

The single-strand DNA–binding protein RPA promotes 5′-strand resection to generate 3′ single strands for homology-dependent DNA double-strand repair.
Abstract
Replication protein A (RPA), the eukaryotic single-strand deoxyribonucleic acid (DNA [ss-DNA])–binding protein, is involved in DNA replication, nucleotide damage repair, mismatch repair, and DNA damage checkpoint response, but its function in DNA double-strand break (DSB) repair is poorly understood. We investigated the function of RPA in homology-dependent DSB repair using Xenopus laevis nucleoplasmic extracts as a model system. We found that RPA is required for single-strand annealing, one of the homology-dependent DSB repair pathways. Furthermore, RPA promotes the generation of 3′ single-strand tails (ss-tails) by stimulating both the Xenopus Werner syndrome protein (xWRN)–mediated unwinding of DNA ends and the subsequent Xenopus DNA2 (xDNA2)–mediated degradation of the 5′ ss-tail. Purified xWRN, xDNA2, and RPA are sufficient to carry out the 5′-strand resection of DNA that carries a 3′ ss-tail. These results provide strong biochemical evidence to link RPA to a specific DSB repair pathway and reveal a novel function of RPA in the generation of 3′ ss-DNA for homology-dependent DSB repair.
Introduction
DNA double-strand breaks (DSBs) represent the most deleterious threat to genome stability. If not properly repaired, DSBs often lead to chromosome deletions or translocations and, consequently, premature cell death or oncogenic transformation (Vilenchik and Knudson, 2003). Three major pathways have been identified to repair DSBs: nonhomologous end joining (NHEJ), homologous recombination (HR), and single-strand annealing (SSA; Baumann and West, 1998; Karran, 2000; Pastink et al., 2001). NHEJ usually polishes and then directly joins DNA ends in an error-prone process. HR repairs DSBs by copying the missing information from a homologous sequence, which is usually the sister chromatid in mitotic cells. SSA can repair a break that occurs between two direct repeats, and the final product effectively retains only one of the two repeats. HR and SSA are both homology based and require the processing of DSB ends into 3′ single-strand tails (ss-tails; Symington, 2002). In HR, the 3′ ss-tail invades the homologous chromosome, whereas in SSA the 3′ ss-tails from the two sides of the break anneal with each other.
Although the general scheme of the major DSB repair pathways has been outlined, many fundamental mechanistic questions remain poorly understood. For example, many human disease proteins, such as Brca1 and Brca2, have been implicated in DSB repair, but their exact mechanistic roles are still ambiguous despite intensive research. Another protein of great importance and the focus of this study is replication protein A (RPA), the eukaryotic single-strand DNA (ss-DNA)–binding protein (SSB; Wold, 1997). Through both ss-DNA binding and specific protein–protein interactions, RPA has been shown to participate in DNA replication, nucleotide excision repair, base excision repair, mismatch repair, and the ataxia telangiectasia and Rad3 related (ATR)–mediated checkpoint activation (Fanning et al., 2006). There is also evidence for RPA to function in DSB repair, in particular homology-dependent DSB repair. RPA interacts with recombination protein RAD51 and promotes the coating of RAD51 onto ss-DNA and strand invasion (Golub et al., 1998; Stauffer and Chazin, 2004; Wang and Haber, 2004). It also interacts with RAD52 and promotes the complementary-strand annealing activity and repair center formation of RAD52 and HR (Mortensen et al., 1996; Park et al., 1996; Sung, 1997; Hays et al., 1998; Shinohara et al., 1998; Sugiyama et al., 1998; Plate et al., 2008). Genetic analyses have suggested that RPA participates in homology-dependent repair between direct repeats, gene conversion, and SSA, but the effect can be either stimulatory or suppressive depending on allele and assay (Firmenich et al., 1995; Smith and Rothstein, 1995; Hays et al., 1998; Umezu et al., 1998). Knockdown of RPA by siRNAs in mammalian cells also suggests that RPA plays an important role in homology-dependent DSB repair (Sleeth et al., 2007). However, the fact that RPA participates in so many DNA transactions complicates a rigorous mechanistic dissection of its role in DSB repair.
Like other biological processes, a thorough understanding of DSB repair should benefit greatly from in vitro systems that can reconstitute the various repair pathways. One powerful in vitro system is the extract derived from the eggs of the frog Xenopus laevis. This system can efficiently join various DNA ends via a Ku-dependent NHEJ mechanism (Thode et al., 1990; Labhart, 1999). A derivative extract, prepared from nuclei reconstituted in total egg extracts and, thus, called nucleoplasmic extract (NPE), can efficiently reconstitute SSA (Yan et al., 2005). More recently, we have further explored NPE for studying the processing of double-strand DNA (ds-DNA) ends, which is the first reaction for both SSA and HR (Toczylowski and Yan, 2006). We have found that ds-DNA ends are degraded in an ATP-dependent manner and in the 5′→3′ direction to generate 3′ ss-tails, as expected of homology-dependent DSB repair pathways. A major (but not the only) pathway can be divided into two steps: the ATP-dependent unwinding of ds-DNA ends and the ATP-independent 5′→3′ degradation of ss-DNA tails. Moreover, we have found that the major helicase for end unwinding is the Xenopus Werner syndrome protein (WRN [xWRN]), whereas a major 5′→3′ single-strand exonuclease is the Xenopus DNA2 (xDNA2; Toczylowski and Yan, 2006; Liao et al., 2008; Wawrousek et al., 2010). This mechanism is remarkably similar to the one suggested for the Escherichia coli RecQ helicase and RecJ 5′→3′ ss-DNA exonuclease (Handa et al., 2009). It is also supported by many observations in yeast and mammalian cells. In budding yeast Saccharomyces cerevisiae, the RecQ-type helicase SGS1 and nucleases DNA2 and EXO1 participate in the 5′-strand processing of DSBs (Mimitou and Symington, 2008; Zhu et al., 2008; Budd and Campbell, 2009). In mammalian cells, the Bloom syndrome protein (BLM), another RecQ-type helicase, and EXO1 act in parallel pathways to promote end processing (Gravel et al., 2008). Studies in S. cerevisiae further suggest that these two pathways act downstream of MRX (MRE11-RAD50-XRS2) and Sae2 (Mimitou and Symington, 2008; Zhu et al., 2008). Notably, homologues of MRX (MRN [MRE11-RAD50-NBS1]) and Sae2 (C-terminal–binding protein–interacting protein [CtIP]) are also important for strand resection in mammalian cells (Jazayeri et al., 2005; Sartori et al., 2007; Chen et al., 2008) and Xenopus egg extracts (You et al., 2009; Wawrousek et al., 2010). Collectively, these studies suggest that the mechanism for 5′-strand resection at DSBs is highly conserved in yeast, Xenopus, and mammals.
The helicase activity of xWRN and the 5′→3′ ss-DNA exonuclease activity of xDNA2 are both dramatically stimulated by RPA (Chen et al., 2001; Liao et al., 2008). This raises an interesting question about RPA’s own role in homology-dependent DSB repair and 5′-strand degradation. In this study, we use Xenopus egg extracts and biochemical characterizations of the relevant purified proteins to investigate these questions. We find that RPA is important for the SSA pathway of DSB repair. Moreover, we find that RPA is important for the degradation of the 5′ strand to generate the 3′ ss-tail. At the mechanistic level, RPA stimulates both the xWRN-mediated unwinding of ds-DNA ends and the subsequent xDNA2-mediated 5′→3′ degradation of ss-DNA. Furthermore, purified xWRN, xDNA2, and RPA are sufficient to degrade DNA that carries a 3′ ss-tail. These results not only provide the first biochemical evidence to link RPA to a specific homology-based DSB repair pathway in an in vitro system that fully reconstitutes SSA but also increase our understanding of the mechanism of 5′-strand degradation at DNA ends.
Results
RPA is required for SSA
We first analyzed the effect of RPA on the homology-dependent SSA pathway of DSB repair. SSA can be efficiently reconstituted in NPE with a linear DNA carrying two direct repeats (Yan et al., 2005). Specific anti-RPA antibodies were used to deplete it from NPE to a level below detection (>99% depletion; Fig. 1 A). The substrate, pRW4*, was a 5.6-kb linear DNA with a 1.2-kb Tet gene on each end (and the two cohesive ends were partially filled in with TTP and deoxy-CTP (dCTP) to block the simple religation of cohesive ends; Yan et al., 2005). In mock-depleted NPE, pRW4* was efficiently repaired by SSA (and NHEJ) into a series of products that increased in size over time as expected (Fig. 1 C, lanes 7–9). SSA was mostly an intermolecular reaction, and the major products were a 10-kb linear DNA (Fig. 1 C, arrow) and higher molecular weight bands (Fig. 1 C, bracket). The 10-kb DNA is formed from two pRW4* with one of the two junction repeats effectively deleted. NHEJ gave rise to three products (Fig. 1 C, asterisks), corresponding to supercoiled circular monomers, relaxed circular monomers, and linear dimers. The higher molecular weight products are linear arrays formed by the continued addition of pRW4*, mostly through SSA. As shown in Fig. 1 C (lanes 4–6), RPA depletion caused a dramatic reduction in the formation of the SSA products. The formation of NHEJ products, in contrast, was little affected. To ensure that this effect was specific, we added the purified RPA back to the RPA-depleted NPE to determine whether it can complement the defect. As shown in Fig. 1 C (lanes 1–3), in the presence of the purified RPA, SSA products were again easily detected (the less than complete rescue was largely caused by the technical difficulty in adding a sufficient amount of RPA to the depleted NPE without over diluting it). Together, these data indicate that RPA is indeed required for the SSA pathway of DSB repair in NPE.
Figure 1.

Effect of RPA depletion on SSA. (A) Western blot of RPA- or mock-depleted NPE. RPA was detected with a rat antibody against the large subunit (p70). The standards for quantitation were extracts loaded at the indicated amounts relative to the depleted extracts. (B) Silver staining of the RPA protein purified from Xenopus egg extracts. 0.5 µg of protein was separated by a 12% SDS-PAGE and then detected by silver staining (Bio-Rad Laboratories). The right lane is the protein size marker (Invitrogen). (C) SSA repair in RPA- and mock-depleted NPE. 12 ng/µl of the pRW4* substrate was incubated in RPA- and mock-depleted NPE at room temperature, and the samples taken at the indicated times were treated with SDS-EDTA–proteinase K, separated on a 1% TAE-agarose gel, and detected by SYBR gold staining. The NHEJ products are marked by the asterisks, and the SSA products are marked by the arrow and the bracket. (n) represents multiple repeats.
RPA is important for DNA end processing
What might be the mechanistic role of RPA in SSA? SSA is initiated by the 5′→3′ processing of ends to generate 3′ ss-tails, which are then annealed, and the resulting gaps and flaps are finally repaired. The function of RPA in complementary-strand annealing and DNA synthesis to fill in the gap is well established, so we focused on the potential effect of RPA on the 5′→3′ processing of ends. NPE contains robust activity for the 5′→3′ processing of ds-DNA ends (Toczylowski and Yan, 2006). The DNA substrate for end processing was prepared by linearizing pUC19 plasmid with EcoRI and then filling in the 3′ recessive ends with [32P]dATP and dideoxy-TTP (ddTTP). This DNA could not engage in either SSA because of the lack of homologous sequences at ends or NHEJ because of the block of ends by a dideoxynucleotide (Yan et al., 2005). When incubated in the mock-depleted NPE, the DNA was gradually degraded as judged by both SYBR gold DNA staining and 32P label (Fig. 2 A). However, in RPA-depleted NPE, the DNA was much more stable. Moreover, this defect could be efficiently complemented by the addition of the purified RPA protein (Fig. 2 A). Notably, the purified RPA protein by itself showed no nuclease activity, indicating that the complementation was not caused by the inadvertent introduction of some contaminating nuclease into the RPA-depleted NPE. An unrelated SSB, the phage T4 gp32 protein, also provided some complementation but was much less efficient than RPA in supporting end processing (Fig. 2 B). The molar amount of gp32 used was four times that of RPA and, thus, sufficient to bind approximately the same amount of ss-DNA as RPA could (Chen et al., 2001). Collectively, these data suggest that RPA stimulates the 5′→3′ processing of DNA ends and does so by mechanisms that appear to involve not only the stabilization of ss-DNA by the coating of SSBs but also the physical interactions between RPA and other end processing proteins.
Figure 2.

Effect of RPA on DNA end processing. (A) 4 ng/µl of linear pUC19 DNA labeled by [32P]dATP and blocked by ddTTP at the 3′ end was incubated in RPA-depleted NPE (supplemented with either 0.25 µM RPA or buffer) or mock-depleted NPE. One additional reaction, pUC19 incubated with the RPA protein only, served as a control. Samples were taken at the indicated times, treated with SDS-EDTA–proteinase K, and separated on a 1% TAE-agarose gel. The gel was first stained with SYBR gold to detect total DNA and then dried for exposure to phosphoimager to detect 32P. The percentages of the substrate remaining were relative to the zero time point. (B) Differential activities of RPA and gp32 in supporting DNA end processing in RPA-depleted NPE. The final concentrations for RPA and gp32 were 0.25 and 1 µM, respectively. Molecular markers are given in kilobases.
RPA stimulates the xWRN-mediated unwinding of DNA ends
What might be the mechanistic role for RPA in DNA end processing? We have previously shown that a major (but not the only) end processing pathway in NPE can be divided into at least two steps: the unwinding of ds-DNA ends and the 5′→3′ degradation of the resulting ss-tails (Toczylowski and Yan, 2006). The major helicase for end unwinding is xWRN (Toczylowski and Yan, 2006). RPA is known to interact physically with xWRN and stimulates its helicase activity (Chen et al., 2001), suggesting that it might stimulate the unwinding step of end processing. We tested this hypothesis by using an assay depicted in Fig. 3 A. The substrate for the unwinding assay was a 48-bp oligonucleotide duplex (thio 5′ duplex). One strand carried a biotin moiety at its 5′ end, and the complementary strand carried 24 normal nucleotides in the 5′ half followed by 21 thionucleotides in the 3′ half and two [32P]dATPs near the 3′ end. The DNA was first coated onto Streptavidin magnetic beads and then incubated in either RPA- or mock-depleted NPE. End processing could only proceed from the 5′ end of the thio-containing strand and then stall at the thionucleotides (Toczylowski and Yan, 2006). If there had been no unwinding, the thio strand would have remained annealed to the biotin strand and, thus, bound to the beads. If there had been unwinding, the thio strand would have been released into the supernatant. As expected, in mock-depleted NPE, the partially degraded thio strand was released into the supernatant (Fig. 3 B). In RPA-depleted NPE, in contrast, a very little amount of the thio strand was released into the supernatant. This defect could be rescued by the purified RPA protein (Fig. 3 C). The T4 gp32 protein was, in contrast, much less efficient in supporting end unwinding (Fig. 3 D). These data suggest that RPA is required for efficient stimulation of the xWRN-mediated unwinding of DNA ends.
Figure 3.

Effect of RPA on end unwinding. (A) Unwinding assay. Thin line, normal nucleotides; thick line, thio nucleotides; *, 32P label. (B) The thio 5′ oligonucleotide duplex precoated onto Streptavidin magnetic beads was incubated in RPA- or mock-depleted NPE. Samples were separated into bead and supernatant fractions and analyzed on a 10% native TAE-PAGE. B, beads; S, supernatant. (C) Rescue of the unwinding defect by the purified RPA protein. The thio 5′ oligonucleotide duplex precoated onto Streptavidin magnetic beads was incubated in RPA-depleted NPE supplemented with either 0.25 µM of the purified RPA protein or buffer. After 30-min incubation, the reactions were terminated and analyzed in the same way as in A. The arrowhead indicates the released product. (D) Differential activities of RPA and gp32 in supporting end unwinding. The final concentrations for RPA and gp32 are 0.25 and 1 µM, respectively.
RPA physically interacts with xDNA2 and stimulates the 5′→3′ ss-DNA exonuclease activity of xDNA2
After the unwinding of DNA ends, the 5′ ss-tail is then degraded by the 5′→3′ single-strand exonuclease activity of xDNA2. We have previously shown that the 5′→3′ ss-DNA exonuclease activity of xDNA2 is dramatically stimulated by RPA (Liao et al., 2008). The substrate used in the study, 48mer-1, might form secondary structures, so it is possible that the role of RPA is simply to keep 48mer-1 in a single-strand state. To test this hypothesis, we also designed an oligonucleotide composed mostly of deoxyadenines (dAs; 48mer-5), which was predicted to be incapable of forming stable secondary structures at room temperature (Zuker, 2003). The two single-strand oligonucleotide substrates carried 32P-labeled dA (at positions 46 and 47 in 48mer-1 and at 20 potential positions distributed between position 22 and position 47 in 48mer-5) and bound to Streptavidin paramagnetic beads via the 3′-terminal biotin-deoxycytidine (dC; see Materials and methods). As shown in Fig. 4 A, with 48mer-1, xDNA2’s 5′→3′ nuclease activity was greatly stimulated by RPA, confirming our previous observation (Liao et al., 2008). The degradation stalled 12–17 nt away from beads, presumably because of steric hindrance. 48mer-5 showed slightly more degradation than 48mer-1 in the absence of RPA, suggesting that the lack of secondary structures does improve degradation by the intrinsic nuclease activity of xDNA2. However, in the presence of RPA, xDNA2’s nuclease activity was still greatly stimulated. These data suggest that efficient stimulation of xDNA’s 5′→3′ nuclease activity depends on not just the single strandedness of DNA but also RPA.
Figure 4.

Functional and physical interactions between RPA and xDNA2. (A) The effect of RPA on xDNA2’s 5′→3′ exonuclease activity against two different single-stranded oligonucleotides. The substrates were labeled with 32P-labeled dA (marked by the asterisks) and attached to Streptavidin paramagnetic beads via the 3′ biotin-dC. After incubation at room temperature for 1 h, the reactions were stopped with SDS-EDTA, boiled for 10 min, and separated on a 10% TAE-PAGE. The percentage of the substrate undegraded was relative to the total signal for each reaction. The sizes of the products were determined by separating on a sequencing gel (not depicted). (B) The effect of RPA and T4 gp32 on the nuclease activity of xDNA2. The substrate, 48mer-1 beads, was incubated with various proteins as indicated at room temperature for 1 h and analyzed similarly to that in A. (C) Coimmunoprecipitation of RPA and xDNA2. The immunoprecipitates were separated on an 8% SDS-PAGE, transferred to a polyvinylidene fluoride membrane, and probed for different proteins by Western blotting. For RPA, a rat antibody against the p70 subunit was used for Western blotting. Untreated cytosol was loaded at the indicated amounts to provide the standard for quantitation. White lines indicate that intervening lanes have been spliced out. (D) Interaction between the purified RPA and xDNA2. FLAG beads were precoated with either recombinant xDNA2 or BSA and then incubated with the purified RPA protein. The beads and supernatant fractions were analyzed similarly to that in C. xRPA, Xenopus RPA. Ab, antibody.
We next determined whether stimulation of xDNA2 is specific to RPA or can be accomplished with other unrelated SSBs. As shown in Fig. 4 B, in contrast to RPA, gp32 displayed no stimulatory effect, even at the 250-nM concentration. This dramatic difference between RPA and gp32 suggests that the stimulation of xDNA2’s 5′→3′ exonuclease activity depends on a direct physical interaction between RPA and xDNA2. To test this hypothesis, we performed a coimmunoprecipitation with anti-RPA antibodies. As shown in Fig. 4 C, anti-RPA antibodies brought down not only RPA but also a fraction of xDNA2 and xWRN (∼5%). However, the converse experiment with anti-xDNA2 antibodies did not bring down RPA, probably because the antibodies disrupted the interaction. To more definitively demonstrate that RPA interacts directly with xDNA2, we expressed xDNA2 as a recombinant protein with a FLAG affinity tag at the C terminus. This protein was precoated onto FLAG agarose beads and then incubated with the purified RPA protein. As shown in Fig. 4 D, the xDNA2 beads efficiently brought down RPA. In contrast, the control beads (preincubated with BSA) brought down only trace amounts of RPA. Collectively, these data show that RPA physically interacts with xDNA2 and suggest that this interaction is required to fully stimulate the 5′→3′ ss-DNA exonuclease activity of xDNA2.
RPA is important for the xDNA2-mediated degradation of 5′ ss-DNA tails
Is the stimulation of xDNA2’s ss-DNA exonuclease activity by RPA relevant to the degradation of 5′ ss-DNA tails in NPE? To address this question, we prepared an ss-DNA substrate by heat denaturing the 3′-labeled linear ds-pUC19 DNA. We have previously shown that ss-DNA was degraded in NPE by 5′→3′ ss-DNA exonucleases, which were mainly xDNA2 (Liao et al., 2008). The denatured ss-DNA was incubated in RPA- or mock-depleted NPE and then analyzed by agarose gel electrophoresis. As shown in Fig. 5 A, although the ss-DNA was rapidly degraded in mock-depleted NPE, it was very stable in RPA-depleted NPE. This effect could be complemented by the purified RPA protein. Interestingly, the mobility of DNA was altered after incubation in RPA-depleted NPE in a way suggesting that most of the ss-DNA had been reannealed into ds-DNA, presumably by the strand-annealing proteins in the extract. RPA might thus facilitate ss-DNA degradation simply by preventing the single strands from reannealing. To test this hypothesis, we determined whether the T4 gp32 protein could substitute for RPA in ss-DNA degradation. As shown in Fig. 5 B, gp32 indeed led to a strong inhibition of strand reannealing, but the ss-DNA was degraded much less efficiently than in the control-depleted extract. Collectively, these data indicate that RPA plays an important role in the 5′ ss-DNA degradation step of end processing. They also suggest that RPA does so both by physically stimulating the ss-DNA exonuclease activity of xDNA2 and by preventing the reannealing of ss-DNA strands.
Figure 5.

Effect of RPA on 5′ ss-tail degradation. (A) Denatured pUC19 DNA labeled with 32P at the 3′ end was incubated in RPA- or mock-depleted NPE supplemented with buffer or the purified RPA protein. Samples were collected at the indicated times, treated with SDS-EDTA–proteinase K, and separated on a 1% TAE-agarose gel. Two additional reactions containing ss-DNA incubated with the purified RPA protein or buffer, but no NPE, served as controls. (B) Differential activities of RPA and gp32 in supporting ss-DNA degradation. The final concentrations for RPA and gp32 are 0.25 and 1 µM, respectively.
xWRN, xDNA2, and RPA are sufficient to degrade DNA that carries a 3′ ss-tail
xWRN, xDNA2, and RPA are all required for the degradation of 5′ strands at DNA ends. An important mechanistic question is whether they are also sufficient for this reaction. As a 3′→5′ DNA helicase, xWRN is incapable of initiating DNA unwinding from blunt ends (Fry, 2002). Indeed, when xWRN, xDNA2, and RPA are incubated with blunt-ended linear DNA, only trace amounts of DNA degradation could be detected (Fig. 6). Studies in yeast have suggested that Sgs1 acts downstream of MRE11 and/or Sae2, which appear to process DNA by unknown mechanisms to a limited extent to provide a 3′ ss-tail for the more extensive processing by Sgs1 and DNA2 (Mimitou and Symington, 2008; Zhu et al., 2008). To test this hypothesis, we prepared a DNA substrate with preformed 3′ ss-tails by limited digestion of the blunt-ended DNA with λ exonuclease. When such preprocessed DNA (carrying ∼200–750 nt of 3′ ss-DNA) exonuclease was incubated with xWRN, xDNA2, RPA, and ATP, it was efficiently processed (Fig. 6). All four components were required for this reaction. In particular, RPA could not be replaced by gp32, again confirming that RPA’s activity in end processing depends on its interaction with xWRN and xDNA2. In the absence of xDNA2, the preprocessed DNA was still gradually unwound, but not degraded, by xWRN and RPA. In the absence of RPA, DNA was stable except that the 3′ label was gradually lost, which was consistent with the weak intrinsic 3′→5′ exonuclease of xWNR (Fry, 2002). Together, these results demonstrate that xWRN, xDNA2, and RPA are not only important but also sufficient for the 5′-strand degradation of preprocessed DNA.
Figure 6.

xWNR, xDNA2, and RPA are sufficient to degrade 5′ preprocessed DNA. Purified xWRN, xDNA2, and RPA or gp32 were incubated with blunt-ended DNA or 5′ preprocessed DNA (labeled on the 3′ end) in various combinations in the presence or absence of ATP. Asterisks indicate the 32P label. Samples were taken at the indicated times, treated with SDS-EDTA–proteinase K, and separated on four 1% TAE-agarose gels. The gels were first stained with SYBR gold and then dried for 32P. Reactions from the same gel are indicated by a bounding box, a dashed bounding box, a gray dashed box, or no bounding box. White lines indicate that intervening lanes have been spliced out. Molecular markers shown on the left are given in kilobases.
Discussion
In this study, we use the Xenopus extract system and enzymatic characterizations to investigate the role of RPA in DSB repair. The major findings are that (a) RPA is required for SSA repair; (b) RPA is important for the 5′→3′ strand-specific degradation at DNA ends; (c) RPA interacts with xDNA2 and stimulates its 5′→3′ ss-DNA exonuclease activity; (d) RPA promotes both the xWRN-mediated unwinding of ends and the subsequent xDNA2-mediated degradation of 5′ ss-tails; (e) RPA’s function in 5′-strand processing cannot be fully replaced by the T4 gp32 SSB; and (f) xWRN, xDNA2, and RPA are sufficient to degrade the 5′ strand of a 5′ preprocessed DNA. As the major SSB in eukaryotic cells, RPA participates in probably every DNA transaction that involves ss-DNA. It has thus been difficult to rigorously determine its role in a particular pathway without concerns over indirect effects from defects in other processes, such as cell cycle progression or transcriptional regulation. Our study using the Xenopus system, which does not suffer from this drawback, provides direct biochemical evidence to link RPA to a specific DSB repair pathway. Moreover, it reveals a novel role for RPA in stimulating the 5′→3′ processing of DNA ends to generate 3′ ss-tails.
The processing of ds-DNA ends into 3′ ss-tails is the first step of homology-dependent DSB repair. RPA is important for the 5′ strand–specific degradation of DNA ends and does so by promoting both the unwinding of ends and the degradation of 5′ ss-tails. This is in contrast to xWRN, which is involved exclusively in the end unwinding step (Toczylowski and Yan, 2006) and xDNA2, which is involved exclusively in the 5′ ss-tail degradation step (Liao et al., 2008). RPA’s role in end unwinding is by stimulating the helicase activity of xWRN. It has long been observed that RPA can physically interact with WRN and stimulate its helicase activity, but the biological significance of this interaction has been unclear (Shen et al., 1998; Brosh et al., 1999; Chen et al., 2001). Our observation that RPA stimulates the xWRN-mediated unwinding of DNA ends for homology-dependent DSB repair provides a rationale for this interaction. RPA can also physically interact with xDNA2 and stimulates its 5′→3′ ss-DNA exonuclease activity. Its role in 5′ ss-tail degradation is thus most likely by stimulating the exonuclease activity of xDNA2. In addition to these two active roles mediated by protein–protein interaction, RPA also possesses a passive role in preventing the reannealing of ss-DNA. This passive role can be substituted by an unrelated SSB, such as the T4 gp32 protein, but the two active roles in stimulating the helicase activity of xWNR and the nuclease activity of xDNA2 cannot. The coupled mechanism of 5′-strand resection thus most likely depends on RPA to physically interact and stimulate both the xWNR-mediated end unwinding step and the xDNA2-mediated 5′ ss-tail degradation step. These three proteins form a unique module to catalyze the 5′→3′ processing of DNA ends for homology-dependent DSB repair. Interestingly, the E. coli RecQ helicase and RecJ nuclease are also stimulated by SSBs, implying that SSBs might play similar stimulatory roles in this 5′-strand resection pathway (Harmon and Kowalczykowski, 2001; Han et al., 2006; Shereda et al., 2007). Similarly, BLM, another RecQ helicase that has recently been shown to participate in end processing, is also stimulated by RPA (Brosh et al., 2000). Thus, using a module consisting of a RecQ-type helicase, a 5′→3′ ss-DNA exonuclease, and an SSB might be a highly conserved mechanism for the 5′-strand degradation of DNA ends (Fig. 7). The relative contribution of WRN and BLM (or other members of the RecQ helicase family) in higher eukaryotes to 5′-strand resection is likely determined by the expression level of each protein. Consistent with this idea, xWRN, which is the dominant RecQ helicase for end processing in NPE, is over five times more abundant than Xenopus BLM (unpublished data).
Figure 7.
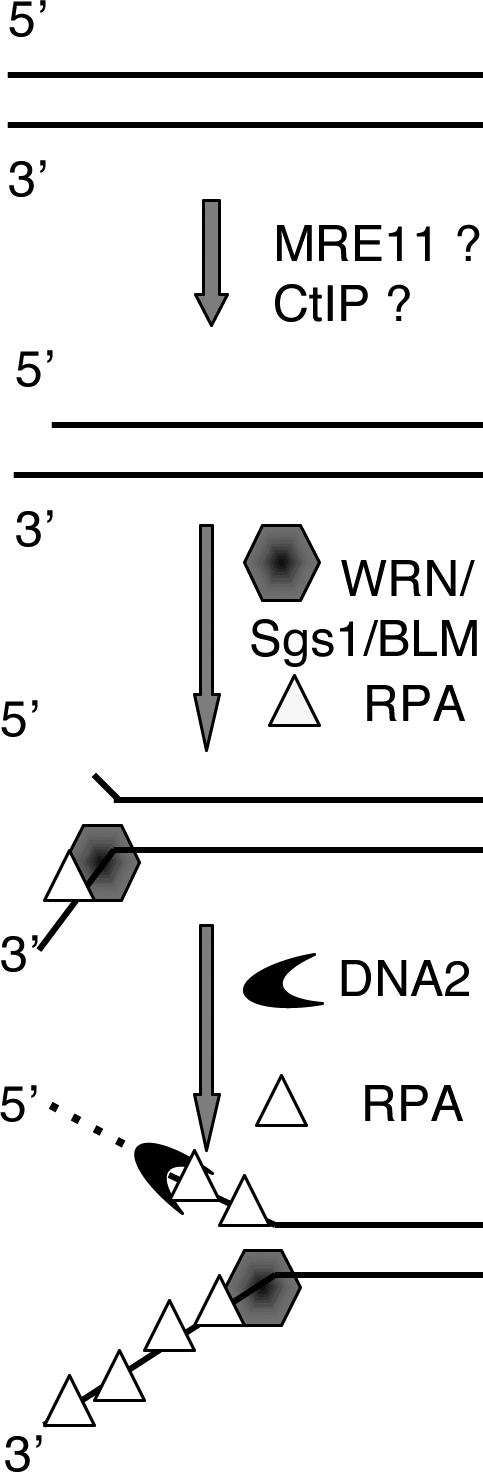
The WRN-DNA2-RPA–mediated DNA end processing pathway. After initial processing by a yet to be defined (probably a MRE11 and CtIP dependent) mechanism, WRN (and other RecQ-type helicases, such as Sgs1 and BLM) unwinds DNA ends. The 5′ ss-tail is then degraded by an ss-DNA exonuclease, such as DNA2. RPA stimulates both reactions.
Although gp32 cannot substitute for RPA to stimulate the purified xWRN and xDNA2, it does provide a partial complementation of end processing in RPA-depleted NPE. This suggests that there might be other end processing factors/pathways that depend on the stabilization of ss-DNA by an SSB, such as RPA or gp32, but do not require physical interactions with RPA. Genetic analyses in yeast S. cerevisiae suggest that EXO1 appears to act in a pathway parallel to Sgs1 and that MRX and Sae2 can also promote some limited end processing in the absence of Sgs1 and EXO1. Consistent with these genetic observations, depletions of neither xWRN nor xDNA2 can completely abolish end processing in Xenopus extracts (Toczylowski and Yan, 2006; Liao et al., 2008; Wawrousek et al., 2010). Our reconstitution experiment showed that xWRN, xDNA2, and RPA cannot degrade a blunt-ended DNA. In addition, Xenopus homologues of MRX (xMRN) and Sae2 (xCtIP) are also important for the formation of ss-DNA in Xenopus egg extracts (You et al., 2009; Wawrousek et al., 2010). Further studies are required to rigorously test how these other factors/pathways are affected by RPA.
Are the in vitro biochemical experiments relevant to DNA end processing on chromosomal DNA in cells? An essential function for RPA in SSA is consistent with genetic analyses in S. cerevisiae. For example, SSA has been characterized in the rfa1-t11 allele and shown to be 8.5-fold less efficient when compared with the wild-type strain. Another allele, rfa1-M2, displays a decreased rate of recombination between direct repeats (Longhese et al., 1994). Allele rfa1-D228Y, which by itself does not have much effect on SSA, can partially rescue the SSA defect in the rad52 mutant (Smith and Rothstein, 1999). There has been no direct in vivo study on the role of RPA in 5′-strand resection in cells, but some experiments do provide indirect evidence for such a role in other organisms. For instance, in S. cerevisiae, the rfa1-D228Y allele is associated with the lack of long single-stranded intermediates in the rad52 mutant background (Smith and Rothstein, 1999). A reasonable explanation is that this mutant RPA is defective in 5′-strand resection. In addition, it has been observed that RPA knockdown inhibits the recruitment of the cell cycle checkpoint kinase ATR to ionizing radiation–induced foci (Zou and Elledge, 2003). ATR is activated by ss-DNA, so this observation is consistent with a role for RPA in stimulating the generation of ss-DNA in addition to its role in directly recruiting ATR through protein–protein interaction with ATR-interacting protein. The insights gained from the Xenopus system should continue to provide detailed understanding of the mechanism responsible for DSB end processing, repair, and checkpoint activation in cells.
Materials and methods
Extract preparation and nuclear reconstitution
Crude interphase extracts, membrane-free cytosol, and demembranated sperm chromatin were prepared according to the published procedures (Smythe and Newport, 1991). NPEs were prepared according to the published protocol (Walter et al., 1998).
Antibody preparation
The following antibodies were used in this study: rabbit anti-RPA, rat anti-RPA1 (the p70 subunit of Xenopus RPA), and rabbit anti-xWRN (amino acids 1–466). The rabbit anti-RPA antibodies were against all three subunits of the native RPA purified from Xenopus egg extracts (Fang and Newport, 1993) and used without further purification. The other two antibodies were raised against the corresponding recombinant GST fusion proteins according to the standard procedure (Goding, 1986). The antibodies were purified on affinity columns constructed with the corresponding recombinant proteins according to a procedure described previously (Yan et al., 1993).
Immunodepletion
Immunodepletion of cytosol was performed by incubating cytosol (40 + 20 µl ELB [10 mM Hepes, pH 7.5, 250 mM sucrose, 2.5 mM MgCl2, 50 mM KCl, and 1 mM DTT]) with 20 µl protein A–Sepharose beads (Sigma-Aldrich) that had been precoated with rabbit anti-RPA serum or control serum (30 µl of serum/20 µl of beads). After incubation (with rotation) at 4°C for 2.5 h, the beads were removed by low speed centrifugation, and the supernatants were treated again with fresh beads. Immunodepletion of NPE was performed in a similar way except that the beads were coated with 40 µl of serum/20 µl of beads for a total of three rounds.
Interaction between RPA and xDNA2
For the coimmunoprecipitation experiment, 10 µl protein A–Sepharose beads coated with 20 µl anti-xDNA2, anti-xRPA, and control sera was incubated with 30 µl cytosol (diluted 1.5× with ELB) at 4°C for 1 h. The beads were washed sequentially with 500 µl ELB, 500 µl ELB + 50 mM NaCl + 0.1% NP-40, and 500 µl ELB. The bound proteins were separated on an 8% SDS-PAGE, transferred to a polyvinylidene fluoride membrane, and probed for RPA, xDNA2, and xWRN by Western blotting. Detection was achieved with chemiluminescence (SuperSignal; Thermo Fisher Scientific).
To detect the interaction between the purified RPA and recombinant xDNA2, the full-length xDNA2 ORF was cloned by PCR using primers derived from the sequence in the database (Liu et al., 2000). After confirmation by sequencing, the xDNA2 ORF was subcloned into pFastBac (Invitrogen) to create a fusion protein with a FLAG tag at the C terminus. SF9 cells expressing the recombinant xDNA2 protein were collected, and nuclear extracts were prepared according to the manufacturer’s instructions (Invitrogen). The extracted proteins were fractionated sequentially by HiTrap Q (peak at 250 mM NaCl) and HiTrap Heparin (peak at 425 mM NaCl) columns. 50 µl of the peak heparin fraction containing recombinant xDNA2 or 5 µg BSA (New England Biolabs, Inc.) was coated onto 10 µl anti-FLAG M2 agarose beads (Sigma-Aldrich). The xDNA2 and BSA beads were then incubated with 20 µl of the purified RPA (80 ng/µl) at 4°C for 1 h and then washed twice with 500 µl of buffer A50 (40 mM Tris-HCl, pH 8, 1 mM EDTA, 10% glycerol, 50 mM NaCl, and 1 mM DTT). Proteins in the beads and supernatant fractions were analyzed by Western blotting to detect RPA and xDNA2.
SSA and end processing assays
These assays were performed essentially as previously described (Yan et al., 2005; Toczylowski and Yan, 2006). The substrate for SSA, pRW4*, was a 5.7-kb linear DNA carrying two 1.2-kb direct repeats at the ends. A typical SSA assay contained 5 µl RPA- or mock-depleted NPE, 0.5 µl of 10× ATP mix (20 mM ATP, 200 mM phosphocreatine, 0.5 mg/ml creatine kinase, and 50 mM DTT), and 12 ng/µl DNA in 7.5 µl of volume. The substrate for end processing, linearized pUC19, carried a 32P-labeled dA and a ddC at the 3′ ends. ss-DNA was prepared by denaturing linear pUC19 at 95°C for 2 min and then immediately chilling it on ice for 5 min. A typical end processing reaction contained 5 µl of depleted NPE, 0.5 µl of 10× ATP mix, and 4 ng/µl DNA in 7.5 µl of volume. The reactions were incubated at room temperature, and samples were taken at the indicated times and mixed with an equal volume of 2% SDS/25 mM EDTA. At the end, the samples were brought up to 10 µl with H2O and supplemented with 1 µl proteinase K (10 mg/ml). After incubation at room temperature for at least 2 h, the samples were analyzed by 1% TAE (Tris–acetic acid–EDTA)-agarose gel electrophoresis. Gels were first stained with SYBR gold (Invitrogen) and then dried and exposed to phosphoimager (BAS-2500; Fujifilm). The RPA protein was purified from cytosol according to a procedure similar to the one previously published (Fang and Newport, 1993). The final concentration of RPA for complementation was ∼0.05 µM in the DSB foci experiment and 0.25 µM in the SSA and end processing assays. The T4 gp32 protein (Affymetrix) was used at a 1-µM final concentration in the SSA and end processing experiments.
DNA unwinding assay
The substrate for the unwinding assay was a 48mer double-stranded oligonucleotide with one strand carrying a 5′ biotin and the other strand carrying 21 thionucleotides on the 3′ half and a 32P label at the 3′ end (Toczylowski and Yan, 2006). The DNA was coated onto Streptavidin magnetic beads (Invitrogen) according to the standard procedure. A typical unwinding reaction contained 5 µl of depleted NPE, 0.5 µl of 10× ATP mix, 0.5 µl DNA beads (0.5 ng/µl), and 1.5 µl ELB buffer. After incubation at room temperature, 3.75 µl of each reaction was withdrawn at the indicated times and mixed with 11.25 µl of washing buffer (10 mM Tris-HCl, pH 8, 1 mM EDTA,1-M NaCl, and 0.05% NP-40). The beads were separated from the supernatants (10 µl of saved), washed with 15 µl of washing buffer, and resuspended in 10 µl of washing buffer. The supernatant and bead fractions were incubated with 3.3 µl of 4% SDS/50 mM EDTA, 6.7 µl H2O, and 2 µl proteinase K (10 mg/ml) at room temperature for 2 h and analyzed by 10% TAE-PAGE.
DNA nuclease assay
Two pairs of oligonucleotides were used to prepare the 32P-labeled, 3′-biotinylated single-stranded oligonucleotides as substrates for nuclease assays: 5′-GGAAACAGCTATGACCATGATTAC-3′/3′-CCTTTGTCGATACTGGTACTAATGCACAACCACCCACAACACACCTTG-5′ and 5′-GAAAGAAGAAAAAGAAAAAGG-3′/3′-CTTTCTTCTTTTTCTTTTTCCTTTCTCCTCCCTTTTTTTTTTTTTTTG-5′. Each pair was annealed, extended with Sequenase 2.0 (Affymetrix) in the presence of [32P]dATP (diluted 15× with cold dATP for the second pair), dGTP, TTP, and biotin-dCTP, coated onto Streptavidin paramagnetic beads, and denatured with NaOH according to the procedure described previously (Toczylowski and Yan, 2006). This resulted in two 48mer single-stranded oligonucleotides bound to beads through the biotin-dC at the 3′ end. The first 48mer (48mer-1) has the sequence of 5′-GGAAACAGCTATGACCATGATTACGTGTTGGTGGGTGTTGTGTGGAAC-3′) and contained two 32P-labeled dA (underlined) near the 3′ terminus. The second 48mer (48mer-5) has the sequence of 5′-GAAAGAAGAAAAAGAAAAAGGAAAGAGGAGGGAAAAAAAAAAAAAAAC-3′ and could be labeled by 32P-dA at 20 potential positions (underlined). A typical nuclease reaction contained 4 µl of proteins to be assayed (in A100 buffer [40 mM Tris-HCl, pH 8, 1 mM EDTA, 10% glycerol, 100 mM NaCl, and 1 mM DTT] or equivalent buffer), 4 µl ELB buffer, and 0.1 µl of oligonucleotide beads (∼0.5 ng/µl). After incubation at room temperature for 60 min with rotation, the reactions were stopped with an equal volume of 2% SDS/25 mM EDTA, heated at 95°C for 10 min, and analyzed by 8% TAE-PAGE.
End processing of 5′ preprocessed DNA
Purified xWRN and xDNA2 have been previously published (Yan et al., 1998; Liao et al., 2008). The DNA substrate with 5′ preprocessed DNA (carrying 3′ single-strand ends) was prepared by treating a 6-kb blunt-ended DNA (digested with BamHI and filled in with dGTP, [32P]dATP, TTP, and ddCTP) with λ exonuclease (New England Biolabs, Inc.; 1 U for 300 ng DNA in 15 µl ELB) at room temperature for 30–120 s. The reaction was terminated with 1.75 µl of 50-mM EDTA, heated at 75°C for 15 min, and supplemented with 1.75 µl of 50-mM MgCl2. Approximately 200–750 nt were removed from the 5′ strand. 1.7 µl of purified xWRN, xDNA2, and RPA (0.05 µM) or gp32 (0.2 µM) in various combinations was incubated with either fully blunt-ended or preprocessed DNA (1 ng/µl of final concentration) in 7.5-µl reactions. For reactions containing ATP, 0.5 µl of the 10× ATP mix was included. Samples were taken at the indicated times, processed as described for the regular end processing assays, and analyzed by 1% TAE-agarose gel electrophoresis.
Acknowledgments
The authors would like to thank Dr. Yoshihiro Matsumoto for reading the manuscript before submission.
This study was supported by a grant from the National Institutes of Health to H. Yan (R01 GM57962-02).
Footnotes
Abbreviations used in this paper:
- ATR
- ataxia telangiectasia and Rad3 related
- BLM
- Bloom syndrome protein
- CtIP
- C-terminal–binding protein–interacting protein
- dA
- deoxyadenine
- dC
- deoxycytidine
- DSB
- double-strand break
- ds-DNA
- double-strand DNA
- HR
- homologous recombination
- NHEJ
- nonhomologous end joining
- NPE
- nucleoplasmic extract
- RPA
- replication protein A
- SSA
- single-strand annealing
- SSB
- ss-DNA–binding protein
- ss-DNA
- single-strand DNA
- ss-tail
- single-strand tail
- WRN
- Werner syndrome protein
- xDNA2
- Xenopus DNA2
- xWRN
- Xenopus WRN
References
- Baumann P., West S.C. 1998. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem. Sci. 23:247–251 10.1016/S0968-0004(98)01232-8 [DOI] [PubMed] [Google Scholar]
- Brosh R.M., Jr, Orren D.K., Nehlin J.O., Ravn P.H., Kenny M.K., Machwe A., Bohr V.A. 1999. Functional and physical interaction between WRN helicase and human replication protein A. J. Biol. Chem. 274:18341–18350 10.1074/jbc.274.26.18341 [DOI] [PubMed] [Google Scholar]
- Brosh R.M., Jr, Li J.L., Kenny M.K., Karow J.K., Cooper M.P., Kureekattil R.P., Hickson I.D., Bohr V.A. 2000. Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its helicase activity. J. Biol. Chem. 275:23500–23508 10.1074/jbc.M001557200 [DOI] [PubMed] [Google Scholar]
- Budd M.E., Campbell J.L. 2009. Interplay of Mre11 nuclease with Dna2 plus Sgs1 in Rad51-dependent recombinational repair. PLoS One. 4:e4267 10.1371/journal.pone.0004267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.Y., Graham J., Yan H. 2001. Evidence for a replication function of FFA-1, the Xenopus orthologue of Werner syndrome protein. J. Cell Biol. 152:985–996 10.1083/jcb.152.5.985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Nievera C.J., Lee A.Y., Wu X. 2008. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem. 283:7713–7720 10.1074/jbc.M710245200 [DOI] [PubMed] [Google Scholar]
- Fang F., Newport J.W. 1993. Distinct roles of cdk2 and cdc2 in RP-A phosphorylation during the cell cycle. J. Cell Sci. 106:983–994 [DOI] [PubMed] [Google Scholar]
- Fanning E., Klimovich V., Nager A.R. 2006. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 34:4126–4137 10.1093/nar/gkl550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firmenich A.A., Elias-Arnanz M., Berg P. 1995. A novel allele of Saccharomyces cerevisiae RFA1 that is deficient in recombination and repair and suppressible by RAD52. Mol. Cell. Biol. 15:1620–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry M. 2002. The Werner syndrome helicase-nuclease—one protein, many mysteries. Sci. Aging Knowledge Environ. 2002:re2. [DOI] [PubMed] [Google Scholar]
- Goding J.W. 1986. Monoclonal Antibodies: Principles and Practice: Production and Application of Monoclonal Antibodies in Cell Biology, Biochemistry, and Immunology. Academic Press, London: 315 pp [Google Scholar]
- Golub E.I., Gupta R.C., Haaf T., Wold M.S., Radding C.M. 1998. Interaction of human rad51 recombination protein with single-stranded DNA binding protein, RPA. Nucleic Acids Res. 26:5388–5393 10.1093/nar/26.23.5388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel S., Chapman J.R., Magill C., Jackson S.P. 2008. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 22:2767–2772 10.1101/gad.503108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han E.S., Cooper D.L., Persky N.S., Sutera V.A., Jr, Whitaker R.D., Montello M.L., Lovett S.T. 2006. RecJ exonuclease: substrates, products and interaction with SSB. Nucleic Acids Res. 34:1084–1091 10.1093/nar/gkj503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa N., Morimatsu K., Lovett S.T., Kowalczykowski S.C. 2009. Reconstitution of initial steps of dsDNA break repair by the RecF pathway of E. coli. Genes Dev. 23:1234–1245 10.1101/gad.1780709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon F.G., Kowalczykowski S.C. 2001. Biochemical characterization of the DNA helicase activity of the Escherichia coli RecQ helicase. J. Biol. Chem. 276:232–243 10.1074/jbc.M006555200 [DOI] [PubMed] [Google Scholar]
- Hays S.L., Firmenich A.A., Massey P., Banerjee R., Berg P. 1998. Studies of the interaction between Rad52 protein and the yeast single-stranded DNA binding protein RPA. Mol. Cell. Biol. 18:4400–4406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazayeri A., Falck J., Lukas C., Bartek J., Smith G.C., Lukas J., Jackson S.P. 2005. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 8:37–45 10.1038/ncb1337 [DOI] [PubMed] [Google Scholar]
- Karran P. 2000. DNA double strand break repair in mammalian cells. Curr. Opin. Genet. Dev. 10:144–150 10.1016/S0959-437X(00)00069-1 [DOI] [PubMed] [Google Scholar]
- Labhart P. 1999. Ku-dependent nonhomologous DNA end joining in Xenopus egg extracts. Mol. Cell. Biol. 19:2585–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao S., Toczylowski T., Yan H. 2008. Identification of the Xenopus DNA2 protein as a major nuclease for the 5′→3′ strand-specific processing of DNA ends. Nucleic Acids Res. 36:6091–6100 10.1093/nar/gkn616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Choe W., Campbell J.L. 2000. Identification of the Xenopus laevis homolog of Saccharomyces cerevisiae DNA2 and its role in DNA replication. J. Biol. Chem. 275:1615–1624 10.1074/jbc.275.3.1615 [DOI] [PubMed] [Google Scholar]
- Longhese M.P., Plevani P., Lucchini G. 1994. Replication factor A is required in vivo for DNA replication, repair, and recombination. Mol. Cell. Biol. 14:7884–7890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou E.P., Symington L.S. 2008. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 455:770–774 10.1038/nature07312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen U.H., Bendixen C., Sunjevaric I., Rothstein R. 1996. DNA strand annealing is promoted by the yeast Rad52 protein. Proc. Natl. Acad. Sci. USA. 93:10729–10734 10.1073/pnas.93.20.10729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M.S., Ludwig D.L., Stigger E., Lee S.H. 1996. Physical interaction between human RAD52 and RPA is required for homologous recombination in mammalian cells. J. Biol. Chem. 271:18996–19000 10.1074/jbc.271.31.18996 [DOI] [PubMed] [Google Scholar]
- Pastink A., Eeken J.C., Lohman P.H. 2001. Genomic integrity and the repair of double-strand DNA breaks. Mutat. Res. 480-481:37–50 [DOI] [PubMed] [Google Scholar]
- Plate I., Hallwyl S.C., Shi I., Krejci L., Müller C., Albertsen L., Sung P., Mortensen U.H. 2008. Interaction with RPA is necessary for Rad52 repair center formation and for its mediator activity. J. Biol. Chem. 283:29077–29085 10.1074/jbc.M804881200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori A.A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., Jackson S.P. 2007. Human CtIP promotes DNA end resection. Nature. 450:509–514 10.1038/nature06337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J.C., Gray M.D., Oshima J., Loeb L.A. 1998. Characterization of Werner syndrome protein DNA helicase activity: directionality, substrate dependence and stimulation by replication protein A. Nucleic Acids Res. 26:2879–2885 10.1093/nar/26.12.2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shereda R.D., Bernstein D.A., Keck J.L. 2007. A central role for SSB in Escherichia coli RecQ DNA helicase function. J. Biol. Chem. 282:19247–19258 10.1074/jbc.M608011200 [DOI] [PubMed] [Google Scholar]
- Shinohara A., Shinohara M., Ohta T., Matsuda S., Ogawa T. 1998. Rad52 forms ring structures and co-operates with RPA in single-strand DNA annealing. Genes Cells. 3:145–156 10.1046/j.1365-2443.1998.00176.x [DOI] [PubMed] [Google Scholar]
- Sleeth K.M., Sørensen C.S., Issaeva N., Dziegielewski J., Bartek J., Helleday T. 2007. RPA mediates recombination repair during replication stress and is displaced from DNA by checkpoint signalling in human cells. J. Mol. Biol. 373:38–47 10.1016/j.jmb.2007.07.068 [DOI] [PubMed] [Google Scholar]
- Smith J., Rothstein R. 1995. A mutation in the gene encoding the Saccharomyces cerevisiae single-stranded DNA-binding protein Rfa1 stimulates a RAD52-independent pathway for direct-repeat recombination. Mol. Cell. Biol. 15:1632–1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J., Rothstein R. 1999. An allele of RFA1 suppresses RAD52-dependent double-strand break repair in Saccharomyces cerevisiae. Genetics. 151:447–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smythe C., Newport J.W. 1991. Systems for the study of nuclear assembly, DNA replication, and nuclear breakdown in Xenopus laevis egg extracts. Methods Cell Biol. 35:449–468 10.1016/S0091-679X(08)60583-X [DOI] [PubMed] [Google Scholar]
- Stauffer M.E., Chazin W.J. 2004. Physical interaction between replication protein A and Rad51 promotes exchange on single-stranded DNA. J. Biol. Chem. 279:25638–25645 10.1074/jbc.M400029200 [DOI] [PubMed] [Google Scholar]
- Sugiyama T., New J.H., Kowalczykowski S.C. 1998. DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc. Natl. Acad. Sci. USA. 95:6049–6054 10.1073/pnas.95.11.6049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung P. 1997. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J. Biol. Chem. 272:28194–28197 10.1074/jbc.272.45.28194 [DOI] [PubMed] [Google Scholar]
- Symington L.S. 2002. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 66:630–670 10.1128/MMBR.66.4.630-670.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thode S., Schäfer A., Pfeiffer P., Vielmetter W. 1990. A novel pathway of DNA end-to-end joining. Cell. 60:921–928 10.1016/0092-8674(90)90340-K [DOI] [PubMed] [Google Scholar]
- Toczylowski T., Yan H. 2006. Mechanistic analysis of a DNA end processing pathway mediated by the Xenopus Werner syndrome protein. J. Biol. Chem. 281:33198–33205 10.1074/jbc.M605044200 [DOI] [PubMed] [Google Scholar]
- Umezu K., Sugawara N., Chen C., Haber J.E., Kolodner R.D. 1998. Genetic analysis of yeast RPA1 reveals its multiple functions in DNA metabolism. Genetics. 148:989–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilenchik M.M., Knudson A.G. 2003. Endogenous DNA double-strand breaks: production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA. 100:12871–12876 10.1073/pnas.2135498100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J., Sun L., Newport J. 1998. Regulated chromosomal DNA replication in the absence of a nucleus. Mol. Cell. 1:519–529 10.1016/S1097-2765(00)80052-0 [DOI] [PubMed] [Google Scholar]
- Wang X., Haber J.E. 2004. Role of Saccharomyces single-stranded DNA-binding protein RPA in the strand invasion step of double-strand break repair. PLoS Biol. 2:E21 10.1371/journal.pbio.0020021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawrousek K.E., Fortini B.K., Polaczek P., Chen L., Liu Q., Dunphy W.G., Campbell J.L. 2010. Xenopus DNA2 is a helicase/nuclease that is found in complexes with replication proteins And-1/Ctf4 and Mcm10 and DSB response proteins Nbs1 and ATM. Cell Cycle. 9:1156–1166 10.4161/cc.9.6.11049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wold M.S. 1997. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 66:61–92 10.1146/annurev.biochem.66.1.61 [DOI] [PubMed] [Google Scholar]
- Yan H., Merchant A.M., Tye B.K. 1993. Cell cycle-regulated nuclear localization of MCM2 and MCM3, which are required for the initiation of DNA synthesis at chromosomal replication origins in yeast. Genes Dev. 7:2149–2160 10.1101/gad.7.11.2149 [DOI] [PubMed] [Google Scholar]
- Yan H., Chen C.Y., Kobayashi R., Newport J. 1998. Replication focus-forming activity 1 and the Werner syndrome gene product. Nat. Genet. 19:375–378 10.1038/1263 [DOI] [PubMed] [Google Scholar]
- Yan H., McCane J., Toczylowski T., Chen C. 2005. Analysis of the Xenopus Werner syndrome protein in DNA double-strand break repair. J. Cell Biol. 171:217–227 10.1083/jcb.200502077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z., Shi L.Z., Zhu Q., Wu P., Zhang Y.W., Basilio A., Tonnu N., Verma I.M., Berns M.W., Hunter T. 2009. CtIP links DNA double-strand break sensing to resection. Mol. Cell. 36:954–969 10.1016/j.molcel.2009.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z., Chung W.H., Shim E.Y., Lee S.E., Ira G. 2008. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 134:981–994 10.1016/j.cell.2008.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L., Elledge S.J. 2003. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 300:1542–1548 10.1126/science.1083430 [DOI] [PubMed] [Google Scholar]
- Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406–3415 10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]