

Abstract
The σ initiation factor σ70 of Escherichia coli acts not only in promoter recognition and DNA strand opening, but also to mediate the transformation of RNA polymerase (RNAP) to an antiterminating form by the phage λ gene Q protein. Q is able to bind and modify RNAP when α70, still present in the initially elongating enzyme, recognizes a repeat of the −10 promoter element and induces a transcription pause. We have isolated mutations in the rpoD gene for σ70 that impair Q function because they reduce the ability of σ70 to recognize the downstream pause site. These mutations identify a locus of σ70 that is important for the formation and stability of open promoter complex, likely because it mediates protein interactions with RNAP core.
Keywords: σ factor, promoter, antitermination
σ factors of bacterial RNA polymerase (RNAP) mediate promoter recognition and opening in transcription initiation (for review, see Gross et al. 1992). They act in the form of a holoenzyme, a complex with the universally conserved RNAP core enzyme E (composed of subunits α2ββ′); thus Eσ70 is the holoenzyme of the primary σ factor of Escherichia coli, σ70. Through conformational changes of an initial closed complex, Eσ70 mediates DNA strand separation to form an open complex that can template RNA chain synthesis (McClure 1985). During in vitro synthesis, σ70 becomes dispensable and can dissociate after transcripts reach 5–10 nucleotides in length (Hansen and McClure 1980), leaving the core to elongate RNA chains; thus, σ70 (and other σs) functions primarily in initiation.
In a variant of this simple initiation sequence, σ70 also acts during an early step of elongation to induce a paused transcription complex that is necessary for engagement of the bacteriophage λ gene Q antiterminator, the positive regulator that ensures lytic development in phage growth (Roberts 1992; Ring et al. 1996). Pausing is induced at RNA nucleotides +16 and +17 of the transcript of phage late gene promoter pR′ when σ70 recognizes a repeat of critical bases of the −10 promoter sequence that is displaced 11 bp downstream (Ring et al. 1996) (Fig. 1A). At both the promoter −10 sequence and the downstream pause-inducing sequence, Eσ70 recognizes primarily the nontemplate strand of DNA (Ring and Roberts 1994; Ring et al. 1996; Roberts and Roberts 1996; Marr and Roberts 1997); both the open and paused complexes are stabilized by this single-strand DNA-Eσ70 interaction. Q binds the paused complex by contacting DNA of the qut site, located between the −35 and −10 promoter elements, as well as unknown portions of RNAP (Fig. 1A). This interaction chases RNAP from the pause, allows incorporation of Q as a component of the elongation complex [at least for the related Q protein of phage 82 (W.S. Yarnell and J.W. Roberts, unpubl.)], and fundamentally alters the elongation property of RNAP so that it reads through terminators. σ70 is released at an unknown stage, but presumably early after Q engagement.
Figure 1.
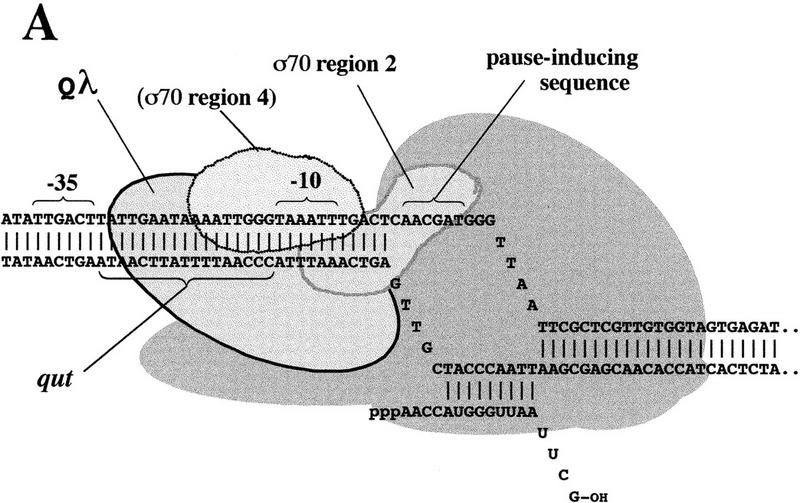

(A) Paused complex of RNAP transcribing the λ late gene promoter. Region 2 of σ70 is placed to interact with the nontemplate strand of the pause-inducing sequence (AACGAT, in which underlining indicates match to the −10 promoter sequence consensus). Qλ is shown bound to the qut site. The location of σ70 region 4 is unknown, but it is placed to suggest interaction with Qλ (H. Sun and J.W. Roberts, unpubl.). The complex is shown in a backtracked form (Reeder and Hawley 1996; Komissarova and Kashlev 1997; Nudler et al. 1997) to account for its sensitivity to cleavage mediated by GreB (W. Yarnell and J.W. Roberts, unpubl.). (B) Map of the σ70 polypeptide, and structure of the region 2.1, 2.2, and 2.4 α-helix bundle of σ70 (Malhotra et al. 1996). Residues T440 and Q437 are believed to make base-specific contacts with promoter DNA of the nontemplate strand, and W433 and W434 to mediate DNA melting; residues L402, N409, and M413 are discussed in the text. (Inset) The structure rotated to view the helices from the left side, nearly end on.
It is not known how σ70 acts to allow Q engagement. The presence of σ70 bound to the pause-inducing sequence is important per se and not simply to induce a pause: RNAP stopped at +16/+17 on a template carrying a mutant pause-inducing sequence, so that σ70 has dissociated or can be easily displaced, is not receptive to Q modification (Yarnell and Roberts 1992). Two nonexclusive possibilities are that σ70 alters the conformation of the rest of the transcription complex to make it receptive to Q modification, and that σ70 recruits Q through direct protein–protein interactions.
This activity provides an opportunity to find elements of σ70 important for Q activity, and, more important, for development of base-specific nontemplate strand DNA contacts in pausing and promoter recognition. For this purpose, we used a genetic screen to identify amino acid substitutions in σ70 that impair Q-mediated antitermination. We find that a major class of these mutations occurs on a surface in region 2.2 of σ70, the most highly conserved region of the polypeptide (Gross et al. 1992). We show that these mutations reduce the promoter proximal pause, an effect that can explain their interference with Q function. Furthermore, as might be expected from the similarity of the paused and open complexes, these mutations also reduce the stability of the open promoter complex. Our results, therefore, identify a surface of σ70 that is important to the structure of the open promoter complex, likely through contacts σ70 makes with core RNAP during the closed- to open-complex transition; this surface also may be part of the σ70-core contacts that form holoenzyme (Malhotra et al. 1996). In addition, the identification of σ70 mutations provides genetic confirmation of the role of σ70 in antitermination and in steps of transcription after initiation of RNA chains.
Results
A genetic screen to identify amino-acid substitutions in σ70 that result in reduced Q antitermination activity
To measure antitermination by Q protein in vivo, and to provide a screen for mutations that alter Q function, we constructed a reporter strain that contains a fusion of pR′, two intrinsic terminators, and lacZ integrated into the E. coli chromosome on a prophage. This strain was transformed with an IPTG-inducible Q expression plasmid, so that IPTG-dependent lacZ expression reports Q function. To search for σ70 mutants that impair Q function, we mutagenized the rpoD gene (encoding σ70) in two halves on the multicopy plasmid pHTσ by PCR random mutagenesis (Zhou et al. 1991), introduced mutagenized plasmids into the reporter strain, and screened by colony color for defective Q function in conditions of lacZ induction. The rpoD gene of pHTσ is also controlled by the lac operator, producing 4–8 times the natural level of σ70 on IPTG induction (D. Ko and J.W. Roberts, unpubl.) The presence of this increased level of wild-type σ70 did not affect Q-dependent expression of lacZ. Note that the mutants recovered could not be dominant lethals for cell growth, and had to compete effectively with the endogenous σ70 for core enzyme to express a phenotype.
We isolated 10 mutations in the carboxy-terminal half of rpoD that support <70% wild-type Q function. We consider here one class of mutations that includes the only two which retain viability without an additional wild-type σ70 gene (L402F and L409D), and thus affect Q function without destroying essential σ70 activity; we include a third (M413T) that is nearby, but has only slight effect in the original reporter assay. Mutations L402F, N409D, and M413T occur in a single stretch of region 2.2 in the atomic structure of residues 114–448 of σ70 (Malhotra et al. 1996), located on one side of a strongly bound three α-helix bundle of σ70 (Fig. 1B). The bundle includes part of helix 12a and helix 12b, which contains region 2.1, believed to be the major core enzyme-binding region (Lesley and Burgess 1989); helix 13, which contains most of region 2.2; and helix 14, which contains parts of regions 2.3 and 2.4. Region 2.4 includes amino acids thought to be involved in base-specific recognition of the −10 element nontemplate DNA strand (Gardella et al. 1989; Kenney et al. 1989; Siegele et al. 1989; Zuber et al. 1989; Roberts and Roberts 1996; Marr and Roberts 1997) and in melting or stabilizing single-stranded DNA (Helmann and Chamberlin 1988; Juang and Helmann 1994). L402, N409, and M413 are on a hydrophobic face of the α-helix 13 (Malhotra et al. 1996) that includes region 2.2 of σ70.
To investigate the influence of the amino acid side chains, we also changed residues 402, 409, and 413 to alanine, to remove important side chain contacts but not introduce new ones (Niu et al. 1994). We also changed to alanine residue Q406, which is the remaining amino acid on the solvent exposed face of helix 13 of region 2.2, but which was not found in the screen. Mutation of the corresponding amino acid (Q80) in σ32 causes a defect in binding to core RNAP (Joo et al. 1997). Like the original mutations, the alanine substitutions support growth in the absence of wild-type σ70 (Fig. 2D and data not shown).
Figure 2.
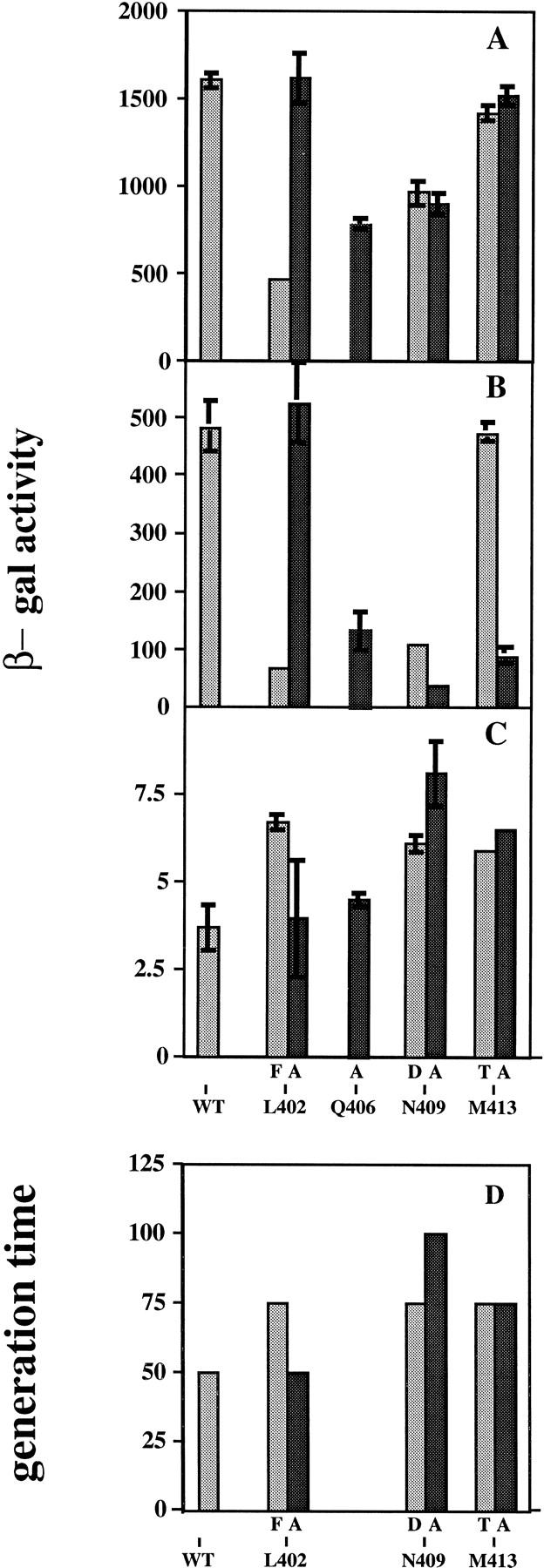
(A, B) Activity of σ70 region 2.2 mutants in Q function; (C) basal expression of pR′; (D) cell growth rate. Each cell contains pHTσ expressing either wild-type or mutant σ70. Q function or basal promoter activity is expressed as β-galactosidase activity from the reporter λ+49 as described. Q is expressed from plasmid pJG100 where it is present. (A) SG20250(pJG100), wild-type σ70 strain background. (B) CAG19284(pJG100), temperature-sensitive lethal σ70 strain background, grown at the restrictive temperature (42°C). (C) CAG19284, at 42°C (no Q present). (D) Growth rate of MTM9973 (recA− version of CAG20176) carrying σ mutant plasmids, in the presence of tryptophan to repress the chromosomal σ70 gene.
The effect of these mutations on Q-dependent β-galactosidase expression in two reporter strains growing uniformly in liquid culture is shown in Figure 2(A,B); expression with wild-type σ70 in the absence of Q is <10 units (Fig. 2C). Figure 2A shows expression levels in the reporter strain used for the selection, in which mutant σ70 competes with wild-type σ70 made from the chromosome; in Figure 2B, the mutants are assayed at 42°C in a strain carrying a temperature-sensitive σ70 (rpoD285), so that only the mutant σ70 is present. First, note that L402F, Q406A, both 409 mutants, and M413A show pronounced defects in Q-dependent lacZ expression in both strains and are more defective in the absence of wild-type σ70 (Fig. 2B). M413T shows slight or no defect in these liquid assays; however, it was visibly mutant in the different physiology of the colony screen, and we show evidence below that it shares defects with L402F and N409D (although more weakly). Note that the L402F, N409D, and M413T mutations show a decreasing severity of defect in this order, and that this relationship is true in other behavior described below.
Because the alanine substitutions at residues 406, 409, and 413 have mutant phenotypes, side-chain atoms beyond the β-carbon are important at these sites. For N409 and M413, the alanine mutations are more severe than the original screened mutations, which presumably retain some favorable interactions. N409D would disrupt a possible hydrogen bond, but might retain other side-chain interactions not possible for alanine; M413T might retain some polar and hydrophobic interactions that would be lost by the change to alanine. Of the four alanine substitutions, only L402A does not decrease Q-activated transcription. In fact, several alternative σ factors, including σ32, have alanine at this amino acid position (Gross et al. 1992). The original mutation obtained in the screen, a change to phenylalanine, introduces a larger side chain that might sterically prevent the normal interaction of this entire surface of σ70.
These mutations could preferentially affect initiation at promoter pR′, or another property of this transcription unit, rather than Q function. However, assay of pR′ in the absence of Q (Fig. 2C) shows that, in contrast to a deficiency, the basal transcription is increased. Furthermore, the increase is related to the severity of the defect in Q function. As discussed below, we ascribe this increased expression to lack of pausing at the promoter proximal site and consequent relief of this partially rate-limiting step in transcription.
The mutations modestly affect the cell growth rate (Fig 2D), thereby increasing the generation time ∼50%. It is most evident for the alanine substitutions that severity of growth defect correlates with deficiency in Q function.
σ70 mutations inhibit phage growth
If the σ70 mutations interfere with Q function, they should impair growth of phage λ. In fact, plaques of phage λc17 grown at 42°C on CG19284 containing pHTσ(L402F) or pHTσ(N409D) are smaller than plaques on CG19284 containing pHTσ+—one-quarter to one-half the diameter.
σ70 mutants reduce Q-activated transcription in vitro
To determine their effect on antitermination in vitro, and to probe the basis of their defects, we purified the mutant σs L402F, N409D, and M413T as His-tagged proteins, reconstituted them into holoenzyme RNAP, and measured terminated versus readthrough transcript from pR′ in the presence and absence of Q protein. In the absence of Q, holoenzyme reconstituted with mutant σ70 gives background terminator readthrough similar to that with wild-type σ70 (2%–3%) (Fig 3). Addition of 100 nm Q to reactions with wild-type Eσ70 produces the expected 50% readthrough, whereas Eσ70 containing mutants L402F, N409D, and M413T gives 8%, 12%, and 17%, respectively (Fig. 3). The relative ability of the three mutant RNAPs to be modified by Q in vitro corresponds to the ability of mutant strains to support Q function in vivo: L402F<N409D<M413T<wild type (Fig. 2).
Figure 3.
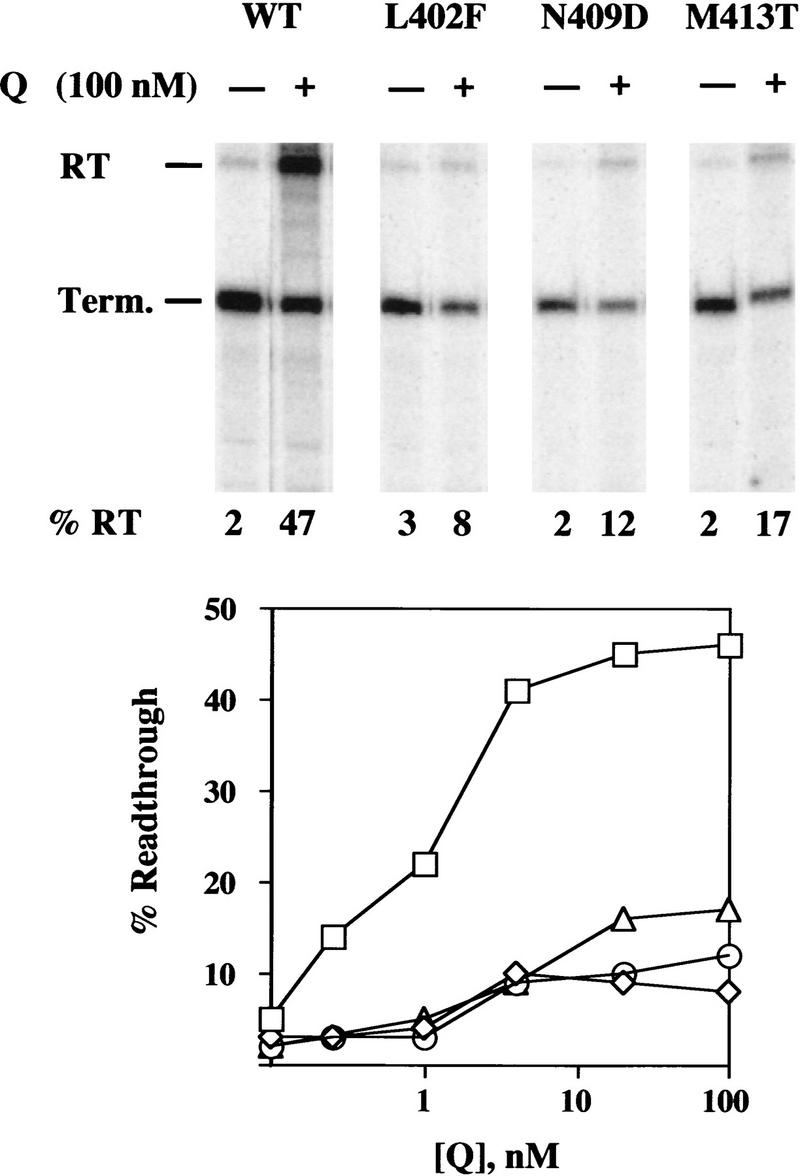
Activity of mutant σ70 proteins in Q-mediated antitermination in 5 min transcription reactions, as described. (□) Wild type; (⋄) L402F; (○) N409D; (▵) M413T.
The antitermination defect of the mutant σs was not overcome by increasing the Q concentration. At Q concentrations between 0.4 nm and 100 nm (saturating for wild type), RNAP reconstituted with mutant σ70 was defective in its ability to be modified by Q (Fig. 3); both wild-type and σ70–mutant RNAP showed about half-maximal Q effect between 1 and 4 nm, despite their different saturation levels. This result suggests that the σ70 mutations do not act by weakening the binding of Q to the paused complex, which should be overcome by higher Q concentration, but instead change the capacity of the transcription complex to be modified.
σ70 mutants reduce promoter-proximal pausing
This reduced capacity for modification by Q might reflect the availability of substrate, namely the paused RNAP. To test this possibility, we analyzed products of single round in vitro transcription of promoter pR′ during a time course of 10 min (Fig 4A). Approximately 73% of wild-type enzyme pauses at the positions +16 and +17, with a half-life of ∼5.9 min. The σ70 mutations reduce both the half-life of the pause—to ∼2 min—and the percentage of RNAP that initially pauses (Fig. 4A). Significantly, the percent capture (percent paused extrapolated to zero time) corresponds to the ability of each mutant protein to support modification by Q. L402F supports the lowest Q activity (17% of wild type) and also has the lowest initial capture rate (30% of wild type), whereas M413T supports the highest Q activity (36% of wild type) and has the highest initial capture rate (75% of wild type) of the three mutants. Thus, these σ70 mutants are deficient in forming and/or maintaining the nontemplate strand binding that stabilizes the pause. The mutant defects follow the same order of severity as before.
Figure 4.
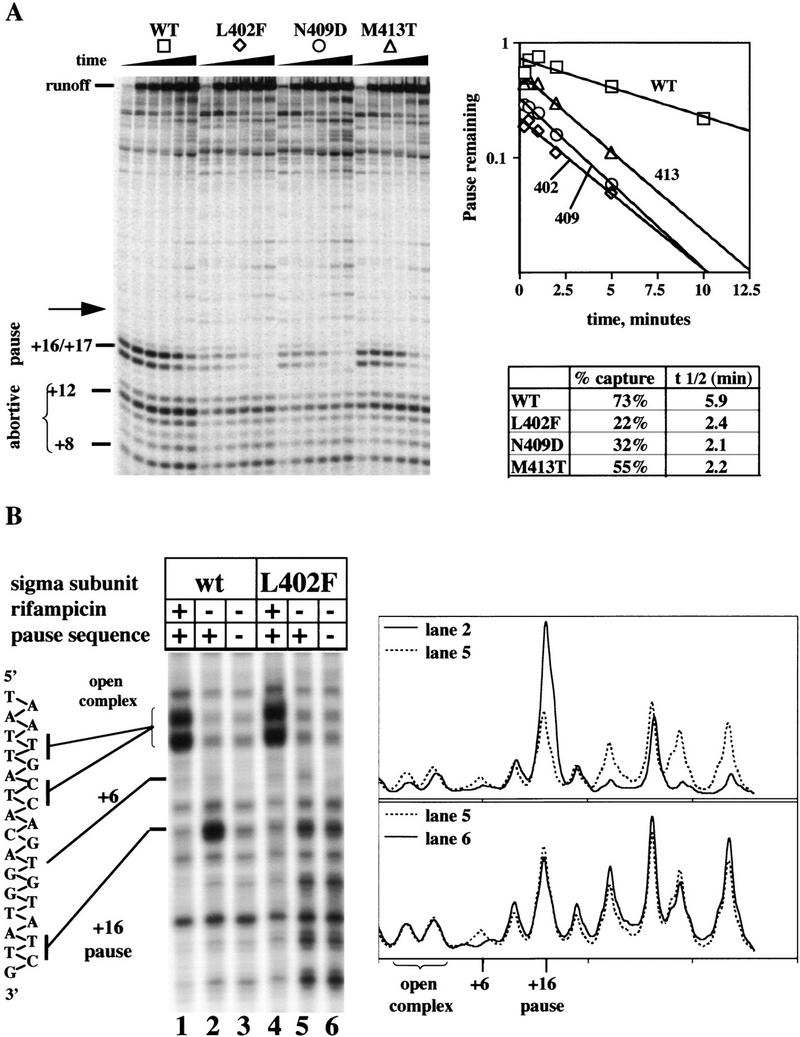
Deficiency of region 2.2 mutants in promoter-proximal pausing. (A) Pausing measured in vitro, in the absence of Q. Transcription reactions constructed as described were sampled at 0.25. 0.5, 1,2,5, and 10 min. (B) Pause detected in vivo by KMnO4 treatment of strain MTM9973 (a recA− derivative of CAG20176) carrying plasmid pBAD33spc rpoD expressing either rpoD+ or rpoDL402F, and a plasmid with pR′ or the nonpausing derivative pR′(+6C). Rifampicin was added 5 min before KMnO4 treatment in lanes 1 and 4.
The same pausing defect is detectable in vivo. Treatment of a growing culture with KMnO4 marks bases in the single-strand transcription bubble, thus revealing the pause (Kainz and Roberts 1992). Figure 4B shows KMnO4 analysis of the nontemplate strand around p-R′ in growing cultures containing wild-type or L402F Eσ70. Lanes 1 and 4 illustrate cultures treated with rifampicin to collect RNAP on the promoter, revealing open complex reactivity at −2 through −8. Lanes 2 and 5 show cultures without rifampicin, revealing pause bubble reactivity at +14 to +16. Comparison of lanes 2 and 3 shows that a +6C mutation destroying the pausing consensus eliminates the +16 pause signal in wild type, as expected. Signal in the pause position is weaker by two- to threefold for σ70 L402F (Fig. 4B, cf. lanes 2 and 5). Unexpectedly, there is more signal downstream of the pause site in σ70 L402F than in wild type; this reactivity represents active transcription complexes, because it is dispersed on incubation with rifampicin (Fig 4B, lane 4). Apparently, σ70 L402F not only fails to engage the pausing signal efficiently, but also induces elongation anomalies farther downstream. Presumably the signal that does appear in L402F at the pause position is part of the generalized downstream reactivity and not pause signal recognition, because it is not reduced by the +6C mutation (Fig. 4B, cf. lanes 5 and 6). Traces of this anomalous pausing may appear in in vitro transcription as well (Fig. 4A, cf. bands at arrow in lane 1 of each set).
L402F and N409D affect the rates of dissociation and association of the open complex
Because pausing at +16/+17 reflects the same binding between nontemplate strand DNA sequence and Eσ70 that also stabilizes the open complex, helix 13 mutations should affect the stability of the open complex. To show this, we used the method of Zhang et al. (1996), who showed that native gel electrophoresis traps open complex and freezes the distribution of free and bound promoter in a reaction mixture. We formed open complex by incubating labeled DNA with excess Eσ70 for 10 min at 37°C and measured dissociation during incubation at 17°C in the presence of excess competitor DNA (Fig. 5); under these conditions, the half-life for dissociation of open complex of wild-type RNAP is ∼30 min (Roberts and Roberts 1996). Open complex made with either L402F or N409D is severalfold less stable than wild-type complex (Fig. 5A,B); L402F is the least stable, consistent with its stronger phenotype for pausing and Q function. M413T is indistinguishable from wild type, consistent with its milder phenotype and with its similar yield of abortive products (see below).
Figure 5.
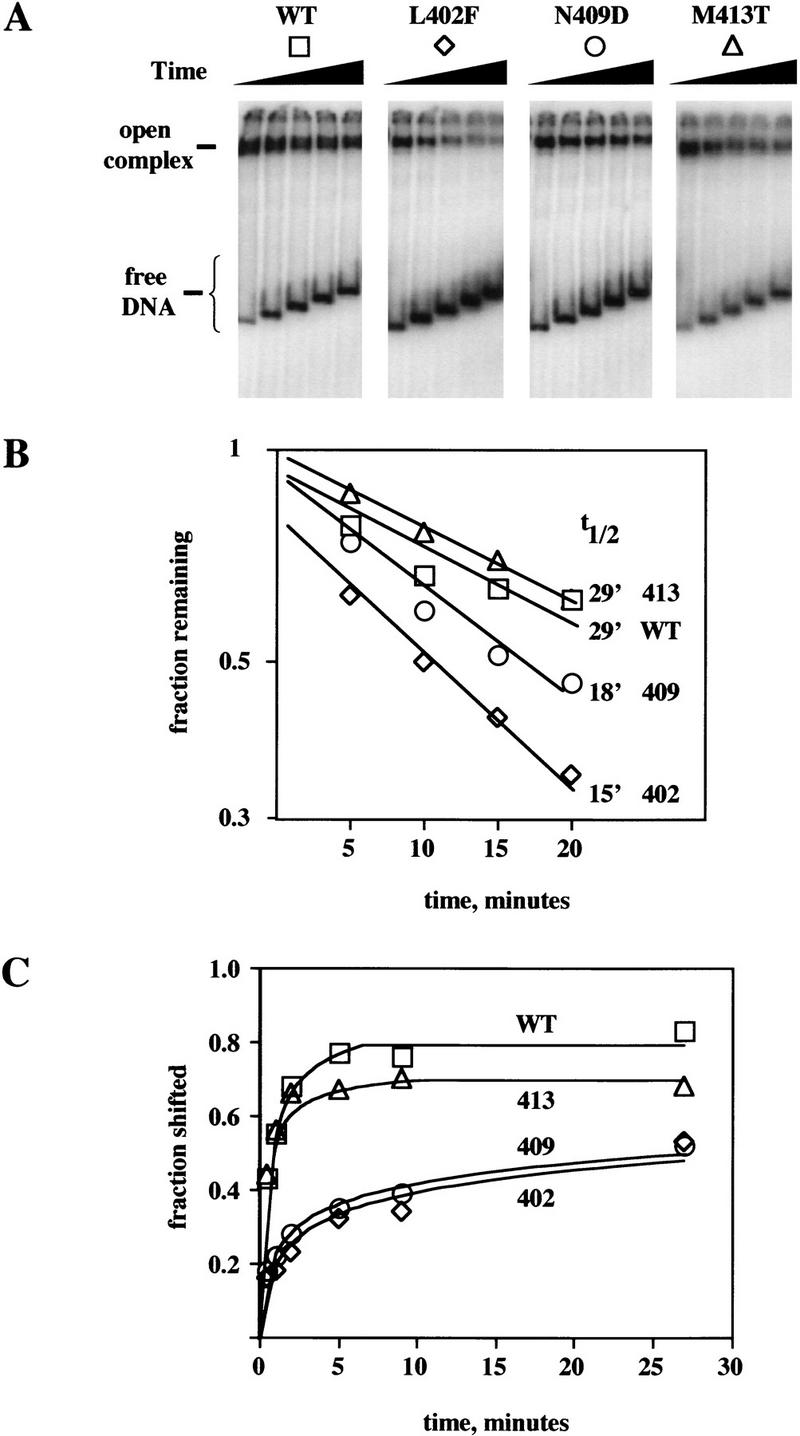
Effect of region 2.2 mutations on rate of open promoter complex dissociation and formation by Eσ70. (A) Gel analysis of dissociation kinetics of preformed complexes on promoter pR′. The DNA fragment was as described, except that it contained a −12 T–C mutation of promoter pR′ to weaken the complex and accentuate the rate of decay. (B) Graphical analysis of A. (C) Association of Eσ70 with DNA containing (wild type) promoter pR′. (□) Wild type; (⋄) L402F; (○) N409D; (▵) M413T.
We also measured the rate of association of Eσ70 and pR′ promoter DNA to form open complex (Fig. 5C). Wild-type and M413T holoenzyme show similar association kinetics, reasonably consistent with the expected first order capture of free DNA into complex at RNAP excess, and saturating by 5 min. In contrast, L402F and N409D give apparently biphasic curves, with a rapid rise in the first minute followed by a much slower rise. Possibly part of the initial closed complex diverts into a nonproductive pathway that is only slowly reversible; whatever the precise explanation, it is clear that L402F and N409D are defective in open complex formation. Similar kinetic anomalies have been found previously for weak RNAP–promoter interactions (Zhang et al. 1996).
L402F and N409D reduce abortive transcription
The yield of abortive transcripts, which are produced by repeated synthesis of oligonucleotides in the open complex (Johnston and McClure 1976), reflects open complex stability, because a less stable complex can escape into productive elongation after fewer cycles of abortive synthesis. This is true, for example, for nontemplate strand promoter nucleotide changes that affect the Eσ70-DNA interaction (Roberts and Roberts 1996). Inspection of Figure 4A shows a deficiency in abortive synthesis by open complexes with mutant σs, particularly evident as the yield of +12 relative to +8. For a different experiment, we measured the yield of 8-mer and 12-mer abortive RNAs for wild-type and σ70 mutant holoenzyme (Fig. 6). Quantitation of abortive and total elongated transcripts (+16/+17 pause and longer) reveals that +12 abortive transcripts are two- to threefold deficient relative to wild type for L402 and N409D, although normal for M413T (Fig. 6). Thus, the two most severe σ70 mutants show a distinct defect in abortive synthesis. The defect in longer (+12) but not shorter (+8) abortive RNAs might reflect failure of the mutant Eσ70–DNA bond to sustain greater strain associated with greater extension of the catalytic center.
Figure 6.
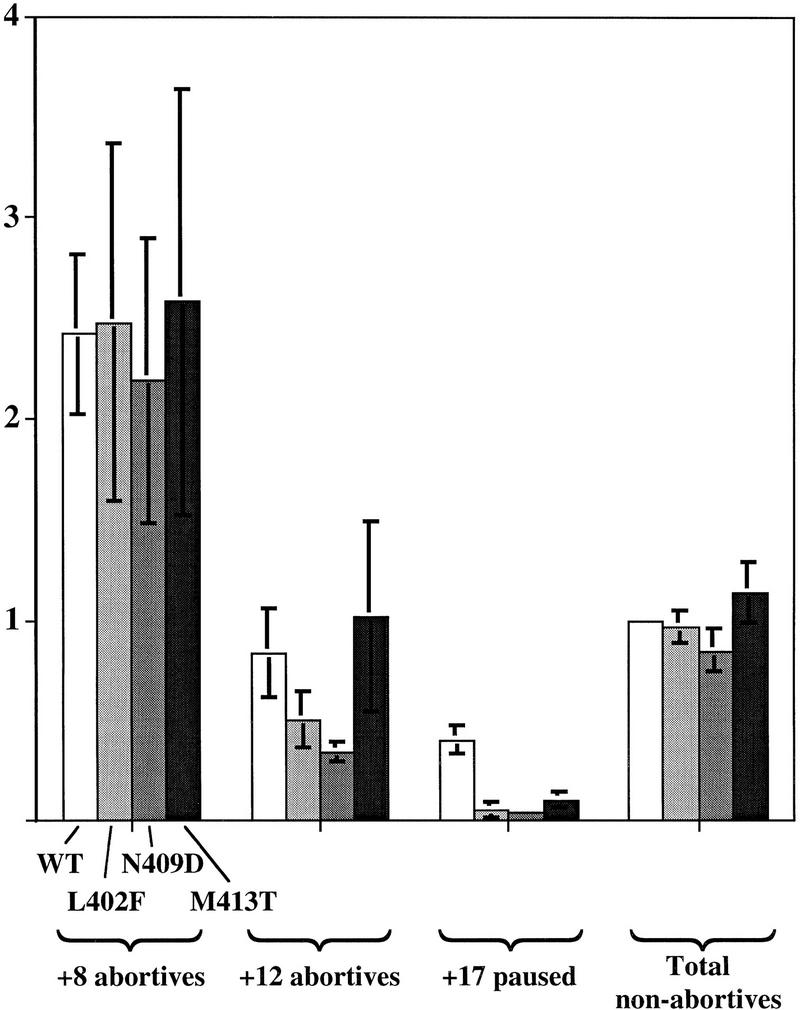
Deficiency of region 2.2 mutant σ70 in abortive initiation. Products of three separate 5′ in vitro synthesis experiments were analyzed by gel, quantified as described, and averaged. (Open bars) WT; (light gray bars) L402F; (dark gray bars) N409D; (solid bars) M413T.
Discussion
We have identified a surface in subregion 2.2 of E.coli σ70 that stabilizes the open promoter complex, and, by its ability to mediate pausing near the start site, is required for the antitermination activity of phage λ Q protein. The region 2.2 mutations we identified appear to act both by impairing formation of the paused complex, allowing more RNAP to transcribe through without being caught, and destabilizing the complex once it is formed. They might more severely affect pausing than promoter function in general if pausing requires relatively fast engagement of Eσ70 into the DNA bound state as it passes the site.
We considered that the pause-reducing phenotype might result from weaker core-σ binding after initiation, allowing release of σ70 before RNAP reaches the pause-inducing sequence. However, if the pause defect were solely the result of early release of σ70, the percent capture would be reduced, but the pause half-life would be unchanged; because all three mutations reduce the pause half-life as well as percentage capture, they must reduce stability of the paused complex, as is also true for the open complex at the promoter.
If the pause limits transcription, which it does slightly for pR′ and much more for a stronger promoter (Kainz and Roberts 1995), then the reduced-pausing σ70 mutations should have higher levels of transcription than wild-type σ70 in transcription not affected by Q. This would account for the higher basal transcription rate of pR′ by holoenzyme containing region 2.2 mutant σ70 relative to wild-type σ70.
Of the four regions of extensive similarity among σ factors, the most conserved is region 2 (Lonetto et al. 1992), encompassing amino acids 371–456 in σ70, and believed to mediate core RNAP binding, DNA melting, and sequence-specific promoter contacts. Until recently, no function had been assigned to subregion 2.2, the most conserved segment among σ factors. Because the exposed surface of region 2.2 helix 13 is hydrophobic and is near the core-binding segment—residues 361–390 of region 2.1—Malhotra et al. (1996) suggest that the helix of region 2.2 also binds core RNAP, or possibly another protein surface. In agreement, point mutation analysis of alternative σ factors (Shuler et al. 1995; Tintut and Gralla 1995) implicates helix 13 in core contacts, and the mutation Q80R in σ32 (corresponding to amino acid 406 in σ70) decreases core binding, according to a sedimentation assay (Joo et al. 1997).
We show that mutations in region 2.2 decrease Q-activated transcription in vivo and in vitro, stimulate basal transcription from pR′ in vivo, decrease promoter-proximal pausing, and impair open complex formation and stability. Our mutations lie on the solvent-exposed surface of α-helix 13 of region 2.2, which contains amino acids 401–418. We suggest the mutations do not affect promoter binding through direct effect on DNA-σ contacts, but instead act indirectly: The DNA-binding surface of region 2.4 and the region 2.2 residues are on opposite sides of a helix bundle and, thus, are unlikely to both contact promoter DNA. Furthermore, the strongest mutation of our set occurs at the solvent-exposed residue L402, which is nonpolar and, thus, is likely to contact another hydrophobic surface.
We suggest that region 2.2 of σ70 makes contacts with the subunits of core RNAP that form and strengthen during the transition from closed to open promoter complex. Conformational changes could be transmitted allosterically to the DNA-binding helix, which is packed against helix 13, thereby explaining the effect of helix 13 mutations on open complex formation and stability (Fig. 7). Region 2.2 may function differently from region 2.1 in core interaction; deletion analysis showed that amino acids downstream of 390, which includes all of region 2.2, are dispensable for core binding, whereas deletions in region 2.1 impair core binding (Lesley and Burgess 1989). Furthermore, the activity of region 2.2 mutants in our screen requires that they still bind core, and initiate efficiently at promoter pR′, to mediate expression that is insensitive to Q function; in addition, mutant σ had to compete effectively against wild-type σ70 expressed from the chromosomal gene in the initial mutant screen.
Figure 7.
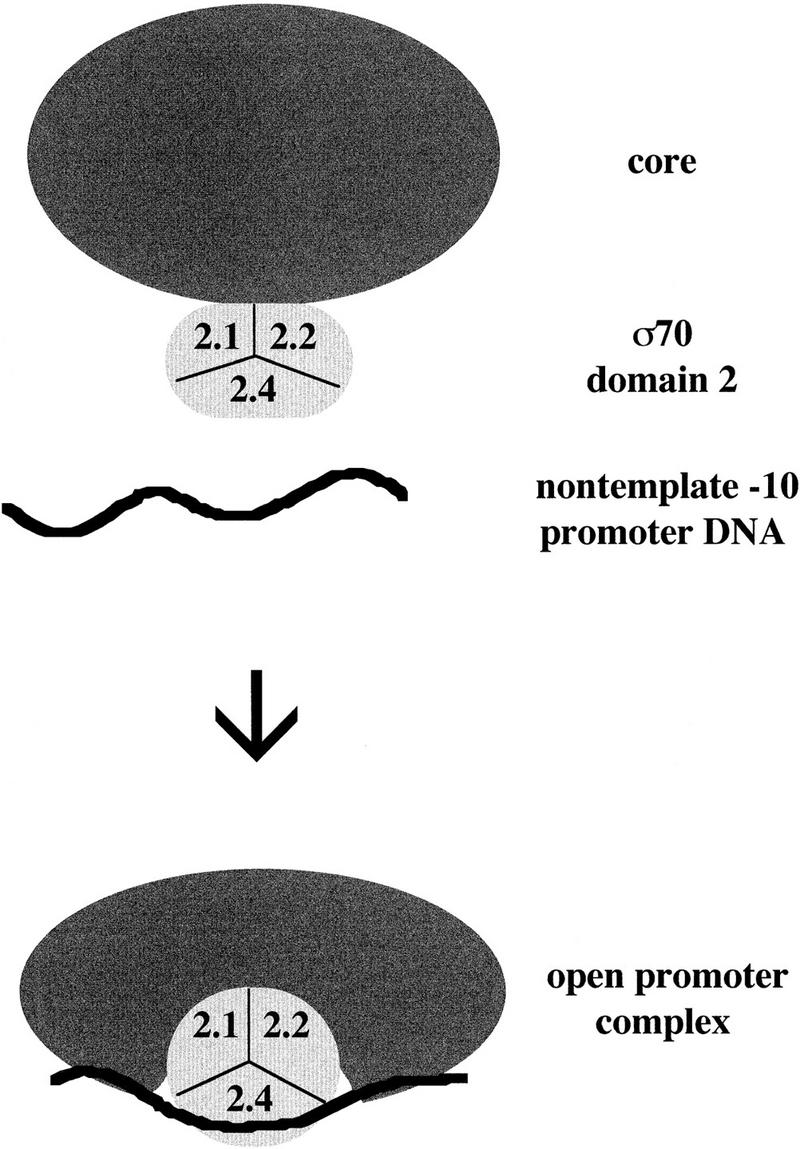
Model of nontemplate −10 region promoter DNA interaction with holoenzyme to form the open promoter complex.
We can estimate roughly how defective the mutant σ70 might be in competing with wild-type σ70 to form holoenzyme. By Western analysis, we found that the plasmid expresses mutant σs at four- to eightfold excess over the wild-type σ70 in SG20250 (λ+49), the strain used for the screen. On the basis of the measurements of Jishage and Ishihama (1995), core should be present in limiting concentration relative to σ70. Taking the activity of each mutant σ70 in supporting Q function in the σ70 temperature-sensitive reporter CG19284 (λ+49) as its intrinsic activity in the absence of competitor, we calculate the theoretical Q function when plasmid-expressed mutant competes with the single copy amount of wild-type σ70 in SG20250 (λ+49), assuming the mutant has no defect in core binding; and we then calculate the severity of the core-binding defect if the predicted and observed numbers do not match (Table 1). For the most severe mutation, L402F, calculation indicates no core-binding defect. Others show small defects, except for the ∼70-fold defect of M413A. We note that the calculated severity of the core-binding defect increases inward along helix 13. Thus, helix 13 appears to have some role in binding free core RNAP in addition to its postulated function in the closed- to open-complex transition.
Table 1.
Estimation of RNAP core-binding defect of σ70 mutants
|
|
Q activity
|
Calculated core binding (% of WT)
|
Calculated binding defect
|
||
|---|---|---|---|---|---|
| mutant alone (%)
|
mutant plus wild type σ70 (%)
|
theoretical, no binding defect (%)
|
|||
| L402F | 14 | 30 | 28 | 88 | 1.1 |
| L402A | 108 | 101 | 107 | 107 | — |
| Q406A | 28 | 49 | 40 | 49 | 2.1 |
| N409D | 23 | 60 | 36 | 22 | 4.6 |
| N409A | 8 | 56 | 23 | 18 | 5.5 |
| M413T | 98 | 89 | 98 | — | — |
| M413A | 19 | 95 | 32 | 1.3 | 76 |
Theoretical Q activity, assuming no core binding defect, was calculated by assuming that 1/6 of Q activity was provided by wild-type (WT) σ70 and 5/6 by mutant σ70, as implied by the measured excess of mutant over wild type (four- to eightfold) due to expression of mutant σ70 from the plasmid. The core-binding ability of each mutant then was calculated as proportional to the fraction of mutant σ70 present that would have to be functional to account for the measured Q activity.
The notion that binding of the exposed face of helix 13 of region 2.2 into core repositions region 2.4 (and possibly regions of core) to bind single-stranded DNA in the open promoter complex implies significant conformational changes in promoter binding; such changes were predicted by kinetic studies of initiation at the promoters lacUV5 and pR. Buc and McClure (1985) and Roe et al. (1985) presented evidence for two distinct closed intermediates (I1 and I2) of λpR: R + P ↔ I1 ↔ I2 ↔ RPo. Roe et al. (1985) proposed that the first isomerization (I1 ↔ I2), which presumably establishes the major contacts that stabilize the open complex, involves a conformational change in RNAP that nucleates DNA melting. Furthermore, they proposed that energetics of this step are dominated by burial of a hydrophobic surface, on the basis of a large and negative heat capacity of the reaction, its lack of salt dependence, and its relative insensitivity to DNA supercoiling. We suggest that L402 is a major component of this hydrophobic surface, and that because of their similar phenotypes, the other mutated amino acids, Q406, N409, and M413, are also buried during the transition from I1 to I2. It is consistent with this assignment that the I1 ↔ I2 transition is rate limiting for both the association and dissociation of open complex (Roe et al. 1985), and that the L402F and N409D mutations affect both steps. It is further consistent that formation of open complex by L402F and N409D mutant Eσ70 is stimulated to wild-type levels (although not to wild-type rates) by high glycerol concentration (D. Ko and J.W. Roberts, unpubl.), which, Roe et al. (1985) suggest, acts to stabilize the conformational change in RNAP.
Materials and methods
Strains and plasmids
The reporter strains were constructed as single-copy lysogens of λ+49, a derivative of λRS45 that contains an operon fusion of λpR′ and two transcription terminators preceding lacZ, as described by Simons and Kleckner (1987) (J. Guo and J.W. Roberts, unpubl.). Bacterial strains were SG20250 (MC4100, F−araD139 Δ (lacIPOZY)U169 strA thi), from S. Gottesman (NCI, NIH, Bethesda, MD); CAG19284 [rpoD285 arg lac thi recA argG75 lacZ49(F′laciQ)], carrying a temperature-sensitive lethal mutation in rpoD, from A. Dombroski; CAG20176 [araD139Δara–leu)7696 galE15 galK16 ΔlacX74 rpsL hsdR2(r−m−) mcrA mcrB::Δcm → trp rpoD:Tn(tet)], a strain in which the endogenous copy of rpoD can be repressed by tryptophan, from C. Gross (UCSF School of Medicine); and MTM9973, a recA−-derivative of CAG20176. The Q-source plasmid pJG100 contains λ gene Q controlled by the lactose operator, and one copy of lacIq (J. Guo and J.W. Roberts, unpubl.). Plasmid pHTσ contains rpoD transcribed from the lac promoter, and was obtained from M. Kainz (University of Wisconsin, Madison). Plasmid pBAD33spc rpoD is a derivative of pBAD33 (Guzman et al. 1995) carrying spectinomycin resistance and rpoD (M.T. Marr and J.W. Roberts, unpubl.). Plasmid M650, the template for PCR reaction to make DNA for in vitro synthesis, contains promoter pR′ fused to terminators tc and to (McDowell et al. 1994) (M. Marr and J.W. Roberts, unpubl.). λc17 phage was from laboratory stock.
PCR mutagenesis and screening
rpoD was mutagenized in two segments, according to Zhou et al. (1991). The segment from −19 to +847 was amplified in six reactions, which were pooled and cloned into the XbaI and SphI sites of pHTσ. The segment from +720 to +1908 was amplified in five reactions, which were pooled and cloned into the SphI and HindIII sites of pHTσ. Ligation mixes were electroporated into the reporter strain SG20250 (λ+49) (pJG100), and grown on MacConkey plates supplemented with ampicillin (100 μg/ml) and spectinomycin (70 μg/ml). Expression of wild-type σ70 from pHTσ gives red color, indicating Q function; putative mutants were picked as white or pink, verified by retransformation, and characterized by β-gal assay. The plasmid was also transformed into CAG19284(λ+49) to measure the effect of the mutations without active endogenous wild-type σ70. Basal expression in the absence of Q was measured in the same strain lacking pJG100. More than half of the original plasmids had multiple mutations, but fragments containing a single mutation to which the phenotype could be ascribed were isolated and recloned. In addition to the viable mutants described here, our screen also yielded σ70 mutants that are lethal as haploids, including sites in DNA recognition regions 2.4 and 4.2 (D. Ko and J.W. Roberts, unpubl.); analysis of these may reveal other functions of σ70 in Q activity.
Cell growth and β-galactosidase assay
Overnight cultures were diluted 1:50 into LB medium supplemented with the appropriate antibiotics and 1 mm IPTG (to induce Q from pJG100), or (for MTM9973) 100 μg/ml tryptophan to repress expression of the chromosomal rpoD and 2 mg/ml arabinose to induce rpoD from the plasmid. For growth of CAG19284 at restrictive temperature, overnight cultures were grown at 32°C and growth after dilution was at 42°C. β-Galactosidase activity was measured at an OD600 of 0.5 in duplicate samples from at least two different cultures; the mean and standard deviation are given.
Site-directed mutagenesis
Alanine substitutions were introduced by PCR overlap mutagenesis (Ring et al. 1996) and verified by sequencing.
Proteins and DNA
Mutations in rpoD were subcloned from pHTσ into a pET28-based σ70 expression vector. Wild-type and mutant 6-His-tagged σ70 were purified as described (Marr and Roberts 1997). RNAP holoenzyme was purified as described (Hager et al. 1990); Bio-Rex 70 was used to remove σ as described (Burgess and Jendrisak 1975). RNAP was reconstituted by incubating wild-type or mutant σ70 at a 25-fold molar excess over core for 30 min at room temperature (23°C). λ Q protein and NusA protein were purified as described (Yarnell and Roberts 1992). DNA template for in vitro transcription and band-shift analysis of open promoter complexes was synthesized by PCR as described (Ring et al. 1996). The 292-bp DNA used for band-shift contained λ promoter pR′ located such that the transcription start point was at 142 bp. The 459-bp DNA used for in vitro transcription to measure Q activity (Fig. 3) contained pR′ located such that the transcription start point was at 88 bp and the release site of terminator tC at 271 bp, giving a terminated RNA of 184 nucleotides and a readthrough RNA of 372 nucleotides. The template DNA used to measure pausing (Fig. 4) gave a runoff RNA of 147 nucleotides and had no terminators.
In vitro transcription
Reactions containing 2 nm template, 10 nm reconstituted holoenzyme, and 100 nm NusA were preincubated at 37°C for 5 min in 20 mm Tris-Cl (pH 8.0), 0.1 mm EDTA, 1 mm DTT, 50 mm KCl, 200 μm ATP, GTP, and CTP, 50 μm UTP, 0.3 μC/μl [α-32P]UTP, and 10%–25% glycerol; when antitermination was measured, λQ was then added at up to 100 nm. Synthesis reactions for experiments of Figures 6 and 7 contained high (23%–25%) glycerol, which equalizes total synthesis between mutant and wild-type enzymes. Synthesis was started by the addition of 2.5 μl of 40 mm MgCl and 100 μg/μl rifampicin to a final reaction volume of 25 μl. Synthesis was stopped by the addition of 125 μl of 0.6 mTris (pH 8.0), 12 mm EDTA, and 80 μg/ml tRNA. Transcription reactions were extracted with 150 μl of phenol/chloroform/isoamyl alcohol (25:24:1) and precipitated with 2.5 volumes of 100% EtOH. Samples were resuspended in 4 μl of 80% formamide, 1× TBE, and 0.05% bromophenol blue, and transcripts were resolved in 12% polyacrylamide gels containing 8 m urea. Bands were visualized and quantified by PhosphorImager (Storm 840) and Image Quant software.
KMnO4 reactivity in vivo
Reactivity was measured in strain MTM9973 carrying pBAD33spc rpoD+ or pBAD33spc rpoDL402F, and pXY306 or pXY306(+6C), as described (Kainz and Roberts 1992), except that growth was in LB supplemented with 0.2% arabinose and 100 μg/ml tryptophan; KMnO4 was used at 30 mm; reactions were quenched with 100 mm Tris (pH 8.0), 100 mm EDTA, 150 mm β-mercaptoethanol, and 100 mm NaCl; and modified pyrimidines were detected by 15 cycles of linear PCR with end-labeled oligonucleotide.
Assay of open complex by native gel electrophoresis
The formation and dissociation of open complex were measured essentially according to Zhang et al. (1996) by use of PCR-synthesized DNA end-labeled with T4 polynucleotide kinase. For measurement of dissociation, reaction mixtures containing 20 mm Tris HCl (pH 8.0), 0.1 mm EDTA, 1 mm DTT, 50 mm KCl, 4 mm MgCl2, 100 μg/ml BSA, 0.1 nm 32P-labeled DNA, and 20 nm reconstituted RNAP were incubated for 10 min at 37°C. Heparin was added to 100 μg/ml and reactions were shifted to 17°C; 5-μl aliquots were removed over time and loaded onto a 5% polyacrylamide/1× TBE gel running at 100 V at room temperature (23°C). After 2.5 hr, the gel was dried and bands visualized and quantified by PhosphorImager. To measure formation of open complex, reaction mixtures constructed as above were incubated at 37°C; 5-μl samples were taken at indicated times and added to 1-μl of heparin to give a final concentration of 100 μg/ml. After 45 sec at room temperature, the entire 6 μl was loaded onto a gel and analyzed as described above.
Acknowledgments
We thank C. Gross and members of the laboratory for criticizing the manuscript; C. Gross, A. Dombrowski, S. Gottesman, and M. Kainz for bacterial strains and plasmids; S. Darst for providing coordinates of the σ70 atomic structure; and the National Institutes of Health for support. D.K. thanks the Cornell Hughes Scholars Program for support.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jwr7@cornell.edu; FAX (607) 255-2428.
References
- Buc H, McClure WR. Kinetics of open complex formation between Escherichia coli RNA polymerase and the lac UV5 promoter. Evidence for a sequential mechanism involving three steps. Biochemistry. 1985;24:2712–2723. doi: 10.1021/bi00332a018. [DOI] [PubMed] [Google Scholar]
- Burgess RR, Jendrisak JJ. A procedure for the rapid, large-scale purification of Escherichia coli DNA-dependent RNA polymerase involving polymin P precipitation and DNA-cellulose chromatography. Biochemistry. 1975;14:4634–4638. doi: 10.1021/bi00692a011. [DOI] [PubMed] [Google Scholar]
- Gardella T, Moyle H, Susskind MM. A mutant Escherichia coli Sigma 70 subunit of RNA polymerase with altered promoter specificity. J Mol Biol. 1989;206:579–590. doi: 10.1016/0022-2836(89)90567-6. [DOI] [PubMed] [Google Scholar]
- Gross C, Lonetto M, Losick R. Bacterial sigma factors. In: McKnight S, Yamamoto K, editors. Transcriptional regulation. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. pp. 129–176. [Google Scholar]
- Guzman L-M, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager DA, Jin DJ, Burgess RR. Use of mono Q high-resolution ion-exchange chromatography to obtain highly pure and active Escherichia coli RNA polymerase. Biochemistry. 1990;29:7890–7894. doi: 10.1021/bi00486a016. [DOI] [PubMed] [Google Scholar]
- Hansen HM, McClure WR. Role of the σ subunit of Escherichia coli RNA polymerase in initiation. II. Release of σ from ternary complexes. J Biol Chem. 1980;255:9564–9570. [PubMed] [Google Scholar]
- Helmann JD, Chamberlin MJ. Structure and function of bacterial sigma factors. Annu Rev Biochem. 1988;57:839–872. doi: 10.1146/annurev.bi.57.070188.004203. [DOI] [PubMed] [Google Scholar]
- Jishage M, Ishihama A. Regulation of RNA plymerase sigma subunit synthesis in Escherichia coli: Intracellular levels of sigma 70 and sigma 38. J Bacteriol. 1995;177:6832–6835. doi: 10.1128/jb.177.23.6832-6835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston DE, McClure WR. Abortive initiation of in vitro RNA synthesis on bacteriophage λ DNA. In: Losick RR, Chamberlin MJ, editors. RNA polymerase. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1976. pp. 413–428. [Google Scholar]
- Joo DM, Ng N, Calendar R. A σ32 mutant with a single amino acid change in the highly conserved region 2.2 exhibits reduced core RNA polymerase affinity. Proc Natl Acad Sci. 1997;94:4907–4912. doi: 10.1073/pnas.94.10.4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juang Y-L, Helmann J. A promoter merting region in the primary σ factor of Bacillus subtilis. Identification of functionally important aromatic amino acids. J Mol Biol. 1994;235:1470–1488. doi: 10.1006/jmbi.1994.1102. [DOI] [PubMed] [Google Scholar]
- Kainz M, Roberts JW. Structure of transcription elongation complexes in vivo. Science. 1992;255:838–841. doi: 10.1126/science.1536008. [DOI] [PubMed] [Google Scholar]
- ————— Kinetics of RNA polymerase initiation and pausing at the lambda late gene promoter in vivo. J Mol Biol. 1995;254:808–814. doi: 10.1006/jmbi.1995.0657. [DOI] [PubMed] [Google Scholar]
- Kenney TJ, York K, Youngman P, Moran CPJ. Genetic evidence that RNA polymerase associated with σA uses a sporulation-specific promoter in Bacillus subtilis. Proc Natl Acad Sci. 1989;86:9109–9113. doi: 10.1073/pnas.86.23.9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komissarova N, Kashlev M. Transcriptional arrest: Escherichia coli RNA polymerase translocates backward, leaving the 3′ end of the RNA intact and extruded. Proc Natl Acad Sci. 1997;94:1755–1760. doi: 10.1073/pnas.94.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesley SA, Burgess RR. Characterization of the Escherichia coli transcription factor σ70: Localization of a region involved in the interaction with core RNA polymerase. Biochemistry. 1989;28:7728–7734. doi: 10.1021/bi00445a031. [DOI] [PubMed] [Google Scholar]
- Lonetto M, Gribskov M, Gross CA. The σ70 family: Sequence conservation and evolutionary relationships. J Bacteriol. 1992;174:3843–3849. doi: 10.1128/jb.174.12.3843-3849.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra A, Severinova E, Darst SA. Crystal structure of a σ70 subunit fragment from E. coli RNA polymerase. Cell. 1996;87:127–136. doi: 10.1016/s0092-8674(00)81329-x. [DOI] [PubMed] [Google Scholar]
- Marr MT, Roberts JW. Promoter recognition as measured by binding of polymerase to nontemplate strand oligonucleotide. Science. 1997;276:1258–1260. doi: 10.1126/science.276.5316.1258. [DOI] [PubMed] [Google Scholar]
- McClure WR. Mechanism and control of transcription initiation in prokaryotes. Annu Rev Biochem. 1985;54:171–204. doi: 10.1146/annurev.bi.54.070185.001131. [DOI] [PubMed] [Google Scholar]
- McDowell JC, Roberts JW, Jin DJ, Gross C. Determination of intrinsic transcription termination efficiency by RNA polymerase elongation rate. Science. 1994;266:822–825. doi: 10.1126/science.7526463. [DOI] [PubMed] [Google Scholar]
- Niu W, Zhou Y, Dong Q, Ebright YW, Ebright RH. Characterization of the activating region of Escherichia coli: Catabolite gene activator protein (CAP): I. Saturation and alanine-scanning mutagenesis. J Mol Biol. 1994;243:595–602. doi: 10.1016/0022-2836(94)90034-5. [DOI] [PubMed] [Google Scholar]
- Nudler E, Mustaev A, Lukhtanov E, Goldfarb A. The RNA-DNA hybrid maintains the register of transcription preventing backtracking of RNA polymerase. Cell. 1997;89:33–41. doi: 10.1016/s0092-8674(00)80180-4. [DOI] [PubMed] [Google Scholar]
- Reeder TC, Hawley DK. Promoter proximal sequences modulate RNA polymerase II by a novel mechanism. Cell. 1996;87:767–777. doi: 10.1016/s0092-8674(00)81395-1. [DOI] [PubMed] [Google Scholar]
- Ring BZ, Roberts JW. Function of a nontranscribed DNA strand site in transcription elongation. Cell. 1994;78:317–324. doi: 10.1016/0092-8674(94)90300-x. [DOI] [PubMed] [Google Scholar]
- Ring BZ, Yarnell WS, Roberts JW. Function of E. coli RNA polymerase σ factor σ70 in promoter-proximal pausing. Cell. 1996;86:485–493. doi: 10.1016/s0092-8674(00)80121-x. [DOI] [PubMed] [Google Scholar]
- Roberts CW, Roberts JW. Base specific recognition of the nontemplate strand of promoter DNA by E. coli RNA polymerase. Cell. 1996;86:495–501. doi: 10.1016/s0092-8674(00)80122-1. [DOI] [PubMed] [Google Scholar]
- Roberts J. Antitermination and the control of transcription elongation. In: McKnight S, Yamamoto K, editors. Transcriptional regulation. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1992. pp. 389–406. [Google Scholar]
- Roe J, Burgess RR, Record MT., Jr Temperature dependence of the rate constants of the Escherichia coli RNA polymerase-λpR promoter interaction: Assignment of kinetic steps corresponding to protein conformational change and DNA opening. J Mol Biol. 1985;184:441–453. doi: 10.1016/0022-2836(85)90293-1. [DOI] [PubMed] [Google Scholar]
- Shuler MF, Tatti KM, Wade KH, Moran CP. A single amino acid substitution in σE affects its ability to bind core RNA polymerase. J Bacteriol. 1995;177:3687–3694. doi: 10.1128/jb.177.13.3687-3694.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegele DA, Hu JC, Walter WA, Gross CA. Altered promoter recognition by mutant forms of the sigma-70 subunit of Escherichia-coli RNA polymerase. J Mol Biol. 1989;206:591–604. doi: 10.1016/0022-2836(89)90568-8. [DOI] [PubMed] [Google Scholar]
- Simons RW, Houman F, Kleckner N. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene. 1987;53:85–96. doi: 10.1016/0378-1119(87)90095-3. [DOI] [PubMed] [Google Scholar]
- Tintut Y, Gralla JD. PCR mutagenesis identifies a polymerase binding sequence of σ54 that includes a σ70 homology region. J Bacteriol. 1995;177:5818–5825. doi: 10.1128/jb.177.20.5818-5825.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarnell WS, Roberts JW. The phage lambda gene Q transcription antiterminator binds DNA in the late gene promoter as it modifies RNA polymerase. Cell. 1992;69:1181–1189. doi: 10.1016/0092-8674(92)90639-t. [DOI] [PubMed] [Google Scholar]
- Zhang X, Reeder T, Schleif R. Transcription activation parameters at ara pBAD. J Mol Biol. 1996;258:14–24. doi: 10.1006/jmbi.1996.0230. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhang X, Ebright RH. Random mutagenesis of gene-sized DNA molecules by use of PCR with Taq DNA polymerase. Nucleic Acids Res. 1991;19:6052. doi: 10.1093/nar/19.21.6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber P, Healy J, Carter HLI, Cutting S, Moran CPJ, Losick R. Mutation changing the specificity of an RNA polymerase sigma factor. J Mol Biol. 1989;206:605–614. doi: 10.1016/0022-2836(89)90569-x. [DOI] [PubMed] [Google Scholar]