

Abstract
We report an entirely new role for the HSP70 chaperone in dissociating 26S proteasome complexes (into free 20S proteasomes and bound 19S regulators), preserving 19S regulators, and reconstituting 26S proteasomes in the first 1-3 hours following mild oxidative stress. These responses, coupled with direct 20S proteasome activation by poly-ADP ribose polymerase in the nucleus and by PA28αβ in the cytoplasm, instantly provides cells with increased capacity to degrade oxidatively damaged proteins and to survive the initial effects of stress exposure. Subsequent adaptive (hormetic) processes (3-24 hours following stress exposure), mediated by several signal transduction pathways and involving increased transcription/translation of 20S proteasomes, immunoproteasomes, and PA28αβ, abrogate the need for 26S proteasome dissociation. During this adaptive period, HSP70 releases its bound 19S regulators, 26S proteasomes are reconstituted, and ATP-stimulated proteolysis is restored. The 26S proteasome-dependent, and ATP-stimulated, turnover of ubiquitinylated proteins is essential for normal cell metabolism, and its restoration is required for successful stress-adaptation.
Keywords: Oxidative stress, Ubiquitin-Proteasome System, Protein Degradation, Protein Oxidation, Stress adaptation
Introduction
We have previously shown that transient adaptation to mild oxidative stress, over a 24 hour period, includes increased proteasome activity, and increased proteasome-dependent degradation of oxidatively modified proteins in mouse embryonic fibroblasts (MEF) [1]. Although much of this increased proteolysis is dependent upon protein synthesis, and increased 20S proteasome expression, significant increases in both proteasome activity and proteolysis during the first hour of stress-exposure are not. Some of the rapid (within 60 min) transcription/translation-independent increase in proteasome activity and proteolysis may be due to direct activation of nuclear 20S proteasome by poly-ADP ribose polymerase (PARP) [2]; some may be due to direct 20S proteasome activation by the PA28αβγ (11S) regulators [1, 3-5]. The degree of proteasome activation however (almost twofold), suggested that other regulatory mechanisms might also be involved [1].
The proteasome exists in multiple forms, and its activity is modulated by multiple regulators. The 20S core proteasome contains all the proteolytic activity and selectively degrades a wide variety of oxidized proteins [1, 2, 6-12], as well as other substrates [13], in an ATP/ubiquitin-independent manner; perhaps aided by the PA28αβ regulator in the cytoplasm [1], and by PARP [2] and/or the PA28γ regulator [14] in the nucleus. When the core 20S proteasome combines with two 19S regulators (one at each end of the 20S cylinder) it forms the 26S proteasome [15]. The 19S regulators of the 26S proteasome give it the ability to bind the polyubiquitin chain of ubiquitinylated proteins. Most protein substrates for the 26S proteasome must first be polyubiquitinylated by the actions of E1, E2, and E3 ubiquitin activating, conjugating, and targeting enzymes [15]. Proteases, unfoldases, and ATPases within the 19S regulators then catalyze de-ubiquitinylation and unfolding of the bound protein substrate, and introduce the denatured protein into the interior of the 20S proteasome for degradation. A large body of research literature indicates that the 20S proteasome is largely responsible for the degradation of oxidized proteins [1, 2, 5-12, 16-19]: in contrast, the 26S proteasome catalyzes the turnover of most normal proteins, and selectively removes many products of nonsense and missense mutations, but possesses extremely poor selectivity for degradation of oxidized proteins [9, 20-22].
Previous work has shown that the 26S proteasome is highly susceptible to inactivation during oxidative stress, whereas the 20S proteasome is relatively stable to a wide range of oxidative stresses [23, 24]. In addition, Taylor et al. [25, 26] have demonstrated that the E1 and E2, ubiquitin activating and conjugating enzymes are very sensitive to oxidative stress, which would be expected to result in a transient shut-down of protein ubiquitinylation, and of ATP-ubiquitin-dependent proteolysis by the 26S proteasome.
We wondered if transcription/translation-independent increases in proteasome activity and proteolysis during the first hour of oxidative stress exposure might (in part) be due to dissociation of 19S regulators from the 26S proteasome, to generate more free 20S proteasome that could immediately degrade oxidized proteins. In preliminary studies, we noticed that ATP-stimulated proteolysis (requiring intact 26S proteasomes) is largely inactivated by oxidative stress, but recovers over a three-to-five hour period. This finding allows for the possibility that 26S proteasomes might actively dissociate and then re-associate in the early responses to oxidative stress. Since heat shock proteins (HSP's) are known to chaperone many proteins during stress [27], we wondered if HSP's might be able to ‘assist’ in such a dissociation/re-association model. Our new findings indicate that oxidative stress causes 26S proteasomes to immediately dissociate into free 20S proteasomes, while 19S regulators bind to HSP70 chaperones. During a three-to-five hour period, however, the 20S proteasomes and 19S regulators re-associate to restore functional ATP/ubiquitin-dependent proteolysis by the reconstituted 26S proteasomes. During that initial three-to-five hour period, however, the additional ‘free’ 20S proteasomes contribute significantly to overall cellular capacity to degrade oxidatively damaged proteins.
Results
Preferential degradation of oxidized proteins in K562 cells by the proteasomal system
Our previous observation of transcription/translation-independent increases during the first hour of adaptation to oxidative stress was made in mouse embryonic fibroblast (MEF). These same MEF cells then continued to increase both proteasome activity and degradation of oxidized proteins, but in a transcription/translation-dependent manner [1]. Human cells also exhibit proteasome-dependent degradation of oxidized proteins [6-9, 11-13, 17-19, 22, 24], and it was important to test whether human cells exhibit a similar proteasomal adaptive response to oxidative stress as do MEF cells.
K562 human hematopoietic cells exhibited a dose-dependent increase in protein degradation after exposure to H2O2 treatment (Fig. 1A), and also exhibited increased proteolysis with other oxidative stresses (Fig. 1B), including the redox cycling agents menadione and paraquat (which generate a constant stream of O2- and H2O2), and SIN-1 (which generates superoxide and nitric oxide, to form peroxynitrite). These results show that both externally applied, and internally generated oxidants cause increased protein degradation. The proteasome inhibitor lactacystin blocked some 50% of protein degradation in untreated cells and almost 70% of proteolysis in oxidatively stressed cells (Fig. 1C), confirming a major role for proteasome in oxidant-induced proteolysis. Oxidative stress more than doubled the basal level of oxidized intracellular proteins (carbonylated proteins), in a dose-dependent manner, but these oxidized proteins were removed within 24 hours (Fig. 1D) in a process that could be partially inhibited (> 65%) by lactacystin (data not shown); these results indicate that proteasome may be largely responsible for increased capacity to degrade oxidized proteins in K562 cells, following adaptation to mild oxidative stress.
Fig. 1. Preferential degradation of oxidized proteins in K562 cells by the proteasomal system.

Panel A. Degradation of metabolically radio-labeled proteins after H2O2 treatment. Endogenous proteins in K562 cells were metabolically radio-labeled with [35S] Met/Cys, in the pulse/chase procedure described in Materials & Methods. Next, 0, 0.5, or 1mM H2O2 was added in PBS, and percent degradation of [35S] cellular proteins was measured after 24 hours. Panel B. Degradation of metabolically radio-labeled proteins after treatment with paraquat, menadione, or SIN-1. Cells were radio-labeled as per Panel A, and percent protein degradation was measured 24 hours after treatment with various oxidant generators; 20 μM paraquat, 20 μM menadione or 1 mM Sin-1, added in PBS. Panel C. Proteasome-dependence of increased proteolysis. The proteasome-dependence of the proteolysis seen in Panels A and B was tested with the proteasome inhibitor, lactacystin. Cells were exposed to 0.5 mM H2O2 as per Panel A, in the presence or absence of lactacystin (LC) at 20μM. Values are means ± SE's, of four experiments with three measurements each. Panel D. Enhanced proteolysis after H2O2 adaptation removes oxidized intracellular proteins. Cells were treated with 0, 0.5, or 1.0 mM H2O2 as per Panel A. Oxidized intracellular proteins were measured by protein carbonyl ELISA (see Materials & Methods) immediately after 0.5 hr of H2O2 treatment, or after 24 hrs. In all panels, oxidant exposures were conducted in 10% of the volume of the original cell suspension and, after 30 min, the remaining 90% of the volume of complete tissue culture medium was re-added. In Panels A, B, and C, all values are means ± SE of four experiments; in Panel D, values are means ± SE of six experiments, each in triplicate.
Proteasome direct activation and proteasome synthesis during oxidative stress survival and adaptation
The mild oxidative stresses utilized in our studies induced a protective adaptive response that enabled adapted cells to survive much higher oxidant challenges. For example, 24 hours following pre-treatment with H2O2, K562 cells exhibited four-fold increased survival of a 5 mM H2O2 challenge compared with non-adapted cells (11.2 ± 0.9% survival for non-adapted cells versus 46.3 ± 5.1% survival for pre-adapted cells, confirmatory data not shown). These results are in good agreement with our previous studies of oxidative stress adaptation in other cell types [1, 28]. As shown in Fig. 2A, K562 cells exhibited a rapid increase in capacity to degrade an oxidatively damaged hemoglobin (Hb) substrate, within one hour of exposure to mild oxidative stress, and a subsequent greater increase in proteolytic capacity over the succeeding 23 hours. Cycloheximide, which effectively inhibited transcription/translation (data not shown), had no effect on the increased capacity to degrade oxidized Hb within one-hour of oxidative stress exposure, but strongly inhibited subsequent increases in proteolysis over the next 23 hours. In fact, in the presence of cycloheximide, proteolytic capacity actually returned slowly to baseline levels (Fig. 2A). These results for oxidized Hb degradation in K562 cells are in very good agreement with our previous finding, that cycloheximide had no effect on increased MEF cell capacity to degrade the proteasome fluorogenic peptide substrate Suc-LLVY-AMC in the first hour of oxidative stress exposure, but strongly inhibited subsequent increases in proteolysis [1], although it is important to note that we did not actually test an oxidized protein in the previous study.
Fig. 2. Both direct activiation and de novo synthesis of proteasome occur during adaptation to oxidative stress.

Panel A. Cycloheximide effects on increased proteolytic capacity. K562 cells were treated with 0.5 mM H2O2 for 30 min, and cycloheximide (100 g/ml) was then added for incubations lasting from 0.5 hrs to 24 hrs. After various time points over 24 hrs, cell extracts were prepared, and their proteolytic capacity to degrade oxidized [3H] hemoglobin was measured by release of acid-soluble counts (liquid scintillation) as described in Materials & Methods. Panel B. The capacity of cell extracts to degrade oxidized [3H] hemoglobin was measured at both 1 hr, and 24 hrs (both without cycloheximide) after treatment with 0.5 mM hydrogen peroxide, 20 μM paraquat, 20 μM menadione or 1 mM SIN-1, as per Panel A. Values in both panels are means ± SE of three experiments, each in triplicate.
Increased proteolytic capacity to degrade oxidized proteins appears to be a general response to low-level oxidative stress, since it was induced by H2O2, by paraquat and menadione, and by SIN-1 (Fig. 2B). The importance of the proteasome in degrading oxidized Hb was again evidenced by inhibition with lactacystin, which was even more effective after oxidant-induced increases in proteolytic capacity (data not shown). Importantly, all the oxidant stressors tested caused increases in proteolytic capacity to degrade an oxidized protein after both one hour and 24 hours (Fig. 2B). In addition to the lack of effect of cycloheximide within one-hour of H2O2 treatment, it should also be kept in mind that transcription/translation are really too slow to account for the 100% - 200% increases in proteolytic capacity observed within one-hour of H2O2 treatment in Fig. 2B, especially considering the high initial cellular proteasome content, and the relatively slow turnover of the enzyme complex [29]. Thus, although de novo proteasome synthesis (and de novo synthesis of activators like PA28) do appear necessary for the adaptive responses to oxidative stress seen at 24 hours, we reasoned that immediate increases in proteasome activity within one-hour of oxidative stress must depend on some direct activation mechanism.
Temporary inhibition of the 26S proteasome in response to oxidative stress
In the absence of any treatment, the degradation of Suc-LLVY-AMC was stimulated four- to five-fold by addition of ATP (Fig. 3A), consistent with proteolysis by the ATP-stimulated 26S proteasome [24, 30]. Addition of increasing concentrations of H2O2 increased ATP-independent Suc-LLVY-AMC degradation by two- to three-fold, but completely abolished any ATP-stimulation of proteolysis. These data might suggest an immediate loss of 26S proteasome activity with H2O2 treatment, accompanied by an increase in 20S proteasome activity. The 26S proteasome is known to be sensitive to inactivation by H2O2 whereas the 20S proteasome appears quite resistant [23, 24]. One possible explanation for the results of Fig. 3A could be dissociation of the two 19S regulatory subunits from the 26S proteasome, to generate more free 20S proteasome (to conduct ATP-independent proteolysis) but abolishing ATP-stimulated proteolysis (which requires association of the 20S proteasome and 19S regulators). A non-denaturing electrophoresis activity gel showed increased Suc-LLVY-AMC degradation in the region of the 20S proteasome after H2O2 treatment, and diminished degradation by the 26S proteasome (Fig. 3B – left two panels). Immunostained gels revealed increased anti 20S proteasome antibody reactivity immediately following H2O2 treatment, whereas 26S proteasome levels (anti-MSS1 antibody) decreased dramatically (Fig. 3B – middle two panels, and right two panels).
Fig. 3. Temporary inhibition of the 26S proteasome in response to oxidative stress.
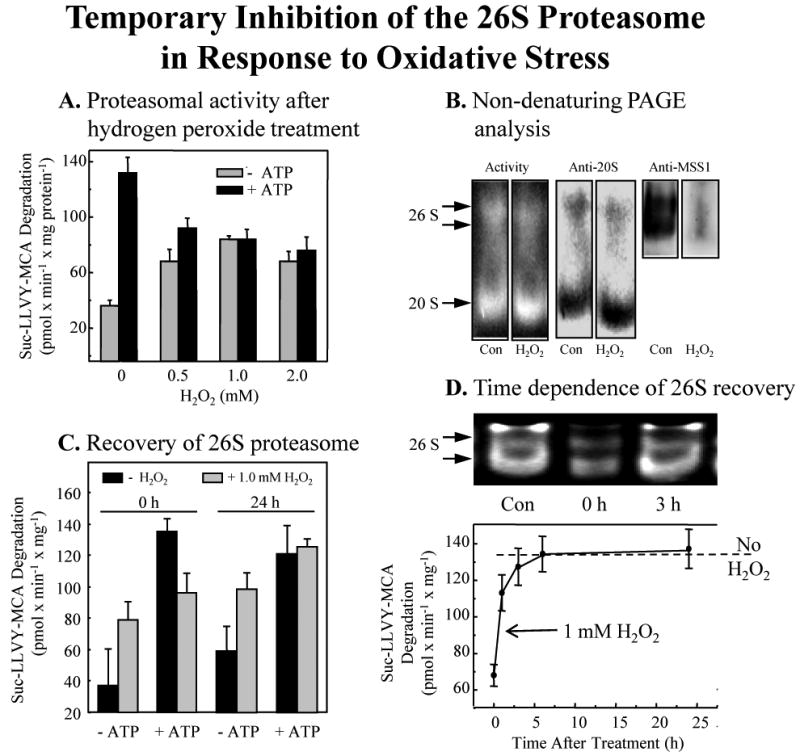
Panel A. Loss of ATP-stimulated, 26S proteasome dependent, activity with H2O2 treatment. Human hematopioetic K562 cells were treated with various concentrations of H2O2 for 30 min, as per Fig. 1. Cells were then immediately harvested and lysed, and the extracts were used to measure capacity to degrade the fluorogenic peptide substrate Suc-LLVY-AMC (a measure of proteasomal chymotrypsin-like activity) in the presence or absence of ATP/Mg2+, as described in Materials & Methods. Data are means ± SE, of six experiments, each in triplicate. Panel B. Proteasome activity gel and Western blots. The panel shows representative non-denaturing proteolytic activity gels using Suc-LLVY-AMC as a substrate (left lanes) [13], immunoblots developed with a (Biomol) anti-20S proteasome antibody (middle lanes), and a (Biomol) anti-MSS1 26S proteasomal-subunit-antibody (right lanes). The lysates were harvested 30min after addition of 1.0 mM H2O2. The activity gel background is black, so proteolysis shows as negative staining (light bands) in the region of the two 26S proteasomal bands and the single 20S proteasome band. The activity gel clearly reveals loss of 26S activity, with increase in 20S activity, after 0.5 hr of H2O2 treatment. Panel C. Recovery of ATP-dependent 26S proteasomal activity 24 hours after H2O2 treatment. Suc-LLVY-AMC degradation, in the presence or absence of ATP/Mg2+ was used to measure the recovery of ATP-dependent 26S proteasomal activity, 0 and 24 hours after H2O2 treatment. Panel D. Time dependence of the recovery of 26S protesomal activity. Experiments were performed as per Panel C. The upper part of the panel represents the 26S proteasome activity in an activity stain of a non-denaturing electrophoresis gel. In the lower part of Panel D, the broken line represents 26S proteasomal activity without H2O2 treatment. The solid line connecting data points reports values for cells treated with 1.0 mM hydrogen peroxide. Values in Panels A and C are means ± SE of four experiments, each in triplicate. Values in Panel D are means ± SE of five experiments, each in triplicate.
26S Proteasome inhibition is accompanied by a loss of 19S RP/20S interaction and recovery of 26S proteasome is independent of protein synthesis
ATP-stimulated (26S proteasome) proteolytic activity returned to pre-oxidation levels within 24 hours (Fig. 3C). Importantly, a careful time course revealed that the restoration was actually complete within 3-5 hours, and coincided with the re-association of 19S regulators with 20S proteasomes to reconstitute 26S proteasomes (Fig. 3D). This reconstitution of 26S proteasome seemed rather rapid and was certainly extensive, perhaps suggesting that it could not be explained by de novo synthesis. To begin to address this question, we metabolically radio-labeled cell proteins, exposed the cells to H2O2, lysed the cells, and then immunoprecipitated the 20S proteasome. Just 30 min. following H2O2 treatment, autoradiography (Fig. 4A) revealed a significant loss of 19S regulator subunit proteins that normally associate with the 20S proteasome to form the 26S proteasome. Some 2.5 hours later, (at 3 hours post-treatment) quantitative analysis of the autoradiograms revealed essentially complete reconstitution of 26S proteasomes. Although H2O2 treatment caused immediate dissociation of 19S regulators from 20S proteasomes, the 19S regulators were not degraded or otherwise destroyed, as evidenced by unchanged levels of the 19S MSS1, S1, and S14 subunits (Fig. 4B). In fact, recovery of 26S proteasome ATP-stimulated activity (Fig. 4C) and reconstitution of 26S proteasomes (not shown) were independent of protein synthesis, as demonstrated by a lack of inhibition by cycloheximide.
Fig. 4. 26S proteasome inhibition is accompanied by a loss of 19SRP/20S interaction and recovery of 26S proteasome is independent of protein synthesis.

Panel A. Immunoprecipitation of the 20S proteasome after H2O2 treatment. Human K562 cells were cultured and metabolically radio-labeled in a pulse-chase procedure, with [35S] -Cys/Met (50 μCi/ml or 1 mCi/107 cells) as described in Materials & Methods. Cells were then treated with 1.0 mM H2O2 and harvested at either 0.5 hr or at 3 hrs after treatment. Immunoprecipitation (see Materials & Methods) of 20S proteasome was performed using an anti-20S proteasome-antibody (Biomol, 2 μl antibody per mg cell lysate). The immunoprecipitate was separated on a 12 % PAGE and analyzed by autoradiography. One representative autoradiogram is shown to the left, and its electropherogram is in the middle of the panel; arrows indicate significant differences between control (dotted line) and H2O2 treated (solid line) samples. Quantification of 20S proteasome and 19S regulator bands from six gels (means ± SE) is shown at the right of Panel A. For quantification of 20S proteasome, we used the upper group of 20S proteasome bands, whereas for 19S regulator quantification we used an anti-MSS1 antibody (Biomol) and measured the 51 kDa co-precipitating band of the MSS1 19S regulator particle. Similar results were obtained using 0.5 mM H2O2 treatment (data not shown). Panel B. Preservation of 19 S regulator subunits MSS1, S1, and S14 after H2O2 treatment. Cells were treated with 1.0 mM H2O2 or used as controls, as in Panel A. After 30 min or 3.0 hrs, the levels of 19S regulator subunits was tested by Western blot. No significant changes in MSS1, S1, or S14 intracellular levels were detected at any point, and this analysis was repeated several times with the same results. Panel C. Recovery of 26S proteasome activity is independent of protein synthesis. K562 cells were treated with 1.0 mM H2O2 as per panels A and B. Cycloheximide (100 μg/ml) was then added to half of the cells and cell extracts were prepared. ATP-stimulated 26S proteasome activity was measured in the extracts by degradation of Suc-LLVY-AMC ± ATP/Mg2+ as per Figs. 3C and 3D. Values are means ± SE of four experiments, each in triplicate.
Integrity of the 19S RP after detachment from the 20S proteasome
Immunoprecipitation of the 19S proteasome regulator MSS1 subunit revealed co-precipitation of (bound) 20S proteasome core subunits in the absence of H2O2 treatment (Fig. 5A). Within 30 min of H2O2 treatment, no 20S proteasome subunits co-precipitated with the 19S regulator MSS1 subunit, but binding was fully restored by 3 hours post H2O2 treatment. In addition to its subunits not being degraded (Fig. 3B), the 19S regulator appears to have remained fully intact following H2O2 treatment, as evidenced by co-precipitation of the 19S S1 and S14 subunits along with the MSS1 subunit (Fig. 5B).
Fig. 5. Integrity of the 19S regulator particle after dissociation from the 20S proteasome.
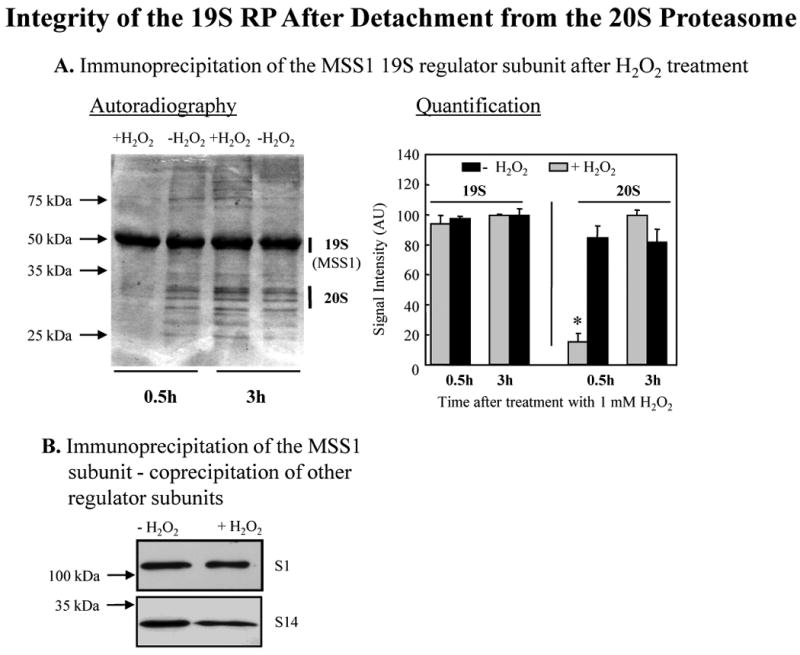
Human K562 cell were cultivated and proteins were metabolically radio-labeled, in a pulse-chase procedure, with [35S] Met/Cys as described in Fig. 1. Panel A. Immunoprecipitation of the MSS1 19S proteasomal regulator after H2O2 treatment. Cells were incubated for 0.5 or 3.0 hrs after treatment with 1.0 mM H2O2 or used as controls. They were harvested and lysed, and immunoprecipitated (see Materials & Methods) with an anti-MSS1 19S proteasomal subunit regulator antibody (Biomol, at 2 μl/mg cell lysate). The immunoprecipitate was analyzed by PAGE, and bands detected by autoradiography. A representative autoradiogram is shown to the left of the panel, in which the MSS1 band is always strong, but the co-precipitation of several 20S proteasome bands is only seen in control samples, and 3 hr after H2O2 treatment. Quantification of results from six gels (means ± SE) is given to the right of the panel where 20S proteasome co-precipitation with MSS1 is clearly seen to be lost 0.5 hr after H2O2 treatment (P < 0.01), but is recovered by 3 hr. Panel B. Co-precipitation of other 19S proteasome regulator subunits with MSS1. Samples from the experiments in Panel A were immunoprecipitated with the MSS1 antibody (Biomol), and then studied by Western blot with antibodies (biomol) against the S1 and S14 subunits of the 19S proteasome regulator.
Induction of HSP70 and binding to 19S
At this point we postulated that a chaperone protein might bind to 19S regulators following H2O2 induced dissociation from 20S core proteasomes, and protect them from proteolytic digestion. We already knew that H2O2 treatment caused increased expression of the HSP70 chaperone [31] so we decided to test if HSP70 might be associating with 19S regulators. As shown in Fig. 6A, when we immunoprecipitated extracts of K562 cells with the MSS1 (19S regulator) antibody and tested for co-precipitation of HSP70 (using an HSP70 antibody) we found that H2O2 treatment caused more than a doubling of 19S regulator bound to HSP70. The actual levels of HSP70 protein were increased by more than two-fold, during the three hours following H2O2 treatment (Fig. 6B).
Fig. 6. Binding of HSP70 to the 19S regulator particle and induction of HSP70.
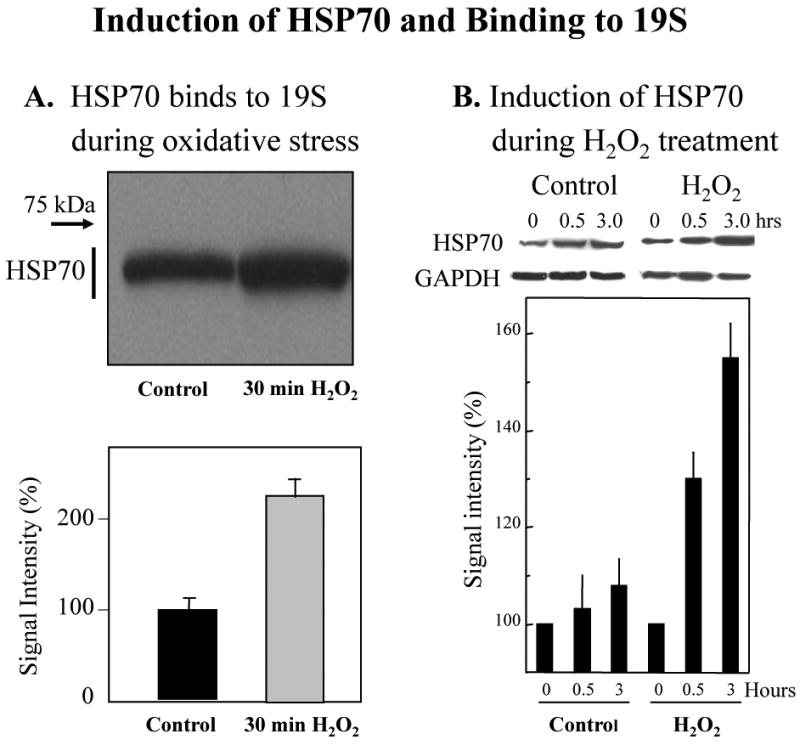
Panel A. Binding of HSP70 to the 19S regulator particle. Human K562 cells were used as controls, or were treated for 30 min with 1.0 mM H2O2 as described above. Cells were then lysed, and the extracts were immunoprecipited (see Materials & Methods) with an antibody (Biomol) against the MSS1 subunit of the 19S proteasome regulator particle. Western blots for co-immunoiprecipitation of HSP70 were then performed. HSP70 Western blots utilized a monoclonal anti-HSP70 (human) antibody from Enzo Life Sciences International, Inc. (Plymouth Meeting, PA), product # ADI-SPA-810. According to the manufacturer, this clone92F3A-5 antibody does not cross-react with the constitutive Hsc70 protein; a representative Western blot is shown at the top of Panel A, and quantification of HSP70 bound to 19S regulators is shown at the bottom of the panel as the average of 6 gels (means ± SE). Panel B. Induction of HSP70 during H2O2 treatment. Cells were treated with 1.0 mM H2O2 and collected after 0.5 or 3.0 hrs, or were used as controls. HSP70 protein levels were measured in cell extracts by quantification of Western blots, and data are means ± SE of six experiments. HSP70 Western blots utilized a monoclonal anti-HSP70 (human) antibody from Enzo Life Sciences International, Inc. (Plymouth Meeting, PA), product # ADI-SPA-810. According to the manufacturer, this clone92F3A-5 antibody does not cross-react with the constitutive Hsc70 protein.
HSP70 is Required to Stabilize 19S RP and Reactivate 26S Proteasome
If HSP70 actually protects 19S regulators immediately following stress, then blocking HSP70 induction should also block reconstitution of 26S proteasomes, and the recovery of ATP-stimulated proteolysis. To test this possibility, we first inhibited the induction of HSP70, following oxidative stress, with antisense oligonucleotides. Pre-treatment with HSP70 antisense both prevented the recovery of ATP-stimulated proteolysis (Fig. 7A) and blocked HSP70 induction (not shown). Next we blocked HSP70 induction with siRNA and, again, the increases in both ATP-stimulated proteolysis and HSP70 levels were lost (Fig. 7B). Finally, we tested the HSP70 inhibitor KNK which also inhibited both the increase in HSP70 levels, and the restoration of (26S proteasome-dependent) ATP-stimulated proteolysis (Fig. 7C).
Fig. 7. HSP70 is required to stabilize 19S RP and reactivate 26S proteasome after oxidative stress.
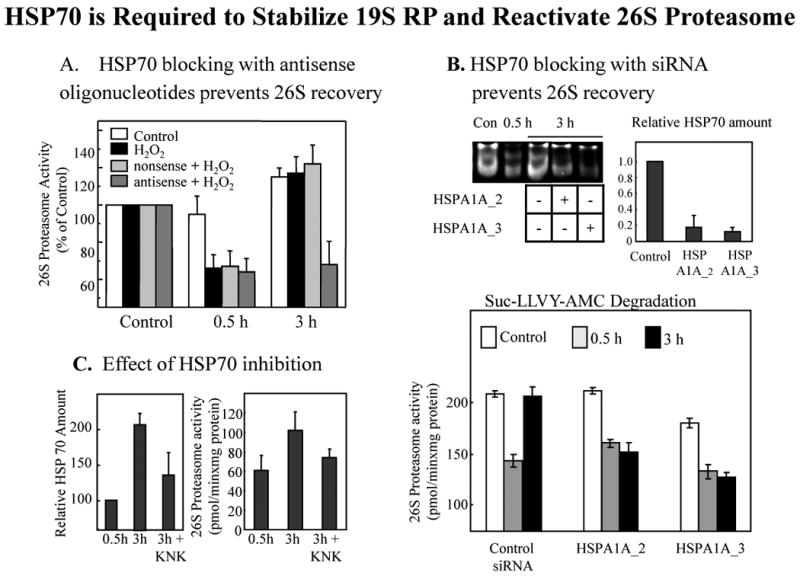
Human K562 cells were treated with 1.0mM H2O2, or were used as controls, as above. Panel A. HSP70 antisense oligonucleotides block recovery of 26S proteasome activity after oxidative stress. K562 cells were pre-incubated with either an HSP70 antisense oligonucleotide (5'-CAC CTT GCC GTG CTG GAA-3') or a nonsense (ns) oligonucleotide, in a final concentration of 10 μM as described by Robertson et al. [50] and as previously employed [13, 16]. After 0, 0.5, or 3.0 hrs of H2O2 treatment, cells were harvested and analyzed for proteasomal activity by suc-LLVY-AMC degradation in the presence of ATP; no changes in proteasome activity in the absence of ATP were observed (data not shown). Values are means ± SE of three experiments, each in triplicate. Panel B. HSP70 siRNA blocks recovery of 26S proteasome activity after oxidative stress. Using procedures previously described [1], K562 cells were transfected, using the HiPerFect Transfection Reagent (#301704), with either of two different siRNA's specific for HSP70: Qiagen product # SI00442967, Hs_HSPA1A_2HP (5'-TCC TGT GTT TGC AAT GTT GAA-3') and Qiagen product #SI00442974, Hs_HSPA1A_3HP siRNA (5'-AGA GAT GAA TTT ATA CTG CCA-3') to a final concentration of 10 μM. Qiagen reports that these siRNA's are specific for the inducible HSP70 and will not affect levels of the constitutive HSC70. The actual HSP70 protein knock-down we achieved with these siRNA treatments was measured by Western blot (as per Fig. 6, relative to GAPDH loading controls) and can be seen in the upper right corner of Panel B (values are means ± SE's of six experiments). The upper left corner of Panel B shows a non-denaturing activity gel of suc-LLVY-AMC degradation [13] in the presence of ATP for control cells, cells treated with H2O2 for 0.5 hr, and cells treated with H2O2 for 3 hrs ± the HSPA1A_2 or HSPA1A_3 HSP70 siRNA's. The gel background is black, so proteolysis shows as negative staining (light bands) in the region of the two 26S proteasomal bands, and clearly reveals loss of 26S activity after 0.5 hr of H2O2 followed by recovery at 3 hr, except when HSP70 induction is blocked with HSPA1A_2 or HSPA1A_3 siRNA's. The lower portion of Panel B shows the results of a fluorescence-based suc-LLVY-AMC degradation assay in the presence of ATP, as a more detailed and quantifiable measure of ATP-stimulated 26S proteasomal activity. Proteolysis was measured by fluorescence emission of free AMC in comparison with standards (see Materials & Methods) after 0, 0.5, or 3.0 hrs of H2O2 treatment, in the presence or absence of the HSPA1A_2 or HSPA1A_3 HSP70 siRNA's; values are means ± SE of three experiments, each in triplicate. Panel C. Effect of the HSP70 inhibitor KNK437 on HSP70 levels and proteasome activity. Cells were pretreated with 100 μM KNK437 for 1 h prior to treatment with 1.0 mM hydrogen peroxide. Cell lysates were analyzed 0.5 h and 3 h after H2O2 treatment by immunoblotting for HSP70 amount (as per Fig. 6) in the left side of the panel, and by AMC fluorescence for 26S proteasomal activity in the presence of ATP in the right side of the panel; all values are means ± SE of three experiments, each in triplicate.
Discussion
Proteasomal responses to mild oxidative stress in human K562 cells are shown to be temporally biphasic. Initial responses (in the first hour) include a doubling of proteolytic capacity to degrade oxidized proteins that is independent of transcription/translation: a direct activation of activity. In contrast, a further doubling of activity occurring over the subsequent 23 hours was blocked by the protein synthesis inhibitor, cycloheximide. Increased proteasome expression over this 24 hour period may well be mediated by the Keap1-Nrf2 pathway, as shown previously for antioxidants [32], and we are currently testing this hypothesis. The native capacity of cells to degrade oxidized proteins, the activation of this activity in the first hour after H2O2 treatment, and the subsequent further increases that relied on transcription/translation were all found to depend on the proteasome, in good agreement with recent studies in MEF cells [1]. Immediately after a mild H2O2 treatment, sufficient to elicit a protective adaptive-response, 20S proteasome (ATP-independent) activity increased whereas 26S proteasome (ATP-stimulated) activity was lost. ATP-stimulated proteolysis, however, returned to pre-treatment levels over the next three hours.
As depicted schematically in Fig. 8, our studies of the proteasome revealed a dissociation of the 26S proteasome into 19S regulators and 20S core proteasomes immediately following H2O2 treatment. The 19S regulators did not dissociate into individual protein subunits, nor were they proteolytically degraded. Instead, intact 19S regulators bound to HSP70 chaperones, whose cellular levels greatly increased. In the first three hours following H2O2 treatment, ATP-stimulated proteolysis was restored and 26S proteasomes were seen to be reconstituted. Reconstitution of 26S proteasomes (i.e. re-association of a core 20S proteasome with two 19S regulators) was unaffected by addition of cycloheximide immediately after 30 min of H2O2 treatment (not prior to treatment), and it should be noted that free HSP70 levels have still been shown to increase even with cycloheximide treatment [33-38]. Importantly blocking HSP70 synthesis by pre-treatment with antisense oligonucleotides, or siRNA added prior to H2O2 and remaining during and after H2O2 exposure (in the absence of cycloheximide) prevented increases in cellular HSP70 levels, blocked the formation of HSP70/19S regulator complexes, prevented the reconstitution of 26S proteasomes, and blocked the restoration of ATP-stimulated 26S proteasome activity. The HSP70 inhibitor KNK also blocked increases in HSP70 and simultaneously prevented the restoration of ATP-stimulated proteolysis. It is worth noting that some HSP70 was bound to 19S regulators even in the absence of (overt) oxidative stress (Fig. 6A). Whether this is the result of accidental oxidation during experimental procedures, or whether it represents a fraction of the 19S regulator population that may be dissociated under physiological conditions, cannot yet be determined. It does seem possible, however, that the association of HSP70 with 19S regulators may be important in a dynamic equilibrium of various proteasomal forms even under normal conditions.
Fig. 8. HSP70-mediated dissociation of the 26S Proteasome during oxidative stress.
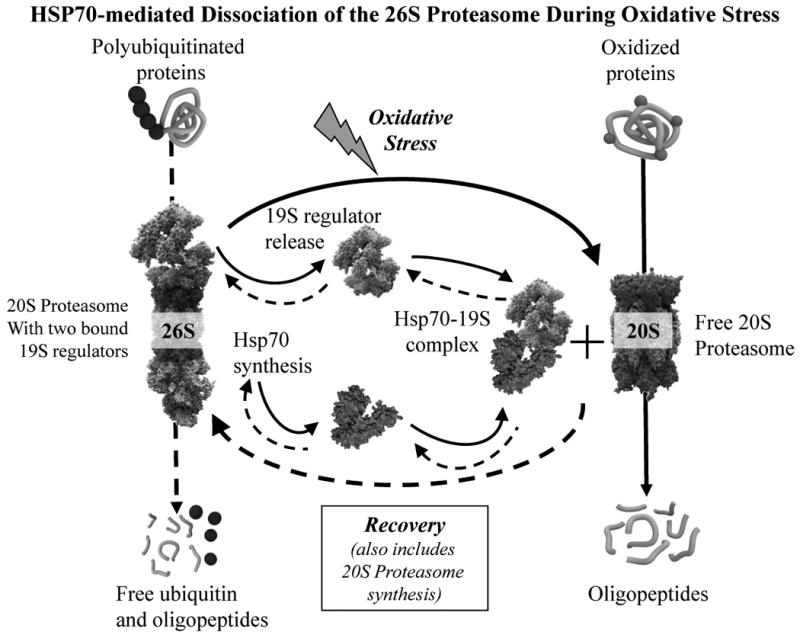
Scematic representation of the major findings of this study. Oxidative stress causes synthesis of HSP70 which forms complexes with 19S regulator proteins of the 26S proteasome, thus generating extra free 20S proteasomes to degrade oxidized proteins as an initial response to oxidative stress. After about three hours, as cells begin to adapt to the stress, the HSP70-19S regulator complexes dissociate and 26S proteasomes reform. If the stress is relatively mild, and cells can mount an adaptive response (hormesis), de novo 20S proteasome synthesis will also begin, as will synthesis of the immunoproteasome and the Pa28αβ (or 11S) proteasome regulator, all of which can contribute to the continued removal of proteins that were oxidized during the stress [1]. In the Scheme, solid arrows indicate oxidative stress effects whereas dashed arrows represent recovery, or unstressed, processes.
While we were revising this paper for publication, an elegant report by Wang et al. [39] provided excellent evidence for the dissociation of 20S proteasomes and 19S regulators during exposure of yeast cells to oxidative stress. Wang et al. [39] found that the proteasome interacting protein, Ecm29 bound preferentially to dissociated 19S regulators, and that this interaction was required for increased degradation of oxidized proteins and yeast survival. Limited data was also provided to show dissociation of 26S proteasomes in human embryonic kidney cells (HEK 293), although no role for Ecm29 was reported in those cells. Whether HSP70 may play a similar role in yeast cells to that found in our work (working in partnership with Ecm29), or whether Ecm29 may also be involved in the dissociation/reassociation of human 26S proteasomes, must now be tested. It should also be noted that the appearance of free 20S proteasomes, upon dissociation of the 26S proteasome, has been reported during coronary occlusion-reperfusion-induced oxidative stress by Bulteau et al. [40]. Thus, dissociation of 26S proteasome into proteolytically active free 20S proteasomes, and 19S regulators (bound to HSP70?) may be of both physiological and pathological significance in eukaryotes.
Our present results, and previous studies [1, 28], suggest a model of transient adaptive oxidative-stress responses in which proteasome plays key roles. Immediately following mild oxidative stress the 26S proteasome dissociates into core 20S proteasomes and 19S regulators that are bound to (and protected by) HSP70 chaperones. ATP/ubiquitin-dependent protein degradation (catalyzed by the 26S proteasome) is thus halted, and cannot be restored until 26S proteasomes reconstitute, some 2-5 hours after H2O2 exposure. These results are entirely consistent with a moderate accumulation of ubiquitinylated proteins (that can't be degraded) immediately following mild oxidative stress [41].
We propose that the rapid liberation of free 20S core proteasomes, by dissociation of 26S proteasomes, provides an almost instantaneous increase in cellular capacity to degrade oxidatively modified proteins, since the 20S proteasome (and not the 26S proteasome) is largely responsible for degrading most oxidized intracellular proteins [1, 2, 6-10, 16-18]. The requirement for HSP70 in this process suggests that it is largely cytoplasmic in nature. When combined with the almost equally rapid and direct activation of nuclear 20S proteasome by poly-ADP ribose polymerase [2], this provides cells with a quick and (relatively) simple way to significantly increase proteolytic capacity to prevent the aggregation and accumulation of oxidized proteins.
Long-term loss of 26S proteasome, however, would abrogate the ubiquitin-proteasome system, causing widespread disruptions in cellular metabolism [42, 43]. It is also hard to imagine how cells could mount a coordinated adaptive response to stress, involving transcription, translation, and turnover of perhaps 100 shock/stress proteins without the ubiquitin-26S proteasome system. Therefore, it may not be surprising that as the 26S proteasomes are reconstituted, 20S proteasomes are actively synthesized de novo and can continue to remove oxidized proteins. In MEF cells, we (BJ 2010) have previously shown that the PA28αβ (or 11S) 20S proteasome activator is synthesized over several hours following oxidative stress, and also contributes to the clearance of oxidized proteins, and to cell survival. In addition, we [1], and others [44-46] have reported the induction of immunoproteasome synthesis following oxidative stress, and we [1] have shown that this inducible proteasome form also contributes both to the clearance of oxidized proteins, and to overall cell survival.
The ability to survive and adapt to various stresses is an important capability of all successful life forms. Previous oxidative stress adaptational studies have largely concentrated on regulation of gene expression and transcription/translation of key proteins over several hours to days, rather than on any (potential) immediate survival responses. We suggest that the initial survival of oxidative stress may require rapid removal of oxidatively damaged cell proteins (to prevent their aggregation and accumulation), which may depend heavily on the immediate doubling of cytoplasmic 20S proteasome capacity by HSP70-mediated dissociation of 26S proteasomes (Fig. 8). This may be augmented by poly-ADP ribose-dependent activation of nuclear proteasome [2], and by 20S proteasome activation by (pre-existing) PA28αβ [1]. Subsequent stress adaptation (hormesis) clearly requires de novo synthesis of 20S proteasome, immunoproteasome, and PA28αβ [1]. Successful adaptation must also require reconstitution of 26S proteasomes, and reactivation of the ATP-ubiquitin proteasome pathway, or normal cell metabolism could not be reestablished. Although we have shown that dissociation and reassociation of the 19S and 20S components of the 26S proteasome requires HSP70, the exact mechanism by which this occurs is not yet clear. Although HSP70 appears necessary for this process, for example, is it also sufficient, or are other mediators such as Ecm29 [39], or perhaps other chaperones also required? Does HSP70 actually conduct dissociation of 26S proteasomes, or is it simply required to protect 19S complexes from proteolytic digestion after dissociation? Does HSP70 have a role in reuniting 19S regulators with 20S proteasome components, or does some other shock or stress protein, or Ecm29 [39] conduct this reunification? We are now trying to test these important questions in various model systems.
Materials & Methods
Cell cultivation
K562 cells (human chronic myelogenous leukaemia) were obtained from American Tissue and Cell Culture (A.T.C.C, CCL243). The cells were cultured in 90% RPMI 1640 medium, supplemented with 10% (v/v) fetal bovine serum. Cells were initially seeded at a density of 0.4 × 106 cells/ml.
Cell harvesting
Cells were washed twice and then lysed by repeated cycles of freezing and thawing, in a solution consisting of 0.25 M sucrose, 25 mM Hepes, 10 mM MgCl2, 1 mM EDTA and 1 mM dithiothreitol.
Protein turnover in intact K562 Cells
Human hematopioetic K562 cells were cultivated under standard conditions in RPMI 1640 until they reached a density of 5 × 106 cells/ml. After washing, cells were metabolically radio-labeled, in a pulse-chase procedure, with [35S] -Cys/Met (50 μCi/ml] for 16 h at a 10 fold higher cell density in Met-free medium. Afterwards the incorporation was stopped by adding a 9 fold volume of complete RPMI (containing ‘cold’ Met) tissue culture medium. This time point was taken as zero. Cells were then centrifuged and either phosphate-buffered saline (PBS), or the indicated concentrations of oxidants in PBS were added. Cells were incubated for 1h to 24 h and the acid-soluble radioactivity was measured by β-scintillation counting after cell lysis and precipitation with an equal volume of 20% trichloroacetic acid (TCA). Percent proteolysis was calculated, as previously [8], as [acid-soluble counts - background counts] × 100 / [total incorporated counts - background counts].
Proteasome activity
To measure the capacity to degrade oxidized hemoglobin, human hematopioetic K562 cells were cultivated in RPMI until they reached a density of 5 × 106 cells/ml. After washing, cells were used as controls, or were treated with 0.5 mM hydrogen peroxide, 20 μM paraquat, 20 μM menadione or 1 mM Sin-1. After incubation for a further 30 min to 24 hours, cells were washed and then lysed by repeated cycles of freezing and thawing, in a solution consisting of 0.25 M sucrose, 25 mM Hepes, 10 mM MgCl2, 1 mM EDTA and 1 mM dithiothreitol. Cell lysates were centrifuged at 14,000 × g for 30 min to remove nonlysed cells, membranes, and nuclei. Supernatants were then incubated in 225 mM Tris buffer (pH 7.8) containing 7.5 mM MgOAc, 7.5 mM MgCl2, 45 mM KCl, and 1 mM dithiothreitol. Hemoglobin was tritium radio-labeled by reductive methylation, oxidized by exposure to H2O2, and then added to cell extracts for measurement of proteolytic capacity, as previously described [8, 13, 16]. Degradation of [3H] Hb was quantified, after TCA precipitation (using albumin as carrier) by liquid scintillation. Percent hydrolysis was calculated as [acid-soluble counts – background counts] × 100 / [total incorporated counts – background counts].
The rate of degradation of fluoregenically labeled Suc-LLVY-AMC was measured as previously described [8, 13, 16]. Briefly, cell lysates were centrifuged at 14,000 × g for 30 min to remove nonlysed cells, membranes, and nuclei. Supernatants were incubated in 225 mM Tris buffer (pH 7.8) containing 7.5 mM MgOAc, 7.5 mM MgCl2, 45 mM KCl, and 1 mM dithiothreitol. The fluorogenic peptide Succinyl-LLVY-7-Amino-4-Methylcoumarin was used as substrate, at a concentration of 200 μM, to measure chymotrypsin-like activity of the proteasome. For all measurements of ATP-stimulated proteolysis, 5 mM MgCl2 and 5 mM ATP were added to reaction mixtures containing ATP-depleted cell lysates. After 30 min of incubation at 37 °C, methyl coumarin liberation was measured with a fluorescence reader (360 nm excitation/485 nm emission) and calculated using free methyl coumarin as a standard.
Protein carbonyl ELISA
Protein carbonyl content was taken as a measure for protein oxidation. Carbonyls were determined in cell lysates (1 mg/ml in lysis buffer with 1 mM butylated hydroxytoluene) by an enzyme-linked immunosorbent assay as introduced by Buss et al. [47] with modifications of Voss et al. [48]. Primary anti-DNP-rabbit-IgG antiserum (Sigma) and a secondary monoclonal anti-rabbit peroxidase-conjugated IgG (Sigma) were used as detection system. Development was performed with o-phenylene diamine and H2O2. Absorbance was measured at 492 nm using a microplate reader (BioTek Instruments EL 340).
Immunoprecipitation
Cell lysates were prepared as described above and immunoprecipitation of proteasomes and MSS1 proteasome 19S regulator subunits was performed according to Zwilling et al. [49]. Briefly, after pre-absorption of the cell lysates with 10% protein A– Sepharose (Amersham) equilibrated 1:1 with 50 mM Tris (pH 8.0), 150 mM NaCl and 0.1% bovine serum albumin (BSA). A primary antibody reaction was performed with 100 μg lysate protein, 100 μL Tris/NaCl buffer (50 mM Tris, 100 mM NaCl, pH 8.0), 0.1% BSA and 4 μg of the antibody directed against the 20S ‘core’ proteasome subunits or the MSS-1 regulator subunit for 2 h at 4°C in a total volume of 200 μL. For secondary antibody reactions, 4 μg anti-rabbit IgG or anti-mouse IgG was added and incubated for 2 h at 4°C. Precipitation was initiated by the addition of 20 μl protein A–Sepharose (equilibrated as above) for 1 h at 4_C. Controls were incubated only with secondary antibody and protein A-Sepharose. The Sepharose was centrifuged (12,000 × g for 30 s) and washed five times with Tris-buffered saline. Sepharose-bound proteins were separated by SDS-PAGE (12% gel) and transblotted on to nitro-cellulose membranes. Membranes were blocked with 5% nonfat dry milk in Tris-buffered saline (TBS) for 90 min with shaking at room temperature. Primary antibodies, either a rabbit polyclonal anti-20S ‘core’ proteasome antibody, a rabbit anti-19S proteasome antibody (MSS-1; Affiniti) or an anti HSP70, were applied overnight at 4 °C. After three washes in TBS supplemented with 0.05% Tween 20, secondary antibody (peroxidase-conjugated IgG) was applied for 1 h. The blots were then washed three times in TBS/ 0.05% Tween 20 and once in TBS, incubated in enhanced chemiluminescence reagent (Supersignal Chemiluminescence Substrate; Pierce), and exposed to photographic film. Control samples that contained secondary antibody plus Sepharose A, or Sepharose A alone, in the immunoprecipitation procedure produced no visible bands in the gels.
Acknowledgments
This research was supported by grant #RO1-ES003598, and by ARRA Supplement 3RO1-ES 003598-22S2, both from the NIH/NIEHS to KJAD.
Abbreviations
- MEF
murine embryonic fibroblasts
- H2O2
hydrogen peroxide
- PA28αβ
a proteasome regulator (also called the 11S regulator)
- PrOxI hypothesis
Protein Oxidation and Immunoproteasome hypothesis of MHC Class I antigen processing (see reference # 45)
- Hb
hemoglobin
- TCA
trichloroacetic acid
- PBS
phosphate-buffered saline
- TCA
trichloroacetic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pickering AM, Koop AL, Teoh CY, Ermak G, Grune T, Davies KJA. The immunoproteasome, the 20S proteasome and the PA28alphabeta proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochem J. 2010;432:585–594. doi: 10.1042/BJ20100878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ullrich O, Reinheckel T, Sitte N, Hass R, Grune T, Davies KJA. Poly-ADP ribose polymerase activates nuclear proteasome to degrade oxidatively damaged histones. Proc Natl Acad Sci U S A. 1999;96:6223–6228. doi: 10.1073/pnas.96.11.6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanaka K, Kasahara M. The MHC class I ligand-generating system: roles of immunoproteasomes and the interferon-gamma-inducible proteasome activator PA28. Immunol Rev. 1998;163:161–176. doi: 10.1111/j.1600-065x.1998.tb01195.x. [DOI] [PubMed] [Google Scholar]
- 4.Ma CP, Slaughter CA, DeMartino GN. Identification, purification, and characterization of a protein activator (PA28) of the 20 S proteasome (macropain) J Biol Chem. 1992;267:10515–10523. [PubMed] [Google Scholar]
- 5.Ahn K, Erlander M, Leturcq D, Peterson PA, Fruh K, Yang Y. In vivo characterization of the proteasome regulator PA28. J Biol Chem. 1996;271:18237–18242. doi: 10.1074/jbc.271.30.18237. [DOI] [PubMed] [Google Scholar]
- 6.Davies KJA, Goldberg AL. Proteins damaged by oxygen radicals are rapidly degraded in extracts of red blood cells. J Biol Chem. 1987;262:8227–8234. [PubMed] [Google Scholar]
- 7.Pacifici RE, Kono Y, Davies KJA. Hydrophobicity as the signal for selective degradation of hydroxyl radical-modified hemoglobin by the multicatalytic proteinase complex, proteasome. J Biol Chem. 1993;268:15405–15411. [PubMed] [Google Scholar]
- 8.Grune T, Reinheckel T, Davies KJA. Degradation of oxidized proteins in K562 human hematopoietic cells by proteasome. J Biol Chem. 1996;271:15504–15509. doi: 10.1074/jbc.271.26.15504. [DOI] [PubMed] [Google Scholar]
- 9.Davies KJA. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83:301–310. doi: 10.1016/s0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- 10.Whittier JE, Xiong Y, Rechsteiner MC, Squier TC. Hsp90 enhances degradation of oxidized calmodulin by the 20 S proteasome. J Biol Chem. 2004;279:46135–46142. doi: 10.1074/jbc.M406048200. [DOI] [PubMed] [Google Scholar]
- 11.Rivett AJ, Levine RL. Metal-catalyzed oxidation of Escherichia coli glutamine synthetase: correlation of structural and functional changes. Arch Biochem Biophys. 1990;278:26–34. doi: 10.1016/0003-9861(90)90226-o. [DOI] [PubMed] [Google Scholar]
- 12.Friguet B, Bulteau AL, Chondrogianni N, Conconi M, Petropoulos I. Protein degradation by the proteasome and its implications in aging. Ann N Y Acad Sci. 2000;908:143–154. doi: 10.1111/j.1749-6632.2000.tb06643.x. [DOI] [PubMed] [Google Scholar]
- 13.Reinheckel T, Grune T, Davies KJA. The measurement of protein degradation in response to oxidative stress. Methods Mol Biol. 2000;99:49–60. doi: 10.1385/1-59259-054-3:49. [DOI] [PubMed] [Google Scholar]
- 14.Mao I, Liu J, Li X, Luo H. REGgamma, a proteasome activator and beyond? Cell Mol Life Sci. 2008;65:3971–3980. doi: 10.1007/s00018-008-8291-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 16.Shringarpure R, Grune T, Mehlhase J, Davies KJA. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J Biol Chem. 2003;278:311–318. doi: 10.1074/jbc.M206279200. [DOI] [PubMed] [Google Scholar]
- 17.Chondrogianni N, Stratford FL, Trougakos IP, Friguet B, Rivett AJ, Gonos ES. Central role of the proteasome in senescence and survival of human fibroblasts: induction of a senescence-like phenotype upon its inhibition and resistance to stress upon its activation. J Biol Chem. 2003;278:28026–28037. doi: 10.1074/jbc.M301048200. [DOI] [PubMed] [Google Scholar]
- 18.Fucci L, Oliver CN, Coon MJ, Stadtman ER. Inactivation of key metabolic enzymes by mixed-function oxidation reactions: possible implication in protein turnover and ageing. Proc Natl Acad Sci U S A. 1983;80:1521–1525. doi: 10.1073/pnas.80.6.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–1156. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- 20.Hiller MM, Finger A, Schweiger M, Wolf DH. ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science. 1996;273:1725–1728. doi: 10.1126/science.273.5282.1725. [DOI] [PubMed] [Google Scholar]
- 21.Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding Q, Dimayuga E, Keller JN. Proteasome regulation of oxidative stress in aging and age-related diseases of the CNS. Antioxid Redox Signal. 2006;8:163–172. doi: 10.1089/ars.2006.8.163. [DOI] [PubMed] [Google Scholar]
- 23.Reinheckel T, Sitte N, Ullrich O, Kuckelkorn U, Davies KJA, Grune T. Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem J. 1998;335(Pt 3):637–642. doi: 10.1042/bj3350637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reinheckel T, Ullrich O, Sitte N, Grune T. Differential impairment of 20S and 26S proteasome activities in human hematopoietic K562 cells during oxidative stress. Arch Biochem Biophys. 2000;377:65–68. doi: 10.1006/abbi.2000.1717. [DOI] [PubMed] [Google Scholar]
- 25.Shang F, Taylor A. Oxidative stress and recovery from oxidative stress are associated with altered ubiquitin conjugating and proteolytic activities in bovine lens epithelial cells. Biochem J. 1995;307(Pt 1):297–303. doi: 10.1042/bj3070297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor A, Davies KJA. Protein oxidation and loss of protease activity may lead to cataract formation in the aged lens. Free Radic Biol Med. 1987;3:371–377. doi: 10.1016/0891-5849(87)90015-3. [DOI] [PubMed] [Google Scholar]
- 27.Jakob U, Muse W, Eser M, Bardwell JC. Chaperone activity with a redox switch. Cell. 1999;96:341–352. doi: 10.1016/s0092-8674(00)80547-4. [DOI] [PubMed] [Google Scholar]
- 28.Wiese AG, Pacifici RE, Davies KJA. Transient adaptation of oxidative stress in mammalian cells. Arch Biochem Biophys. 1995;318:231–240. doi: 10.1006/abbi.1995.1225. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka K, Ichihara A. Half-life of proteasomes (multiprotease complexes) in rat liver. Biochem Biophys Res Commun. 1989;159:1309–1315. doi: 10.1016/0006-291x(89)92253-5. [DOI] [PubMed] [Google Scholar]
- 30.Powell SR, Davies KJA, Divald A. Optimal determination of heart tissue 26S-proteasome activity requires maximal stimulating ATP concentrations. J Mol Cell Cardiol. 2007;42:265–269. doi: 10.1016/j.yjmcc.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jornot L, Mirault ME, Junod AF. Differential expression of hsp70 stress proteins in human endothelial cells exposed to heat shock and hydrogen peroxide. Am J Respir Cell Mol Biol. 1991;5:265–275. doi: 10.1165/ajrcmb/5.3.265. [DOI] [PubMed] [Google Scholar]
- 32.Kwak MK, Wakabayashi N, Greenlaw JL, Yamamoto M, Kensler TW. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol Cell Biol. 2003;23:8786–8794. doi: 10.1128/MCB.23.23.8786-8794.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manejwala FM, Logan CY, Schultz RM. Regulation of hsp70 mRNA levels during oocyte maturation and zygotic gene activation in the mouse. Dev Biol. 1991;144:301–308. doi: 10.1016/0012-1606(91)90423-z. [DOI] [PubMed] [Google Scholar]
- 34.Baler R, Welch WJ, Voellmy R. Heat shock gene regulation by nascent polypeptides and denatured proteins: hsp70 as a potential autoregulatory factor. J Cell Biol. 1992;117:1151–1159. doi: 10.1083/jcb.117.6.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mizuno S, Ishii A, Murakami Y, Akagawa H. Stress dose-dependent suppression of heat shock protein gene expression by inhibiting protein synthesis during heat shock treatment. Cell Struct Funct. 1997;22:7–13. doi: 10.1247/csf.22.7. [DOI] [PubMed] [Google Scholar]
- 36.Siriani D, Mitsiou DJ, Alexis MN. Heat-induced degradation of overexpressed glucocorticoid receptor Separate protective roles of hsp90 and hsp70. J Steroid Biochem Mol Biol. 2005;94:93–101. doi: 10.1016/j.jsbmb.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 37.Mambula SS, Calderwood SK. Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J Immunol. 2006;177:7849–7857. doi: 10.4049/jimmunol.177.11.7849. [DOI] [PubMed] [Google Scholar]
- 38.Lee YJ, Erdos G, Hou ZZ, Cho JM. Cycloheximide Decreases Nascent Polypeptides Level and Affects up-Regulation of Hsp70 Gene-Expression. Journal of Thermal Biology. 1994;19:327–333. [Google Scholar]
- 39.Wang X, Yen J, Kaiser P, Huang L. Regulation of the 26S proteasome complex during oxidative stress. Sci Signal. 3:ra88. doi: 10.1126/scisignal.2001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bulteau AL, Lundberg KC, Humphries KM, Sadek HA, Szweda PA, Friguet B, Szweda LI. Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J Biol Chem. 2001;276:30057–30063. doi: 10.1074/jbc.M100142200. [DOI] [PubMed] [Google Scholar]
- 41.Figueiredo-Pereira ME, Yakushin S, Cohen G. Disruption of the intracellular sulfhydryl homeostasis by cadmium-induced oxidative stress leads to protein thiolation and ubiquitination in neuronal cells. J Biol Chem. 1998;273:12703–12709. doi: 10.1074/jbc.273.21.12703. [DOI] [PubMed] [Google Scholar]
- 42.Layfield R, Lowe J, Bedford L. The ubiquitin-proteasome system and neurodegenerative disorders. Essays Biochem. 2005;41:157–171. doi: 10.1042/EB0410157. [DOI] [PubMed] [Google Scholar]
- 43.Wang X, Guerrero C, Kaiser P, Huang L. Proteomics of proteasome complexes and ubiquitinated proteins. Expert Rev Proteomics. 2007;4:649–665. doi: 10.1586/14789450.4.5.649. [DOI] [PubMed] [Google Scholar]
- 44.Ferrington DA, Hussong SA, Roehrich H, Kapphahn RJ, Kavanaugh SM, Heuss ND, Gregerson DS. Immunoproteasome responds to injury in the retina and brain. J Neurochem. 2008;106:158–169. doi: 10.1111/j.1471-4159.2008.05345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ding Q, Reinacker K, Dimayuga E, Nukala V, Drake J, Butterfield DA, Dunn JC, Martin S, Bruce-Keller AJ, Keller JN. Role of the proteasome in protein oxidation and neural viability following low-level oxidative stress. FEBS Lett. 2003;546:228–232. doi: 10.1016/s0014-5793(03)00582-9. [DOI] [PubMed] [Google Scholar]
- 46.Kotamraju S, Matalon S, Matsunaga T, Shang T, Hickman-Davis JM, Kalyanaraman B. Upregulation of immunoproteasomes by nitric oxide: potential antioxidative mechanism in endothelial cells. Free Radic Biol Med. 2006;40:1034–1044. doi: 10.1016/j.freeradbiomed.2005.10.052. [DOI] [PubMed] [Google Scholar]
- 47.Buss H, Chan TP, Sluis KB, Domigan NM, Winterbourn CC. Protein carbonyl measurement by a sensitive ELISA method. Free Radic Biol Med. 1997;23:361–366. doi: 10.1016/s0891-5849(97)00104-4. [DOI] [PubMed] [Google Scholar]
- 48.Voss P, Horakova L, Jakstadt M, Kiekebusch D, Grune T. Ferritin oxidation and proteasomal degradation: protection by antioxidants. Free Radic Res. 2006;40:673–683. doi: 10.1080/10715760500419357. [DOI] [PubMed] [Google Scholar]
- 49.Zwilling S, Konig H, Wirth T. High mobility group protein 2 functionally interacts with the POU domains of octamer transcription factors. Embo J. 1995;14:1198–1208. doi: 10.1002/j.1460-2075.1995.tb07103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robertson JD, Datta K, Biswal SS, Kehrer JP. Heat-shock protein 70 antisense oligomers enhance proteasome inhibitor-induced apoptosis. Biochem J. 1999;344(Pt 2):477–485. [PMC free article] [PubMed] [Google Scholar]