

Abstract
Negative allosteric modulators (NAMs) of metabotropic glutamate receptor subtype 5 (mGlu5) have remained attractive to researchers as potential therapies for a number of central nervous system related diseases, including anxiety, pain, gastresophageal reflux disease (GERD), addiction, Parkinson’s disease (PD), and fragile X syndrome (FXS). In addition to the many publications with supportive preclinical data with key tool molecules, recent positive reports from the clinic have bolstered the confidence in this approach. During the 2 year time span from 2009 through 2010, a number of new mGlu5 NAM chemotypes have been disclosed and discussed in the primary and patent literature. A summary of several efforts representing many diverse chemotypes are presented here, along with a discussion of representative structure–activity relationships (SAR) and synthetic approaches to the templates where possible.
Keywords: Metabotropic glutamate receptor 5, negative allosteric modulator, anxiety, pain, gastresophageal reflux disease, addiction, Parkinson’s disease, fragile X syndrome
Glutamate (l-glutamic acid), the major excitatory transmitter in the mammalian central nervous system (CNS), exerts its effects through both ionotropic and metabotropic glutamate receptors, thereby generating the fast excitatory synaptic responses at the majority of CNS synapses. While ionotropic glutamate receptors are ligand-gated ion channels responsible for mediation of glutamate fast transmission, the metabotropic glutamate receptors (mGlus) belong to family C of the G-protein-coupled receptors (GPCRs) and modulate the strength of synaptic transmission. The mGlus are characterized by a seven transmembrane (7TM) α-helical domain connected via a cysteine rich-region to a large bilobed extracellular amino-terminal domain. While the orthosteric binding site is contained in the amino-terminal domain, currently known allosteric binding sites reside in the 7TM domain. The eight mGlus discovered to date have been divided according to their structure, preferred signal transduction mechanisms, and pharmacology (group I: mGlu1 and mGlu5; group II: mGlu2 and mGlu3; group III: mGlu4, mGlu6, mGlu7, and mGlu8). Group I mGlus are located postsynaptically and are coupled via Gq to the activation of phospholipase C, which leads to the elevation of intracellular calcium (Ca2+) and activation of protein kinase C (PKC). Both group II and group III mGlus are located presynaptically and are coupled via Gi/Go to the inhibition of adenylyl cylase activity.1,2
The mGlus have long been considered attractive therapeutic targets for a variety of neurological and psychiatric disorders. As such, several small molecule mGlu agonists and antagonists that compete with glutamate for the orthosteric binding site have been developed and studied.3 Still, selectively targeting a specific mGlu through the use of an orthosteric ligand has proven difficult due to the highly conserved nature of that binding site. Furthermore, many orthosteric agents are glutamate derivatives or analogues of glutamate and thus lack the pharmacokinetic or CNS penetration properties desirable in a small molecule probe. A strategy that has proven more effective for achieving selectivity has been the design and use of allosteric modulators of the target. Allosteric modulators that do not activate the receptor directly but instead potentiate the activation induced by glutamate are known as positive allosteric modulators (PAMs). Conversely, molecules that bind to an allosteric site and act as noncompetitive antagonists are termed negative allosteric modulators (NAMs). In addition to the aforementioned selectivity advantages, the location of known allosteric binding sites in the transmembrane domain of the receptor has allowed for the synthesis of molecules with the balance of physiochemical properties required for penetration of the CNS.4−6
The study of mGlu5 and its relation to disease has been aided tremendously by the identification and characterization of two related tool compounds that function as noncompetitive antagonists of that receptor. These molecules are the simple 1,2-diarylalkynes 2-methyl-6-(phenylethynyl) pyridine (MPEP)7 and 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine (MTEP)8 (Figure 1). These important tool compounds have exhibited efficacy in numerous preclinical models of disease, including pain,9 anxiety,10−14 gastresophageal reflux disease (GERD),15,16 and fragile X syndrome (FXS).17,18 In addition, substantial work with these compounds using numerous animal models has furthered the understanding of the link between mGlu5 and drug addiction. These compounds have demonstrated an ability to attenuate various cocaine seeking behaviors in mice,19,20 rats,21−26 and squirrel monkeys.27,28 Further work established their efficacy in animal models with other drugs of abuse, including nicotine,26,29 morphine,30 methamphetamine,31 and alcohol.32−36 Finally, both MPEP and MTEP were recently found to have an antidyskinetic effect in cynomolgus monkeys treated with 4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a compound that causes rapid onset of Parkinsonism.37 Such results indicate that antagonism of mGlu5 might constitute an effective therapy for Parkinson’s disease levodopa-induced dyskinesia (PD-LID), which is a complication that arises following dopamine-replacement therapy. The body of work surrounding the preclinical in vivo characterization of MPEP and MTEP is substantial and bolsters the promise of small molecule antagonists of mGlu5 as therapeutics. As such, research directed toward the discovery of novel mGlu5 NAMs suitable for evaluation in the clinic has been abundant.38−40
Figure 1.

mGlu5 NAMs MPEP, MTEP, and fenobam. General chemical series of clinical compounds from Addex and Novartis.
Further indications that a noncompetitive antagonist of mGlu5 may translate into an effective therapeutic have been noted in recent years through multiple disclosures from the clinic. Addex Pharmaceuticals has reported positive data from phase II clinical studies with the mGlu5 NAM ADX10059 (general chemical series41 shown in Figure 1) in both GERD42 and acute migraine.43,44 FRAXA Research Foundation and Neuropharm have been exploring the potential of fenobam, a compound discovered and evaluated as an anxiolytic in the 1970s,45 for treating FXS. In fact, Neuropharm received Orphan Drug Designation for fenobam to treat FXS in 2006. The early results from these studies in patients have been disclosed and were encouraging. Using prepulse inhibition as an outcome measure, 50% of patients (4/6 males and 2/6 females) responded according to the predefined criteria of efficacy.46 More recent reports from Novartis with their mGlu5 antagonist AFQ056 (general chemical series47 shown in Figure 1) detailed their efforts directed toward identification of the first approved treatment for PD-LID.48 The link between mGlu5 antagonism and PD-LID was further bolstered by a recent communication from Addex describing the efficacy of both ADX10059 as well as their second generation mGlu5 antagonist ADX48621 (general chemical series41 shown in Figure 1) in a nonhuman primate model of PD-LID.49,50 While clinical evaluation of ADX48621 continues, further development of ADX10059 was halted in late 2009 due to concerns over elevated alanine transaminase (ALT) levels in phase IIb studies in GERD patients.51 Novartis also recently released information from a small trial with AFQ056 in adult FXS patients. Although the trial was insufficient in length to measure effects on basic intelligence and details surrounding the trial have yet to be published, noted improvements in certain aberrant behaviors were reported.52 AstraZeneca is evaluating two different mGlu5 NAMs, AZD2066 and AZD2516, in multiple clinical studies in a variety of patients, including those who suffer from neuropathic pain and GERD.53 Phase II studies with Roche’s mGlu5 NAM RG7090 in FXS and treatment resistant depression are also ongoing.54 Seaside Therapeutics recently completed investigational new drug enabling studies with their mGlu5 NAM STX107, which was in-licensed from Merck. Enrollment of a phase II study of STX107 in FXS patients was slated for 2010.55 Details related to the chemical structures of the Seaside, Roche, and AstraZeneca compounds are not currently in the public domain.
With so much preclinical and now clinical support for noncompetitive antagonism of mGlu5 as a therapeutic approach, activity in this area remains plentiful. Since the advanced compounds where chemical structures are known contain an alkyne functional group, most current efforts are focused on alternative chemotypes. New advancements in this field occur regularly, and several recent reviews highlighting progress in the discovery efforts centered on mGlu5 antagonists have been published.38−40 As such, this Review will focus on publications of nonalkyne containing series from the primary and patent literature that have occurred during the years 2009 through 2010. In deciding what information to highlight from lengthy patent applications, particular effort was made to include data for those compounds for which in vivo data, either pharmacokinetic or behavioral, was disclosed.
AstraZeneca
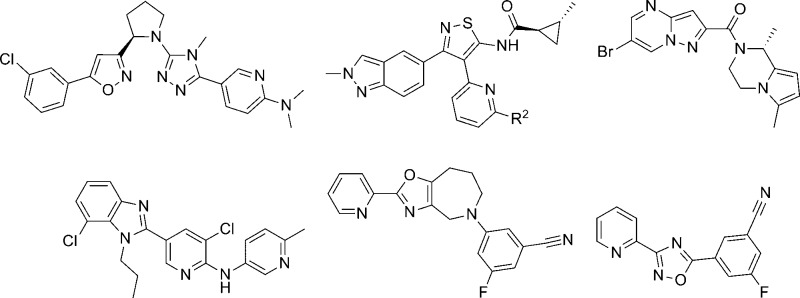
AstraZeneca published several separate patent applications on the same day (April 30, 2009), disclosing similar 1,2,4-triazole antagonists of mGlu5 (Table 1).56−58 One application highlighted compounds that possessed a chiral ether linkage between the 5-aryl-1,2,4-triazole and a second heterocyclic ring (1–3).56 Compounds were characterized in a functional assay that measured their ability to block calcium mobilization in the presence of an agonist in cells expressing human mGlu5. Further information was provided describing the brain to plasma ratio in rats following administration of the compound. The highlighted chiral ether linked compounds were quite potent; however, brain penetration was poor. Another application presented molecules with an amide linkage between the two heteroaryl rings (4, 5).57 These molecules were less potent; however, brain penetration was improved. Significantly more data was presented in the third application, which focused on alkyl amine linkages (6–11).58 Brain penetration remained generally poor with these compounds; however, it could be improved somewhat as evidenced by N-methylpyridone 9. The improvement may have been due to the removal of the hydrogen bond donor in the eastern aromatic ring. Similar brain penetration was observed with a more potent 3-chlorophenyl analogue 10 in the context of an eastern pyrimidine ring, which also lacked a hydrogen bond donor. Also of note is the negative effect on brain penetration observed with the oxadiazole 11 compared to isoxazole 9. The synthetic route for isoxazole 10 (Scheme 1) is representative of the series (yields shown where provided).58,59 The route began with 3-chloroacetophenone 12, which was reacted with sodium hydride and subsequently diethyl oxalate to afford dioxobutanoate 13. Conversion to isoxazole 14 was accomplished through treatment with hydroxylamine hydrochloride. Reduction of the ester functionality was performed with lithium aluminum hydride to give alcohol 15, which was subsequently converted to mesylate 16 using standard conditions. Reaction with the sodium anion of aminotriazole 17 gave the final compound 10. The intermediate 17 was prepared in low yield via a single step through the reaction of acid chloride 18 with aminoguanidine 19. Such a route is readily amenable to rapid variation of the eastern aromatic ring portion of the chemotype.
Table 1. mGlu5 Potency and Rat Brain Penetration SAR in 1,2,4-Triazole Series.

| entry | series | R1 | X | Y | R2 | R3 | mGlu5 IC50 (nM) | rat B/P ratio |
|---|---|---|---|---|---|---|---|---|
| 1 | I | Cl | CHMe (R) | O | Me | A (Z = CH) | 8 | 0.03 |
| 2 | III | Cl | CHMe (R) | O | Me | A (Z = CH) | 6 | 0.01 |
| 3 | III | Cl | CHMe (R) | O | Me | A (Z = N) | 5 | 0.075 |
| 4 | II | Cl | C=O | NMe | Me | B | 37 | 0.26 |
| 5 | II | Cl | C=O | NMe | Me | C (Z = N; R4 = H) | 62 | 0.19 |
| 6 | II | Cl | CH2 | NMe | Et | C (Z = N; R4 = H) | 13 | <0.01 |
| 7 | II | Me | CH2 | NEt | Me | C (Z = N; R4 = H) | 45 | 0.01 |
| 8 | II | Cl | CH2 | NEt | Me | C (Z = CH; R4 = Me) | 65 | <0.01 |
| 9 | II | Me | CH2 | NEt | Me | C (Z = CH; R4 = Me) | 112 | 0.22 |
| 10 | II | Cl | CH2 | NMe | Me | D | 20 | 0.16 |
| 11 | I | Me | CH2 | NEt | Me | C (Z = CH; R4 = Me) | 75 | <0.01 |
Scheme 1.

(i) NaH, DMF, (EtO2C)2, 0 to 80 °C, 67%; (ii) NH2OH·HCl, MeOH, 80 °C, 71%; (iii) LiAlH4, THF, 75%; (iv) NEt3, MsCl, CH2Cl2; (v) NaH, DMF, 79%; (vi) pyridine, −15 to 125 °C, 20%.
An additional six applications were filed on the aforementioned date and described a concept based on the incorporation of a conformational restraint into the previously described alkyl amine linked analogues (Figure 2).60−65 The concept involved a carbon–carbon bond formation between the N-alkyl group at the 3-position of the 1,2,4-triazole and the methlyene linker on the isoxazole, thus generating a cyclic tertiary amine. As before, potency and rat brain penetration data was disclosed for selected compounds in each application. The majority of the compounds for which such data was presented contained an isoxazole ring bound to the cyclic amine (Table 2). In the context of a pyrrolidine constraint with an R-configuration at the lone stereocenter, potent analogues were seen with carboxamide 20(62) and aminopyridine 21.63 Both compounds also demonstrated low rat brain to plasma ratios; however, analogous to the prior series, brain penetration could be improved by removal of the hydrogen bond donors on the eastern ring as evidenced by dimethylaminopyridine 22.63 Although, the dimethylation of 21 improved the brain to plasma ratio by over 3-fold, it also resulted in more than a 10-fold decrease in potency. One application focused on thiophene replacements for the western phenyl ring of the template (23–25).64 Also contained in this application were some 2-azabicyclo[3.1.0] derivatives such as 24. Such a modification to the pyrrolidine core appears to have little effect on potency while demonstrating some improvement in brain penetration (compare 24 to 23). Again, the compound with the highest brain to plasma ratio (25) lacked a hydrogen bond donor. The remaining application provided additional 2-azabicyclo[3.1.0] examples (26, 27) as well as 3-azabicyclo[3.1.0] analogues such as 28.65 Potency remains similar across these examples, but brain to plasma ratios were not reported for any of the 3-azabicyclo[3.1.0] derivatives. Of note is the fact that 5-chlorothiophene 24 demonstrated higher brain to plasma ratio than its 3-chlorophenyl direct comparator 26 without compromising potency. Although specific yields are not reported for most steps, the synthesis of 22 (Scheme 2) is representative for these analogues.63 The sequence began with the chiral pyrrolidine-2-carbaldehyde 29 which was subjected to a one-pot, three-step procedure66 to afford isoxazole 30. Removal of the t-butyl carbamate protecting group was accomplished upon treatment with trifluoroacetic acid, and the resultant amine was reacted with methyl isothiocyanate to provide thiourea 31. Treatment with sodium tert-butoxide and methyl iodide gave methylated product 32, which was subsequently reacted with hydrazide 33 in a microwave assisted process to provide final compound 22 in low yield.
Figure 2.

Constrained analogue concept (shown with isoxazole).
Table 2. mGlu5 Potency and Rat Brain Penetration SAR in Constrained Cyclic Amine 1,2,4-Triazole Analogues.

| entry | series | R1 | R2 | mGlu5 IC50 (nM) | rat B/P ratio |
|---|---|---|---|---|---|
| 20 | I | 3-chlorophenyl | A (Z = CH; R3 = CO2NH2) | 11 | <0.01 |
| 21 | I | 3-chlorophenyl | A (Z = N; R3 = NH2) | <3 | 0.085 |
| 22 | I | 3-chlorophenyl | A (Z = N; R3 = NMe2) | 39 | 0.285 |
| 23 | I | 5-chlorothiophen-3-yl | B (Z = N; R3 = H) | 39 | <0.01 |
| 24 | II | 5-chlorothiophen-3-yl | B (Z = N; R3 = H) | 34 | 0.06 |
| 25 | I | 5-chlorothiophen-3-yl | B (Z = N; R3 = Me) | 61 | 0.16 |
| 26 | II | 3-chlorophenyl | B (Z = N; R3 = H) | 32 | <0.01 |
| 27 | II | 3-chlorophenyl | B (Z = CH; R3 = Me) | 39 | <0.01 |
| 28 | III | 3-chlorophenyl | B (Z = N; R3 = H) | 46 | not reported |
Scheme 2.

(i) NaOH, NH2OH·HCl, tBuOH/H2O (1:1), then TsN(Cl)Na·H2O, then CuSO4·5H2O, NaHCO3, 1-chloro-3-ethynylbenzene; (ii) F3CCO2H, CH2Cl2; (iii) MeN=C=S, CH2Cl2; (iv) NaOtBu, THF, MeI; (v) iPrOH, pyridine, microwave, 150 °C, 2 h, 6%.
Nearly a year after the simultaneous disclosure of these series, another application describing a series of related sulfide bridged analogues was published (Table 3).67 The selected compounds within this application where mGlu5 functional activity and rat brain to plasma ratios were reported were closely related chiral analogues having a methyl group (R configuration) appended to the carbon bridging the western heteroaryl ring to the sulfur atom. In cases where the eastern ring was a pyridone, the activities of the tetrazole and oxadiazole analogues were similar (compare 34 to 37 and 35 to 38); however, the corresponding isoxazoles 40 and 41 were noticeably less potent. Interestingly, modification of the eastern ring to a pyridazinone restores the loss in potency (compare 42 to 36 and 39). Most of these analogues demonstrated very low brain to plasma ratios; however, as was seen previously, removal of the hydrogen bond donor improved brain penetration in certain cases (compare 35 to 34 and 38 to 37). The synthetic route for isoxazole 42 (Scheme 3) is representative of the series. The synthetic sequence began with ester 43, and reaction with methyl Grignard afforded the methyl ketone 44. An enantioselective borane reduction using a chiral oxazaborolidine catalyst68 was used to provide the chiral secondary alcohol 45. Mesylate 46 was prepared using standard methods and reacted with thione 47 under basic conditions to afford final compound 42. Thione 47 was prepared from the reaction of carbohydrazide 48 with methyl isothiocyanate.
Table 3. mGlu5 Potency and Rat Brain Penetration SAR in Sulfide Bridged Analogues.

| entry | series | A | R1 | mGlu5 IC50 (nM) | rat B/P ratio |
|---|---|---|---|---|---|
| 34 | I | CH | H | 19 | <0.01 |
| 35 | I | CH | Me | 87 | 0.10 |
| 36 | I | N | H | 18 | <0.01 |
| 37 | II | CH | H | 24 | 0.09 |
| 38 | II | CH | Me | 30 | 0.16 |
| 39 | II | N | H | 41 | 0.04 |
| 40 | III | CH | H | 146 | <0.01 |
| 41 | III | CH | Me | 380 | 0.02 |
| 42 | III | N | H | 19 | 0.26 |
Scheme 3.

(i) NEt3, MeMgBr, PhMe, THF, 0 °C, 46%; (ii) (R)-1-methyl-3,3-diphenylhexahydro-pyrrolo[1,2-c][1,3,2]oxazaborole, BH3·SMe2, PhMe, THF, 0 °C, 84%; (iii) NEt3, MsCl, CH2Cl2; (iv) DIEA, DMSO, 90 °C, 60%; (v) MeN=C=S, H2O, NaOH, 50 °C.
Gedeon Richter
Work from Gedeon Richter recently described a hit to lead effort in 1,2,4-oxadiazole and tetrazole series that were structurally related to the AstraZeneca compounds outlined above.69 High throughput screening (HTS) of their internal compound collection led to the identification of oxadiazole hit 49 as an mGlu5 NAM with modest potency (Figure 3). Two different in vitro assays were used to characterize compounds. The first assay (mGlu5Ki) was a cell-based binding assay that measured affinity of the compound for the MPEP binding site using the radioligand [3H]-2-((3-methoxyphenyl)ethynyl)-6-methylpyridine (M-MPEP), a derivative of MPEP.70 The second assay was a functional cell-based assay that measured the ability of the compound to block calcium mobilization in the presence of the orthosteric agonist (S)-3,5-dihydroxyphenylglycine. As another means of assessing the quality of compounds generated in the hit to lead process, both size independent ligand efficiency (SILE)71 and lipophilic ligand efficiency metrics (LELP)72 were monitored. LELP was introduced by this group as a measure of the price of ligand efficiency paid in logP. As such, higher LELP values are indicative of less druglike compounds. Evaluation of this scaffold was conducted in three separate areas of the chemotype: phenyl ring substituent(s), cyclic amine, and the amide.
Figure 3.

1,2,4-oxadiazole hit and SAR plan.
Parallel synthesis was used to prepare over 650 oxadiazole analogues and over 600 tetrazole analogues that met criteria (>85% purity by LC-MS) for testing. The 2-piperidinyl, 2-pyrrolidinyl, and 4-thiazolidinyl derivatives were found most active, while 3-piperidinyl analogues were less potent. Selected compounds from these efforts are depicted here (Table 4). In the oxadiazole series, functional activity can be improved by modification of the aryl substituent as evidenced by the enhanced potency of 3-methylphenyl analogue 50 and 3-methoxyphenyl analogue 51 relative to hit compound 49. The 2-pyrrolidine derivatives 52 and 53 were both similar in binding affinity to the 2-piperidine compounds; however, these compounds were significantly weaker in the functional assay. 4-Thiazolidine analogue 54 does not exhibit a similar discrepancy between the two assays. The tetrazole compounds demonstrated enhanced potency and binding affinity relative to their oxadiazole counterparts (compare 55 to 49 and 56 to 50). While all of the oxadiazole compounds were prepared and tested as racemates, tetrazole 55 was prepared as its individual pure enantiomers. Testing revealed that (R)-55 (mGlu5Ki = 58 nM) displayed enhanced binding affinity relative to (S)-55 (mGlu5Ki = 741 nM). The authors point out that this trend of preference for the (R)-enantiomer held up across multiple analogues. Interestingly, the 2-piperidine to 2-pyrrolidine modification does not result in the same dramatic discrepancy between the functional potency and binding affinity seen with the oxadiazoles (compare 58 to 53). Analysis of their efficiency metrics led the authors to conclude that 56 represented an improved lead relative to the original hit 49. The synthetic route employed to access tetrazole analogues such as these is shown here (Scheme 4) in the context of the 2-piperidyl amides. The route began by conversion of Boc-protected piperidine-2-carboxylic acid 59 into its corresponding Weinreb’s amide. Subsequent treatment with lithium aluminum hydride afforded aldehyde 60, which was reacted with tosylhydrazide to give hydrazone 61. Commercially available anilines 62 were converted into their corresponding diazonium salts 63 and then reacted with hydrazone 61 to provide the tetrazoles 64. Deprotection under acidic conditions gave the free amines, which were coupled with various commercially available carboxylic acids in a parallel synthesis paradigm to yield the final products 55–57. The incorporation of the substituted phenyl rings at a late stage and the acid monomers in the final step makes the route particularly amenable to the parallel synthesis approach employed in this endeavor.
Table 4. mGlu5 Affinity and Potency in the 1,2,4-Triazole and Tetrazole Series.

| entry | series | R1 | X | R2 | mGlu5 IC50 (nM) | mGlu5Ki (nM) | SILE | LELP |
|---|---|---|---|---|---|---|---|---|
| 49 | I | 3-Cl | CH2CH2 | cyclobutyl | 1170 | 204 | 3.91 | 3.80 |
| 50 | I | 3-Me | CH2CH2 | cyclobutyl | 490 | 126 | 4.26 | 3.02 |
| 51 | I | 3-OMe | CH2CH2 | cyclobutyl | 501 | 182 | 3.93 | 3.09 |
| 52 | I | 3-Me | CH2 | -CH2OMe | 30 200 | 186 | 4.15 | 1.77 |
| 53 | I | 3-Cl | CH2 | 5-methylthiophen-2-yl | 6610 | 123 | 3.86 | 5.05 |
| 54 | I | 3-Cl | S | 5-methylthiophen-2-yl | 759 | 83 | 3.79 | 5.70 |
| 55 | II | 3-Cl | CH2CH2 | cyclobutyl | 338 | 78 | 3.96 | 3.42 |
| 56 | II | 3-Me | CH2CH2 | cyclobutyl | 224 | 72 | 4.17 | 2.87 |
| 57 | II | 3-Cl | CH2CH2 | 5-bromofuran-2-yl | 213 | 48 | 3.78 | 4.31 |
| 58 | II | 3-Cl | CH2 | 5-methylthiophen-2-yl | 251 | 89 | 3.78 | 4.72 |
Scheme 4.

(i) MeN(OMe)H·HCl, DIEA, DMAP, DCC; (ii) LiAlH4, THF, 0 °C, 85% (2 steps); (iii) TsNHNH2, EtOH, 90%; (iv) NaNO2, HCl, H2O, 0 °C; (v) NaOH, aq. EtOH; (vi) HCl, EtOAc, 80–90%; (vii) R2CO2H, EDC, NEt3, CH2Cl2.
Another series of mGlu5 NAMs emerged from the HTS of the Gedeon Richter compound collection and was also described recently.73 The carbamoyloxime hit 65 demonstrated an affinity for the known MPEP binding site and behaved as a noncompetitive antagonist in the functional cell-based assay (Figure 4). Following resynthesis of 65 and separation of the E and Z isomers, it was established that the E isomer 66 was the more active compound. An SAR plan was implemented that centered on four separate portions of the template, which were identification of replacements for the imidazole ring, potential alternatives to the cyclohexane ring, optimization of the linker between the cyclohexane ring and aniline, and substitution of the aniline ring. Tolerance for these modifications was limited with the linker and cycloalkyl ring, and most active compounds maintained the carbamoyl oxime linked to the cyclohexane. Substitution of the aniline ring with small substituents such as chloro at the meta-position improved affinity and enhanced activity. The majority of SAR was developed through exploring potential replacements for the imidazole ring, and selected examples are described here (Table 5). Introduction of the 3-chloro substituent onto the aniline in the context of the imidazole ring provided 67, which demonstrated enhanced activity relative to 66. Compound 67 was separated into its individual enantiomers by preparative HPLC, and it was determined that the activity of 67 resulted primarily from a single enantiomer ((+)-67 mGlu5Ki = 8.8 nM; (−)-67 mGlu5Ki > 1000 nM). Compound 67 was examined in the Vogel punished drinking test74 in rats and demonstrated significant efficacy at doses as low as 5 mg/kg using intraperitoneal (IP) dosing. Several replacements were tolerated in place of the imidazole ring and provided analogues with similar activity to 67. Examples include 2-thiophenyl analogue 68, phenyl analogue 73, and 3-fluorophenyl analogue 74. On the other hand, the 3-thiophenyl analogue 69 had reduced activity relative to 67 while the saturated 4-morpholinyl analogue 70 had no demonstrated affinity in the binding assay. Enhanced activity was observed with 2-pyridyl analogue 71 and 3-pyridyl analogue 72. In fact, 72 was the most potent compound in the functional assay disclosed in this paper. Several active analogues were also tested for their ability to functionally inhibit mGlu1 and mGlu2 and demonstrated good selectivity for mGlu5. The synthesis of 67 is representative of the series and shown here (Scheme 5). Readily prepared ketone 75 was reacted with hydroxylamine to generate the oxime. The E and Z isomers were separated by either crystallization or chromatography to afford the pure E isomer 76. Treatment with isocyanate 77 provided the final compound 67.
Figure 4.

Carbamoyloxime HTS hit and preferred E-isomer.
Table 5. mGlu5 Affinity and Potency in the Carbamoyloxime Series.
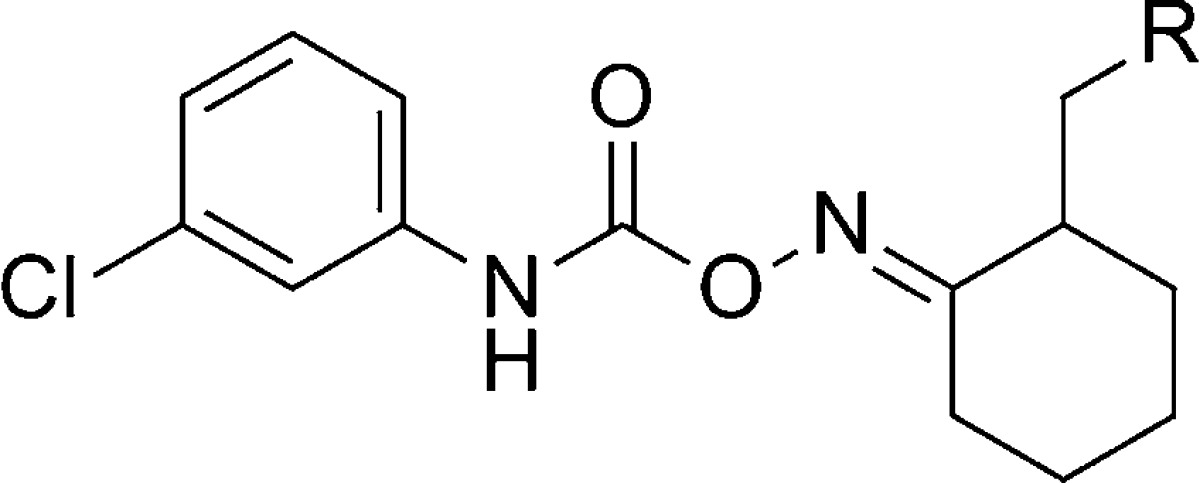
| entry | R | mGlu5 IC50 (nM) | mGlu5Ki (nM) |
|---|---|---|---|
| 67 | 1-imidazolyl | 117 | 11 |
| 68 | 2-thiophenyl | 109 | 9.1 |
| 69 | 3-thiophenyl | 477 | 14 |
| 70 | 4-morpholinyl | >1000 | |
| 71 | 2-pyridyl | 65 | 40 |
| 72 | 3-pyridyl | 15 | 3 |
| 73 | phenyl | 151 | 16 |
| 74 | 3-fluorophenyl | 127 | 40 |
Scheme 5.

(i) NH2OH·HCl, NaOAc, MeOH, H2O, 45 °C, then separation of E and Z isomers, 50–80%; (ii) CH2Cl2, 40–70%.
GlaxoSmithKline
A novel thiazolotriazole mGlu5 NAM chemotype was discovered through HTS of the GlaxoSmithKline compound collection.75 With relatively low molecular weight, high polarity, ligand efficiency, and selectivity over other mGlus, the chemotype was deemed worth further exploration. A synthetic route was employed (Scheme 6) that allowed for variation of four distinct portions of the scaffold. Substituted 1,2,4-triazolethiones 78 were reacted with haloketones 79 to provide 5-carbonyl substituted thiazolotriazoles 80. Reduction of the carbonyl group was accomplished upon treatment with sodium borohydride to afford alcohols 81. Conversion to the carbamate final compounds 82–91 was accomplished by reaction either with the appropriate isocyanates or with carbonyl diimidazole and the appropriate primary amines. SAR developed within this template using a cell-based calcium mobilization assay revealed that mGlu5 activity was sensitive to small structural modifications (Table 6). For instance, while neither the 6-methyl group on the thiazole (83) nor the methyl group on the sp3 carbon bridging the heterocycle and carbamate (84) provides much improvement relative to unsubstituted analogue 82, the combination of both substituents provided potent analogue 85. Hypothesizing that free brain concentration would be critical to achieving occupancy in vivo, the fraction unbound (fub) in rat brain homogenates was monitored for key compounds.76 Researchers sought to balance functional potency with free brain concentration by maximizing a parameter termed pIC50eff (pIC50 + log10[fub]). While saturation of the aniline ring in the form of cyclohexyl analogue 86 resulted in a loss of potency, substitution at the meta-position of the aniline ring with either chloro (87) or fluoro (88) improved potency slightly. Unfortunately the fraction unbound was considerably reduced with 87 and 88 relative to 85, resulting in a lower pIC50eff value. Conversely, pyridyl analogue 89, while less potent than 85, had a similar pIC50eff value due to its larger fraction unbound. Substitution at the 2-position of the thiazolotriazole with cyclopropyl (90) or aryl groups such as thiophene (91) also improved potency. Fraction unbound with 91 was the lowest reported, making the compound less interesting for further studies. The noncompetitive nature of 85 was verified through in vitro studies, and it was also shown to bind to the known MPEP binding site using [3H]-MPEP. Separation of 85 into its respective enantiomers by HPLC revealed a pronounced preference for a single enantiomer ((R)-85 mGlu5 IC50 = 40 nM; (S)-85 mGlu5 IC50 > 10,000 nM). Evaluation of (R)-85 in a rat pharmacokinetic study demonstrated that the compound was both bioavailable and CNS penetrant (F = 28%; AUC brain:blood = 2.0). Finally, evaluation of (R)-85 in the mouse marble burying model of anxiety established its efficacy at a dose as low as 5 mg/kg when dosed orally.77
Scheme 6.

(i) EtOH, μw, 130 °C, 1 h; (ii) NaBH4; (iii) R4NCO, μw, 80 °C, 20 min or R4NH2, CDI, μw, 120 °C, 20 min.
Table 6. mGlu5 Potency and Protein Binding in the Thiazolotriazole Series.
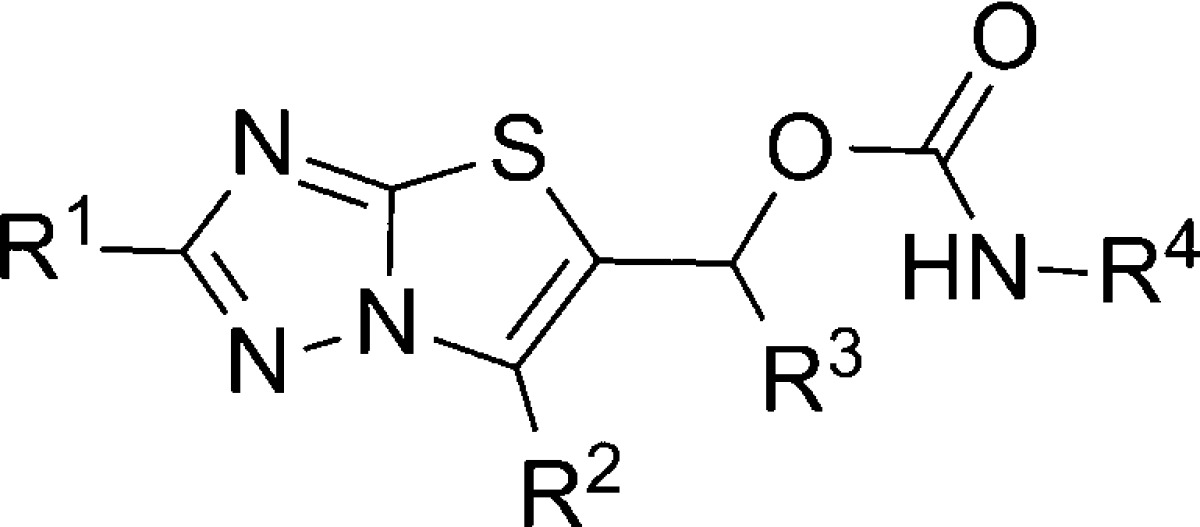
| entry | R1 | R2 | R3 | R4 | mGlu5 IC50 (nM) | fub | pIC50 eff |
|---|---|---|---|---|---|---|---|
| 82 | H | H | H | phenyl | >30 000 | ||
| 83 | H | Me | H | phenyl | >30 000 | ||
| 84 | H | H | Me | phenyl | 10 000 | ||
| 85 | H | Me | Me | phenyl | 63 | 6.8 | 6.0 |
| 86 | H | Me | Me | cyclohexyl | 3200 | ||
| 87 | H | Me | Me | 3-chlorophenyl | 40 | 0.9 | 5.4 |
| 88 | H | Me | Me | 3-fluorophenyl | 50 | 1.7 | 5.5 |
| 89 | H | Me | Me | 3-pyridyl | 316 | 28 | 5.9 |
| 90 | cyclopropyl | Me | Me | phenyl | 13 | ||
| 91 | thiophen-2-yl | Me | Me | phenyl | 7.9 | <0.1 | <5.0 |
Eli Lilly
A relatively narrow patent application from Eli Lilly recently described a series of isothiazole mGlu5 NAMs.78 Variation in this template focused on either hydrogen or small alkyl substituents on the cyclopropyl ring (R1) and indazole nitrogen (R3) (Figure 5). Claimed substituents at the 6-position of the pyridine (R2) were broader, but exemplified compounds were generally small alkyl, cycloalkyl, alkylamino, and alkoxy substituents. Synthesis of a key exemplar compound 100 is outlined here (Scheme 7). The route was initiated with a two step kilogram scale synthesis of aminoisothiazole 93 from thioamide 92. Reaction of 93 with acid chloride 94 provided amide 95. Treatment of 95 with bromine and aqueous sodium hydroxide induced a deacylation/bromination reaction to afford 96. The Suzuki coupling of 96 with the custom prepared boron reagent 97 gave intermediate 98, which was reacted with arylstannane 99 in a Stille coupling to afford the final compound 100 (yield not provided). Compounds exemplified in this patent were tested in a functional calcium mobilization assay and found to have IC50 values less than 75 nM. Analogue 100 was determined to have an IC50 equal to 9.5 nM. The hydrochloride salt of 100 was examined in a stress-induced hyperthermia (SIH) rat model, which is a well-known model of anxiety.79 Rats were dosed orally at 0.3, 1.0, 3.0, and 10 mg/kg and immediately placed in their home cage in a dark room. After the 60 min pretreatment, the animals were individually relocated to a brightly lit room and the first core body temperature (T1) was determined. The second core body temperature reading (T2) was made 10 min later. The difference between T2 and T1 constituted the SIH response. Compound 100 produced a 35% reduction in the SIH response relative to vehicle at a dose of 3.0 mg/kg.
Figure 5.

Isothiazole mGlu5 NAMs.
Scheme 7.

(i) AcCl, pyridine; (ii) Br2, AcOH, 40 °C, 65% (2 steps); (iii) NEt3, CH2Cl2, 82%; (iv) aq. NaOH, Br2, dioxane, 5–10 °C, 85%; (v) 2 N aq. Na2CO3, DME, PdCl2(PPh3)2, 83 °C, 51%; (vi) PdCl2(PPh3)2, THF, reflux.
Lundbeck
A patent application from Lundbeck was recently published describing a series of adamantyl diamides.80 The application contains over 250 exemplified compounds from this general series. Preparation of symmetrical diamides was readily accomplished through coupling of adamantine-1,3-diamine 101 with suitable carboxylic acids or acid chlorides (Scheme 8). For example, a standard coupling reaction with acid 102 afforded diamide 103, and reaction with acid chloride 104 provided diamide 105. In the case of unsymmetrical diamides, several different synthetic routes were employed. In an example of one method, adamantine-1,3-diamine 106 was first converted to amide 107 in low yield and subsequently reacted with acid chloride 108 to afford diamide 109 (yield not provided) (Scheme 9). A longer, more scalable route with higher yields was exemplified by the synthesis of analogue 116 (Scheme 10). The route began with 1-adamantanecarboxylic acid 110, which was transformed via a Ritter reaction to acetamide 111. Cleavage of the acetamide with concentrated hydrochloric acid was followed by conversion of the intermediate to methyl ester 112. Reaction with acid chloride 104 afforded amide 113. Hydrolysis of the methyl ester followed by a Curtius rearrangement gave amine 114, which was then coupled with carboxylic acid 115 to yield diamide 116. Related analogues 117 and 118 were also prepared via the same route.
Scheme 8.

(i) DIEA, EDC, CH2Cl2, 80%; (ii) DIEA, CH2Cl2, 50%.
Scheme 9.

(i) DIEA, BOP, CH2Cl2, 20%; (ii) DIEA, CH2Cl2.
Scheme 10.

(i) HNO3, H2SO4, CH2Cl2, 74%; (ii) conc HCl, 95 °C; (iii) MeOH, SOCl2, 60 °C, 72% (2 steps); (iv) NEt3, CH2Cl2, 97%; (v) LiOH, H2O, THF; (vi) (PhO)2P(O)N3, NEt3, PhMe, 90 °C, 86% (2 steps); (vii) pyBOP, NEt3, CH2Cl2, 74%.
Although, compounds in the application were characterized in both a radioligand binding assay using [3H]-3-methoxy-5-(pyridin-2-ylethynyl)pyridine (mPEPy),81 a close structural analogue of MPEP, as well as a calcium mobilization assay in HEK293 cells that expressed rat mGlu5, data provided for specific compounds was limited. Binding affinity values were reported for analogues 103 (mGlu5Ki = 6.7 nM) and 105 (mGlu5Ki = 40 nM). While the lack of specific in vitro data makes determination of SAR difficult, of particular interest was the reported in vivo activity of selected compounds (Table 7). Compounds were evaluated for their anxiolytic effects in a mouse marble burying model77 as well as a modified Geller–Seifter conflict test in rats.82 While the mouse studies were conducted with subcutaneous dosing, the rat studies used oral dosing. Without functional activity and exposure data, it is not possible to draw correlations to the efficacy data; however, several compounds demonstrated efficacy in the 10–30 mg/kg dose range.
Table 7. Adamantyl Diamide Efficacy in Behavioral Assays.
| statistically significant active dose(s) (mg/kg) |
||
|---|---|---|
| entry | mouse marble burying (SC dosing) | rat conflict test (PO dosing) |
| 103 | 3, 10, 30 | 50 |
| 105 | 30 | 20 |
| 109 | 10, 30 | 30 |
| 116 | 10, 30 | 10, 20 |
| 117 | 10, 30 | 10, 30 |
| 118 | 3, 10, 30 | 20 |
Merz
A recent patent application from Merz contained over 300 exemplified compounds from a 6-halopyrazolo[1,5-a]pyrimidine series.83 Although biological data was not provided for the majority of these compounds, selected compounds were characterized in two cell-based functional assays as well as a radioligand binding assay with [3H]-M-MPEP (Table 8). Both functional assays were calcium mobilization assays that measured the compounds' ability to potentiate the response to the orthosteric agonist l-quisqualic acid. One functional assay was run in stably transfected Chinese hamster ovary (CHO-K1) cells that express human mGlu5, while the second was run in primary astrocytes prepared from cortices of newborn rats. In general, there was good agreement between the three assays, and the SAR trends were similar. Tetrahydrocinnoline amides 119 and 120 (Series I) do not contain a chiral center; however, the tetrahydropyrrolopyrazine amides 121–126 were substituted with a methyl group at the 1-position of the ring, resulting in a chiral center. Potency and binding affinity of these analogues were enhanced by the replacement of the 6-methyl substituent on the tetrahydropyrrolopyrazine ring with a chloro or bromo substituent (compare 123 and 125 to 121 as well as 124 and 126 to 122). Also of note is the improved potency observed with the pure enantiomers (R)-122 and (R)-124 relative to their respective racemates. The synthesis of (R)-122 is shown here and is indicative of the general routes to the pyrazolopyrimidine analogues discussed here. The synthesis of carboxylic acid 130 began with the Fischer esterification and subsequent reduction of nitropyrazole acid 127 to provide aminopyrazole 128 (Scheme 11).84 Treatment of 128 with concentrated hydrochloric acid in the presence of 2-bromomalonaldehyde afforded pyrazolopyrimidine 129. Hydrolysis of the methyl ester under acidic conditions gave carboxylic acid 130, which was coupled with amine 131 under standard conditions to provide final compound (R)-122 (yield not provided). The synthesis of chiral amine 131 was accomplished in two steps beginning with the acid mediated reaction of ethylenediamine with furan 132 to yield pyrrolopyrazine 133. Asymmetric transfer hydrogenation of 133 with chiral ruthenium catalyst 134 gave 131 in near quantitative yield.
Table 8. Functional mGlu5 Activity and Binding Affinity in the Pyrazolopyrimidine Series.

| entry | series | X | R | chirality | CHO-K1 mGlu5 IC50 (nM) | astrocytes mGlu5 IC50 (nM) | mGlu5Ki (nM) |
|---|---|---|---|---|---|---|---|
| 119 | I | Cl | 166 | 82 | 247 | ||
| 120 | I | Br | 189 | 64 | 281 | ||
| 121 | II | Cl | Me | (±) | 327 | 87 | 275 |
| 122 | II | Br | Me | (±) | 223 | 50 | 219 |
| (R)-122 | II | Br | Me | R | 118 | 34 | 74 |
| 123 | II | Cl | Cl | (±) | 91 | 22 | 42 |
| 124 | II | Br | Cl | (±) | 84 | 23 | 45 |
| (R)-124 | II | Br | Cl | R | 25 | 7.8 | not reported |
| 125 | II | Cl | Br | (±) | 34 | 17 | 73 |
| 126 | II | Br | Br | (±) | 75 | 15 | 55 |
Scheme 11.

(i) MeOH, SOCl2, reflux; (ii) H2, Pd/C, THF, AcOH, 88% (2 steps); (iii) conc HCl, EtOH, BrCH(CHO)2, 18%; (iv) aq. H2SO4, heat; (v) EDC, HOBt, DMF, 50 °C; (vi) H2NCH2CH2NH2, aq. HCl, reflux, 70%; (vii) Et3N·HCO2H, MeCN, 99%.
National Institute on Drug Abuse
A group of researchers at NIDA has previously reported on a rational design strategy in an aryl amide series of mGlu5 NAMs.85,86 Further information regarding continued effort within this general chemotype and additional characterization of selected molecules has been published recently.87 The synthesis of many of the compounds in this paper began with 6-methyl-2-aminopyridine 135 (Scheme 12). Coupling of 135 with aryl carboxylic acids or acid chlorides afforded amides 136–140. In cases where R1 (139) or R2 (140) was a bromide, those analogues were further reacted with aryl boronic acids using Suzuki coupling conditions to generate additional analogues 141–147. Compounds were characterized in a calcium mobilization assay (mGlu5 IC50) run in HEK293 cells that expressed rat mGlu5 as well as a radioligand binding assay (mGlu5 Ki) with [3H]MPEP (Table 9). In general, there was good agreement between these assays; however, there were some exceptions. Prior work in this series had shown that simple exchange of a chloro substituent (136) for a cyano group (137) produced a near 4-fold improvement in potency.85 The further introduction of a 5-fluoro substituent to provide analogue 138 further enhanced activity by over 20-fold. Replacement of the cyano group with a 3-pyridyl ring (141) was not well tolerated; however, introduction of a phenyl ring at the R2 position of 137 gave biaryl analogue 142, which was among the most potent of analogues tested. Interestingly, the cyano group was shown to be essential as analogue 143 demonstrated dramatically reduced affinity in the radioligand binding assay. Fluorinated analogues of 142 (144–145) were similarly potent; however, there was a disparity between the binding affinity and functional potency observed with 145 that was not seen with 144. Replacement of the phenyl group with a 1-naphthyl (146) or a 3-pyridyl (147) ring was not favorable. Compounds were also evaluated in mGlu subtype functional assays, and none demonstrated more than 70% inhibition of phosphatidylinositol (PI) hydrolysis at a concentration of 10 μM. Further, compounds 142 and 144 demonstrated only weak activity in off target assays measuring interactions with the dopamine, serotonin, and norepinephrine transporters.
Scheme 12.

(i) ArCO2H, CDI, pyridine or ArCOCl, pyridine, NEt3, CH2Cl2, 50–75%; (ii) ArB(OH)2, Pd(PPh3)4, 2N aq. Na2CO3, PhMe, 110 °C or DME/H2O (3:1), 80 °C, 75–80%.
Table 9. Functional mGlu5 Activity and Binding Affinity in the Aryl Amide Series.
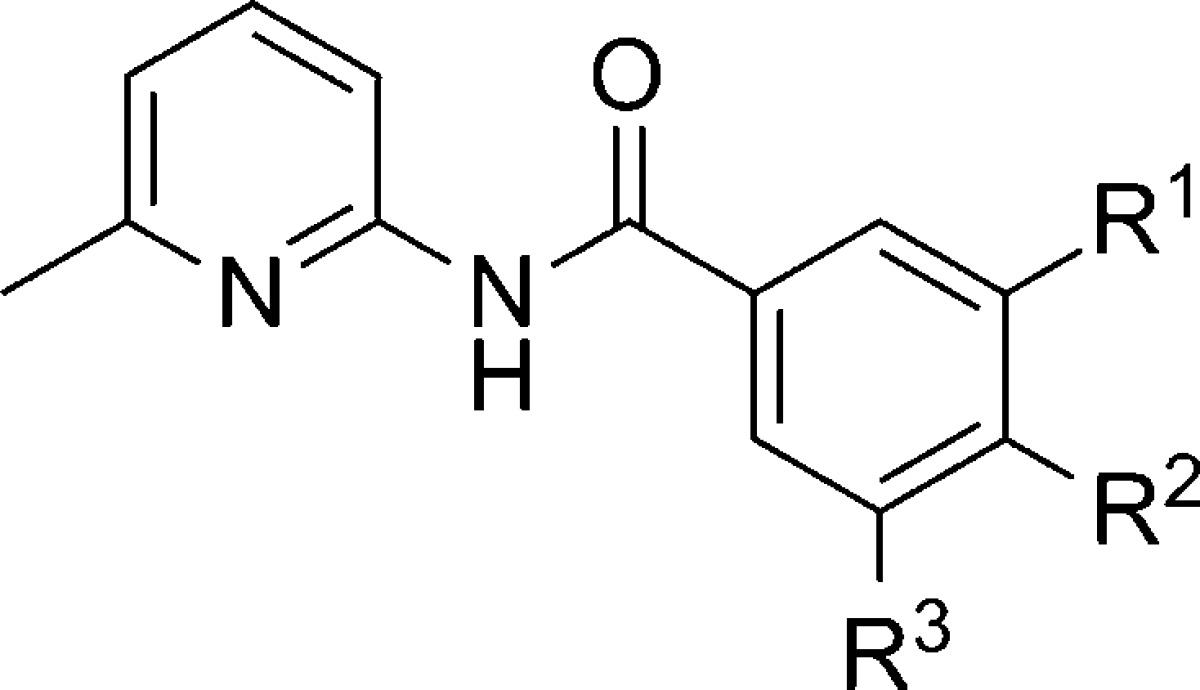
| entry | R1 | R2 | R3 | mGlu5 IC50 (nM) | mGlu5Ki (nM) |
|---|---|---|---|---|---|
| 136 | Cl | H | H | 1870 | 1730 |
| 137 | CN | H | H | 490 | 330 |
| 138 | CN | H | F | 22 | 66 |
| 141 | 3-pyridyl | H | F | not tested | 702 |
| 142 | CN | Ph | H | 14 | 9.8 |
| 143 | H | Ph | H | not tested | 7000 |
| 144 | CN | 3-fluorophenyl | H | 25 | 22 |
| 145 | CN | 4-fluorophenyl | H | 4.6 | 134 |
| 146 | CN | 1-naphthyl | H | 640 | 72 |
| 147 | CN | 3-pyridyl | H | >1000 | 2040 |
Concurrent to their work in the aforementioned aryl amides, NIDA reported on similar efforts in several biaryl chemotypes, including 7-substituted quinolines.88 As the biaryl quinoline template was among the most promising, work was continued in that chemotype and recently disclosed.89 New 7-phenylquinoline analogues were prepared by the synthetic route shown here (Scheme 13). Bromination of 3-chlorobenzonitrile 148 with 1,3-dibromo-5,5-dimethylhydantoin 149 according to the literature procedure afforded aryl bromide 150.90 A Suzuki coupling with boronic acids 151–153 gave the biaryl products 154–156. A second Suzuki coupling between the aryl chloride and boronate ester 158 was accomplished in the presence of phosphine ligand 157 to afford the final products 159–161. A modified synthetic approach was used to access 7-(pyridin-3-yl)quinoline analogues (Scheme 14). The route began with either 5-bromonicotinonitrile 162 or 5-bromo-2-chloronicotinonitrile 163. Suzuki coupling with boronate ester 158 afforded biaryls 164 and 165. A second Suzuki coupling of 165 with phenyl boronic acid 151 using phosphine ligand 157 provided analogue 166. The profile of quinoline analogues in both the functional and radioligand binding assays is shown here (Table 10). Unsubstituted analogue 167 was prepared and reported previously by NIDA,87 and it was also independently prepared and discussed previously by a group at Pfizer.91 The addition of phenyl (159) or 4-fluorophenyl (161) substituents led to compounds with similar binding affinity to 167; however, potency in the functional assay was reduced significantly. Introduction of a 3-pyridyl (160) group alleviated the discrepancy between the two assays, but both affinity and potency were compromised relative to 167. Alteration of the scaffold to introduce a pyridyl nitrogen in the internal ring was more successful, as both unsubstituted analogue 164 and phenyl substituted analogue 166 were similar in activity to 167. Intermediate chloropyridine 165 was also tested, and although it was weak in both assays, the compound was a clear partial antagonist in the functional assay. In this case, the concentration response curve with 165 reached only a 50% inhibition of the EC80 glutamate response. Subtle SAR between full and partial antagonists has been previously noted within alkyne chemotypes in the literature.92,93
Scheme 13.

(i) H2SO4, F3CCO2H, 76%; (ii) Pd(PPh3)4, Na2CO3, DME, H2O, 70 °C, 40–75%; (iii) Pd(OAc)2, K3PO4, 157, dioxane, H2O, 105 °C, 33–42%.
Scheme 14.

(i) PdCl2(dppf), Na2CO3, 158, dioxane, H2O, μw, 140 °C, 65–80%; (ii) Pd(OAc)2, 151, K3PO4, 157, dioxane, H2O, μw, 140 °C, 30%.
Table 10. Functional mGlu5 Activity and Binding Affinity in the Quinoline Series.
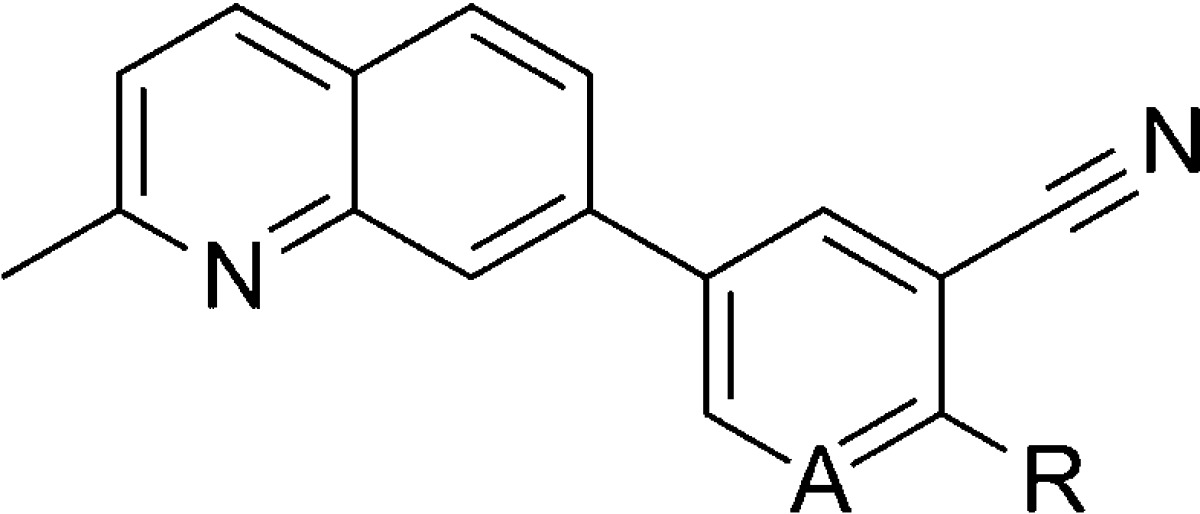
| entry | A | R | mGlu5 IC50 (nM) | mGlu5Ki (nM) |
|---|---|---|---|---|
| 167 | CH | H | 29 | 110 |
| 159 | CH | phenyl | 1250 | 97 |
| 160 | CH | 3-pyridyl | 1340 | 730 |
| 161 | CH | 4-fluorophenyl | 692 | 64 |
| 164 | N | H | 68 | 100 |
| 165 | N | Cl | 3310 | >10 000 |
| 166 | N | phenyl | 81 | 97 |
Novartis
HTS based on the ability of compounds to inhibit agonist induced elevation of intracellular calcium (Ca2+) was used by Novartis to identify a novel nicotinamide series of mGlu5 NAMs. A recent publication described a thorough evaluation of the SAR within this chemotype.94 Two general synthetic routes were used to access compounds within this template (Scheme 15). One route allowed for the introduction of the eastern portion of the chemotype as the final step. Acid chlorides 168 were reacted with amines to yield amides 169. Subsequent SNAr reaction with aryl amines 170 under acidic conditions afforded the final compounds 171–183. A second route used an amide coupling of the hydrolyzed esters of 185 as the final step. The esters 185 were prepared from a Buchwald coupling of aryl amines 170 with 2-chloropyridines 184. The HTS hit and starting point for SAR development was piperidinyl amide 171 (Table 11). Evaluation of other secondary amine amides indicated that piperidinyl (171) and azepanyl (173) amides were superior to pyrrolidinyl (172) and azocanyl (174) amides, and the majority of the remaining SAR was developed in the context of piperidinyl amides. The importance of the 4-chloro substituent on the aniline moiety was illustrated with unsubstituted aniline analogue 175. Potency could be partially restored with other 4-substituents (176–178), though not to the level seen with 171. A 3-fold improvement in potency relative to 171 was seen with the introduction of a chloro group at the 3-position of the pyridine core (180). A similar 5-fold improvement was observed when comparing 181 to 179.
Scheme 15.

(i) NEt3, HNR1R2, CH2Cl2, 0 °C; (ii) AcOH, H2O (3:7), 95 °C; (iii) Pd(OAc)2, BINAP, K2CO3, PhMe, 70 °C; (iv) 2 N aq. NaOH, MeOH, 60 °C; (v) HATU, DIEA, DME.
Table 11. Functional mGlu5 Activity in the Nicotinamide Series.
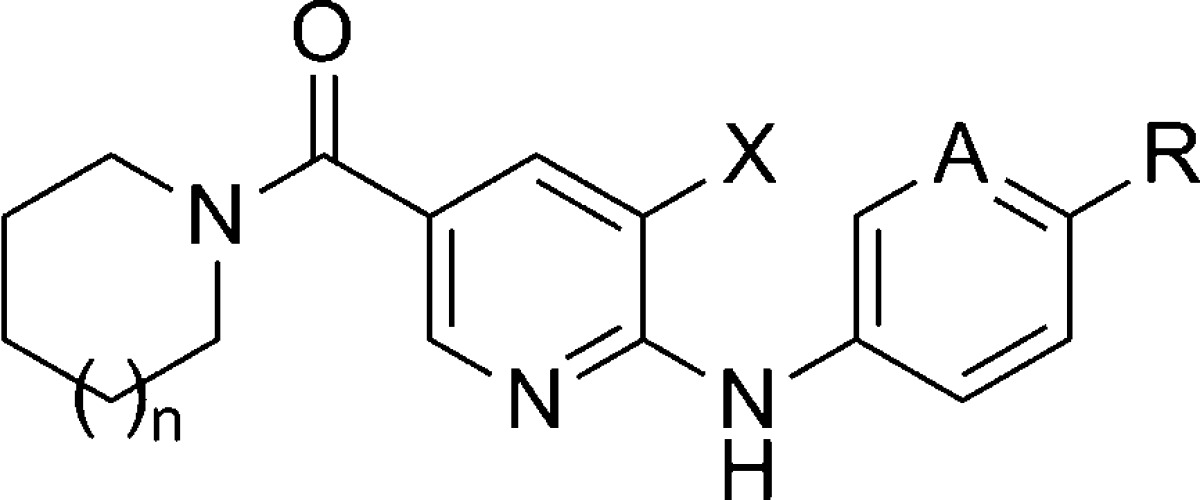
| entry | n | X | A | R | mGlu5 IC50 (nM) |
|---|---|---|---|---|---|
| 171 | 1 | H | CH | Cl | 310 |
| 172 | 0 | H | CH | Cl | 5400 |
| 173 | 2 | H | CH | Cl | 250 |
| 174 | 3 | H | CH | Cl | 2300 |
| 175 | 1 | H | CH | H | ∼10 000 |
| 176 | 1 | H | CH | F | 2300 |
| 177 | 1 | H | CH | Me | 2300 |
| 178 | 1 | H | CH | OMe | 1800 |
| 179 | 1 | H | N | Me | 420 |
| 180 | 1 | Cl | CH | Cl | 95 |
| 181 | 1 | Cl | N | Me | 87 |
Using the 5-amino-2-methylpyridine analogue 181 as a starting point, further derivatization of the piperidinyl ring was made (Table 12). Compounds were also evaluated for their solubility as well as their stability in rat and human liver microsomes. While incorporation of an ethyl group at the 3-position of the piperidinyl ring improved potency with both enantiomers (182), metabolic stability was reduced. Also of note was the difference in solubility observed between the two enantiomers. In the case of the 2-ethyl piperidinyl ring, there was a significant difference in potency between the two enantiomers, with a 12-fold preference for the R-configuration relative to the S-configuration (183). Having determined that (R)-183 offered the optimal balance of properties, the compound was studied in a rat pharmacokinetic study. The compound demonstrated moderate clearance (37.2 mL/min/kg) and good bioavailability (54%). Importantly, CNS penetration with (R)-183 was excellent with a brain to plasma ratio around 1.3 to 1. Given such promising data, analogue (R)-183 was studied in three separate rodent behavioral models of anxiety. The SIH model was evaluated in mice,79,95 and (R)-183 was efficacious at 3 and 10 mg/kg using oral dosing. Further evaluation in rats was conducted using the Vogel conflict test74,96 and the fear-potentiated startle test (FPS).97,98 Dose dependent efficacy was observed with oral dosing in both experiments with significance at 10 and 30 mg/kg in the Vogel test and 3, 10, and 30 mg/kg in the FPS test.
Table 12. mGlu5 Potency, Solubility, and Intrinsic Clearance in the Nicotinamide Series.
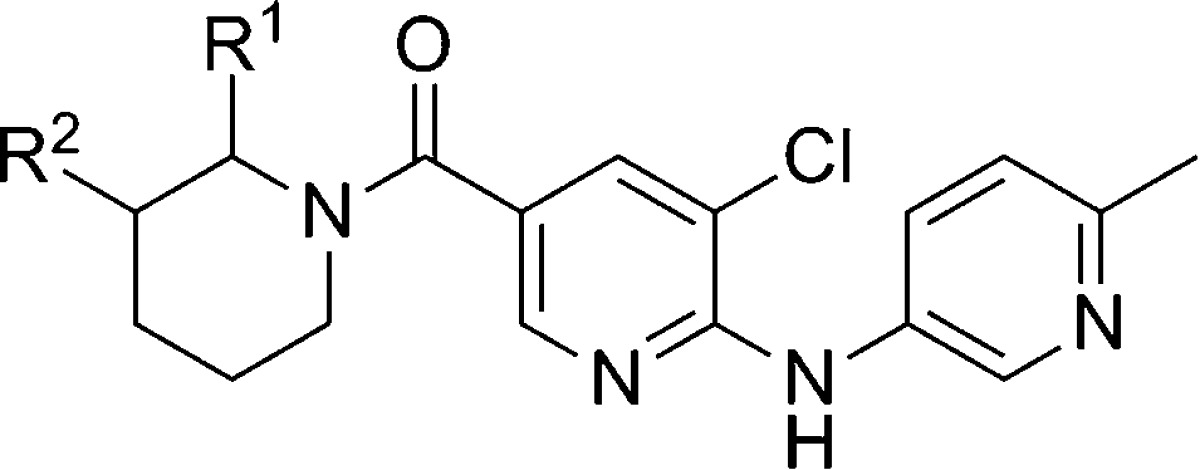
| entry | R1 | R2 | mGlu5 IC50 (nM) | solubility at pH 6.8 (mg/mL) | rat ClINT (μL/min/mg) | human ClINT (μL/min/mg) |
|---|---|---|---|---|---|---|
| 181 | H | H | 87 | 150 | 48 | 33 |
| (R)-182 | H | R-ethyl | 40 | 95 | 643 | 322 |
| (S)-182 | H | S-ethyl | 40 | <3 | 687 | 174 |
| (R)-183 | R-ethyl | H | 32 | 58 | 87 | 89 |
| (S)-183 | S-ethyl | H | 390 | 56 | 190 | 216 |
Novartis researchers continued further optimization efforts in the (R)-183 series and published results from that effort recently.99 Metabolite identification studies revealed that the piperidine moiety of (R)-183 was a major site of oxidative metabolism. As such, medicinal chemistry was focused on the identification of a suitable bioisosteric replacement. Several heterocyclic alternatives were explored including oxadiazoles, triazoles, and isoxazoles and shown to lack suitable activity for further work. Fortunately a 1-alkyl-2-benzimidazolyl group was identified as a promising alternative and SAR exploration was conducted in that area (Table 13). n-Propyl analogue 186, ethyl analogue 187, and isobutyl compound 189 were similar in activity, while the n-butyl analogue 188 was less active. The metabolic stability profiles of 186 and 187 were preferable to those for 188 and 189. Installation of a chloro substituent at each of the positions on the benzimidazole ring (190–193) maintained or reduced intrinsic clearance in both rat and human liver microsomes relative to 186, with the intrinsic clearance of 191 in rat liver microsomes representing the lone exception. The improved potency and encouraging metabolic stability of 193 led to further study of that compound. Radioligand binding assays with [3H]-ABP688100 showed that 193 demonstrated a high affinity for the known allosteric binding site (mGlu5Ki = 2.4 nM). Compound 193 was also shown to be selective against other mGlus (IC50 > 10 μM for mGlu1, mGlu2, and mGlu7) and ionotropic glutamate receptors. Although the intrinsic clearance in rat liver microsomes for 193 and (R)-183 was nearly identical, the in vivo profile for 193 was superior. Clearance was lower (15 mL/min/kg), and bioavailability remained good (41%). Furthermore, CNS penetration remained excellent with a brain to plasma ratio around 1.7 to 1. Evaluation of 193 in the FPS model97,98 using oral dosing demonstrated efficacy in a dose dependent manner with significant responses at both 1 and 3 mg/kg. A four-step synthesis of 193 was employed (Scheme 16). A palladium catalyzed coupling of 2,3-dichloropyridine 194 with amine 195 gave ester 196, which was hydrolyzed to afford acid 197. Reaction with 1,2-phenylenediamine 198 in hot polyphosphoric acid (PPA) yielded benzimidazole 199. Alkylation with n-propyl iodide gave the final compound 193 in low yield following separation from its regioisomer 190.
Table 13. mGlu5 Potency and Intrinsic Clearance in 2-Benzimidazolyl Analogues.
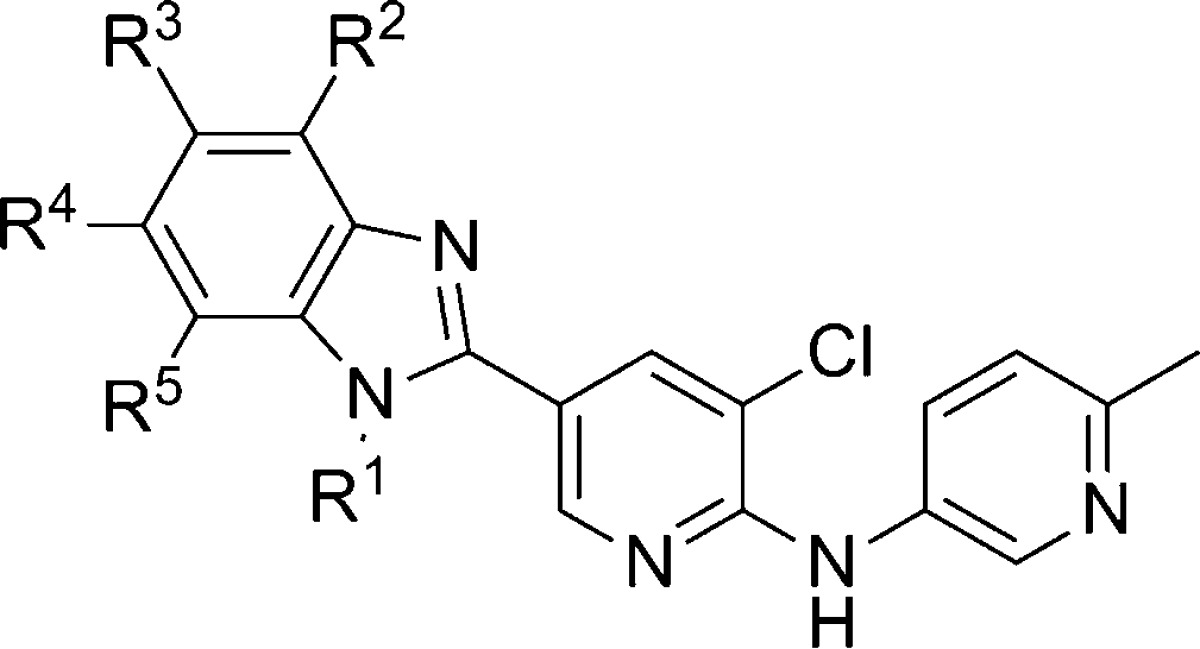
| entry | R1 | R2 | R3 | R4 | R5 | mGlu5 IC50 (nM) | rat ClINT (μL/min/mg) | human ClINT (μL/min/mg) |
|---|---|---|---|---|---|---|---|---|
| 186 | nPr | H | H | H | H | 100 | 83 | 158 |
| 187 | Et | H | H | H | H | 110 | 120 | 118 |
| 188 | nBu | H | H | H | H | 360 | 347 | 279 |
| 189 | iBu | H | H | H | H | 110 | 55 | 826 |
| 190 | nPr | Cl | H | H | H | 200 | 74 | 69 |
| 191 | nPr | H | Cl | H | H | >500 | 400 | 34 |
| 192 | nPr | H | H | Cl | H | 130 | 56 | 33 |
| 193 | nPr | H | H | H | Cl | 24 | 83 | 112 |
Scheme 16.

(i) Pd2(dba)3, K2CO3, BINAP, PhMe, 120 °C, 16 h, 56%; (ii) 1 N aq. NaOH, MeOH, 95%; (iii) PPA, 200 °C, 16 h, 74%; (iv) NaH, nPrI, DMF, 10%.
Sepracor
A rational design approach to new mGlu5 NAMs was employed by researchers at Sepracor, and results from that effort were recently disclosed.101 Their approach centered on novel methods for tethering two aryl groups as alternatives to traditional linkers such as alkynes, amides, and azoles. Several potential scaffolds were prepared before settling on oxazolopiperidine 200 (Figure 6). Compound 200 was a moderately potent mGlu5 NAM in their functional assay, which was run in an inducible cell line expressing human mGlu5. SAR exploration established the 2-pyridyl ring as optimal for the western portion of the template, and the 3-cyanophenyl ring was similarly optimal for the eastern portion. Additional SAR was established in the interior of the scaffold in the context of these substituents (Table 14). Maintaining the oxazolopiperidine core of 200 in the context of the optimized peripheral groups gave analogue 201, which was a potent antagonist. The isomeric oxazole 202 was less potent than 201. The installation of a 5-fluoro group on the phenyl ring boosted potency in both instances (203 and 204). Imidazole 205 and thiazole 206 were weak antagonists. While imidazole 207 and triazole 208 were also potent, both were inferior to oxazole 203. A final modification that further enhanced potency was enlargement of the saturated ring by one carbon to afford azepines 209 and 210. Radioligand binding assays with 203 and 210 using [3H]-MPEP confirmed their interaction with the known allosteric binding site. Both molecules were also examined for off target activity and were found to have quite clean profiles. With a good correlation between the binding and functional assays, both molecules were examined in occupancy studies in rats using [3H]-mPEPy (Table 15). Compounds were dosed IP, and [3H]-mPEPy was dosed by tail-vein injection 1 h later. Both compounds showed good CNS penetration; however, 210 was a superior compound, demonstrating an occupancy ED50 of 0.9 mg/kg. Compound 210 was next taken into rat and monkey bioavailability studies (rat: Clp = 46.5 mL/min/kg; F = 31%; t1/2 = 0.62 h; monkey: Clp = 16.3 mL/min/kg; F = 10%; t1/2 = 0.67 h). The authors believe such results are less than desirable and plan to continue to work to improve the pharmacokinetics of analogues within this scaffold. The synthesis of 210 is described here (Scheme 17). Piperidinone 211 was brominated to yield bromoketone 212. Reaction with ethyl diazoacetate in an Eistert homologation afforded azepine 213. Hydrolysis and decarboxylation was accomplished under acidic conditions. Standard conditions were then used to access azide 214, which was treated with lithium aluminum hydride to give the amino alcohol 215. Amide formation through coupling with picolinic acid was followed by a Dess–Martin oxidation to provide ketone 216. Treatment with Burgess’ reagent under microwave conditions gave oxazoloazepine 217. Removal of the tosyl protecting group and subsequent Buchwald coupling of amine 218 with 3-bromo-5-fluorobenzonitrile provided the final product 210 in low yield.
Figure 6.

Hit oxazolopiperidine 200.
Table 14. mGlu5 Potency in Tetrahydroazolopyridines and Tetrahydroazoloazepines.
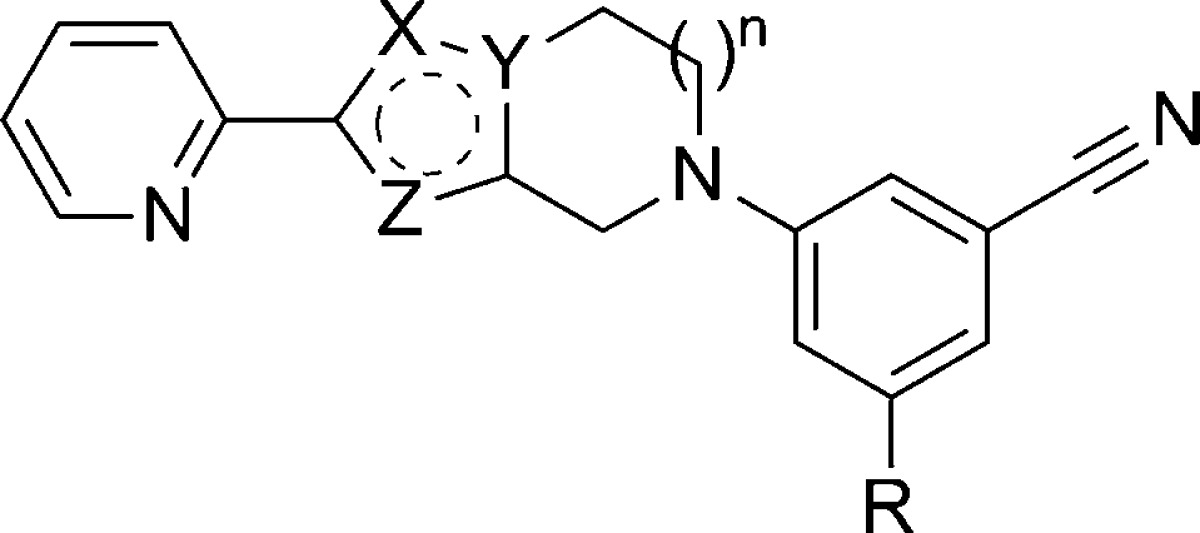
| entry | n | X | Y | Z | R | mGlu5 IC50 (nM) |
|---|---|---|---|---|---|---|
| 201 | 1 | O | C | N | H | 60 |
| 202 | 1 | N | C | O | H | 290 |
| 203 | 1 | O | C | N | F | 28 |
| 204 | 1 | N | C | O | F | 54 |
| 205 | 1 | N | C | NH | F | 34% at 10 μM |
| 206 | 1 | N | C | S | F | 52% at 10 μM |
| 207 | 1 | C | N | N | F | 153 |
| 208 | 1 | N | N | N | F | 346 |
| 209 | 2 | O | C | N | H | 65 |
| 210 | 2 | O | C | N | F | 16 |
Table 15. Receptor Occupancy and Exposures for 203 and 210 in Rats.
| entry | mGlu5 IC50 (nM) | mGlu5Ki (nM) | % RO | plasma levels (nM) | brain levels (nM) | RO50 (mg/kg) |
|---|---|---|---|---|---|---|
| 203 | 28 | 17 | 45 | 2490 | 1290 | >30 |
| 210 | 16 | 17 | 82 | 1430 | 1100 | 0.9 |
Scheme 17.

(i) Br2, CH2Cl2, −5 to −2 °C, 98%; (ii) ethyl diazoacetate, BF3·OEt2, CH2Cl2, −5 to 0 °C, 72%; (iii) 3 N aq. HCl, dioxane, 100 °C, 90%; (iv) NaN3, DMF, 80%; (v) LiAlH4, THF, 0 °C, 52%; (vi) picolinic acid, EDC, HOBt, NEt3, CH2Cl2, 80%; (vii) Dess–Martin periodinane, CH2Cl2, 92%; (viii) Burgess’ reagent, THF, 150 °C, μw, 45 min, 78%; (ix) 48% aq. HBr, 100 °C, 74%; (x) 3-bromo-5-fluorobenzonitrile, Pd2(dba)3, XANTPHOS, KOtBu, PhMe, μw, 100 °C, 22%.
Vanderbilt
The Vanderbilt Program in Drug Discovery has used a functional cell-based HTS of a collection of 160 000 compounds to identify 345 confirmed noncompetitive antagonists of mGlu5. The initial report of the SAR within three distinct series that were identified through this screen was reported in early 2009102 and has been discussed in a prior review.38 Further efforts with different chemical scaffolds that were discovered through this HTS effort have been communicated since this initial publication. One such effort focused on a series of 6-substituted-4-anilinoquinazolines.103 Analogues within this series were prepared through reaction of commercially available anilines and the requisite 4-chloroquinazolines using microwave-assisted organic synthesis (MAOS).104 SAR demonstrated that 4-anilinoquinazolines were superior to the comparator 4-anilinoquinolines and 1-anilinoquinolines, and that the secondary amine was the optimal linker between the quinazoline and the phenyl ring. The majority of SAR evaluation was centered on the 6-position of the quinazoline and on substitution of the aniline ring (Table 16). Potency data for compounds was determined using a functional assay that measured the ability of compound to block the mobilization of calcium by an EC80 concentration of glutamate in HEK293 cells expressing rat mGlu5. The screening hit in this series was 6-bromoquinazoline 219. Substitution of the aniline was critical to potency as evidenced by unsubstituted analogue 220. Tolerance for substitution was limited although both aryl bromide 222 and methyl analogue 223 were similar in potency to the hit compound. A similar trend was noted with regard to substitution at the 6-position on the quinazoline ring, where chloro (225 and 229) and fluoro (226 and 230) analogues were also potent antagonists. In this case, a 6-nitroquinazoline 227 was also similar in potency to the hit compound. Additional SAR was conducted in context of the 6-fluoroquinazoline (230–234), and SAR was somewhat shallow. Analogues 219, 229, and 230 were determined to interact with the known allosteric binding site through radioligand binding studies with [3H]-mPEPy. Further, when the same three compounds were tested against other mGlus, all were inactive up to 10 μM, except for mGlu1, where potency was submicromolar in each case.
Table 16. mGlu5 Potency in the Quinazoline Series.
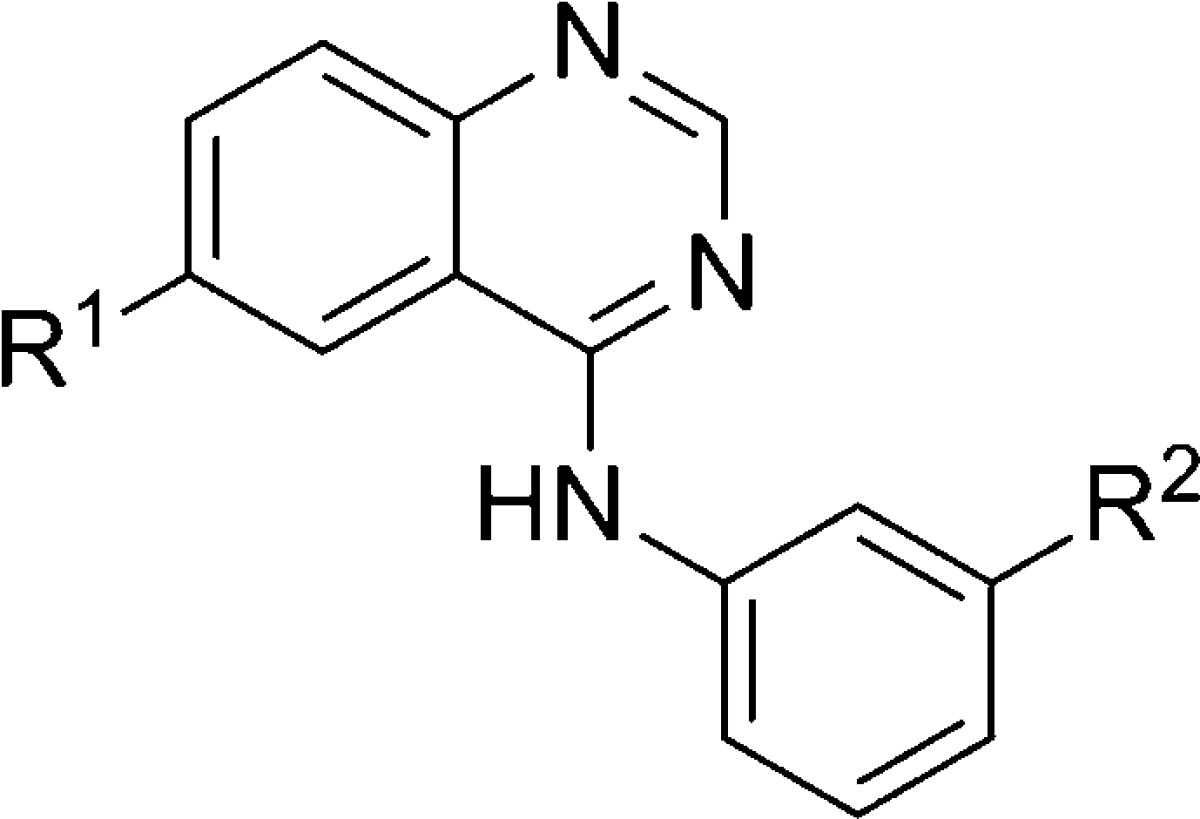
| entry | R1 | R2 | mGlu5 IC50 (nM) |
|---|---|---|---|
| 219 | Br | Cl | 256 |
| 220 | Br | H | >10 000 |
| 221 | Br | F | 1970 |
| 222 | Br | Br | 174 |
| 223 | Br | Me | 246 |
| 224 | Br | OMe | >10 000 |
| 225 | Cl | Cl | 130 |
| 226 | F | Cl | 311 |
| 227 | NO2 | Cl | 274 |
| 228 | OMe | Cl | 1510 |
| 229 | Cl | Br | 124 |
| 230 | F | Br | 96 |
| 231 | F | Me | 1350 |
| 232 | F | CF3 | >10 000 |
| 233 | F | CN | 1370 |
| 234 | F | OMe | >10 000 |
A third publication from the Vanderbilt group describing the HTS follow-up recently appeared.105 Although the publication describes three distinct chemotypes discovered in the HTS, the majority of the SAR discussion centers on an N,N′-(1,3-phenylene)diamide series. The preparation of these compounds begins with commercially available 3-nitroaniline 235 (Scheme 18). Amide formation with either 2-methoxy or 3-chlorobenzoyl chloride was accomplished under standard conditions to afford 236 and 237, respectively. The reduction of the nitro group via Raney nickel hydrogenation gave anilines 238 and 239. A parallel synthesis, library approach was used in reactions with acid chlorides and with the aid of resin bound scavengers to provide final compounds 240–255 (Table 17). The hit compound in this series was dibenzamide 240, which was confirmed to bind at the MPEP binding site through studies with [3H]-mPEPy (mGlu5Ki = 1100 nM). Multiple examples were prepared with a methoxy group at R1 and substituted phenyl rings at R2. Tolerance for substitution in these cases was limited to the 3-position (compare 241 and 242 to 240). Other substituents at the 3-position failed to yield compounds with superior potency to hit 240 (243–246). Installation of nonbenzamide groups at R2 led to multiple inactive compounds such as benzyl amide 247. Choosing to hold the 3-chlorobenzamide constant led to a second library of compounds without improvement on hit 240. Still, several moderately potent compounds were obtained with small cycloalkyl amides (252 and 253). Potency quickly fell off if the cycloalkyl amide was too large as noted with cyclopentyl amide 251. Also of note were isonicotinamide 254 (EC50 = 1870 nM; Glumax = 49%) and nicotinamide 255 (EC50 = 5540 nM; Glumax = 56%), which acted as PAMs. Such an example is particularly interesting, as it constitutes an example of pharmacology mode switching in a nonalkyne mGlu5 series.
Scheme 18.

(i) 2-Methoxybenzoyl chloride or 3-chlorobenzoyl chloride, DIEA, DMAP, DMF, 64–89%; (ii) H2, Raney nickel, EtOH, EtOAc, 89–94%; (iii) R2COCl, PS-DMAP, PS-DIEA, then PS-isocyanate and PS-trisamine.
Table 17. mGlu5 Potency in the N,N′-(1,3-Phenylene)Diamide Series.
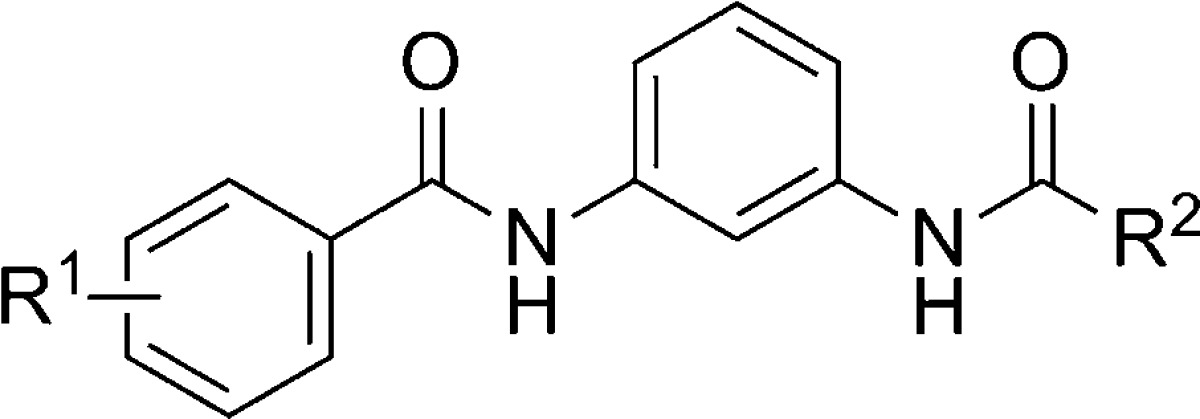
| entry | R1 | R2 | mGlu5 IC50 (nM) |
|---|---|---|---|
| 240 | 2-OMe | 3-chlorophenyl | 880 |
| 241 | 2-OMe | 2-chlorophenyl | >30 000 |
| 242 | 2-OMe | 4-chlorophenyl | >30 000 |
| 243 | 2-OMe | 3-fluorophenyl | 6480 |
| 244 | 2-OMe | 3-methylphenyl | 1350 |
| 245 | 2-OMe | 3-(CF3)phenyl | 10,600 |
| 246 | 2-OMe | 3-cyanophenyl | 1800 |
| 247 | 2-OMe | benzyl | >30 000 |
| 248 | 3-Cl | 2-chlorophenyl | 2620 |
| 249 | 3-Cl | 3-chlorophenyl | >30 000 |
| 250 | 3-Cl | 2-furyl | 4510 |
| 251 | 3-Cl | cyclopentyl | >30 000 |
| 252 | 3-Cl | cyclobutyl | 5270 |
| 253 | 3-Cl | cyclopropyl | 2230 |
| 254 | 3-Cl | isonicotinamide | PAM |
| 255 | 3-Cl | nicotinamide | PAM |
Follow up work to the HTS screen yielded a fourth publication in 2010 focused on developing SAR around an oxadiazole hit compound 256 (Table 18).106 The oxadiazole series was previously disclosed as mGlu5 NAMs in a patent application from NPS Pharmaceuticals;107 however, Vanderbilt continued work in this series with the goal of developing a new, useful tool compound in a series without an alkyne or amide moiety. Compounds in this series can be prepared in a single step through a condensation reaction of 2-pyridylamidoxime with substituted benzoic acids. Although most new analogues (257–259) failed to achieve improved potency relative to hit compound 256, 3-cyano-5-fluorophenyl analogue 260 was a notable improvement and was selected for extensive evaluation as a potential tool compound. Radioligand binding studies revealed that 260 was fully competitive with [3H]-mPEPy (mGlu5Ki = 17 nM), suggesting an interaction with the known allosteric binding site. Further studies with the neutral ligand 5MPEP93 confirmed this interaction. Compound 260 also demonstrated an ability to induce rightward shifts of the glutamate concentration response curve while decreasing the maximal response to glutamate, a profile consistent with noncompetitive inhibition. Compound 260 was determined to be selective over other mGlus. Evaluation of 260 in a modified version of the Geller–Seifter conflict paradigm model12,82 in rats revealed that the compound dose dependently increases punished responding relative to vehicle at 3 and 10 mg/kg (IP dosing) and significantly decreased unpunished responding a the 10 mg/kg dose. Compound 260 was also efficacious in the mouse marble burying model of anxiety,77 demonstrating a significant decrease in marble burying relative to vehicle at the 10 mg/kg dose (IP dosing). The anxiolytic effects observed here with 260 were comparable to MTEP, which was run as a positive control. A particularly interesting experiment presented here examined 260 and MTEP in a PCP-induced hyperlocomotion model in rats, a model of psychotomimetic-like activity. Both compounds were dosed IP at 10 mg/kg, and while MTEP produced a robust potentiation of PCP induced hyperlocomotion, no such activity was observed with 260.
Table 18. mGlu5 Potency in Oxadiazole Series.
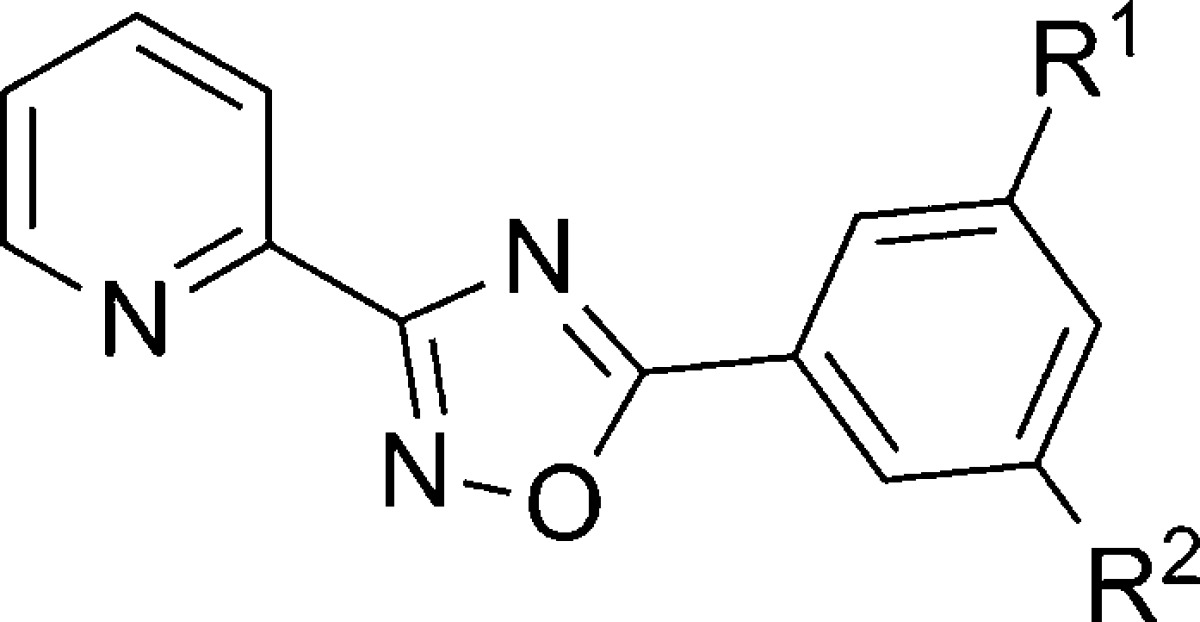
| entry | R1 | R2 | mGlu5 IC50 (nM) |
|---|---|---|---|
| 256 | OMe | OMe | 62 |
| 257 | H | CF3 | 2360 |
| 258 | H | Cl | 240 |
| 259 | H | Br | 215 |
| 260 | F | CN | 24 |
In addition to pursuing new mGlu5 NAM scaffolds based on their HTS results, Vanderbilt has also been investigating rational design approaches. One such effort was published recently and described SAR in a series of 3-cyano-5-fluoro-N-arylbenzamides.109 Recognizing the prevalence of 3-cyano-5-fluorophenyl rings found across known mGlu5 NAM scaffolds (see 138, 203, 210, and 260 for examples), chemists used a library approach to rapidly generate new amide analogues prepared from 3-cyano-5-fluorobenzoic acid according to standard methods (Table 19). Several heteroaryl amides were prepared, and most new analogues were only weak antagonists as was observed with compound 262. The 6-methylpyridine analogue 138 was previously described by NIDA (Table 9) and demonstrated the expected potent activity. Isosteric 4-methylthiazole analogue 261 had not been previously described and was equally potent. Tolerance for substitution of the thiazole was limited as exemplified by compound 263. Noting the weak antagonist activity observed with simple aniline analogue 264, a series of substituted aniline analogues were prepared. Anilines substituted with small 3-substituents (265 and 266) demonstrated substantially enhanced potency relative to 264. Also of interest were the results with regioisomeric analogues 267 and 268. Although their IC50 values were nearly identical, like most of the potent compounds reported here, 267 was a full antagonist; however, 268 was a clear partial antagonist. In the case of partial antagonists, the complete response curve plateaus whereas full antagonists bring the curve near to zero. In this case, at a 30 μM top concentration of 268, the amplitude of response as a percentage of maximal response (100 μM glutamate) plateaus at 43%. Radioligand binding studies with [3H]-mPEPy verified the interaction of four compounds (138, 261, 265, and 266) with the known allosteric binding site. Following evaluation of the same four compounds in microsomal stability assays, both 261 and 265 were evaluated in rat pharmacokinetic studies using IP dosing at 10 mg/kg doses. The CNS penetration was quite good with both compounds (brain to plasma ratios = 4.1 (261) and 1.4 (265)). Furthermore, protein binding studies with each compound (rat plasma = 92.4% bound (261) and 93.7% bound (265)) indicated that ample free fraction is available to make both potentially interesting tool compounds. Current efforts are focused in that area.
Table 19. mGlu5 Potency in 3-Cyano-5-fluoro-N-arylbenzamide Series.
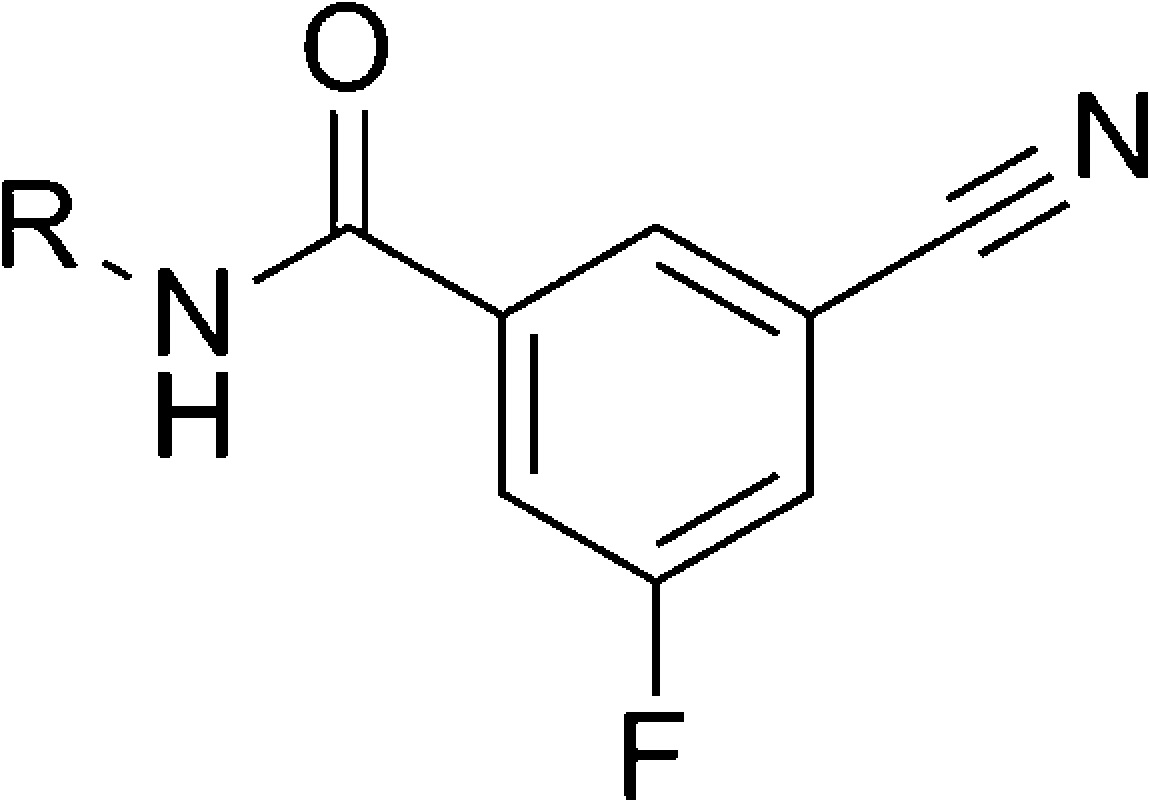
| entry | R | mGlu5 IC50 (nM) |
|---|---|---|
| 138 | 6-methylpyridin-2-yl | 65 |
| 261 | 4-methylthiazol-2-yl | 59 |
| 262 | 1-methyl-1H-pyrazol-3-yl | >10 000 |
| 263 | 4-cyclopropylthiazol-2-yl | 900 |
| 264 | phenyl | 5440 |
| 265 | 3-chlorophenyl | 45 |
| 266 | 3-methylphenyl | 122 |
| 267 | 3-chloro-2-fluorophenyl | 347 |
| 268 | 3-chloro-4-fluorophenyl | 377 |
Conclusion
Considering the aforementioned cases of positive clinical reports with mGlu5 NAMs and the immense effort from groups across the globe directed toward the discovery and optimization of many diverse chemotypes, the likelihood of a molecule making it to market in the future would seem high. Extensive effort has identified a number of templates that contain the alkyne moiety found in earlier compounds such as MTEP and MPEP. Other than the previously discovered compound fenobam, current clinical compounds where structures (or generic structures) are in the public domain also contain this functionality. Whether the newer chemotypes described here offer advantages to the alkyne containing compounds remains to be determined. Of particular note is that despite the diversity of chemical scaffolds that have been disclosed, where reported, the interaction of these compounds appears to be with the known allosteric (MPEP) binding site. The discovery of a second allosteric binding site and compounds that interact with it would be a notable advancement and would no doubt spur further research. In any case, the current excitement for this target and approach to new therapeutics should continue to result in new and interesting discoveries for several years to come. Hopefully, such discoveries will also soon translate into useful therapeutics for patients.
Abbreviations
- 7TM
seven transmembrane
- Ac
acetyl
- ALT
alanine transaminase
- Ar
aryl
- AUC
area under the curve
- BINAP
2,2′-bis(diphenylphosphino)-1,1′-binaphthyl
- BOP
benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium hexafluorophosphate
- CDI
carbonyldiimidazole
- CHO
Chinese hamster ovary
- CNS
central nervous system
- dba
dibenzylideneacetone
- DCC
N,N′-dicyclohexylcarbodiimide
- DIEA
N,N-diisopropylethylamine
- DMAP
4-dimethylaminopyridine
- DME
dimethoxyethane
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- dppf
1,1′-bis(diphenylphosphanyl) ferrocene
- EDC
1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
- Et
ethyl
- FPS
fear-potentiated startle test
- FXS
fragile X syndrome
- GERD
gastresophageal reflux disease
- GPCR
G-protein-coupled receptors
- HATU
2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate methanaminium
- HEK
human embryonic kidney
- HOBt
hydroxybenzotriazole
- HPLC
high performance liquid chromatography
- HTS
high throughput screening
- iBu
isobutyl
- IP
intraperitoneal
- iPr
isopropyl
- LELP
lipophilic ligand efficiency metrics
- Me
methyl
- mGlu
metabotropic glutamate receptor
- M-MPEP
2-((3-methoxyphenyl)ethynyl)-6-methylpyridine
- MPEP
2-methyl-6-(phenylethynyl) pyridine
- mPEPy
3-methoxy-5-(pyridin-2-ylethynyl)pyridine
- Ms
methanesulfonyl
- MTEP
3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine
- NAM
negative allosteric modulator
- NIDA
National Institute on Drug Abuse
- nBu
n-butyl
- nPr
n-propyl
- PAM
positive allosteric modulator
- PCP
phencyclidine
- PD
Parkinson’s disease
- PD-LID
Parkinson’s disease levodopa-induced dyskinesia
- Ph
phenyl
- PI
phosphatidylinositol
- PKC
protein kinase C
- PPA
polyphosphoric acid
- PS
polystyrene
- pyBOP
(benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate
- SAR
structure–activity relationship
- SIH
stress-induced hyperthermia
- SILE
size independent ligand efficiency
- SNAr
nucleophilic aromatic substitution
- tBu
tert-butyl
- THF
tetrahydrofuran
- Ts
para-toluenesulfonic acid
- XANTPHOS
4,5-bis(diphenylphosphino)-9,9-dimethylxanthene
The author thanks NIDA (RO1 DA023947) and Seaside Therapeutics (VUMC33842) for their support of our programs at the Vanderbilt Center for Neuroscience Drug Discovery focused on the development of negative allosteric modulators of mGlu5.
Author Contributions
K.A.E. researched the patent and primary literature, wrote the manuscript, and prepared all figures, schemes, and tables.
Funding Statement
National Institutes of Health, United States
References
- Schoepp D. D.; Jane D. E.; Monn J. A. (1999) Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology 38, 1431–1476. [DOI] [PubMed] [Google Scholar]
- Conn P. J.; Pin J.-P. (1997) Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 37, 205–237. [DOI] [PubMed] [Google Scholar]
- Knöpfel T.; Kuhn R.; Allgeier H. (1995) Metabotropic glutamate receptors: Novel targets for drug development. J. Med. Chem. 38, 1417–1426. [DOI] [PubMed] [Google Scholar]
- Niswender C. M.; Jones C. K.; Conn P. J. (2005) New therapeutic frontiers for metabotropic glutamate receptors. Curr. Top. Med. Chem. 5, 847–857. [DOI] [PubMed] [Google Scholar]
- Ritzén A.; Mathiesen J. M.; Thomsen C. (2005) Molecular pharmacology and therapeutic prospects of metabotropic glutamate receptor allosteric modulators. Basic Clin. Pharmacol. Toxicol. 97, 202–213. [DOI] [PubMed] [Google Scholar]
- Kew J. N. C. (2004) Positive and negative allosteric modulation of metabotropic glutamate receptors: emerging therapeutic potential. Pharmacol. Ther. 104, 233–244. [DOI] [PubMed] [Google Scholar]
- Gasparini F.; Lingenhöhl K.; Stoehr N.; Flor P. J.; Heinrich M.; Vranesic I.; Biollaz M.; Allgeier H.; Heckendorn R.; Urwyler S.; Varney M. A.; Johnson E. C.; Hess S. D.; Rao S. P.; Sacaan A. I.; Santori E. M.; Veliocelebi G.; Kuhn R. (1999) Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systematically active mGlu5 receptor antagonist. Neuropharmacology 38, 1493–1503. [DOI] [PubMed] [Google Scholar]
- Cosford N. D.; Tehrani L.; Roppe J.; Schweiger E.; Smith N. D.; Anderson J.; Bristow L.; Brodkin J.; Jiang X.; McDonald I.; Rao S.; Washburn M.; Varney M. A. (2003) 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine: a potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J. Med. Chem. 46, 204–206. [DOI] [PubMed] [Google Scholar]
- Zhu C. Z.; Wilson S. G.; Mikusa J. P.; Wismer C. T.; Gauvin D. M.; Lynch J. J.; Wade C. L.; Decker M. W.; Honore P. (2004) Assessing the role of metabotropic glutamate receptor 5 in multiple nociceptive modalities. Eur. J. Pharmacol. 506, 107–118. [DOI] [PubMed] [Google Scholar]
- Nicolas L. B.; Kolb Y.; Prinssen E. P. M. (2006) A combined marble burying-locomoter activity test in mice: A practical screening test with sensitivity to different classes of anxiolytics and antidepressants. Eur. J. Pharmacol. 547, 106–115. [DOI] [PubMed] [Google Scholar]
- Pietraszek M.; Sukhanov I.; Maciejak P.; Szyndler J.; Gravius A.; Wislowska A.; Plaznik A.; Bespalov A. Y.; Danysz W. (2005) Anxiolytic-like effects of mGlu1 and mGlu5 receptor antagonists in rats. Eur. J. Pharmacol. 514, 25–34. [DOI] [PubMed] [Google Scholar]
- Busse C. S.; Brodkin J.; Tattersall D.; Anderson J. J.; Warren N.; Tehrani L.; Bristow L. J.; Varney M. A.; Cosford N. D. P. (2004) The behavioral profile of the potent and selective mGlu5 receptor antagonist 3-(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine (MTEP) in rodent models of anxiety. Neuropsychopharmacology 29, 1971–1979. [DOI] [PubMed] [Google Scholar]
- Klodzinska A.; Tatarczynska E.; Chojnacka-Wójcik E.; Nowak G.; Cosford N. D. P.; Pilc A. (2004) Anxiolytic-like effects of MTEP, a potent and selective mGlu5 receptor agonist does not involve GABAA signaling. Neuropharmacology 47, 342–350. [DOI] [PubMed] [Google Scholar]
- Spooren W. P. J. M.; Vassout A.; Neijt H. C.; Kuhn R.; Gasparini F.; Roux S.; Porsolt R. D.; Gentsch C. (2000) Anxiolytic-like effects of the prototypical metabotropic glutamate receptor 5 antagonist 2-methyl-6-(phenylethynyl)pyridine in rodents. J. Pharmacol. Exp. Ther. 295, 1267–1275. [PubMed] [Google Scholar]
- Jensen J.; Lehmann A.; Uvebrant A.; Carlsson A.; Jerndal G.; Nilsson K.; Frisby C.; Blackshaw L. A.; Mattsson J. P. (2005) Transient lower esophageal sphincter relaxations in dogs are inhibited by a metabotropic glutamate receptor 5 antagonist. Eur. J. Pharmacol. 519, 154–157. [DOI] [PubMed] [Google Scholar]
- Frisby C. L.; Mattsson J. P.; Jensen J. M.; Lehmann A.; Dent J.; Blackshaw L. A. (2005) Inhibition of transient lower esophageal sphincter relaxation and gastroesophageal reflux by metabotropic glutamate receptor ligands. Gastroenterology 129, 995–1004. [DOI] [PubMed] [Google Scholar]
- de Vrij F. M. S.; Levenga J.; van der Linde H. C.; Koekkoek S. K.; De Zeeuw C. I.; Nelson D. L.; Oostra B. A.; Willemsen R. (2008) Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol. Dis. 31, 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q. J.; Rammal M.; Tranfaglia M.; Bauchwitz R. P. (2005) Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 49, 1053–1066. [DOI] [PubMed] [Google Scholar]
- McGeehan A. J.; Olive M. F. (2003) The mGluR5 antagonist MPEP reduces the conditioned rewarding effects of cocaine but not other drugs of abuse. Synapse 47, 240–242. [DOI] [PubMed] [Google Scholar]
- Chiamulera C.; Epping-Jordan M. P.; Zocchi A.; Marcon C.; Cottiny C.; Tacconi S.; Corsi M.; Orzi F.; Conquet F. (2001) Reinforcing and locomotor stimulant effects of cocaine are absent in mGluR5 null mutant mice. Nat. Neurosci. 4, 873–874. [DOI] [PubMed] [Google Scholar]
- Martin-Fardon R.; Baptista M. A. S.; Dayas C. V.; Weiss F. (2009) Dissociation of the effects of MTEP [3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]piperidine] on conditioned reinstatement and reinforcement: Comparison between cocaine and a conventional reinforcer. J. Pharmacol. Exp. Ther. 329, 1084–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaresan V.; Yuan M.; Yee J.; Famous K. R.; Anderson S. M.; Schmidt H. D.; Pierce R. C. (2009) Metabotropic glutamate receptor 5 (mGluR5) antagonists attenuate cocaine priming- and cue-induced reinstatement of cocaine seeking. Behav. Brain Res. 202, 238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckstrom P.; Hyytiä P. (2006) Ionotropic and metabotropic glutamate receptor antagonism attenuates cue-induced cocaine seeking. Neuropsychopharmacology 31, 778–786. [DOI] [PubMed] [Google Scholar]
- Iso Y.; Grajkowska E.; Wroblewski J. T.; Davis J.; Goeders N. E.; Johnson K. M.; Sanker S.; Roth B. L.; Tueckmantel W.; Kozikowski A. P. (2006) Synthesis and structure-activity relationships of 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]pyridine analogues as potent, noncompetitive metabotropic glutamate receptor subtype 5 antagonists; search for cocaine medications. J. Med. Chem. 49, 1080–1100. [DOI] [PubMed] [Google Scholar]
- Kenny P. J.; Boutrel B.; Gasparini F.; Koob G. F.; Markou A. (2005) Metabotropic glutamate 5 receptor blockade may attenuate cocaine self-administration by decreasing brain reward function in rats. Psychopharmacology 179, 247–254. [DOI] [PubMed] [Google Scholar]
- Tessari M.; Pilla M.; Andreoli M.; Hutcheson D. M.; Heidbreder C. A. (2004) Antagonism at metabotropic glutamate 5 receptors inhibits nicotine- and cocaine-taking behaviours and prevents nicotine-triggered relapse to nicotine-seeking. Eur. J. Pharmacol. 499, 121–133. [DOI] [PubMed] [Google Scholar]
- Platt D. M.; Rowlett J. K.; Spealman R. D. (2008) Attenuation of cocaine self-administration in squirrel monkeys following repeated administration of the mGluR5 antagonist MPEP: comparison with dizocilpine. Psychopharmacology 200, 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B.; Platt D. M.; Rowlett J. K.; Adewale A. S.; Spealman R. D. (2005) Attenuation of behavioral effects of cocaine by the metabotropic glutamate receptor 5 antagonist 2-methyl-6-(phenylethynyl)-pyridine in squirrel monkeys: Comparison with dizocilpine. J. Pharmacol. Exp. Ther. 312, 1232–1240. [DOI] [PubMed] [Google Scholar]
- Tronci V.; Vronskaya S.; Montgomery N.; Mura D.; Balfour D. J. K. (2010) The effects of the mGluR5 receptor antagonist 6-methyl-2-(phenylethynyl)-pyridine (MPEP) on behavioural responses to nicotine. Psychopharmacology 211, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlinska J.; Bochenski M. (2007) Comparison of the effects of mGluR1 and mGluR5 antagonists on the expression of behavioral sensitization to the locomotor effect of morphine and the morphine withdrawal jumping in mice. Eur. J. Pharmacol. 558, 113–118. [DOI] [PubMed] [Google Scholar]
- Gass J. T.; Osborne M. P. H.; Watson N. L.; Brown J. L.; Olive M. F. (2009) mGluR5 antagonism attenuates methamphetamine reinforcement and prevents reinstatement of methamphetamine-seeking behavior in rats. Neuropsychopharmacology 34, 820–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams C. L.; Short J. L.; Lawrence A. J. (2010) Cue-conditioned alcohol seeking in rats following abstinence: Involvement of metabotropic glutamate 5 receptors. Br. J. Pharmacol. 159, 534–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besheer J.; Grondin J. J. M.; Salling M. C.; Spanos M.; Stevenson R. A.; Hodge C. W. (2009) Interoceptive effects of alcohol require mGlu5 receptor activity in the nucleus accumbens. J. Neurosci. 29, 9582–9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass J. T.; Olive M. F. (2009) Role of protein kinase C epsilon (PKCε) in the reduction of ethanol reinforcement due to mGluR5 antagonism in the nucleus accumbens shell. Psychopharmacology 204, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder J. P.; Spanos M.; Stevenson J. R.; Besheer J.; Salling M.; Hodge C. W. (2008) Cue-induced reinstatement of alcohol-seeking behavior is associated with increased ERK1/2 phosphorylation in specific limbic brain regions: Blockade by the mGluR5 antagonist MPEP. Neuropharmacology 55, 546–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lominac K. D.; Kapasova Z.; Hannun R. A.; Patterson C.; Middaugh L. D.; Szumlinski K. K. (2006) Behavioral and neurochemical interactions between group 1 mGluR antagonists and ethanol: Potential insight into their anti-addictive properties. Drug Alcohol Depend 85, 142–156. [DOI] [PubMed] [Google Scholar]
- Morin N.; Grégoire L.; Gomez-Mancilla B.; Gasparini F.; Di Paolo T. (2010) Effect of the metabotropic glutamate receptor type 5 antagonists MPEP and MTEP in Parkinsonian monkeys. Neuropharmacology 58, 981–986. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W.; Emmitte K. A. (2009) Recent progress in the discovery and development of negative allosteric modulators of mGluR5. Curr. Opin. Drug Discovery Dev. 12, 446–457. [PubMed] [Google Scholar]
- Gasparini F.; Bilbe G.; Gomez-Mancilla B.; Spooren W. (2008) mGluR5 antagonists: Discovery, characterization and drug development. Curr. Opin. Drug Discovery Dev. 11, 655–665. [PubMed] [Google Scholar]
- Jaeschke G.; Wettstein J. G.; Nordquist R. E.; Spooren W. (2008) mGluR5 receptor antagonists and their therapeutic potential. Expert Opin. Ther. Pat. 18, 123–142. [Google Scholar]
- Rocher J.-P. (2010) Discovery of novel series of selective mGluR5 negative allosteric modulators. Metabotropic glutamate receptors: Translation from discovery to clinical trials, New York, NY, February 23, 2010; The New York Academy of Sciences, New York. [Google Scholar]
- Keywood C.; Wakefield M.; Tack J. (2009) A proof-of-concept study evaluating the effect of ADX10059, a metabotropic glutamate receptor-5 negative allosteric modulator, on acid exposure and symptoms in gastroesophageal reflux disease. Gut 58, 1192–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin J. C. A.; Goadsby P. J. (2010) Glutamatergic fine tuning with ADX-10059: A novel therapeutic approach for migraine?. Exp. Opin. Invest. Drugs 19, 555–561. [DOI] [PubMed] [Google Scholar]
- Goadsby P. J., and Keywood C. G. (2009) Investigation of the role of mGluR5 inhibition in migraine: A proof of concept study of ADX10059 in acute treatment of migraine. Abstracts of Papers, 61st annual meeting of the American Academy of Neurology, Seattle, WA, Abstract P06.006, American Academy of Neurology, Saint Paul, MN. [Google Scholar]
- Pecknold J. C.; McClure D. J.; Appeltauer L.; Wrzesinski L.; Allan T. (1982) Treatment of anxiety using fenobam (a nonbenzodiazepine) in a double-blind standard (diazepam) placebo-controlled study. J. Clin. Psychopharmacol. 2, 129–132. [PubMed] [Google Scholar]
- Berry-Kravis E. M.; Hessl D.; Coffey S.; Hervey C.; Schneider A.; Yuhas J.; Hutchinson J.; Snape M.; Tranfaglia M.; Nguyen D. V.; Hagerman R. (2009) A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J. Med. Genet. 46, 266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini F. (2009) mGluR5 antagonists as potential symptomatic therapy for Fragile X Disorder. Abstracts of Papers, 239th ACS national meeting, San Francisco, CA, Abstract MEDI 12031, American Chemical Society: Washingtion, DC. [Google Scholar]
- Berg D., Godau J., Trenkwalder C., Eggert K., Csoti I., Storch A., Gasparini F., Hariry S., Johns D., and Gomez-Mancilla B. (2009) A randomized, controlled trial evaluating AFQ056 in reducing levodopa-induced dyskinesia in patients with Parkinson’s disease. Abstracts of Papers, 13th International Congress of Parkinson’s Disease and Movement Disorders, Paris, France, Abstract LB-05, The Movement Disorder Society: Milwaukee, WI. [Google Scholar]
- Addex Pharmaceuticals: Addex ADX10059 has potential for Parkinson’s disease levodopa induced dyskinesia (PD-LID). Press Release (2009) September 14.
- Addex Pharmaceuticals: Addex ADX48621 positive primate Parkinson’s data. Press Release (2009) November 24.
- Addex Pharmaceuticals: Development of ADX10059 ended for long-term use. Press Release (2009) December 15.
- Harris G.Promise seen in drug for retardation syndrome. (April 30, 2010) New York Times, p A1. [Google Scholar]
- AstraZeneca PLC: AstraZeneca Development Pipeline. Pipeline Summary (2009) July 30.
- F. Hoffman-LaRoche, Ltd.: Roche Pipeline 2010. Pipeline Summary (2010) April 15. [Google Scholar]
- Seaside Therapeutics: Novel therapies for patients with autism and Fragile X Syndrome. Corporate Fact Sheet (2010) Quarter 1.
- Dove P., Granberg K., Isaac M., Någård M., and Slassi A. (2009) 1,2,4-triazole ether derivatives as modulators of mGluR5. WO 2009/054785 A1.
- Bratt E., Granberg K., Isaac M., Någård M., and Slassi A. (2009) Amide linked heteroaromatic derivatives as modulators of mGluR5. WO 2009/054790 A1.
- Isaac M., and Wållberg A. (2009) Amino 1,2,4-triazole derivatives as modulators of mGluR5. WO 2009/054794 A1.
- Wensbo D., Xin T., Stefanac T., Arora J., Edwards L., Isaac M., Slassi A., Stormann T. M., McLeod D. A., Kers A., Malmberg J., Oscarsson K., Gyback H., Johansson M., Minidis A., Waldman M., Yngve U., and Osterwall C.. New compounds. WO 2004/014881 A2.
- Granberg K., and Wållberg A. (2009) 1,2,4-Triazole aryl N-oxides derivatives as modulators of mGluR5. WO 2009/054786 A1.
- Bratt E., Granberg K. (2009) 1,2,3-Triazole pyrrolidine derivatives as modulators of mGluR5. WO 2009/054789 A1.
- Granberg K., Slassi A., Stefanac T., and Wållberg A. (2009) 1,2,4-Triazole carboxylic acid derivatives as modulators of mGluR5. WO 2009/054787 A1.
- Granberg K., and Holm B. (2009) Aminopyridine derivatives as modulators of mGluR5. WO 2009/054792 A1.
- Granberg K., Holm B., and Någård M. (2009) Thiophene 1,2,4-triazole derivatives as modulators of mGluR5. WO 2009/054793 A1.
- Granberg K., and Holm B. (2009) Fused pyrrolidine 1,2,4-triazole derivatives as modulators of mGluR5. WO 2009/054791 A1.
- Hansen T. V.; Wu P.; Fokin V. V. (2005) One-pot copper(I)-catalyzed synthesis of 3,5-disubstituted isoxazoles. J. Org. Chem. 70, 7761–7764. [DOI] [PubMed] [Google Scholar]
- Granberg K., and Holm B. (2010) Sulphide bridged derivatives as modulators of mGluR5. WO 2010/123451 A1.
- Corey E. J.; Bakshi R. K.; Shibata S. (1987) Highly enantioselective borane reduction of ketones catalyzed by chiral oxazaborolidines. Mechanism and synthetic implications. J. Am. Chem. Soc. 109, 5551–5553. [Google Scholar]
- Wágner G.; Wéber C.; Nyéki O.; Nógrádi K.; Bielik A.; Molnár L.; Bobok A.; Horvath A.; Kiss B.; Kolok S.; Nagy J.; Kurkó D.; Gál K.; Greiner I.; Szombathelyi Z.; Keserü G. M.; Domány G. (2010) Hit to lead optimization of disubstituted oxadiazoles and tetrazoles as mGluR5 NAMs. Bioorg. Med. Chem. Lett. 20, 3737–3741. [DOI] [PubMed] [Google Scholar]
- Gasparini F.; Andres H.; Flor P. J.; Heinrich M.; Inderbitzin W.; Lingenhöhl K.; Müller H.; Munk V. C.; Omilusik K.; Stierlin C.; Stoehr N.; Vranesic I.; Kuhn R. (2002) [3H]-M-MPEP, a potent, subtype-selective radioligand for the metabotropic glutamate receptor subtype 5. Bioorg. Med. Chem. Lett. 12, 407–409. [DOI] [PubMed] [Google Scholar]
- Nissink J. W. M. (2009) Simple size-independent measure of ligand efficiency. J. Chem. Inf. Model. 49, 1617. [DOI] [PubMed] [Google Scholar]
- Keserű G. M.; Makara G. M. (2009) The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discovery 8, 203–212. [DOI] [PubMed] [Google Scholar]
- Galambos J.; Wágner G.; Nógrádi K.; Bielik A.; Molnár L.; Bobok A.; Horváth A.; Kiss B.; Kolok S.; Nagy J.; Kurkó; Bakk M. L.; Vastag M.; Sághy K.; Gyertyán I.; Gál K.; Greiner I.; Szombathelyi Z.; Keserű G. M.; Domány G. (2010) Carbamoyloximes as novel non-competitive mGlu5 receptor antagonists. Bioorg. Med. Chem. Lett. 20, 4371–4375. [DOI] [PubMed] [Google Scholar]
- Vogel J. R.; Beer B.; Clody D. E. (1971) A simple and reliable conflict procedure for testing anti-anxiety agents. Psychopharmacology 21, 1–7. [DOI] [PubMed] [Google Scholar]
- Pilla M.; Andreoli M.; Tessari M.; Delle-Fratte S.; Roth A.; Butler S.; Brown F.; Shah P.; Bettini E.; Cavallini P.; Benedetti R.; Minick D.; Smith P.; Tehan B.; D’Alessandro P. D.; Lorthioir O.; Ball C.; Garzya V.; Goodacre C.; Watson S. (2010) The identification of novel orally active mGluR5 antagonist GSK2210875. Bioorg. Med. Chem. Lett. 20, 7521–7524. [DOI] [PubMed] [Google Scholar]
- Ferrari L.; Crestan V.; Sabattini G; Vinco F.; Fontana S.; Gozzi A. (2010) Brain penetration of local anaesthetics in the rat: Implications for experimental neuroscience. J. Neurosci. Methods 186, 143–149. [DOI] [PubMed] [Google Scholar]
- Njung’e K.; Handley S. L. (1991) Evaluation of marble-burying behavior as a model of anxiety. Pharmacol., Biochem. Behav. 38, 63–67. [DOI] [PubMed] [Google Scholar]
- Britton T. C., Dehlinger V., Fivush A. M., Hollinshead S. P., and Vokits B. P. (2009) 3-Indazolyl-4-pyridylisothiazoles. WO2009/123855 A1.
- Dallmann R.; Steinlechner S.; von Hörsten S.; Karl T. (2006) Stress-induced hyperthermia in the rat: Comparison of classical and novel recording methods. Lab. Anim. 40, 186–193. [DOI] [PubMed] [Google Scholar]
- Jimenez H. N., Li G., Doller D., Grenon M., White A. D., Guo M., and Ma G. (2010) Adamantyl diamide derivatives and uses of same. WO 2010/011570 A1.
- Cosford N. D. P.; Roppe J.; Tehrani L.; Schweiger E. J.; Seiders T. J.; Chaudary A.; Rao S.; Varney M. A. (2003) [3H]-Methoxymethyl-MTEP and [3H]-methoxy-PEPy: potent and selective radioligands for the metabotropic glutamate subtype 5 (mGlu5) receptor. Bioorg. Med. Chem. Lett. 13, 351. [DOI] [PubMed] [Google Scholar]
- Moore N. A.; Rees G.; Sanger G.; Tye N. C. (1994) Effects of olanzapine and other antipsychotic agents on responding maintained by a conflict schedule. Behav. Pharmacol. 5, 196–202. [DOI] [PubMed] [Google Scholar]
- Henrich M., Weil T., Müller S., Nagel J., Gravius A., Kauss V., Zemribo R., and Erdmane E. (2009) Pyrazolopyrimidines, a process for their preparation and their use as medicine. WO 2009/095254 A1.
- Danysz W., Dekundy A., Hechenberger M., Henrich M., Jatzke C., Nagel J., Parsons C. G. R., Weil T., Fotins J., Gutcaits A., Kalvinsh I., Zemribo R., and Kauss V. (2008) Pyrazolopyrimidines, a process for their preparation and the use as a medicine. WO 2008/015271 A1.
- Kulkarni S. S.; Newman A. H. (2007) Design and synthesis of novel heterobiaryl amides as metabotropic glutamate receptor subtype 5 antagonists. Bioorg. Med. Chem. Lett. 17, 2074–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni S. S.; Nightingale B.; Dersch C. M.; Rothman R. B.; Newman A. H. (2006) Design and synthesis of noncompetitive metabotropic glutamate receptor subtype 5 antagonists. Bioorg. Med. Chem. Lett. 16, 3371–3375. [DOI] [PubMed] [Google Scholar]
- Kulkarni S. S.; Zou M.-F.; Cao J.; Deschamps J. R.; Rodriguez A. L.; Conn P. J.; Newman A. H. (2009) Structure–activity relationships comparing N-(6-methylpyridin-yl)-substituted aryl amides to 2-methyl-6-(substituted-arylethynyl)pyridines or 2-methyl-4-(substituted-arylethynyl)thiazoles as novel metabotrobic glutamate receptor subtype 5 antagonists. J. Med. Chem. 52, 3563–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni S. S.; Newman A. H. (2007) Discovery of heterobicyclic templates for novel metabotropic glutamate receptor subtype 5 antagonists. Bioorg. Med. Chem. Lett. 17, 2987–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P.; Zou M.-F.; Rodriguez A. L.; Conn P. J.; Newman A. H. (2010) Structure–activity relationships in a novel series of 7-substituted-aryl quinolines and 5-substituted-aryl benzothiazoles at the metabotropic glutamate receptor subtype 5. Bioorg. Med. Chem. 18, 3026–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boice G. N.; Savarin C. G.; Murry J. A.; Conrad K.; Matty L.; Corley E. G.; Smitrovich J. H.; Hughes D. (2004) An efficient synthesis of a highly functionalized 4-arylpiperidine. Tetrahedron 60, 11367–11374. [Google Scholar]
- Milbank J. B. J.; Knauer C. S.; Augelli-Szafran C. E.; Sakkab-Tan A. T.; Lin K. K.; Yamagata K.; Hoffman J. K.; Zhuang N.; Thomas J.; Galatsis P.; Wendt J. A.; Mickelson J. W.; Schwarz R. D.; Kinsora J. J.; Lotarski S. M.; Stakich K.; Gillespie K. K.; Lam W. W.; Mutlib A. E. (2007) Rational design of 7-arylquinolines as non-competitive metabotropic glutamate receptor subtype 5 antagonists. Bioorg. Med. Chem. Lett. 17, 4415–4418. [DOI] [PubMed] [Google Scholar]
- Sharma S.; Rodriguez A. L.; Conn P. J; Lindsley C. W. (2008) Synthesis and SAR of a mGluR5 allosteric partial antagonist lead: Unexpected modulation of pharmacology with slight structural modifications to a 5-(phenylethynyl)pyrimidine scaffold. Bioorg. Med. Chem. Lett. 18, 4098–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A. L.; Nong Y.; Sekaran N. K.; Alagille D.; Tamagnan G. D.; Conn P. J. (2005) A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts as a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol. Pharmacol. 68, 1793–1802. [DOI] [PubMed] [Google Scholar]
- Spanka C.; Glatthar R.; Desrayaud S.; Fendt M.; Orain D.; Troxler T.; Vranesic I. (2010) Piperidyl amides as novel, potent and orally active mGlu5 receptor antagonists with anxiolytic-like activity. Bioorg. Med. Chem. Lett. 20, 184–188. [DOI] [PubMed] [Google Scholar]
- Bouwknecht J. A.; Olivier B.; Paylor R. E. (2007) The stress-induced hyperthermia paradigm as a physiological animal model for anxiety: A review of pharmacological and genetic studies in the mouse. Neurosci. Biobehav. Rev. 31, 41–59. [DOI] [PubMed] [Google Scholar]
- Millan M. J.; Brocco M. (2003) The Vogel conflict test: Procedural aspects, γ-aminobutyric acid, glutamate and monoamines. Eur. J. Pharmacol. 463, 67–96. [DOI] [PubMed] [Google Scholar]
- Davis M.; Falls W. A.; Campeau S.; Kim M. (1993) Fear-potentiated startle: A neural and pharmacological analysis. Behav. Brain Res. 58, 175–198. [DOI] [PubMed] [Google Scholar]
- Davis M. (1986) Pharmacological and anatomical analysis of fear conditioning using the fear-potentiated startle paradigm. Behav. Neurosci. 100, 814–824. [DOI] [PubMed] [Google Scholar]
- Carcache D.; Vranesic I.; Blanz J.; Desrayaud S.; Fendt M.; Glatthar R. (2011) Benzimidazoles as potent and orally active mGlu5 receptor antagonists with an improved PK profile. ACS Med. Chem. Lett. 2, 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintermann S.; Vranesic I.; Allgeier H.; Brülisauer A.; Hoyer D.; Lemaire M.; Moenius T.; Urwyler S.; Whitebread S.; Gasparini F.; Auberson Y. (2007) ABP688, a novel selective and high affinity ligand for the labeling of mGlu5 receptors: Identification, in vitro pharmacology, pharmacokinetic and biodistribution studies. Bioorg. Med. Chem. 15, 903–914. [DOI] [PubMed] [Google Scholar]
- Burdi D. F.; Hunt R.; Fan L.; Hu T.; Wang J.; Guo Z.; Huang Z.; Wu C.; Hardy L.; Detheux M.; Orsini M. A.; Quinton M. S.; Lew R.; Spear K. (2010) Design, synthesis, and structure–activity relationships of novel bicyclic azole-amines as negative allosteric modulators of metabotropic glutamate receptors. J. Med. Chem. 53, 7107–7118. [DOI] [PubMed] [Google Scholar]
- Rodriguez A. L.; Williams R.; Zhou Y.; Lindsley S. R.; Le U.; Grier M. D.; Weaver C. D.; Conn P. J.; Lindsley C. W. (2009) Discovery and SAR of novel mGluR5 non-competitive antagonists not based on an MPEP chemotype. Bioorg. Med. Chem. Lett. 19, 3209–3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts A. S.; Saleh S. A.; Le U.; Rodriguez A. L.; Weaver C. D.; Conn P. J.; Lindsley C. W.; Emmitte K. A. (2009) Discovery and SAR of 6-substituted-4-anilinoquinazolines as non-competitive antagonists of mGlu5. Bioorg. Med. Chem. Lett. 19, 6623–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipe W. D.; Wolkenberg S. E.; Lindsley C. W. (2005) Accelerating lead development by microwave-enhanced medicinal chemistry. Drug Discovery Today: Technol. 2, 155. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Rodriguez A. L.; Williams R.; Weaver C. D.; Conn P. J.; Lindsley C. W. (2009) Synthesis and SAR of novel, non-MPEP chemotype mGluR5 NAMs identified by functional HTS. Bioorg. Med. Chem. Lett. 19, 6502–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A. L.; Grier M. D.; Jones C. K.; Herman E. J.; Kane A. S.; Smith R. L.; Williams R.; Zhou Y.; Marlo J. E.; Days E. L.; Blatt T. N.; Jadhav S.; Menon U. N.; Vinson P. N.; Rook J. M.; Stauffer S. R.; Niswender C. M.; Lindsley C. W.; Weaver C. D.; Conn P. J. (2010) Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol. Pharmacol. 78, 1105–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wagenen B. C., Stormann T. M., Moe S. T., Sheehan S. M., McLeod D. A., Smith D. L., Isaac M. B., Slassi A. (2001) Heteropolycyclic compounds and their use as metabotropic glutamate receptor antagonists. WO 01/12627 A1.
- Felts A. S.; Lindsley S. R.; Lamb J. P.; Rodriguez A. L.; Menon U. N.; Jadhav S.; Jones C. K.; Conn P. J.; Lindsley C. W.; Emmitte K. A. (2010) 3-Cyano-5-fluoro-N-arylbenzamides as negative allosteric modulators of mGlu5: Identification of easily prepared tool compounds with CNS exposure in rats. Bioorg. Med. Chem. Lett. 20, 4390–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]