

Abstract
Glutamate is the major excitatory transmitter in the mammalian central nervous system (CNS), exerting its effects through both ionotropic and metabotropic glutamate receptors. The metabotropic glutamate receptors (mGlus) belong to family C of the G-protein-coupled receptors (GPCRs). The eight mGlus identified to date are classified into three groups based on their structure, preferred signal transduction mechanisms, and pharmacology (group I: mGlu1 and mGlu5; group II: mGlu2 and mGlu3; group III: mGlu4, mGlu6, mGlu7, and mGlu8). Noncompetitive antagonists, also known as negative allosteric modulators (NAMs), of mGlu5 offer potential therapeutic applications in diseases such as pain, anxiety, gastresophageal reflux disease (GERD), Parkinson’s disease (PD), fragile X syndrome, and addiction. The development of structure−activity relationships (SAR) in a (3-cyano-5-fluorophenyl)biaryl series using our functional cell-based assay is described in this communication. Further characterization of a selected compound, 3-fluoro-5-(2-methylbenzo[d]thiazol-5-yl)benzonitrile, in additional cell based assays as well as in vitro assays designed to measure its metabolic stability and protein binding indicated its potential utility as an in vivo tool. Subsequent evaluation of the same compound in a pharmacokinetic study using intraperitoneal dosing in mice showed good exposure in both plasma and brain samples. The compound was efficacious in a mouse marble burying model of anxiety, an assay known to be sensitive to mGlu5 antagonists. A new operant model of addiction termed operant sensation seeking (OSS) was chosen as a second behavioral assay. The compound also proved efficacious in the OSS model and constitutes the first reported example of efficacy with a small molecule mGlu5 NAM in this novel assay.
Keywords: mGlu5, negative allosteric modulator, noncompetitive antagonist, addiction
Glutamate (l-glutamic acid) is the major excitatory transmitter in the mammalian central nervous system, acting through both ionotropic and metabotropic glutamate receptors. The metabotropic glutamate receptors (mGlus) belong to family C (also known as family 3) of the G-protein-coupled receptors (GPCRs). The mGlus are characterized by a seven transmembrane (7TM) α-helical domain that is connected via a cysteine-rich region to a large bilobed extracellular amino-terminal domain. The location of the orthosteric binding site is in the extracellular domain; however, all of the known allosteric binding sites are located in the transmembrane domain. The eight mGlus discovered to date have been classified according to their structure, preferred signal transduction mechanisms, and pharmacology. Group I receptors (mGlu1 and mGlu5) are coupled to Gαq, a process that results in an increase in intracellular calcium. Group II receptors (mGlu2 and mGlu3) and group III receptors (mGlu4, mGlu6, mGlu7, and mGlu8) are coupled to Gαi, which leads to decreases in cyclic adenosine monophosphate (cAMP) levels. Group I receptors are predominately located postsynaptically and typically enhance postsynaptic signaling. In contrast, the group II and III receptors are located presynaptically and typically have inhibitory effects on neurotransmitter release.1,2
A common issue with orthosteric ligands as potential therapeutics has been poor selectivity among the various mGlus due to a highly conserved binding site. A potential solution to such selectivity issues was discovered through the development of allosteric modulators.3,4 An area of study within this field that has garnered significant attention has been the design of small molecule negative allosteric modulators (NAMs), also known as noncompetitive antagonists, of mGlu5.5−8 Most of the early mGlu5 NAM preclinical in vivo work was conducted with two structurally related tool compounds, 2-methyl-6-(phenylethynyl) pyridine (MPEP)9 and 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine (MTEP).10 These compounds have demonstrated efficacy in numerous preclinical models of disease, including pain,11 anxiety,12−16 gastresophageal reflux disease (GERD),17,18 Parkinson’s disease levodopa induced dyskinesia (PD-LID),19 and fragile X syndrome.20,21 Following several years of discovery and development work by many organizations, some positive clinical reports with mGlu5 NAMs have emerged. Addex Pharmaceuticals has disclosed positive data from phase II clinical studies with the mGlu5 NAM ADX10059 in GERD22 and acute migraine.23 FRAXA Research Foundation and Neuropharm have been exploring the potential of the agent fenobam for treating fragile X syndrome, and early results in patients have been encouraging.24 Finally, Novartis has recently reported positive results from a study designed to examine the efficacy, safety, and tolerability of AFQ056 in the management of PD-LID.25 Such clinical validation with mGlu5 antagonists has further increased interest in the target and encouraged continued research in the area.
In addition to the diseases outlined above, extensive work with MPEP and MTEP has established their utility in numerous animal models of drug addiction. An ability to attenuate various cocaine seeking behaviors in mice,26,27 rats,28−33 and squirrel monkeys34,35 with these compounds has been noted. Such a body of evidence provides a compelling case for an mGlu5 NAM as a treatment for cocaine addiction. The intensely addictive properties of cocaine have been well established. Furthermore, the risk of relapse among addicts is high, even after long periods of abstinence. Potentially severe medical complications associated with cocaine abuse include cardiac arrest, seizures, stroke, and coma.36 There are currently no FDA approved medications for the treatment of cocaine addiction, although there are some compounds under investigation in clinical trials.37 In spite of the success observed with MTEP and MPEP in preclinical models of cocaine addiction, examples with other structurally distinct mGlu5 NAMs in addiction models are lacking.
We have been interested in the identification of new chemotypes for the design of mGlu5 noncompetitive antagonists and have recently reported some of the results from this effort.38−40 Our previously described work was based on the development of hits identified using a functional cell-based high-throughput screen of a collection of 160 000 compounds. We have also focused a portion of our mGlu5 NAM effort on rational design approaches and have recently communicated the initial results from that effort.41 One area of interest to us centered on the development of structure−activity relationships (SARs) in a (3-cyano-5-fluorophenyl)biaryl series and is the subject of this communication. An interesting new compound has emerged from this effort, 3-fluoro-5-(2-methylbenzo[d]thiazol-5-yl)benzonitrile. Herein we describe the profile of this compound in multiple cell based assays as well as in vitro assays designed to measure its metabolic stability and protein binding. Exposure of the compound in both plasma and brain samples following intraperitoneal dosing in mice indicated the suitability of the molecule for use as an in vivo tool compound. The compound was evaluated in a mouse marble burying assay, since that model has been established as a useful tool for the assessment of mGlu5 NAM activity. A new operant model of addiction termed operant sensation seeking (OSS) was chosen as a second behavioral assay. The compound also proved efficacious in the OSS model and constitutes the first reported example of efficacy with a small molecule mGlu5 NAM in this novel assay.
Results and Discussion
An examination of some of the primary literature describing the SARs of various mGlu5 NAM chemotypes revealed some common structural features.42−45 One such feature was the presence of a 3-cyano-5-fluorophenyl ring in several of the most potent analogues across multiple chemical series (Figure 1). We developed a chemical plan in order to build around this common structural motif. Significant effort has been detailed by the referenced research groups around the phenyl portion of their respective templates. Our plan was to hold this phenyl ring constant with 3-cyano-5-fluoro substituents and to prepare new aryl and heteroaryl groups at position one. We had successfully employed a similar approach in the design of 3-cyano-5-fluoro-N-arylbenzamides mGlu5 antagonists.41 One of the advantages of such an approach was that analogues could be prepared in a single step, in this case through the Suzuki coupling of commercially available 3-cyano-5-fluorophenylboronic acid with readily available aryl and heteroaryl halides (Scheme 1). All syntheses were carried out using one of three different reaction conditions using microwave-assisted organic synthesis (MAOS).46 MAOS in combination with a high-throughput preparative LC/MS system allowed for the rapid purification of compounds and generation of timely SAR.47
Figure 1.

3-Cyano-5-fluorophenyl ring containing noncompetitive antagonists of mGlu5.
Scheme 1.

(i) Pd(tBu3)2, 1 M Cs2CO3/THF (1:1), μw, 150 °C, 10 min. (ii) Pd(PPh3)4, 1 M Cs2CO3/THF (1:1), μw, 120 °C, 20 min. (iii) PdCl2(dppf), 1 M Na2CO3/DMF (1:3), μw, 140 °C, 20 min.
One of the areas of obvious interest was 6,6-fused heterocycles, and we thus prepared 10 examples within this class of compounds (Table 1). 2-Methyl-7-arylquinoline 3 was described as a potent mGlu5 NAM in the literature44 and was very potent in our functional assay as well. Our cell-based assay measures the ability of the compound to block the mobilization of calcium by an EC80 concentration of glutamate in HEK293A cells expressing rat mGlu5. The 2-methyl substituent in 3 proved a significant boost to activity as evidenced by the 19-fold drop in potency seen with unsubstituted quinoline 5. 1,8-Naphthyridine 6 was similar in potency to 5. SAR around mGlu5 antagonism for a series of 1,8-naphthyridines was previously described in the literature,48 although 6 was not reported in that publication. While introduction of a nitrogen atom into chemotype 5 to afford 6 was well tolerated, a similar modification to provide quinoxaline 7 resulted in a compound inactive up to the highest concentration tested (30 μM). Quinoxaline 8 demonstrated only moderate potency; however, the compound was unique among this set of compounds in that its activity could be classified as a partial antagonist. Partial mGlu5 antagonists have been reported and well characterized in the MPEP chemotype.49,50 Such molecules only partially block the response to glutamate, even with increasing concentrations of the antagonist. Compound 8 appears to fall into the partial antagonist class of inhibitors, with a maximum antagonism of 80%. Introduction of a methyl substituent at the 2-position of 8 to afford analogue 9, a modification that likely has substantial effects on the conformation of the biaryl, reduced potency. Quinazolinone derivatives 10 and 11 were inactive and weak antagonists, respectively. Finally, in order to evaluate saturated analogues of quinoline 5, we prepared tetrahydroquinoline 12 and benzoxazine 13. Compound 12 was inactive up to 30 μM, while compound 13 was only a weak antagonist.
Table 1. SAR of 6,6-Fused Ring Heterocycles.
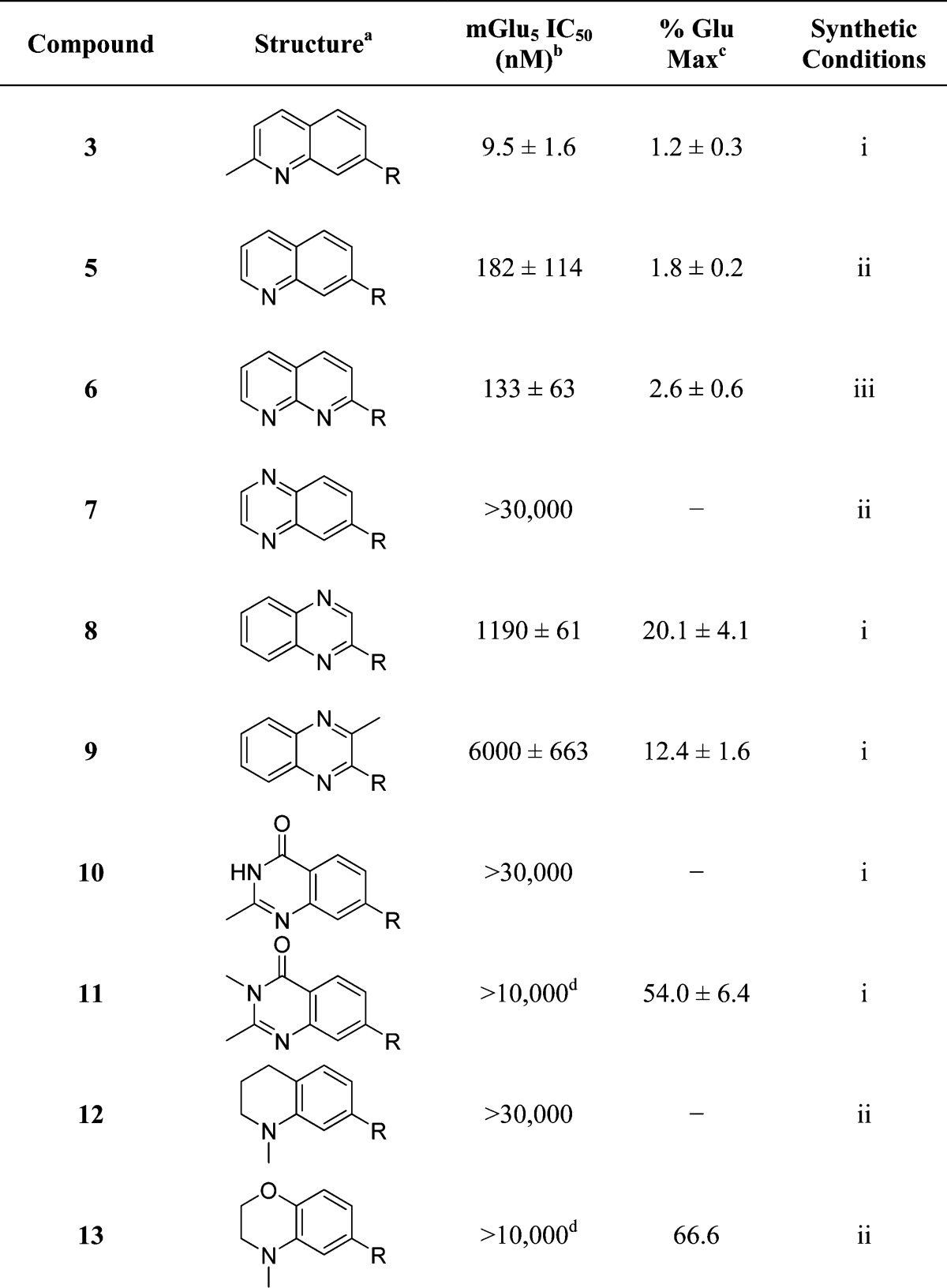 |
R = 3-Cyano-5-fluorophenyl.
Calcium mobilization mGlu5 assay; values are average of n ≥ 3.
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); values are average of n ≥ 3.
CRC does not plateau.
The second area of interest was 5,6-fused heterocycles, and we prepared several new analogues within this class of compounds (Table 2). Benzoxazole 14 was inactive up to 30 μM; however, modification of the 2-substituent from methyl to ethyl afforded 15, a partial antagonist with moderate potency. Further modification of this group to cyclopropyl gave weak antagonist 16. Benzthiazole 17 lacked activity up to 30 μM; however, introduction of a methyl group at the 2-position afforded compound 18, which was quite potent. The des-fluoro analogue of 18 was prepared and tested previously by the NIDA research group;51 however, it demonstrated only moderate affinity (Ki = 2.1 μM) in their radioligand binding assay. Benzoxadiazole 19 and benzthiadiazole 20 both were inactive up to the highest concentration tested. Indazole 21 was a weak antagonist that was subsequently methylated under standard conditions. The resultant regioisomers 22 and 23 were readily separable by flash chromatography. While compound 22 was devoid of activity, compound 23 demonstrated moderate potency.
Table 2. SAR of 5,6-Fused Ring Heterocycles.
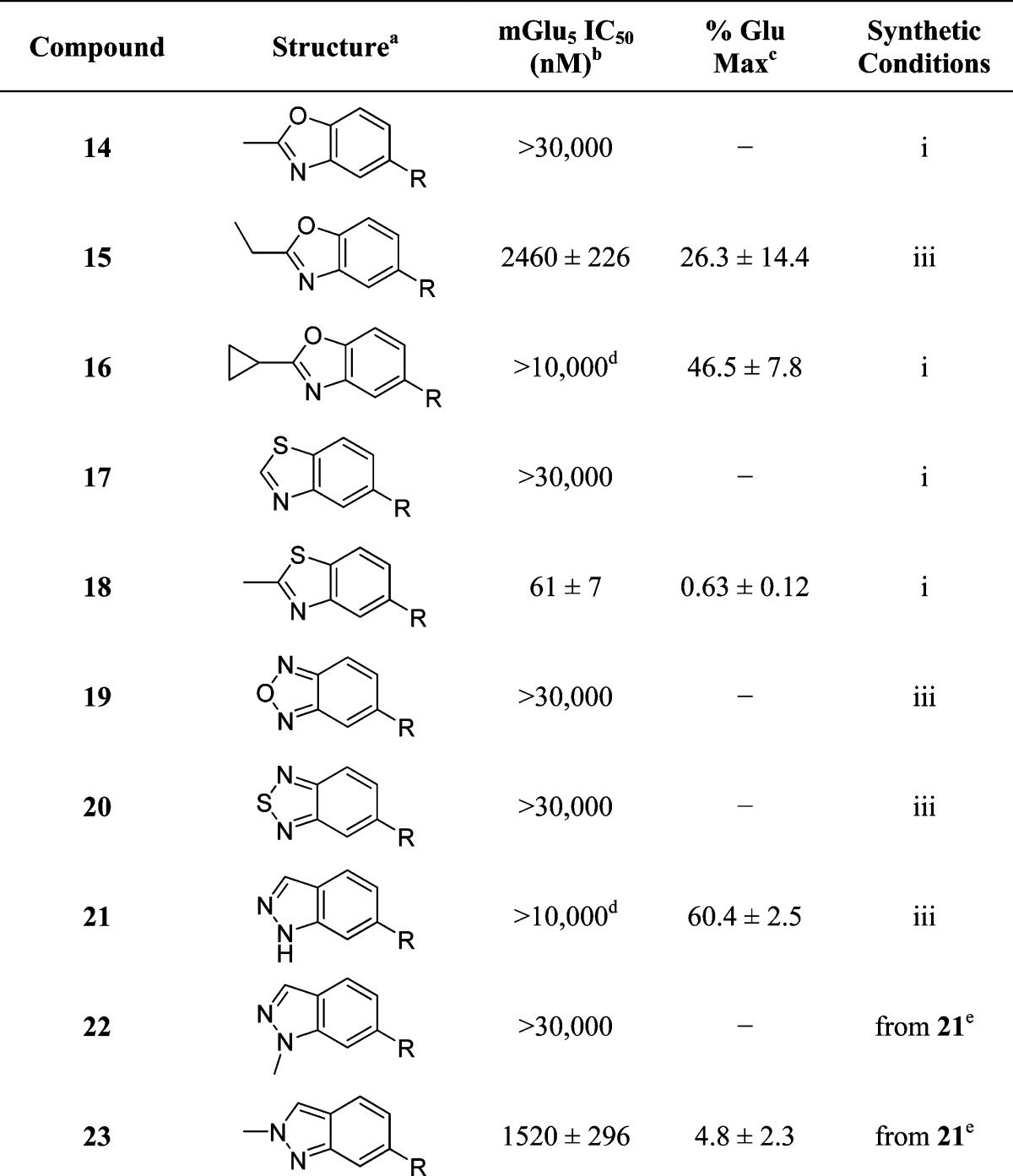 |
R = 3-Cyano-5-fluorophenyl.
Calcium mobilization mGlu5 assay; values are average of n ≥ 3.
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); values are average of n ≥ 3.
CRC does not plateau.
Reaction of 21 with K2CO3 and MeI in DMF afforded a separable mixture of 22 and 23
As a supplement to the 5,6-fused heterocycles described previously, we also prepared several analogues around a benzimidazole scaffold (Table 3). While the unsubstituted analogue 24 lacked activity at 30 μM, as we have seen before, installation of a methyl substituent at either the 1- or 2-position improved activity, affording weak antagonists 25 and 26. Like compound 24, imidazopyridine 27 was inactive up to 30 μM; however, in this case, addition of a 2-methyl substituent in the form of analogue 28 provided no potency improvement. On the other hand, addition of a 3-methyl substituent gave analogue 29, which possessed good potency. Such dramatic potency changes due to subtle or minor structural modifications are typical of allosteric modulators of mGlu5 and other GPCR targets. Interestingly, dimethyl analogue 30 was inactive up to 30 μM, indicating that the 2-methyl substituent is actually not tolerated in the case of these imidazopyridine analogues.
Table 3. SAR of Benzimidazole Analogues.
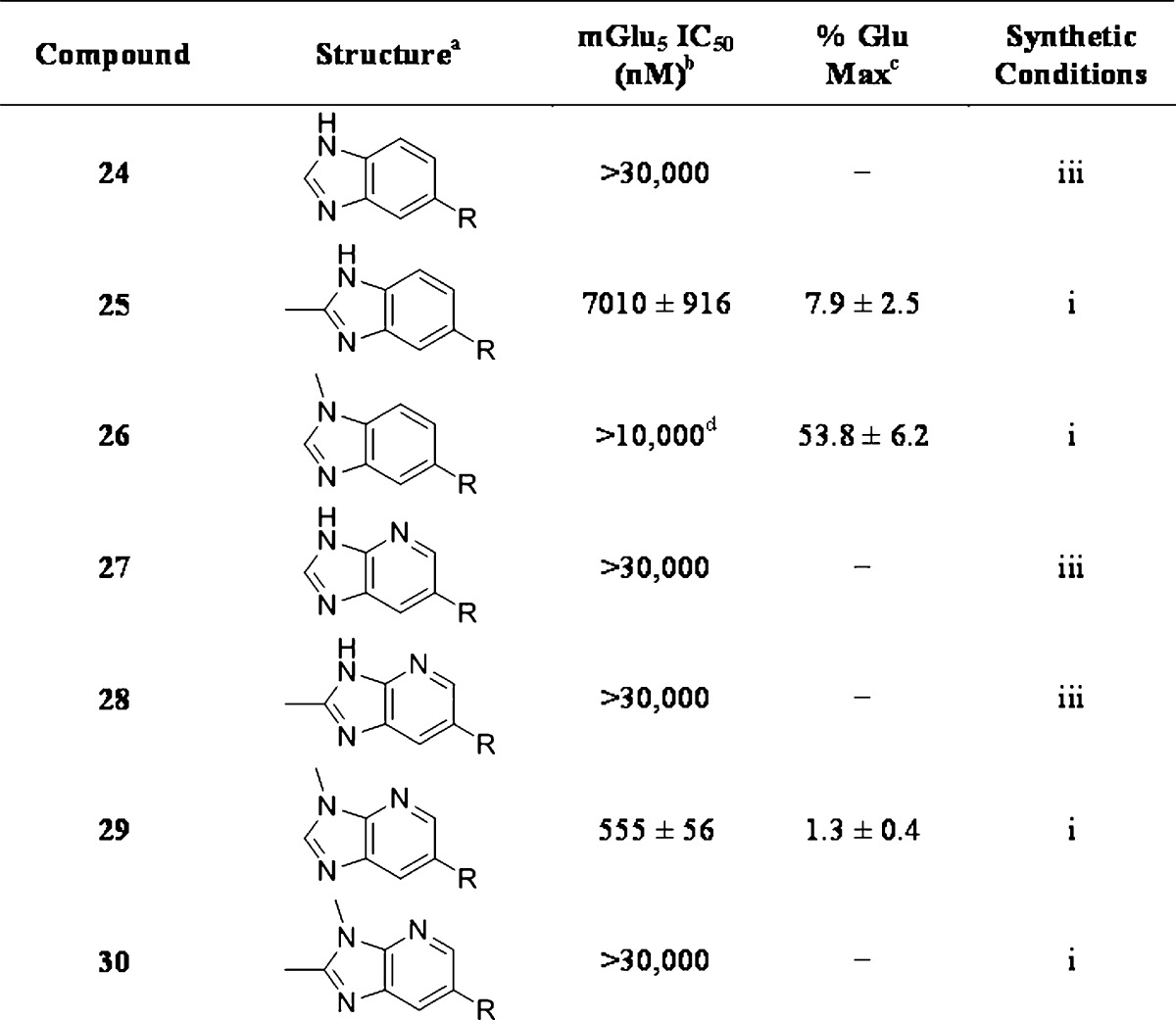 |
R = 3-Cyano-5-fluorophenyl.
Calcium mobilization mGlu5 assay; values are average of n ≥ 3.
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); values are average of n ≥ 3.
CRC does not plateau.
Having identified a new, potent noncompetitive antagonist of mGlu5 in the form of benzthiazole 18, we decided to further profile this compound. A binding affinity determination measuring the ability of the compound to compete with the equilibrium of [3H]3-methoxy-5-(pyridin-2-ylethynyl)pyridine,52 a close structural analogue of MPEP, confirmed the interaction of 18 with the known mGlu5 allosteric binding site (Figure 2A). The Ki value for binding was approximately 8-fold less than the potency in the functional assay; however, at the highest concentration, the molecule is able to almost completely block the binding of the radioligand. The results of the binding assay may indicate that although compound 18 clearly interacts with portions of the MPEP binding site, the binding sites for the two molecules may not be identical. Rat cortical astrocytes have been reported to predominantly express mGlu5 and offer an attractive native system for the characterization of modulators of this receptor.53 As such, we decided to examine the effect of a fixed concentration (10 μM) of 18 on the response to increasing concentrations of glutamate in these cultured cells. Not surprisingly, we observed a near complete blockade of the glutamate response under these conditions (Figure 2B). Compound 18 was also examined in cell based functional assays for its selectivity versus additional mGlus and was determined to be inactive against mGlu1−4 and mGlu7−8.
Figure 2.

Radioligand binding and blockade of glutamate response in rat cortical astrocytes. (A) Compound 18 potently inhibits binding of [3H]3-methoxy-5-(pyridin-2-ylethynyl)pyridine. (B) 10 μM of compound 18 produces a near complete blockade of the response to glutamate in rat cortical astrocytes.
We were interested in evaluating the potential utility of compound 18 for use as an in vivo tool compound and therefore determined its stability in mouse and human liver microsomes (Table 4). The metabolic stability in both species was rather poor, indicating that a dosing route other than oral would likely be necessary in order to avoid a high first pass metabolism. The compound was also evaluated for the degree to which it is bound to relevant proteins. Binding to both mouse and human plasma proteins was similarly high. Additionally, the compound was highly bound to mouse brain homogenates. Being highly bound to protein can potentially limit the ability of the free drug available to interact with the receptor. Although this was obviously a concern with 18, it was not viewed as prohibitive to its further progression as its exposure in the brain would ultimately determine its utility. We next evaluated 18 in a mouse pharmacokinetic study (10 mg/kg) using intraperitoneal dosing (Figure 3). Exposure in the systemic plasma (AUC0−6 h = 801 ng·h/mL) was good, and exposure in the brain (AUC0−6 h = 1530 ng·h/g) was nearly 2-fold higher than that in plasma. While the maximum concentration in the brain was achieved at the initial time point (15 min) and clearance was rapid, exposure remained above 500 ng/g until the 1 h time point. Such a profile indicated that the molecule would most likely only be useful in an in vivo assay over that time frame.
Table 4. In Vitro DMPK Profile of 18.
| metabolic stability in liver microsomes | |
|---|---|
| species | % parent remaining |
| mouse | 18 |
| human | 20 |
| protein binding | |
|---|---|
| sample | % bound |
| mouse brain homogenate | 99.9 |
| mouse plasma | 99.5 |
| human plasma | 99.4 |
Figure 3.
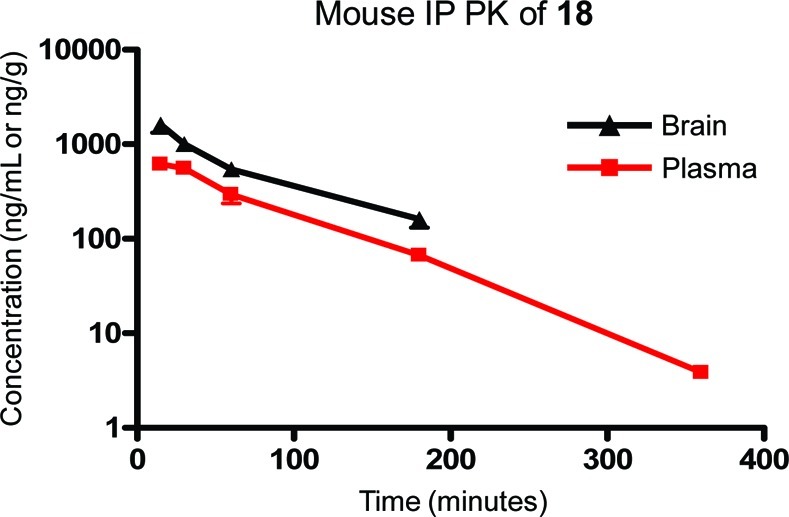
Pharmacokinetics of 18 following intraperitoneal dosing in mice demonstrated good exposure in the brain out to 1 h.
Prior to evaluation of a new tool compound such as 18 in a novel behavioral assay, it was desirable to ensure its effectiveness in an assay known to be sensitive to other mGlu5 antagonists. It is well-known that mice will bury foreign objects such as glass marbles in deep bedding.54 Low doses of anxiolytic benzodiazepines have been demonstrated to inhibit this behavior.55,56 Moreover, the known mGlu5 NAMs MPEP and fenobam are effective in this model.15,16 These facts along with the relative convenience of this assay make it a useful in vivo screening tool. We examined both compound 18 as well as MTEP (positive control) in this assay using a 15 min pretreatment with both compounds (Figure 4). The 15 mg/kg dose of MTEP produced a significant effect as expected. Significant inhibition of marble burying was also observed with 18 at 30 mg/kg. Evaluation of these results in the context of the prior pharmacokinetic study was considered potentially useful. In the pharmacokinetic study, the average brain concentration of 18 at 30 min post dose was 3.75 μM. The marble burying assay was conducted between the 15 and 45 min time points post dose so the 30 min brain concentration is a relevant concentration to consider. If one assumed a dose linear increase in exposure, a 30 mg/kg dose should lead to brain exposures in excess of 10 μM. In considering the reasons that these relatively high brain concentrations of 18 are required for efficacy, one explanation may lie in the highly bound nature of the compound, which restricts the availability of free drug to engage the receptor.
Figure 4.

Inhibition of marble burying by compound 18 in mice.
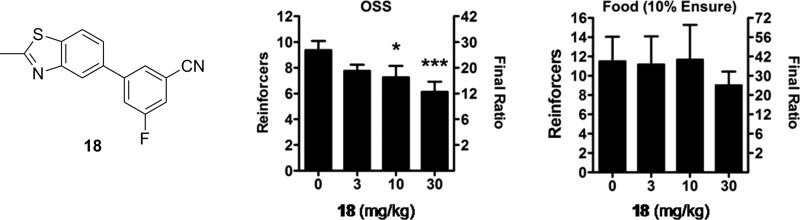
The association between novelty and sensation seeking with elevated drug intake has been documented in humans57,58 as well as rodents,59 which suggests overlap in the pathways mediating such behaviors. Novel stimuli and drugs of abuse both have been shown to increase dopamine levels in the nucleus accumbens shell but not in the core.60 In contrast, natural reinforcers such as food only increase dopamine within the core.61 A number of operant models for the study of addiction have been described that measure the ability of various reinforcers to influence behavior.62 A new model was recently described demonstrating that C57Bl/6J mice readily acquired operant responding to varied visual and auditory stimuli without prior training, a phenomenon termed operant sensation seeking (OSS).63,64 In this assay, mice will “self-administer” visual cues in the form of flashing lights of random duration in combination with an auditory stimulus. Disruption of dopamine signaling with low doses of the dopamine antagonist cis-flupenthixol increased responding for OSS stimuli, similar to effects seen with cocaine self-administration.65 Moreover, knockout mice lacking the D1 dopamine receptor failed to acquire operant responding to OSS stimuli,63 while acquisition of operant behavior with food has been noted.66 Mice lacking mGlu5 also fail to acquire OSS despite having normal acquisition of food self-administration,67 suggesting that OSS can be used as an in vivo screening tool for mGlu5 antagonism. A previous report found that mGlu5 knockout mice did not self-administer cocaine,68 providing further evidence that the reinforcing effects of OSS may be more similar to psychostimulants than food. We were interested in examining the effects of the mGlu5 NAM 18 in this model, as a small molecule antagonist of this receptor had yet to be examined in this assay. Compound 18 was found to dose dependently reduce progressive ratio responding for OSS stimuli; however, there was no significant effect observed with food reinforcer (Figure 5). When the experiments were repeated using the known mGlu5 NAM MTEP, we found the same effect, that OSS was dose-dependently reduced while food self-administration was not (Figure 6). Results with MTEP reinforce the conclusion that the observed effects are due to antagonism of mGlu5. These results also indicate that OSS may be an additional useful model for screening small molecule antagonists of mGlu5, particularly for their evaluation as a treatment for drug addiction.
Figure 5.

Dose dependent reduction of progressive ratio responding for OSS stimuli, but not for food, by compound 18 in mice. OSS, n = 8; food, n = 6; *p < 0.05; ***p < 0.001.
Figure 6.

Dose dependent reduction of progressive ratio responding for OSS stimuli, but not for food, by MTEP in mice. OSS, n = 8; Food, n = 8; *p < 0.05.
In summary, we have discovered and characterized a new mGlu5 NAM tool compound using a rational drug design approach based on common features of known antagonists. Compound 18 potently inhibited the mobilization of calcium by an EC80 concentration of glutamate in HEK293A cells expressing rat mGlu5. A 10 μM concentration of 18 resulted in a near complete blockade of the glutamate response in rat cortical astrocytes. Its interaction with the known allosteric binding site was also confirmed with a radioligand binding assay. In spite of a relatively low stability in mouse liver microsomes and a high level of protein binding, exposure of 18 in mouse brains was supportive of further in vivo studies. Efficacy was observed in a marble burying model of anxiety as well as an operant model of addiction. While multiple mGlu5 NAM compounds have previously been shown to inhibit marble burying, the experiments detailed herein with compound 18 and the well-known tool MTEP constitute the first mGlu5 NAMs reported to be efficacious in the OSS model. Compound 18 is an attractive tool compound, as it can be readily prepared in a single step synthesis from commercially available starting materials. Further studies with 18 will be reported in the near future.
Methods
Synthesis and Characterization of 3-Fluoro-5-(2-methylbenzo[d]thiazol-5-yl)benzonitrile
To 10 separate microwave vials were added 5-bromo-2-methylbenzo[d]thiazole (0.250 g, 1.10 mmol), bis(tri-t-butylphosphine)palladium(0) (0.0560 g, 0.110 mmol), and 3-cyano-5-fluorophenylboronic acid (0.181 g, 1.10 mmo1) each. Aqueous cesium carbonate solution (1 M, 7.5 mL, 7.5 mmol) and tetrahydrofuran (THF; 7.5 mL) were added to each vial. Each reaction vial was microwaved for 10 min at 150 °C. The reaction mixtures were allowed to separate into two layers. Each organic layer (top layer) was removed, and all were combined. The combined organics were filtered through 0.20 μm nylon filters and washed with 5% methanol in dichloromethane (DCM). The filtrate was placed in a separatory funnel and washed with water. The organic layer was dried over MgSO4, filtered, and concentrated in vacuo. The residue was dissolved in 50% methanol in DCM and filtered through a plug of silica gel. The filtrate was diluted with DCM and filtered through filter paper to remove silica gel. The filtrate was concentrated in vacuo. The resultant solid was recrystallized from methanol to afford 1.50 g (51%) of the desired product. Prior to use in vivo, the particle size of the material was reduced using a jet mill. 1H NMR (400 MHz, CDCl3) δ 8.14 (s, 1H), 7.96 (d, J = 8.3 Hz, 1H), 7.76 (s, 1H), 7.61 (d, J = 9.4 Hz, 1H), 7.55 (dd, J = 8.3, 1.4 Hz, 1H), 7.39 (d, J = 7.6 Hz, 1H), 2.94 (s, 3H). HRMS (ESI) m/z 269.0549 [M + H]+ (269.0549 calculated for C15H10N2SF).
Calcium Mobilization Assay
HEK 293A cells stably expressing mGlu5 were plated in black-walled, clear-bottomed, poly-d-lysine coated 384-well plates (BD Biosciences, San Jose, CA) in 20 μL assay medium (DMEM containing 10% dialyzed FBS, 20 mM HEPES, and 1 mM sodium pyruvate) at a density of 20K cells/well. The cells were grown overnight at 37 °C in the presence of 6% CO2. The next day, medium was removed and the cells incubated with 20 μL of 2 μM Fluo-4, AM (Invitrogen, Carlsbad, CA) prepared as a 2.3 mM stock in DMSO and mixed in a 1:1 ratio with 10% (w/v) pluronic acid F-127 and diluted in assay buffer (Hank’s balanced salt solution, 20 mM HEPES, and 2.5 mM Probenecid (Sigma-Aldrich, St. Louis, MO)) for 45 min at 37 °C. Dye was removed, 20 μL of assay buffer was added, and the plate was incubated for 10 m at room temperature. Ca2+ flux was measured using the Functional Drug Screening System (FDSS6000, Hamamatsu, Japan). Compounds were serially diluted 1:3 into 10 point concentration response curves (30 μM to 1 nM final) and transferred to daughter plates using the Echo acoustic plate reformatter (Labcyte, Sunnyvale, CA). Compounds were diluted into assay buffer to a 2× stock using a Thermo Fisher Combi (Thermo Fisher, Waltham, MA) which was applied to cells at t = 3 s. Cells were incubated with the test compounds for 140 s and then stimulated with an EC20 concentration of glutamate; 74 s later an EC80 concentration of glutamate was added and readings taken for an additional 40 s. Data were collected at 1 Hz. Concentration response curves were generated using a four point logistical equation with XLfit curve fitting software for Excel (IDBS, Guildford, U.K.).
Radioligand Binding Assay
Membranes were prepared from rat mGlu5 HEK293A cells. Compounds were diluted in assay buffer (50 mM Tris/0.9% NaCl, pH 7.4) to a 5× stock, and 100 μL of test compound was added to each well of a 96 deep-well assay plate. Then 300 μL aliquots of membranes diluted in assay buffer (40 μg/well) were added to each well. After that, 100 μL of [3H]methoxyPEPy (2 nM final concentration) was added and the reaction was incubated at room temperature for 1 h with shaking. After the incubation period, the membrane-bound ligand was separated from free ligand by filtration through glass-fiber 96-well filter plates (Unifilter-96, GF/B, PerkinElmer Life and Analytical Sciences, Boston, MA). The contents of each well were transferred simultaneously to the filter plate and washed three to four times with assay buffer using a cell harvester (Brandel Cell Harvester, Brandel Inc., Gaithersburg, MD). An amount of 40 μL of scintillation fluid was added to each well, and the membrane-bound radioactivity determined by scintillation counting (TopCount, PerkinElmer Life and Analytical Sciences). Nonspecific binding was estimated using 5 μM MPEP. Concentration response curves were generated using a four parameter logistical equation in GraphPad Prism (GraphPad Software, Inc., La Jolla, CA).
Rat Cortical Astrocytes Assay
Rat astrocytes (ScienCell, San Diego, CA, cat# R1800) were thawed and plated into poly-l-lysine coated T-75 flasks (500K cells per flask) in astrocyte medium (ScienCell, San Diego, CA, cat# 1801) and grown overnight at 37 °C in the presence of 6% CO2. Astrocyte medium was changed after 16 h and then every other day until cells reached confluence (approximately 1 week after thaw). Cells were plated in black-walled, clear-bottomed, poly-l-lysine hand-coated 384-well tissue culture plates (Greiner Bio-One, Monroe, NC) in 20 μL of assay medium (DMEM containing 10% dialyzed FBS, 20 mM HEPES, and 1 mM sodium pyruvate) at a density of 20K cells/well. Calcium assays were run as described above for HEK293A cells except that astrocytes were instead incubated in the FDSS with the test compounds for 140 s and then stimulated with a range of concentrations of glutamate and readings taken for an additional 40 s.
Stability in Liver Microsomes
The test compounds (1 μM) were incubated for 15 min at 37 °C with shaking, in medium containing human/rodent liver microsomes, phosphate buffer, and the cofactor NADPH. Following incubation, the samples were extracted using ice-cold acetonitrile containing 0.1% formic acid and 50 ng/mL of an internal standard. The extracts were analyzed by means of HPLC/MS/MS using a ThermoFinnigan TSQ Quantum Ultra (Thermo Fisher Scientific, Waltham, MA) mass spectrometer in the positive ion mode, by selective reaction monitoring. The chromatographic separation was achieved on an Acquity UPLC BEH C18 column (1.7 μm; 2.1 × 50 mm) at a flow rate of 0.8 mL/min. A gradient program was used with the mobile phase, combining solvent A (95:5 0.1% formic acid in water/acetonitrile) and solvent B (95:5 acetonitrile/0.1% formic acid in water) as follows: 20% B up to 0.5 min, ramped from 20−100% B by 1 min, and held at 100% B until 2 min. The composition was returned to 20% B by 2.2 min. The total run time was 5 min. The column temperature was maintained at 50 °C. The software Xcalibur version 2.2 was used to control the instrument and collect data. The electrospray ionization source was fitted with a stainless steel capillary (100 μm i.d.). Nitrogen was used as both the sheath gas and the auxiliary gas. The ion transfer tube temperature was maintained at 350 °C. The spray voltage, tube lens voltage, and pressure of sheath gas and auxiliary gas were optimized to achieve maximal response using the test compounds mixing with the mobile phase A (50%) and B (50%) at a flow rate of 0.8 mL/min. Collision-induced dissociation was performed on test compounds and internal standard under 1.5 mTorr of argon. The compounds were optimized individually for their optimal conditions analysis conditions using QuickQuan software (version 2.3). Percent test compound remaining following incubation was calculated based on the amount of compound in the incubated samples compared to similarly prepared unincubated controls.
Plasma Protein and Brain Homogenate Binding Assays
A 96-well rapid equilibrium dialysis (RED) apparatus (Thermo Scientific) was used to determine the free fraction in the blood and brain for compound. Mouse plasma and brain tissues were obtained fresh on the day of the experiment. A total of 6−8 brain tissues were homogenized with PBS to a final composition of 1:3 (w/w) brain/PBS using a sonic dismembrator (Fisher Scientific) in an ice bath. Neat plasma and diluted brain homogenate were spiked with compound at 1000 ng/g concentrations, and 200 μL aliquots (n = 3 replicate determinations) were loaded into the sample chambers of the RED plate. Dialysis versus PBS (350 μL) was carried out for 4 h in a temperature-controlled incubator at 37 °C using a shaker at 130 revolutions/min. At the end of the incubation period, 50 μL aliquots of blood, brain homogenate, or PBS were transferred to a 96 deep-well plate, and the composition in each well was balanced with control fluid, such that the volume of PBS to blood or brain was the same. Sample extraction was performed by the addition of 300 μL of acetonitrile containing an internal standard. Samples were vortex mixed for 5 min and then centrifuged for 10 min and supernatants injected onto LC/MS/MS. The unbound fraction in plasma was determined as the ratio of the peak area in buffer to that in plasma. The unbound fraction in brain was determined as the ratio of the peak area in buffer to that in brain, with correction for dilution factor according to eq 1,69
| 1 |
where D is the dilution factor in brain homogenate and fu (apparent) is the measured free fraction of diluted brain tissue.
Mouse Pharmacokinetic Study
Compound 18 was formulated as a 10% Tween 80 microsuspension in sterile water at the concentration of 0.5 mg/mL and administered intraperitoneally to male CD-1 mice weighing around 30 g at the dose of 10 mg/kg. The volume of administration used was 20 mL/kg. The mice blood and brain samples were collected at 15, 30, 60, 180, and 360 min after dose administration. Animals were euthanized and decapitated, and the brains were removed, thoroughly washed in cold PBS, and immediately frozen on dry ice. Blood (cardiac puncture) was collected in EDTA Vacutainer tubes, and plasma was separated by centrifugation and stored at −80 °C until analysis. Three animals were used for each time point. On the day of analysis, frozen whole brains were weighed and then homogenized in 1:5 (w/w) volumes of ice-cold PBS (pH 7.4). The sample extraction of plasma (100 μL) and brain homogenate (100 μL) was performed by a method based on protein precipitation, using three volumes of cold acetonitrile containing 0.1% formic acid and an internal standard having a final concentration of 50 ng/mL. Extracts were vortex mixed for 5 min followed by centrifugation at 14 000 rpm for 10 min. The supernatants of plasma and brain homogenate extracts were analyzed by means of HPLC/MS/MS, using a ThermoFinnigan TSQ Quantum Ultra (Thermo Fisher Scientific, Waltham, MA) mass spectrometer in positive ion mode. The chromatographic separation was achieved on an Acquity UPLC BEH C18 column (1.7um; 2.1 × 50 mm) at a flow rate of 0.8 mL/min. The gradient program was used with the mobile phase, combining solvent A (95:5 0.1% formic acid in water/acetonitrile) and solvent B (95:5 acetonitrile/0.1% formic acid in water) as follows: 20% B (0.5 min), 20−95% B (0.5 min), 95% B (1 min), 95−20% B (0.2 min), 20% B (2.8 min). The column temperature was set at 50 °C. The software Xcalibur version 2.0 was used to control the instrument and collect data. The electrospray ionization source was fitted with a stainless steel capillary (100 μm i.d.). Nitrogen was used as both the sheath gas and the auxiliary gas. The ion transfer tube temperature was 300 °C. The spray voltage, tube lens voltage, and pressure of sheath gas and auxiliary gas were optimized to achieve maximal response using the test compounds mixing with the mobile phase A (50%) and B (50%) at a flow rate of 0.8 mL/min. Collision-induced dissociation was performed on compound and internal standard under 1.0 mTorr of argon. Selected reaction monitoring was carried out using the transitions from m/z 269.3 to 184.3 for test compound at a collision energy of 36 eV, and m/z 310−223 for internal standard at a collision energy of 25 eV. The calibration curves were constructed and linear response was obtained in the range of 10−2000 ng/mL by spiking known amounts of test compound in blank brain homogenates and plasma. Brain concentrations were corrected for dilution in PBS. The final PK parameters were calculated by noncompartmental analysis using WinNonlin software (version 5.1, Pharsight Inc.).
Marble Burying Experiment
Compounds
The mGlu5 NAMs MTEP and 18 (prepared in-house) were dissolved in 10% Tween 80, vortexed vigorously, heated gently with a Master heat gun (Master Appliance Corp., Racine, WI), and sonicated at 37 °C for 30 min. The pH was checked using 0−14 EMD strips and adjusted to approximately 7. All doses were administered at 10 mL/kg ip.
Five Dose Groups
Vehicle, 15 mg/kg MTEP (positive control), 3 mg/kg 18, 10 mg/kg 18, and 30 mg/kg 18.
Subjects
This study was conducted using male Harlan CD-1 mice (Harlan Sprague−Dawley, Indianapolis, IN), weighing 30−35 g. Subjects were housed in a large colony room under a 12-h light/dark cycle (lights on at 6:00 a.m.) with food and water provided ad libitum. Test sessions were performed between 10:00 a.m. and 4:00 p.m. All dose groups consisted of 12 mice. All experiments were conducted in accordance with the National Institute of Health regulations of animal care covered in Principles of Laboratory Animal Care (National Institutes of Health publication 85−23, revised 1985) and were approved by the Institutional Animal Care and Use Committee.
Procedure
Eight small Plexiglas cages (32 × 17 × 14 cm) were arranged in two rows of four cages on top of a large, round table. Mice were transported from the colony room to the testing room and allowed to habituate for 30 min. Mice were pretreated with a dose of MTEP or 18 for 15 min and individually placed in the cages in which 12 black glass marbles (14 mm diameter) had been evenly distributed (spaced 6.4 cm vertically and 4.25 cm horizontally from each other and the walls of the cage) on top of 2.5 cm Diamond Soft Bedding (Harlan Teklad, Madison, WI). The compound and comparator were evaluated in a counterbalanced design, in which all doses of compounds were tested in each session. Mice receiving the same dose were placed in cages on opposite sides of the table to control for effects of lighting and context. Clear, perforated plastic lids were set on top of each cage, and the amount of marble burying was recorded over a 30 min interval. The mice were then removed from the cages, and the number of buried marbles was counted using the criteria of greater than 2/3 covered by bedding. Each session was videotaped with a Sony MiniDV camcorder equipped with a Sony wide-angle lens mounted on a 1.5 m tripod.
Data Analysis
The data for the dose−response studies were analyzed by a between-group analysis of variance. If there was a main effect of dose, then each dose group was compared with the vehicle control group using a Dunnett’s comparison. The calculations were performed using JMP IN 8 (SAS Institute, Cary, NC) statistical software and graphed using SigmaPlot9 (Sasgua, MA).
Operant Sensation Seeking (OSS) Experiments
Animal Care
Male C57Bl/6J mice (3−5 weeks old) were housed in Vanderbilt Animal Care Facilities in groups of four with lights on from 3:00 p.m. to 3:00 a.m. Food and water was available ad libitum for the duration of the experiments. All procedures were approved by the Animal Care and Use Committee at Vanderbilt University. Experiments took place between 9:00 a.m. and 2:00 p.m. and began after 3 days of handling.
Operant Chambers
Operant sensation seeking (OSS) was performed as previously reported.63,64,67 Operant training chambers are housed inside sound-attenuating cubicles containing an exhaust fan (MED Associates). Chambers (21.6 × 17.8 × 12.7 cm3) are equipped with two levers, one on each side of the right wall as described.70−72 Levers are mounted 2.2 cm above the grid floor, with cue lamps (yellow LEDs) mounted 2 cm above them, and a house lamp mounted on the opposite wall. At the beginning of each session, the house light is illuminated and the exhaust fan is turned on. For OSS, a compound visual/auditory stimulus is presented after completion of the required ratio, while presses on the inactive lever are counted but have no programmed consequence. The stimulus is a presentation of flashing cue lights with random duration of 1, 2, 4, or 8 s and random flash rate of 0.625, 1.25, 2.5, or 5 Hz. Each light flash is randomly on the right or left side of the chamber, and the house light is turned off during the visual stimuli. The auditory stimulus is activation of an infusion pump located within the cubicle (no infusion is made).
Fixed Ratio (FR)
Parallel experiments using separate mice (n = 8 per group) were run using either OSS or food reinforcer. Experiments began with daily 1 h operant sessions without any prior training. OSS mice received varied visual and auditory stimuli as a reinforcer, while food mice received diluted vanilla Ensure (∼40 μL of a 10% solution) as a reinforcer. Experiments began with 10−14 sessions using an FR-1 schedule of reinforcement. Mice that did not meet criteria (≥20 active lever presses, ≥60% lever accuracy for the final two sessions) within the first 14 sessions continued FR-1 training until meeting criteria or until the maximum of 25 sessions had been run. One animal (food reinforced) was excluded for not meeting criteria after 25 sessions.
Progressive Ratio (PR)
After FR training, mice responded for reinforcers on a progressive ratio (PR) in 2 h sessions. The schedule of reinforcement was increased in the following pattern: 1, 2, 4, 6, 9, 12, 16, 20, 25, 30, and so forth.63,67 To facilitate acquisition of PR, only the active lever was available. Mice had five initial PR sessions prior to the drug testing phase of the experiment. During these five sessions, mice were habituated to the injection procedure (20 mL/kg saline, i.p., 10 min prior to sessions).
Testing Phase
Testing of the compound was done on a PR schedule of reinforcement as described above. Compound 18 was prepared and given 10 min prior to operant sessions (vehicle: 10% Tween-80, 20 mL/kg i.p.). Mice received each dose (0, 3, 10, 30 mg/kg) 48 h apart using a within-subjects Latin square design, with two nondrug sessions (saline pretreatment) between each dose. Operant responding (reinforcers earned) following drug treatment was analyzed by one-way repeated measures ANOVA followed by Dunnett’s post hoc tests comparing vehicle to each drug dose. Mice were excluded if they did not earn ≥3 reinforcers following vehicle injection. One mouse (food reinforced group) was excluded for this reason. Testing was done as described above with the exception that all animals received 12 FR sessions prior to the five PR sessions. MTEP-HCl (Ascent Scientific, Princeton, NJ) was prepared and given 10 min prior to operant sessions (vehicle: 0.9% saline, 20 mL/kg i.p.), and mice received each dose (0, 1.5, 5, 15 mg/kg) as described for compound 18. Data were analyzed by one-way repeated measures ANOVA followed by Dunnett’s post hoc tests comparing vehicle to each drug dose. One data point from the final day of experiments was removed due to injury during injection.
Acknowledgments
Matt Mulder, Chris Denicola, and Sichen Chang are thanked for the purification of compounds using the mass-directed HPLC system. The authors thank Daryl Venable for technical support with the mGlu5 cell-based functional assay.
Abbreviations
mGlu, metabotropic glutamate receptor; GPCR, G-protein-coupled receptor; NAM, negative allosteric modulator; GERD, gastroesophageal reflux disease; PD, Parkinson’s disease; 7TM, seven transmembrane; cAMP, cyclic adenosine monophosphate; MPEP, 2-methyl-6-(phenylethynyl) pyridine; MTEP, 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine; PDLID, Parkinson’s disease levodopa induced dyskinesia; MAOS, microwave-assisted organic synthesis; LC, liquid chromatography; MS, mass spectrometry; SAR, structure−activity relationship(s); NIDA, National Institute on Drug Abuse; FDA, Food and Drug Administration; OSS, operant sensation seeking; THF, tetrahydrofuran; DCM, dichloromethane; DMEM, Dulbecco's modified Eagle's medium; FBS, fetal bovine serum; HEPES, N′-2- hydroxyethylpiperazine-N′-2 ethanesulfonic acid; DMSO, dimethylsulfoxide; Tris, tris(hydroxymethyl)aminomethane; methoxyPEPy, 3-methoxy-5-(pyridin-2-ylethynyl)pyridine; FDSS, Functional Drug Screening System; NADPH, nicotinamide adenine dinucleotide phosphate; HPLC, high performance liquid chromatography; RED, rapid equilibrium dialysis; PBS, phosphate buffered saline; Tween 80, polyoxyethylene (20) sorbitan monooleate; EDTA, ethylenediaminetetraacetic acid; PK, pharmacokinetics; LED, light-emitting diode; FR, fixed ratio; PR, progressive ratio; ANOVA, analysis of variance; RMANOVA, repeated measures analysis of variance; dppf, 1,1'-bis(diphenylphosphino)ferrocene.
Author Contributions
K.A.E. and C.W.L. oversaw and designed the chemistry. B.S.B. performed synthetic chemistry work. P.J.C. oversaw and designed the molecular pharmacology experiments. A.L.R. oversaw, designed, and performed the molecular pharmacology experiments. S.B.J. performed the in vivo PK and the mouse plasma protein and brain homogenate binding studies, including the analytical chemistry for these experiments. U.N.M. performed the liver microsomal stability studies and the human plasma protein binding study, including the analytical chemistry for these experiments. C.K.J. oversaw and interpreted the data from the marble burying behavioral study. A.S.K. performed the marble burying behavioral study. D.G.W. and C.M.O. oversaw, designed, performed, and interpreted the data from the OSS study.
C.W.L. receives funding from NIH, NIMH, NIDA, the Alzheimer’s Association, the Michael J. Fox Foundation, Seaside Therapeutics, and Johnson&Johnson. C.K.J. receives funding from NIMH and Tennessee Valley Healthcare System (U.S. Department of Veteran’s Affairs). P.J.C. receives funding from NIH, NIMH, NIDA, the Michael J. Fox Foundation, Seaside Therapeutics, and Johnson&Johnson. C.M.O. receives funding from NIDA. D.G.W. receives funding from NIH, NIDA, NIAAA, and NIMH. K.A.E. receives funding from NIH. The authors thank NIDA (RO1 DA023947-01) and Seaside Therapeutics (VUMC33842) for their support of our programs in the development of noncompetitive antagonist of mGlu5. The authors also thank NIDA (K99 DA026994) for supporting the development and characterization of OSS.
Funding Statement
National Institutes of Health, United States
References
- Schoepp D. D.; Jane D. E.; Monn J. A. (1999) Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology 38, 1431–1476. [DOI] [PubMed] [Google Scholar]
- Conn P. J.; Pin J.-P. (1997) Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 37, 205–237. [DOI] [PubMed] [Google Scholar]
- Ritzén A.; Mathiesen J. M.; Thomsen C. (2005) Molecular pharmacology and therapeutic prospects of metabotropic glutamate receptor allosteric modulators. Basic Clin. Pharmacol. Toxicol. 97, 202–213. [DOI] [PubMed] [Google Scholar]
- Kew J. N. C. (2004) Positive and negative allosteric modulation of metabotropic glutamate receptors: emerging therapeutic potential. Pharmacol. Ther. 104, 233–244. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W.; Emmitte K. A. (2009) Recent progress in the discovery and development of negative allosteric modulators of mGluR5. Curr. Opin. Drug Discovery Dev. 12, 446–457. [PubMed] [Google Scholar]
- Gasparini F.; Bilbe G.; Gomez-Mancilla B.; Spooren W. (2008) mGluR5 antagonists: Discovery, characterization and drug development. Curr. Opin. Drug Discovery Dev. 11, 655–665. [PubMed] [Google Scholar]
- Jaeschke G.; Wettstein J. G.; Nordquist R. E.; Spooren W. (2008) mGlu5 receptor antagonists and their therapeutic potential. Expert. Opin. Ther. Pat. 18, 123–142. [Google Scholar]
- Rodriguez A. L.; Williams R. (2007) Recent progress in the development of allosteric modulators of mGluR5. Curr. Opin. Drug Discovery Dev. 10, 715–722. [PubMed] [Google Scholar]
- Gasparini F.; Lingenhöhl K.; Stoehr N.; Flor P. J.; Heinrich M.; Vranesic I.; Biollaz M.; Allgeier H.; Heckendorn R.; Urwyler S.; Varney M. A.; Johnson E. C.; Hess S. D.; Rao S. P.; Sacaan A. I.; Santori E. M.; Veliocelebi G.; Kuhn R. (1999) Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systematically active mGlu5 receptor antagonist. Neuropharmacology 38, 1493–1503. [DOI] [PubMed] [Google Scholar]
- Cosford N. D.; Tehrani L.; Roppe J.; Schweiger E.; Smith N. D.; Anderson J.; Bristow L.; Brodkin J.; Jiang X.; McDonald I.; Rao S.; Washburn M.; Varney M. A. (2003) 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine: a potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J. Med. Chem. 46, 204–206. [DOI] [PubMed] [Google Scholar]
- Zhu C. Z.; Wilson S. G.; Mikusa J. P.; Wismer C. T.; Gauvin D. M.; Lynch J. J.; Wade C. L.; Decker M. W.; Honore P. (2004) Assessing the role of metabotropic glutamate receptor 5 in multiple nociceptive modalities. Eur. J. Pharmacol. 506, 107–118. [DOI] [PubMed] [Google Scholar]
- Pietraszek M.; Sukhanov I.; Maciejak P.; Szyndler J.; Gravius A.; Wislowska A.; Plaznik A.; Bespalov A. Y.; Danysz W. (2005) Anxiolytic-like effects of mGlu1 and mGlu5 receptor antagonists in rats. Eur. J. Pharmacol. 514, 25–34. [DOI] [PubMed] [Google Scholar]
- Busse C. S.; Brodkin J.; Tattersall D.; Anderson J. J.; Warren N.; Tehrani L.; Bristow L. J.; Varney M. A.; Cosford N. D. P. (2004) The behavioral profile of the potent and selective mGlu5 receptor antagonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]-pyridine (MTEP) in rodent models of anxiety. Neuropsychopharmacology 29, 1971–1979. [DOI] [PubMed] [Google Scholar]
- Klodzinska A.; Tatarczynska E.; Chojnacka-Wójcik E.; Nowak G.; Cosford N. D. P.; Pilc A. (2004) Anxiolytic-like effects of MTEP, a potent and selective mGlu5 receptor agonist does not involve GABAA signaling. Neuropharmacology 47, 342–350. [DOI] [PubMed] [Google Scholar]
- Spooren W. P. J. M.; Vassout A.; Neijt H. C.; Kuhn R.; Gasparini F.; Roux S.; Porsolt R. D.; Gentsch C. (2000) Anxiolytic-like effects of the prototypical metabotropic glutamate receptor 5 antagonist 2-methyl-6-(phenylethynyl)pyridine in rodents. J. Pharmacol. Exp. Ther. 295, 1267–1275. [PubMed] [Google Scholar]
- Nicolas L. B.; Kolb Y.; Prinssen E. P. M. (2006) A combined marble burying-locomoter activity test in mice: A practical screening test with sensitivity to different classes of anxiolytics and antidepressants. Eur. J. Pharmacol. 547, 106–115. [DOI] [PubMed] [Google Scholar]
- Jensen J.; Lehmann A.; Uvebrant A.; Carlsson A.; Jerndal G.; Nilsson K.; Frisby C.; Blackshaw L. A.; Mattsson J. P. (2005) Transient lower esophageal sphincter relaxations in dogs are inhibited by a metabotropic glutamate receptor 5 antagonist. Eur. J. Pharmacol. 519, 154–157. [DOI] [PubMed] [Google Scholar]
- Frisby C. L.; Mattsson J. P.; Jensen J. M.; Lehmann A.; Dent J.; Blackshaw L. A. (2005) Inhibition of transient lower esophageal sphincter relaxtation and gastroesophageal reflux by metabotropic glutamate receptor ligands. Gastroenterology 129, 995–1004. [DOI] [PubMed] [Google Scholar]
- Morin N.; Grégoire L.; Gomez-Mancilla B.; Gasparini F.; Di Paolo T. (2010) Effect of the metabotropic glutamate receptor type 5 antagonists MPEP and MTEP in parkinsonian monkeys. Neuropharmacology 58, 981–986. [DOI] [PubMed] [Google Scholar]
- de Vrij F. M. S.; Levenga J.; van der Linde H. C.; Koekkoek S. K.; De Zeeuw C. I.; Nelson D. L.; Oostra B. A.; Willemsen R. (2008) Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol. Dis. 31, 127–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q. J.; Rammal M.; Tranfaglia M.; Bauchwitz R. P. (2005) Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 49, 1053–1066. [DOI] [PubMed] [Google Scholar]
- Keywood C.; Wakefield M.; Tack J. (2009) A proof-of-concept study evaluating the effect of ADX10059, a metabotropic glutamate receptor-5 negative allosteric modulator, on acid exposure and symptoms in gastro-oesophageal reflux disease. Gut 58, 1192–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goadsby P. J., Keywood C. G.Investigation of the role of mGluR5 inhibition in migraine: A proof of concept study of ADX10059 in acute treatment of migraine. Abstracts of Papers, 61st annual meeting of the American Academy of Neurology, Seattle, WA, April 25-May 2, 2009; American Academy of Neurology: Saint Paul, MN, 2009; P06.006. [Google Scholar]
- Berry-Kravis E. M.; Hessl D.; Coffey S.; Hervey C.; Schneider A.; Yuhas J.; Hutchinson J.; Snape M.; Tranfaglia M.; Nguyen D. V.; Hagerman R. (2009) A pilot open label, single dose trial of fenobam in adults with fragile X syndrome. J. Med. Genet. 46, 266–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg D., Godau J., Trenkwalder C., Eggert K., Csoti I., Storch A., Gasparini F., Hariry S., Johns D., Gomez-Mancilla B.. Abstracts of Papers, 13th International Congress of Parkinson’s Disease and Movement Disorders, Paris, France, June 7−11, 2009; The Movement Disorder Society: Milwaukee, WI, 2009; LB-05. [Google Scholar]
- McGeehan A. J.; Olive M. F. (2003) The mGluR5 antagonist MPEP reduces the conditioned rewarding effects of cocaine but not other drugs of abuse. Synapse 47, 240–242. [DOI] [PubMed] [Google Scholar]
- Chiamulera C.; Epping-Jordan M. P.; Zocchi A.; Marcon C.; Cottiny C.; Tacconi S.; Corsi M.; Orzi F.; Conquet F. (2009) Reinforcing and locomoter stimulant effects of cocaine are absent in mGluR5 null mutant mice. Nat. Neurosci. 4, 873–874. [DOI] [PubMed] [Google Scholar]
- Martin-Fardon R.; Baptista M. A. S.; Dayas C. V.; Weiss F. (2009) Dissociation of the effects of MTEP [3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine] on conditioned reinstatement and reinforcement: Comparison between cocaine and a conventional reinforcer. J. Pharmacol. Exp. Ther. 329, 1084–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaresan V.; Yuan M.; Yee J.; Famous K. R.; Anderson S. M.; Schmidt H. D.; Pierce R. C. (2009) Metabotropic glutamate receptor 5 (mGluR5) antagonists attenuate cocaine priming- and cue-induced reinstatement of cocaine seeking. Behav. Brain Res. 202, 238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckstrom P.; Hyytiä P. (2006) Ionotropic and metabotropic glutamate receptor antagonism attenuates cue-induced cocaine seeking. Neuropsychopharmacology 31, 778–786. [DOI] [PubMed] [Google Scholar]
- Iso Y.; Grajkowska E.; Wroblewski J. T.; Davis J.; Goeders N. E.; Johnson K. M.; Sanker S.; Roth B. L.; Tueckmantel W.; Kozikowski A. P. (2006) Synthesis and structure-activity relationships of 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine analogues as potent, noncompetitive metabotropic glutamate receptor subtype 5 antagonists; search for cocaine medications. J. Med. Chem. 49, 1080–1100. [DOI] [PubMed] [Google Scholar]
- Kenny P. J.; Boutrel B.; Gasparini F.; Koob G. F.; Markou A. (2005) Metabotropic glutamate 5 receptor blockade may attenuate cocaine self-administration by decreasing brain reward function in rats. Psychopharmacology 179, 247–254. [DOI] [PubMed] [Google Scholar]
- Tessari M.; Pilla M.; Andreoli M.; Hutcheson D. M.; Heidbreder C. A. (2004) Antagonism at metabotropic glutamate 5 receptor inhibits nicotine- and cocaine-taking behaviours and prevents nicotine-triggered relapse to nicotine-seeking. Eur. J. Pharmacol. 499, 121–133. [DOI] [PubMed] [Google Scholar]
- Platt D. M.; Rowlett J. K.; Spealman R. D. (2008) Attenuation of cocaine self-administration in squirrel monkeys following repeated administration of the mGluR5 antagonist MPEP: comparison with dizocilpine. Psychopharmacology 200, 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B.; Platt D. M.; Rowlett J. K.; Adewale A. S.; Spealman R. D. (2005) Attenuation of behavioral effects of cocaine by the metabotropic glutamate receptor 5 antagonist 2-methyl-6-(phenylethynyl)-pyridine in squirrel monkeys: comparison with dizocilpine. J. Pharmacol. Exp. Ther. 312, 1232–1240. [DOI] [PubMed] [Google Scholar]
- National Institute on Drug Abuse (2009) Research Report Series, Cocaine: Abuse and Addiction, NIH Publication No. 09-4166, U.S. Department of Health and Human Services, National Institute of Health, Rockville, MD. [Google Scholar]
- Karila L.; Gorelick D.; Weinstein A.; Noble F.; Benyamina A.; Coscas S.; Blecha L.; Lowenstein W.; Martinot J. L.; Reynaud M.; Lépine J. P. (2009) New treatments for cocaine dependence: a focused review. Int. J. Neuropsychopharmacology 11, 425–438. [DOI] [PubMed] [Google Scholar]
- Felts A. S.; Saleh S. A.; Le U.; Rodriguez A. L.; Weaver C. D.; Conn P. J.; Lindsley C. W.; Emmitte K. A. (2009) Discovery and SAR of 6-substituted-4-anilinoquinazolines as non-competitive antagonists of mGlu5. Bioorg. Med. Chem. Lett. 19, 6623–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Rodriguez A. L.; Williams R.; Weaver C. D.; Conn P. J.; Lindsley C. W. (2009) Synthesis and SAR of novel, non-MPEP chemotype mGluR5 NAMs identified by functional HTS. Bioorg. Med. Chem. Lett. 19, 6502–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A. L.; Williams R.; Zhou Y.; Lindsley S. R.; Le U.; Grier M. D.; Weaver C. D.; Conn P. J.; Lindsley C. W. (2009) Discovery and SAR of novel mGluR5 non-competitive antagonists not based on an MPEP chemotype. Bioorg. Med. Chem. Lett. 19, 3209–3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts A. S.; Lindsley S. R.; Lamb J. P.; Rodriguez A. L.; Menon U. N.; Jadhav S.; Jones C. K.; Conn P. J.; Lindsley C. W.; Emmitte K. A. (2010) 3-Cyano-5-fluoro-N-arylbenzamides as negative allosteric modulators of mGlu5: Identification of easily prepared tool compounds with CNS exposure in rats. Bioorg. Med. Chem. Lett. 20, 4390–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill T. G.; Krause S.; Ryan C.; Bonnefous C.; Govek S.; Seiders T. J.; Cosford N. D. P.; Roppe J.; Kamenecka T.; Patel S.; Gibson R. E.; Sanabria S.; Riffel K.; Eng W.; King C.; Yang X.; Green M. D.; O’Malley S. S.; Hargreaves R.; Burns H. D. (2005) Synthesis, characterization, and first successful monkey imaging studies of metabotropic monkey imaging studies of metabotropic glutamate receptor subtype 5 (mGluR5) PET radiotracers. Synapse 56, 205–216. [DOI] [PubMed] [Google Scholar]
- Kulkarni S. S.; Zou M.-F.; Cao J.; Deschamps J. R.; Rodriguez A. L.; Conn P. J.; Newman A. H. (2009) Structure-activity relationships comparing N-(6-methylpyridin-yl)-substituted aryl amides to 2-methyl-6-(substituted-arylethynyl)pyridines or 2-methyl-4-(substituted-arylethynyl)thiazoles as novel metabotropic glutamate receptor subtype 5 antagonists. J. Med. Chem. 52, 3563–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milbank J. B. J.; Knauer C. S.; Augelli-Szafran C. E.; Sakkab-Tan A. T.; Lin K. K.; Yamagata K.; Hoffman J. K.; Zhuang N.; Thomas J.; Galatsis P.; Wendt J. A.; Mickelson J. W.; Schwarz R. D.; Kinsora J. J.; Lotarski S. M.; Stakich K.; Gillespie K. K.; Lam W. W.; Mutlib A. E. (2007) Rational design of 7-arylquinolines as non-competitive metabotropic glutamate receptor subtype 5 antagonists. Bioorg. Med. Chem. Lett. 17, 4415–4418. [DOI] [PubMed] [Google Scholar]
- Tehrani L. R.; Smith N. D.; Huang D.; Poon S. F.; Roppe J. R.; Seiders T. J.; Chapman D. F.; Chung J.; Cramer M.; Cosford N. D. P. (2005) 3-[Substituted]-5-(5-pyridin-2-yl-2H-tetrazol-2-yl)benzonitriles: Identification of highly potent and selective metabotropic glutamate subtype 5 receptor antagonists. Bioorg. Med. Chem. Lett. 15, 5061–5064. [DOI] [PubMed] [Google Scholar]
- Shipe W. D.; Wolkenberg S. E.; Lindsley C. W. (2005) Accelerating lead development by microwave-enhanced medicinal chemistry. Drug Discovery Today: Technol. 2, 155–161. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W.; Weaver D.; Jones C.; Marnett L.; Conn P. J. (2007) Preclinical drug discovery research and training at Vanderbilt. ACS Chem. Biol. 2, 17–20. [DOI] [PubMed] [Google Scholar]
- Galatsis P.; Yamagata K.; Wendt J. A.; Connolly C. J.; Mickelson J. W.; Milbank J. B. J.; Bove S. E.; Knauer C. S.; Brooker R. M.; Augelli-Szafran C. E.; Schwarz R. D.; Kinsora J. J.; Kilgore K. S. (2007) Synthesis and SAR comparison of regioisomeric aryl naphthyridines as potent mGlu5 receptor antagonists. Bioorg. Med. Chem. Lett. 17, 6525–6528. [DOI] [PubMed] [Google Scholar]
- Sharma S.; Rodriguez A. L.; Conn P. J.; Lindsley C. W. (2008) Synthesis and SAR of a mGluR5 allosteric partial antagonist lead: Unexpected modulation of pharmacology with slight structural modifications to a 5-(phenylethynyl)pyrimidine scaffold. Bioorg. Med. Chem. Lett. 18, 4098–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A. L.; Nong Y.; Sekaran N. K.; Alagille D.; Tamagnan G. D.; Conn P. J. (2005) A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts as a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol. Pharmacol. 68, 1793–1802. [DOI] [PubMed] [Google Scholar]
- Kulkarni S. S.; Newman A. H. (2007) Discovery of heterocyclic templates for novel metabotropic glutamate receptor subtype 5 antagonists. Bioorg. Med. Chem. Lett. 17, 2987–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosford N. D. P.; Roppe J.; Tehrani L.; Schweiger E. J.; Seiders T. J.; Chaudary A.; Rao S.; Varney M. A. (2003) [3H]-Methoxymethyl-MTEP and [3H]-methoxy-PEPy: potent and selective radioligands for the metabotropic glutamate subtype 5 (mGlu5) receptor. Bioorg. Med. Chem. Lett. 13, 351–354. [DOI] [PubMed] [Google Scholar]
- Peavy R. D.; Sorensen S. D.; Conn P. J. (2002) Differential regulation of metabotropic glutamate receptor 5-mediated phosphoinositide hydrolysis and extracellular signal-regulated kinase responses by protein kinase C in cultured astrocytes. J. Neurochem. 83, 110–118. [DOI] [PubMed] [Google Scholar]
- Deacon R. M. J. (2006) Digging and marble burying in mice: simple methods for in vivo identification of biological impacts. Nat. Protoc. 1, 122–124. [DOI] [PubMed] [Google Scholar]
- Njung’e K.; Handley S. L. (1991) Effects of 5-HT uptake inhibitors, agonists, and antagonists on the burying of harmless objects by mice: a putative test for anxiolytic agents. Br. J. Pharmacol. 104, 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broekkamp C. L.; Rijk H. W.; Joly-Gelouin D.; Lloyd K. L. (1986) Major tranquilizers can be distinguished from minor tranquilizers on the basis of effects on marble burying and swim-induced grooming in mice. Eur. J. Pharmacol. 126, 223–229. [DOI] [PubMed] [Google Scholar]
- Cloninger C. R. (1987) Neurogenetic adaptive mechanisms in alcoholism. Science 236, 410–416. [DOI] [PubMed] [Google Scholar]
- Zuckerman M. (1986) Sensation seeking and the endogenous deficit theory of drug abuse. NIDA Res. Monogr. 74, 59–70. [PubMed] [Google Scholar]
- Piazza P. V.; Deminiere J. M.; Le Moal M.; Simon H. (1989) Factors that predict individual vulnerability to amphetamine self-administration. Science 245, 1511–1513. [DOI] [PubMed] [Google Scholar]
- Rebec G. V.; Christensen J. R.; Guerra C.; Bardo M. T. (1997) Regional and temporal differences in real-time dopamine efflux in the nucleus accumbens during free-choice novelty. Brain Res. 776, 61–67. [DOI] [PubMed] [Google Scholar]
- Mitchell M. B.; Gratton A. (1992) Partial dopamine depletion of the prefrontal cortex leads to enhanced mesolimbic dopamine release elicited by repeated exposure to naturally reinforcing stimuli. J. Neurosci. 12, 3609–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien C. P.; Gardner E. L. (2005) Critical assessment of how to study addiction and its treatment: human and non-human animal models. Pharmacol. Ther. 108, 18–58. [DOI] [PubMed] [Google Scholar]
- Olsen C. M.; Winder D. G. (2009) Operant sensation seeking engages similar neural substrates to operant drug seeking in C57 mice. Neuropsychopharmacology 34, 1685–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen C. M., Winder D. G. (2010) Operant sensation seeking in the mouse. J. Visualized Exp. 45, Nov 10, 2010, DOI: 10.3791/2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettenberg A.; Pettit H. O.; Bloom F. E.; Koob G. F. (1982) Heroin and cocaine intravenous self-administration in rats: mediation by separate neural systems. Psychopharmacology 78, 204–209. [DOI] [PubMed] [Google Scholar]
- Caine S. B.; Thomsen M.; Gabriel K. I.; Berkowitz J. S.; Gold L. H.; Koob G. F.; Tonegawa S.; Zhang J.; Xu M. (2007) Lack of self-administration of cocaine in dopamine D1 receptor knock-out mice. J. Neurosci. 27, 13140–13150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen C. M.; Childs D. S.; Stanwood G. D.; Winder D. G. (2010) Operant sensation seeking requires metabotropic glutamate receptor 5 (mGluR5). PLoS One 5, e15085.; DOI: 10.1371/journal.pone.0015085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiamulera C.; Epping-Jordan M. P.; Zocchi A.; Marcon C.; Cottiny C.; Tacconi S.; Corsi M.; Orzi F.; Conquet F. (2001) Reinforcing and locomoter stimulant effects of cocaine are absent in mGluR5 null mutant mice. Nat. Neurosci. 9, 873–874. [DOI] [PubMed] [Google Scholar]
- Kalvass J. C.; Maurer T. S. (2002) Influence of non-specific brain and plasma bindin on CNS exposure: implications for rational drug discovery. Biopharm. Drug Dispos. 23, 327–338. [DOI] [PubMed] [Google Scholar]
- Olsen C. M.; Winder D. G. (2006) A method for single-session cocaine self-administration in the mouse. Psychopharmacology 187, 13–21. [DOI] [PubMed] [Google Scholar]
- Schramm-Sapyta N. L.; Olsen C. M.; Winder D. G. (2006) Cocaine self-administration reduces excitatory responses in mouse nucleus accumbens shell. Neuropsychopharmacology 31, 1444–1451. [DOI] [PubMed] [Google Scholar]
- Grueter B. A.; Gosnell H. B.; Olsen C. M.; Schramm-Sapyta N. L.; Mathews G. C.; Landreth G. E.; Winder D. G. (2006) Extracellular-signal regulated kinase 1-dependent subtype 5 metabotropic glutamate receptor induced long-term depression in the bed nucleus of the stria terminalis is disrupted by cocaine administration. J. Neurosci. 26, 3210–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]