, Charkravarti D, Heyman RA, Buck J, Evans RM. Novel RAR ligands in Xenopus embryos. Proc Natl Acad Sci. 1996;93:4873–4878. doi: 10.1073/pnas.93.10.4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg B, Kang H, Bolado J, Jr, Chen H, Craig AG, Moreno TA, Umesono K, Perlmann T, De Robertis EM, Evans RM. BXR, an embryonic orphan nuclear receptor activated by a novel class of endogenous benzoate metabolites. Genes & Dev. 1998;12:1269–1277. doi: 10.1101/gad.12.9.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger H-J, Schuetz JD, Schuetz EG, Guzelian PS. Paradoxical transcriptional activation of rat liver cytochrome P-450 3A1 by dexamethasone and the antiglucocorticoid pregnenolone 16α-carbonitrile: Analysis by transient transfection into primary monolayer cultures of adult rat hepatocytes. Proc Natl Acad Sci. 1992;89:2145–2149. doi: 10.1073/pnas.89.6.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholerton S, Daly AK, Idle JR. The role of individual human cytochromes P450 in drug metabolism and clinical response. Trends Pharmacol Sci. 1992;13:434–439. doi: 10.1016/0165-6147(92)90140-2. [DOI] [PubMed] [Google Scholar]
- Denison MS, Whitlock JP., Jr Xenobiotic-inducible transcription of cytochrome P450 genes. J Biol Chem. 1995;270:18175–18178. doi: 10.1074/jbc.270.31.18175. [DOI] [PubMed] [Google Scholar]
- Devereaux J, Haeberli P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards OM, Courtenay-Evans RJ, Galley JM, Hunter J, Tait AD. Changes in cortisol metabolism following rifampicin therapy. Lancet. 1974;2:548–551. [PubMed] [Google Scholar]
- Fernandez-Salguero P, Gonzalez FJ. The CYP2A gene subfamily: Species differences, regulation, catalytic activities and role in chemical carcinogenesis. Pharmacogenetics. 1995;5:S123–128. doi: 10.1097/00008571-199512001-00013. [DOI] [PubMed] [Google Scholar]
- Forman BM, Umesono K, Chen J, Evans RM. Unique response pathways are established by allosteric interactions among nuclear hormone receptors. Cell. 1995;81:541–550. doi: 10.1016/0092-8674(95)90075-6. [DOI] [PubMed] [Google Scholar]
- Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors a and d. Proc Natl Acad Sci. 1997;94:4312–4317. doi: 10.1073/pnas.94.9.4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez FJ. Human cytochromes P450: problems and prospects. Trends Pharmacol Sci. 1992;13:346–352. doi: 10.1016/0165-6147(92)90107-h. [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ, Song B-J, Hardwick JP. Pregnenolone 16α-carbonitrile-inducible P-450 gene family: Gene conversion and differential regulation. Mol Cell Biol. 1986;6:2969–2976. doi: 10.1128/mcb.6.8.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlicher M, Widmark E, Li Q, Gustafsson JA. Fatty acids activate a chimera of the clofibric acid-activated receptor and glucocorticoid receptor. Proc Natl Acad Sci. 1992;89:4653–4657. doi: 10.1073/pnas.89.10.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich FP. Metabolism of 17α-ethynylestradiol in humans. Life Sci. 1990;47:1981–1988. doi: 10.1016/0024-3205(90)90431-p. [DOI] [PubMed] [Google Scholar]
- Hankinson O. The aryl hydrocarbon receptor complex. Ann Rev Pharmacol Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- Herman RJ, Nakamura K, Wilkinson GR, Wood AJ. Induction of propranolol metabolism by rifampicin. Br J Clin Pharmacol. 1983;16:565–569. doi: 10.1111/j.1365-2125.1983.tb02218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, Thompson EB, Rosenfeld MG, Evans RM. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature. 1985;318:635–641. doi: 10.1038/318635a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenberg SM, Giguere V, Segui P, Evans RM. Colocalization of DNA-binding and transcriptional activation functions in the human glucocorticoid receptor. Cell. 1987;49:39–46. doi: 10.1016/0092-8674(87)90753-7. [DOI] [PubMed] [Google Scholar]
- Holtbecker N, Fromm MF, Kroemer HK, Ohnhaus EE, Heidemann H. The nifedipine–rifampin interaction. Evidence for induction of gut wall metabolism. Drug Metab Dispos. 1996;24:1121–1123. [PubMed] [Google Scholar]
- Horst RL, Reinhardt TA. Vitamin D metabolism. In: Feldman D, Glorieux FH, Pike JW, editors. Vitamin D. San Diego, CA: Academic Press; 1997. pp. 14–15. [Google Scholar]
- Kliewer SA, Sundseth SS, Jones SA, Brown PJ, Wisely GB, Koble CS, Devchand P, Wahli W, Willson TM, Lenhard JM, Lehmann JM. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator–activated receptors alpha and gamma. Proc Natl Acad Sci. 1997;94:4318–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Moore JT, Wade L, Staudinger JL, Jones MA, McKee DD, Oliver BM, Willson TM, Zetterstrom RH, Perlmann T, Lehmann J. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- Kolars JC, Awni WM, Merion RM, Watkins PB. First-pass metabolism of cyclosporin by the gut. Lancet. 1991;338:1488–1490. doi: 10.1016/0140-6736(91)92302-i. [DOI] [PubMed] [Google Scholar]
- Kolars JC, Schmiedlin-Ren P, Schuetz JD, Fang C, Watkins PB. Identification of rifampin-inducible P450IIIA4 (CYP3A4) in human small bowel enterocytes. J Clin Invest. 1992;90:1871–1878. doi: 10.1172/JCI116064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriazopoulou V, Vagenakis AG. Abnormal overnight dexamethasone suppression test in subjects receiving rifampicin therapy. J Clin Endocrinol Metab. 1992;75:315–317. doi: 10.1210/jcem.75.1.1619024. [DOI] [PubMed] [Google Scholar]
- Kyriazopoulou V, Parparousi O, Vagenakis AG. Rifampicin-induced adrenal crisis in Addisonian patients receiving corticosteroid replacement therapy. J Clin Endocrinol Metab. 1984;59:1204–1206. doi: 10.1210/jcem-59-6-1204. [DOI] [PubMed] [Google Scholar]
- Ladias JAA, Karathanasis SK. Regulation of the apolipoprotein AI gene by Arp-1, a novel member of the steroid receptor superfamily. Science. 1991;251:561–565. doi: 10.1126/science.1899293. [DOI] [PubMed] [Google Scholar]
- Lee KH, Shin JG, Chong WS, Kim S, Lee JS, Jang IJ, Shin SG. Time course of the changes in prednisolone pharmacokinetics after co-administration or discontinuation of rifampin. Eur J Clin Pharmacol. 1993;45:287–289. doi: 10.1007/BF00315399. [DOI] [PubMed] [Google Scholar]
- Li C, Evans RM. Ligation independent cloning irrespective of restriction site compatibility. Nucleic Acids Res. 1997;25:4165–4166. doi: 10.1093/nar/25.20.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonning PE, Bakke P, Thorsen T, Olsen B, Gulsvik A. Plasma levels of estradiol, estrone, estrone sulfate and sex hormone binding globulin in patients receiving rifampicin. J Steroid Biochem. 1989;33:631–635. doi: 10.1016/0022-4731(89)90052-6. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: The second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister WA, Thompson PJ, Al-Habet SM, Rogers HJ. Rifampicin reduces effectiveness and bioavailability of prednisolone. Br Med J. 1983;286:923–925. doi: 10.1136/bmj.286.6369.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molowa DT, Schuetz EG, Wrighton SA, Watkins PB, Kremers P, Mendez-Picon G, Parker A, Guzelian PS. Complete cDNA sequence of a cytochrome P-450 inducible by glucocorticoids in human liver. Proc Natl Acad Sci. 1986;83:5311–5315. doi: 10.1073/pnas.83.14.5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndanusa BU, Mustapha A, Abdu-Aguye I. The effect of single doses of rifampicin on the pharmacokinetics of oral nifedipine. J Pharm Biomed Anal. 1997;15:1571–1575. doi: 10.1016/s0731-7085(97)00044-7. [DOI] [PubMed] [Google Scholar]
- Norman AW, Litwack G. Hormones. 2nd ed. San Diego, CA: Academic Press; 1997. [Google Scholar]
- Ogura T, Evans RM. A retinoic acid-triggered cascade of HOXB1 gene activation. Proc Natl Acad Sci. 1995;92:387–391. doi: 10.1073/pnas.92.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnhaus EE, Breckenridge AM, Park BK. Urinary excretion of 6 β-hydroxycortisol and the time course measurement of enzyme induction in man. Eur J Clin Pharmacol. 1989;36:39–46. doi: 10.1007/BF00561021. [DOI] [PubMed] [Google Scholar]
- Perlmann T, Rangarajan PN, Umesono K, Evans RM. Determinants for selective RAR and TR recognition of direct repeat HREs. Genes & Dev. 1993;7:1411–1422. doi: 10.1101/gad.7.7b.1411. [DOI] [PubMed] [Google Scholar]
- Rendic S, Di Carlo FJ. Human cytochrome P450 enzymes: A status report summarizing their reactions, substrates, inducers and inhibitors. Drug Metab Rev. 1997;29:413–580. doi: 10.3109/03602539709037591. [DOI] [PubMed] [Google Scholar]
- Russell DW, Wilson JD. Steroid 5α-reductase: Two genes/two enzymes. Ann Rev Biochem. 1994;63:25–61. doi: 10.1146/annurev.bi.63.070194.000325. [DOI] [PubMed] [Google Scholar]
- Schuetz EG, Guzelian PS. Induction of cytochrome P-450 by glucocorticoids in rat liver: II. Evidence that glucocorticoids regulate induction of cytochrome P-450 by a nonclassical receptor mechanism. J Biol Chem. 1984;259:2007–2012. [PubMed] [Google Scholar]
- Selye H. Hormones and resistance. J Pharm Sci. 1971;60:1–28. doi: 10.1002/jps.2600600102. [DOI] [PubMed] [Google Scholar]
- Staden R. The current status and portability of our sequence handling software. Nucleic Acids Res. 1986;14:217–231. doi: 10.1093/nar/14.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer KA, Cetnarowski AB, Goldfarb A, Lebsack ME, Chang T, Sedman AJ. Enhanced pirmenol elimination by rifampin. J Clin Pharmacol. 1988;28:1094–1097. doi: 10.1002/j.1552-4604.1988.tb05721.x. [DOI] [PubMed] [Google Scholar]
- Sucov HM, Murakami KK, Evans RM. Characterization of an autoregulated response element in the mouse retinoic acid receptor type beta gene. Proc Natl Acad Sci. 1990;87:5392–5396. doi: 10.1073/pnas.87.14.5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzolo M, Borretta G, Ali A, Cesario F, Magro G, Boccuzzi A, Reimondo G, Angeli A. Misdiagnosis of Cushing’s syndrome in a patient receiving rifampicin therapy for tuberculosis. Horm Metab Res. 1995;27:148–150. doi: 10.1055/s-2007-979927. [DOI] [PubMed] [Google Scholar]
- Umesono K, Murakami KK, Thompson CC, Evans RM. Direct repeats as selective response elements for the thyroid hormone, retinoic acid and vitamin D3 receptors. Cell. 1991;65:1255–1266. doi: 10.1016/0092-8674(91)90020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins PB, Murray SA, Winkelman LG, Heuman DM, Wrighton SA, Guzelian PS. The erythromycin breath test as an assay of glucocorticoid-inducible liver cytochromes P-450. J Clin Invest. 1989;83:688–697. doi: 10.1172/JCI113933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wietholtz H, Marschall HU, Sjovall J, Matern S. Stimulation of bile acid 6 alpha-hydroxylation by rifampin. J Hepatol. 1996;24:713–718. doi: 10.1016/s0168-8278(96)80268-6. [DOI] [PubMed] [Google Scholar]
- Willy PJ, Umesono K, Ong ES, Evans RM, Heyman RA, Mangelsdorf DJ. LXR, a nuclear receptor that defines a distinct retinoid-response pathway. Genes & Dev. 1995;9:1033–1045. doi: 10.1101/gad.9.9.1033. [DOI] [PubMed] [Google Scholar]
- Wurtz J-M, Bourguet W, Renaud J-P, Vivat V, Chambon P, Moras D, Gronenmeyer H. A canonical structure for the ligand-binding domain of nuclear receptors. Nat Struct Biol. 1996;3:87–94. doi: 10.1038/nsb0196-87. [DOI] [PubMed] [Google Scholar]
- Zawawi TH, al-Hadramy MS, Abdelwahab SM. The effects of therapy with rifampicin and isoniazid on basic investigations for Cushing’s syndrome. Ir J Med Sci. 1996;165:300–302. doi: 10.1007/BF02943098. [DOI] [PubMed] [Google Scholar]