

Abstract
The Rac and Cdc42 GTPases share several regulators and effectors, yet perform distinct biological functions. The factors determining such specificity in vivo have not been identified. In a mutational screen in Drosophila to identify Rac-specific signaling components, we isolated 11 alleles of myoblast city (mbc). mbc mutant embryos exhibit defects in dorsal closure, myogenesis, and neural development. DOCK180, the mammalian homolog of Mbc, associates with Rac, but not Cdc42, in a nucleotide-independent manner. These results suggest that Mbc is a specific upstream regulator of Rac activity that mediates several morphogenetic processes in Drosophila embryogenesis.
Keywords: Rac, signal transduction, dorsal closure, embryogenesis, GTPases, morphogenesis
The Rho family of GTPases, which includes Rho, Rac, and Cdc42, regulate a variety of cellular processes including cytoskeletal reorganization, endocytosis, cell cycle progression, and transcriptional activity (for review, see van Aelst and D’Souza-Schorey 1997). In fibroblasts, activation of Cdc42, Rac, or Rho leads to particular rearrangements of the actin cytoskeleton resulting in filopodia, lamellipodia, and stress fiber formation, respectively. Moreover, evidence suggests that each of these GTPases can be activated by distinct extracellular stimuli. Several proteins have now been identified that interact selectively with either Rho, Rac, or Cdc42, and it seems likely that the specificity of these interactions in vitro accounts for at least some of the signaling specificity among these GTPases in vivo (Hall 1998). However, the various Rho family members have also been found to share several regulators and effector targets in in vitro studies (Hall 1998), thereby complicating a thorough mechanistic understanding of the signaling specificity that is observed in vivo.
In Drosophila, distinct requirements for closely related homologs of the Rho, Rac, and Cdc42 GTPases have been identified in the various morphogenetic events associated with embryogenesis. For example, Rho1 is required for gastrulation (Barrett et al. 1997); Rho1, Rac1, and Cdc42 are required for dorsal closure and tissue polarity (Harden et al. 1995; Eaton et al. 1996; Strutt et al. 1997); and Rac1 and Cdc42 have been implicated in neural development and myogenesis (Luo et al. 1994). Using Drosophila genetics, we identified Myoblast city (Mbc), a homolog of mammalian DOCK180 (Erickson et al. 1997) and Caenorhabditis elegans CED-5 (Wu and Horvitz 1998) as a specific mediator of Rac1 activity in several morphogenetic processes during Drosophila embryogenesis, including myogenesis, neural development, and dorsal closure.
Results and Discussion
Overexpression of Rho, Rac, and Cdc42 GTPases in the fly eye causes distinct developmental defects
Previously, we described developmental eye defects caused by overexpression of the wild-type Drosophila Rho1 GTPase in transgenic flies (Hariharan et al. 1995). To determine whether the related Rac and Cdc42 GTPases could similarly disrupt eye development, we generated transgenic flies in which wild-type Rac1, Rac2, and Cdc42 GTPases are expressed in the developing eye under the control of the synthetic GMR promoter (Hay et al. 1994). Flies harboring a single copy of the rac1 transgene exhibit an externally rough eye (Fig. 1, D vs. A), and retinal sections revealed a loss of pigment cells and a disruption of the normal ommatidial morphology, with occasional loss of photoreceptors (Fig. 1, E vs. B). With two copies of the GMR–rac1 transgene, a complete disruption of normal eye structure is observed; a similar phenotype is observed in GMR–rac2 transgenic flies (data not shown).
Figure 1.
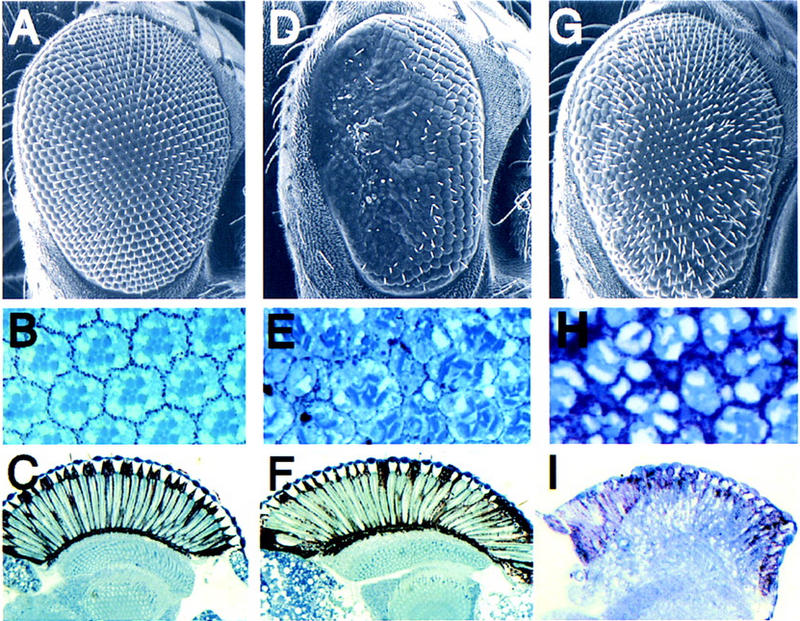
GMR–rac and GMR–cdc42 transgenic flies exhibit distinct rough eye phenotypes. Scanning electron micrographs (A,D,G), transverse (B,E,H), and longitudinal (C,F,I) retinal sections of wild-type (A–C), GMR–rac17a (D–F), and GMR–cdc42 (G–I).
The GMR–cdc42 transgenic flies exhibit externally rough eyes distinct from those seen in the rac1 and rac2 transgenics (Fig. 1G). Retinal sectioning revealed missing photoreceptors and a disruption of ommatidial morphology (Fig. 1H). Although the cdc42-induced eye phenotype somewhat resembles the GMR–rho1 phenotype (Hariharan et al. 1995), the cdc42 transgenics also exhibit an abnormal rhabdomere morphology. We also examined the postmitotic elongation event that establishes the depth of the retina. Overexpression of either rho1 (Barrett et al. 1997) or cdc42, but not rac1, disrupts the normal elongation of all retinal cells (Fig 1C,F,I). Thus, each of these members of the Rho GTPase family, when overexpressed, induces distinct alterations of normal eye development.
Identification of suppressors of the rac1-induced rough eye
To identify specific components of a Rac1 signaling pathway in Drosophila, rac1 transgenic flies were used to screen for dominant mutations that specifically suppress the rac1-induced rough eye phenotype but not that caused by GMR–cdc42 or GMR–rho1. First, we determined that chromosomal deficiences that cover either rac1 or rac2 each suppress the GMR–rac1 eye defect, confirming that the phenotype is sensitive to the levels of endogenous Rac activity and that Rac1 and Rac2 are normally expressed during eye development (data not shown). To identify rac1-suppressing mutations, mutagenized wild-type males were mated with GMR–rac1 females and the resulting F1 progeny were examined for suppression of the rough eye phenotype. A total of 23,000 F1 flies were screened, and 36 dominantly suppressing mutations were identified. Three complementation groups were established on the basis of lethality, and a single complementation group of 11 alleles is described hereafter as Suppressor of rac1 [Su(rac)1]. Each of the Su(rac)1 alleles dominantly suppresses the GMR–rac1-induced rough eye surface (Fig. 2A,G) as well as the underlying retinal morphology (Fig. 2D,J), rescuing the percentage of normal appearing ommatidia from 3% in GMR–rac flies to 97% in GMR–rac1/Su(rac)1 flies. Each of these alleles also suppresses the GMR–rac2-mediated defect (data not shown), although none suppresses a GMR–rho1 phenotype [no normal appearing ommatidia in flies with or without Su(rac)1 alleles] or a GMR–cdc42-induced phenotype [4% normal appearing ommatida with or without Su(rac)1 mutations] (Fig. 2B; E vs. H, K, and C; F vs. I and L). These data suggest that Su(rac)1 encodes a specific component of a Rac-mediated signaling pathway.
Figure 2.
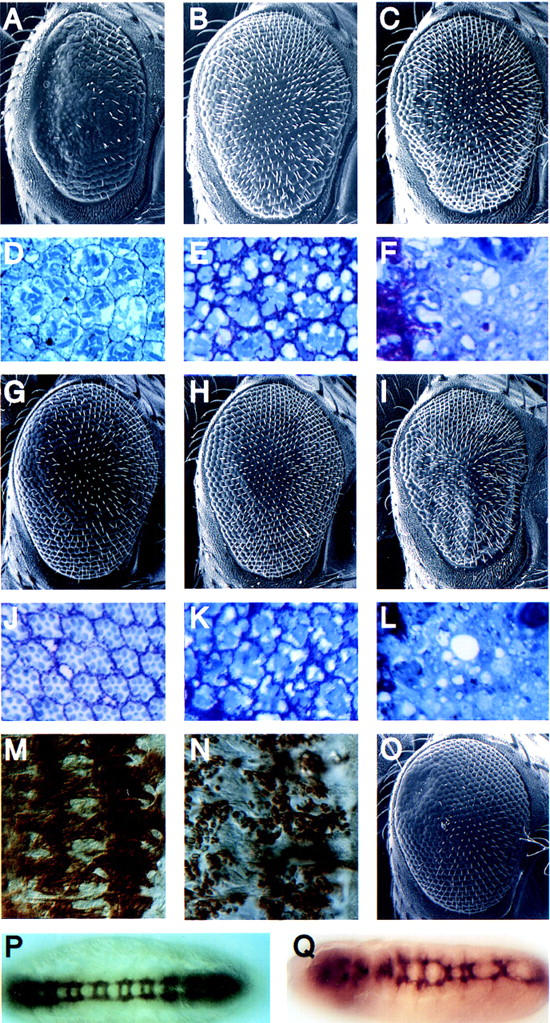
Su(rac)1 specifically suppresses the rac1-induced rough eye and is allelic to mbc. Scanning electron micrographs (A–C,G–I,O) and tranverse retinal sections (D–F,J–L) of GMR–rac17a/iso3 (A,D), GMR–rac17a/Su(rac)12.35 (G,J), GMR–cdc42-33b/iso3 (B,E), GMR–cdc42-33b/Su(rac)13.5 (H,K), GMR–rho11rho13/iso3 (C,F), and GMR–rho11rho13/Su(rac)12.35 (I,L) flies. Wild-type (M) and Su(rac)12.35/Su(rac)14.25 (N) stage 15 embryos (dorsal right) stained with antibodies against myosin heavy chain. Wild-type (P) and mbc11.63/mbc14.25 (Q) stage 15 embryos stained with the BP102 antibody.
Su(rac)1 is allelic to the gene myoblast city
Because a specific requirement for Rac activity, but not that of Cdc42, has been demonstrated in the fusion of myoblasts during muscle development (Luo et al. 1994), we examined the musculature of Su(rac)1 mutants. Myoblast fusion is normally completed by stage 15 (Fig. 2M; Campos-Ortega and Hartenstein 1989); however, in stage 15 Su(rac)1 mutants, myoblasts are largely unfused (Fig. 2N). Meiotic mapping localized Su(rac)1 alleles to a chromosomal region similar to that of a previously reported gene, mbc, that is also associated with a loss-of-function myoblast fusion defect (Rushton et al. 1995; Erickson et al. 1997). We determined that null alleles of mbc fail to complement the lethality and myoblast fusion phenotype of several alleles of Su(rac)1. Moreover, mbc alleles also suppress the GMR–rac1 phenotype (Fig. 2, A vs. O). Together, these results indicate that Su(rac)1 is allelic to mbc. mbc encodes a protein that is highly homologous throughout its length to the mammalian protein DOCK180, which has been implicated in the regulation of cell morphology (Hasegawa et al. 1996; Erickson et al. 1997). Although the role of Rac in myoblast fusion is unknown, these results suggest the Mbc mediates the activity of Rac in this morphogenetic process in which actin rearrangements have been implicated previously (Sanger et al. 1971).
Next we examined other phenotypes that would be consistent with aberrant Rac signaling. Drosophila Rac1 has been implicated in axonal outgrowth (Luo et al. 1994), and we found that mbc mutants exhibit a low penetrance defect in the fasciculation of axons of the ventral nerve cord neurons (Fig. 2, P vs. Q). Specifically, some mbc mutant embryos exhibit improper spacing between commissures and, in extreme cases, a lack of fasciculation of the longitudinal connectives, possibly because of abnormal migration of the central nervous system (CNS) neurons across the ventral surface. In support of a role for Mbc in cell migration is the recent observation that mutations in ced-5, the C. elegans homolog of mbc, result in defective migration of the distal tip cells of the gonad (Wu and Horvitz 1998). Additionally, mutations in mig-2, a C. elegans gene encoding a Rac-like GTPase, also affect distal tip cell migration and axon outgrowth (Zipkin et al. 1997). Moreover, the mammalian Rac GTPase appears to regulate the motility of cultured fibroblasts (Keely et al. 1997). It is possible that a pathway mediated by both Rac and Mbc regulates neuronal migration and axon growth, and may explain the CNS defects observed in mbc mutant embryos.
Jun kinase activation is not substantially reduced in mbc mutant embryos
In addition to the defects in myoblast fusion and CNS development, we found that Su(rac)1 alleles exhibit a dorsal closure defect similar to that reported previously for mbc mutants (data not shown) and embryos expressing dominant-negative rac1 (Harden et al. 1995). During dorsal closure, two symmetric epithelial monolayers coordinately migrate from their lateral position to fuse along the dorsal midline. The row of cells along the dorsal apical edge, known as the leading up edge (LE) cells, elongate first and remain morphologically distinct from the more ventral cells until the two sheets have nearly met at the midline (Campos-Ortega and Hartenstein 1989). Recently, a Rac-mediated signaling pathway that regulates this process has been elucidated. Rac1 appears to activate the c-Jun amino (N)-terminal kinase (JNK) pathway, which leads to decapentaplegic (dpp) expression in the LE cells of the dorsal epidermis, and several JNK pathway mutants associated with reduced dpp expression exhibit similar dorsal closure defects, including hemipterous (hep; Jun kinase kinase), basket (Jun kinase), Djun, and kayak (c-Fos) (Glise and Noselli 1997; Hou et al. 1997; Riesgo-Escovar and Hafen 1997a,b).
To determine whether Mbc mediates the activity of Rac in the activation of JNK during dorsal closure, we examined the expression of dpp mRNA in mbc mutant embryos. In wild-type embryos, dpp is expressed predominantly in the visceral mesoderm and the LE of the dorsal epidermis, as described previously (Fig. 3A,B). In mbc mutant embryos (Fig. 3C,D), 50% of which exhibit dorsal closure defects, dpp is expressed at normal levels in the majority of embryos but appears to be mildly reduced specifically in the LE cells of some of these embryos. This is in contrast to hep mutant embryos, in which dpp expression in LE cells is clearly absent (Fig. 3E,F). This result suggests that Mbc is not absolutely required for JNK pathway activation and may play a distinct role in dorsal closure. However, we cannot exclude the possiblity that Mbc contributes to the activation of JNK in the LE cells, but the effects of its absence are masked by a redundant function of Cdc42, which is also capable of activation of JNK in the LE cells (Glise and Noselli 1997).
Figure 3.
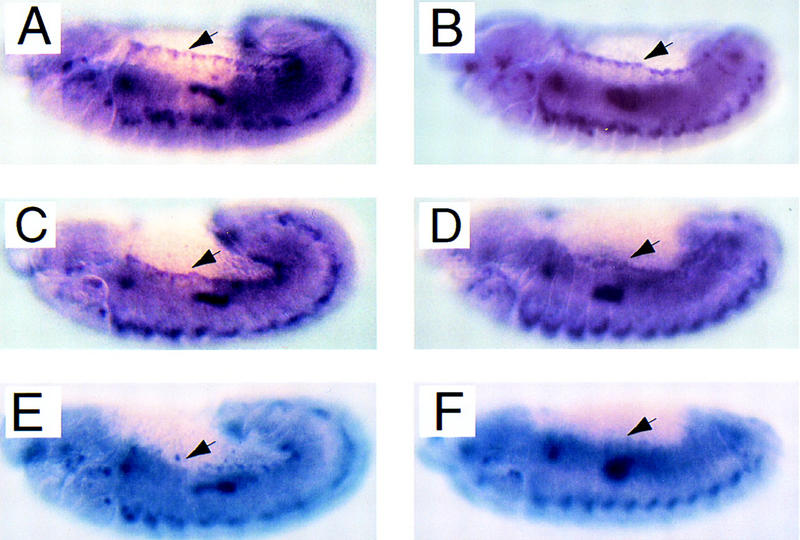
Mutations in mbc do not substantially reduce levels of dpp expression in the leading edge cells of the dorsal epidermis. In situ hybridizations of an anti-sense dpp probe to stage 12 (A,C,E) and 13 (B,D,F) wild-type (A,B), mbc3.5/Df(3R)mbc-f5.3 (C,D), and hep1/hep1 (E,F) embryos, reveals a loss of dpp expression in hep1, but not mbc mutant embryos (E to C vs. F to D) in the row of leading edge cells (indicated by arrows).
In mammalian cells, activation of the JNK cascade by Rac and Cdc42 is distinct from the GTPase-induced cytoskeletal rearrangements (Joneson et al. 1996; Lamarche et al. 1996; Westwick et al. 1997), indicating that these functions of Rac and Cdc42 are separable. Because mutations in mbc do not substantially alter dpp expression in the LE, despite causing a dorsal closure defect, it seems likely that Mbc functions in regulating the cytoskeletal changes that drive dorsal closure. It has been demonstrated that mbc mutant embryos exhibit alterations in the morphology of the LE cells, including mislocalization of fasciclin III to the dorsal side of the LE cells prior to closure, as well as reductions in polymerized actin throughout the epidermis at the time of dorsal closure (Erickson et al. 1997).
DOCK180 associates with Rac but not with Cdc42
Because our genetic data support a role for Mbc in Rac-mediated signaling, we examined the possibility that these proteins interact biochemically. Mbc is highly homologous to DOCK180 (Erickson et al. 1997), a protein identified by its interaction with the adaptor protein Crk (Hasegawa et al. 1996), and to the C. elegans protein CED-5 (Wu and Horvitz 1998). Expression of DOCK180 in C. elegans partially rescues the ced-5 phenotype, indicating that the function of these proteins has been evolutionarily conserved (Wu and Horvitz 1998). Moreover, a role for DOCK180 in regulating cell morphology has been suggested by the observation that targeting of DOCK180 to the plasma membrane results in a dramatic shape change in NIH-3T3 cells, potentially implicating Rac (Hasegawa et al. 1996). Therefore, we tested the possibility that DOCK180 physically associates with Rac in cotransfected cells. DOCK180 can be detected specifically in a complex with Rac but not with Cdc42 (Fig. 4A), and this binding appears to be nucleotide independent, as DOCK180 is detected in a complex with both activated (predominantly GTP loaded) and dominant-negative (predominantly GDP associated) forms of Rac. As a control for binding specificity and nucleotide dependence, the Rac/Cdc42 effector target, p65 PAK, was tested similarly in parallel. These results confirm that p65 PAK binds specifically to the active forms of Rac and Cdc42 (Manser et al. 1994; Fig. 4A, bottom), and substantiate the observation that DOCK180 exhibits nucleotide-independent, specific binding to Rac, consistent with the Rac-specific genetic interaction of mbc observed in vivo.
Figure 4.
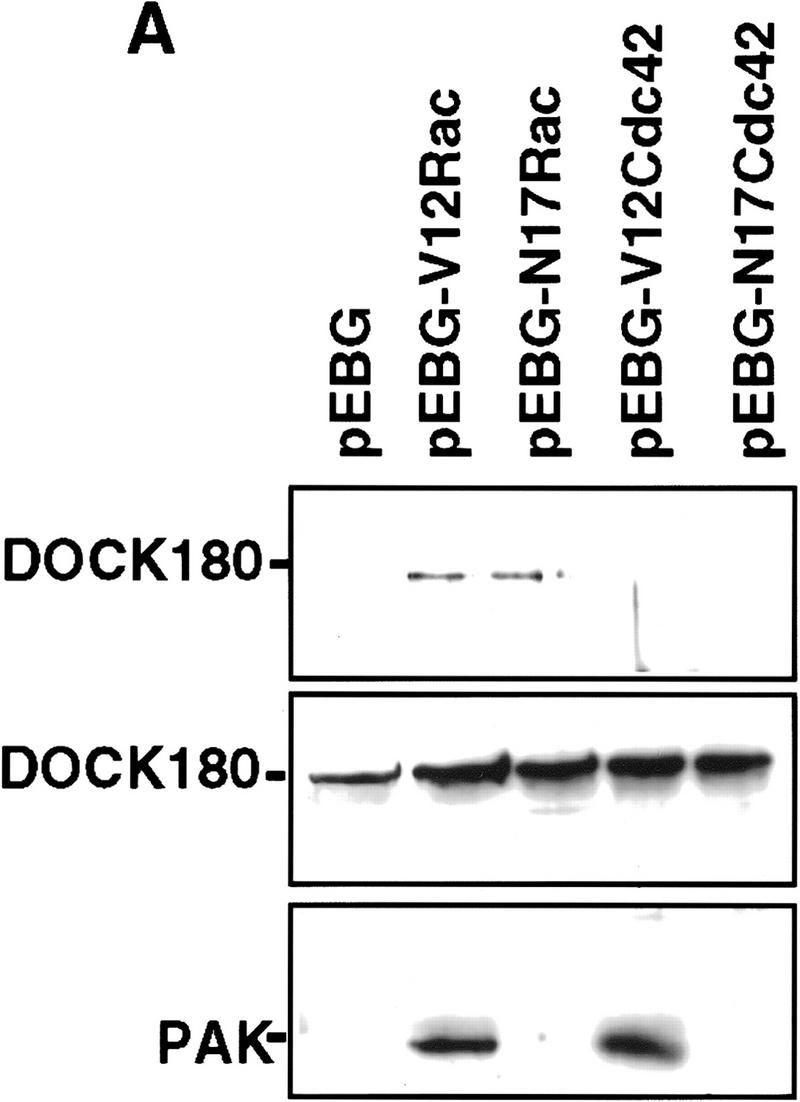


DOCK180 functions upstream of Rac. (A) Cos cells transfected with DOCK180, together with empty vector (pEBG), mammalian GST expression plasmids encoding RacV12, RacN17, Cdc42V12, or Cdc42N17, were subjected to precipitation by glutathione–Sepharose beads (top) and analyzed by SDS-PAGE and anti-DOCK180 immunoblotting. The relative levels of DOCK180 expression are indicated (middle), Cos cells were transfected with Flag-tagged PAK (see above for details) and analyzed by SDS-PAGE and anti-Flag immunoblotting (bottom) (B) Lysates from BOSC cells transfected with either Flag-tagged DOCK180 (left) or Flag-tagged PRK2 (right) were subjected to precipitation by glutathione–Sepharose beads bound to bacterially produced GST–Rac or GST–Rho fusion proteins, either without nucleotide or bound to GDP or GTPγS, and analyzed by SDS-PAGE followed by anti-Flag immunoblotting. The relative levels of GST fusion proteins are indicated (bottom), as revealed by anti-GST immunoblotting. (C) Immunoblot of lysates from Cos cells transfected with c-Jun (lane 1), c-Jun plus anisomycin treatment (lane 2), c-Jun, together with RacV12 (lane 3), DOCK180 (lane 4), and DOCK180 and RacN17 (lane 5). Phosphorylation of c-Jun was detected by immunoblotting with an antibody directed against S63–phospho–c-Jun.
The observation that DOCK180 associates equally well with activated or dominant-negative forms of Rac suggests that, like the guanine nucleotide exchange actors (GEFs), this interaction may be mediated by the nucleotide-free form of the GTPase (Hart et al. 1996). To test this possibility, we examined the interaction of DOCK180 with bacterially expressed GST–Rac in the presence or absence of nucleotide. The interaction of DOCK180 with Rac is blocked by the addition of either GDP or GTPγS (Fig. 4B), suggesting that DOCK180 preferentially forms a complex with the nucleotide-free form of Rac. Additionally, no association between DOCK180 and nucleotide-free Rho was observed. The interaction of the Rho/Rac effector PRK2 with Rac is detected only in the presence of active, GTPγS-bound Rac (Fig. 4B), as reported previously (Vincent and Settleman 1997), confirming that nucleotide-dependent binding can be detected in this assay.
The observation that DOCK180 can be found in a complex with nucleotide-free Rac strongly suggests that Mbc/DOCK180 is functioning upstream of Rac. In cultured cells expressing activated Rac, an increase in JNK activity can be readily detected (Coso et al. 1995). Therefore, to determine whether DOCK180 could contribute to Rac activation, we examined JNK activity in DOCK180-transfected mammalian cells. Cos cells were transfected with c-Jun together with RacV12, DOCK180, or DOCK180 and RacN17, and c-Jun phosphorylation was measured by immunoblotting with an anti-phospho-c-Jun antibody (Fig. 4C). As reported previously, anisomycin strongly induces phosphorylation of c-Jun. DOCK180 stimulates JNK activity to a similar extent as RacV12, and this stimulation of JNK activity is blocked by coexpression of RacN17. This result indicates that DOCK180 is likely to function upstream of Rac.
Because Mbc/DOCK180/CED-5 do not contain the Dbl-homology domain found in all known GEFs for the Rho family of GTPases (Whitehead et al. 1997), it is unlikely that they function directly as a Rac activators. Our Rac–DOCK180-binding results do not exclude the presence of additional components that bridge this interaction. DOCK180 binds to the adaptor protein Crk (Hasegawa et al. 1996), which associates directly with the Rac GEFs, Sos and Vav (Matsuda et al. 1994; Smit et al. 1996), via its amino-terminal SH3 domain. Although the interaction of DOCK180 with Crk requires this same SH3 domain, implying that these are mutually exclusive complexes, it was reported recently that p130 CAS (Crk-associated substrate), which contains multiple SH2-dependent Crk-binding sites (Sakai et al. 1994), regulates cell migration in a Rac-dependent manner (Klemke et al. 1998). Possibly, CAS serves as a scaffold for multiple Crk complexes, some that include DOCK180 and others that include Rac GEFs, and this complex facilitates the interaction of Rac with the Rac GEFs, thereby leading to Rac activation. Interestingly, both Crk and CAS localize to membrane ruffles in migratory cells (Klemke et al. 1998), raising the possibility that a complex containing Crk and CAS as well as a Rac GEF and DOCK180 leads to subcellularly localized Rac activation. As targeting of DOCK180 to the plasma membrane causes morphological changes that resemble those seen in response to Rac activation (Hasegawa et al. 1996), it is possible that the role of DOCK180 in such a complex is to facilitate localization of these proteins to cell membranes.
In light of these biochemical observations, there are two models that most simply explain the role of Mbc in dorsal closure. Possibly, Mbc is required for Rac activation in the LE cells during dorsal closure, but some functional redundancy for JNK regulation is provided by Cdc42. In this scenario, Mbc is still required for Rac-dependent cytoskeletal changes, reflecting a more stringent requirement for Rac activity in regulating cell morphology than in regulating transcription. Consistent with such a possibility, we find that only a small fraction of mbc mutant embryos that exhibit a dorsal closure defect exhibit any detectable reduction in dpp expression. Although this result suggests a lesser role for Mbc in JNK activation than in cytoskeletal regulation, we observed that overexpression of DOCK180 leads to activation of JNK in transfected mammalian cells, suggesting that Mbc can potentially play a role in activating JNK in vivo. Alternatively, two separate pools of Rac, with different subcellular localizations, may be utilized for distinct biological processes. In this scenario, Mbc promotes activation of a pool of Rac that regulates reorganization of the actin cytoskeleton but does not substantially affect the pool of Rac required for JNK activation. Consistent with this model, we find that DOCK180 colocalizes with Rac in membrane ruffles (data not shown), raising the possibility that Rac, and perhaps other GTPases, can regulate distinct biological processes within a single cell by virtue of subcellularly localized activation.
Materials and methods
Fly stocks
Full-length Drosophila rac1, rac2, and cdc42 coding sequences (Luo et al. 1994; Hariharan et al. 1995) were subcloned into the pGMR vector (Hay et al. 1994) and transformed into Drosophila as described previously (Hariharan et al. 1995). GMR–rac17a flies harbor one copy of the transgene and GMR–cdc42 33b flies harbor three copies of the transgene. w; iso2; iso3 is isogenized for the second and third chromosomes and was used as the wild-type strain. mbc alleles used for phenotype analysis are 1.63, 2.35, 3.5, and 4.25. Each allele suppresses the rac1-induced rough eye to a similiar extent, although the dorsal closure phenotype of these alleles in combination with Df(3R)mbc-f5.3 is variably penetrant, indicating that they are hypomorphic. Df(3R)mbc-f5.3 was provided by S. Abmayr (Erickson et al. 1997) and mbczz351 by A. Michelson (Harvard Medical School, Boston, MA). hep mutants were provided by S. Noselli (Centre National de la Recherche Scientifique, Toulouse, France). For mutagenesis, w; iso2; iso3 males were fed 25 mm ethylmethane sulfonate overnight and mated to w; GMR–rac17a females. The F1 progeny were examined by light microscopy for suppression of the rac1-induced eye phenotype. Four of the Su(rac)1 alleles were meiotically mapped with the markers th, st, c, sr, e, and ca, and the mutations were found to lie between e (3–70.7) and ca (3–100.7).
Scanning electron microscopy and histology
Retinal eye sections were prepared from fly heads embedded in Durcapan resin (Fluka Chemical) as described previously (Tomlinson and Ready 1987). Scanning electron micrographs of eyes were carried out as described previously (Kimmel et al. 1990).
Immunohistochemistry and in situ hybridization of embryos
Antibody staining was carried out on embryos as described previously (Patel 1994) with anti-myosin heavy chain antiserum [provided by D. Kiehart (Duke University, Chapel Hill, NC)], BP102 monoclonal antibody (obtained from the Developmental Studies Hybridoma Bank), or anti-β-galactosidase (Promega) to detect the presence of the TM3 balancer, which carries a P-element insertion with a lacZ marker. Whole-mount in situ hybridization was performed as described (Van Vactor and Kopczynski 1998) using digoxygenin-labeled probes (Genius II kit, Boehringer Mannheim) synthesized from a plasmid containing dpp cDNA (provided by E. Hafen, Riesgo-Escovar and Hafen 1997a,b). A lacZ RNA probe was used to mark the embryos with the balancer chromosome. Embryos were staged according to anatomical characteristics as described previously (Campos-Ortega and Hartenstein 1985a).
Transfections
Cos-7 and BOSC cells were maintained at 37°C in Dulbecco’s modified Eagle medium supplemented with 10% heat-inactivated fetal bovine serum. For transfections, Cos cells in 10-cm plates were incubated with 5 μg of plasmid DNA, mixed with DEAE–dextran for 4 hr, and subjected to a 45-sec shock with 10% dimethylsulfoxide. BOSC transfections were carried out by calcium phosphate precipitation. Cells were collected for protein analysis 48 hr after transfection.
GTPase binding assays
Plasmids encoding mammalian GST fusions of Rac1 and Cdc42 (provided by J. Blenis; Harvard Medical School, Boston, MA) were cotransfected with either Flag-tagged-PAK or DOCK180 (Hasegawa et al. 1996) into Cos cells. Cells were lysed in a solution containing 50 mm HEPES (pH 7.4), 150 mm NaCl, 1.5 mm MgCl2, 5 mm EGTA, 1% Triton X-100, 10% glycerol, and protease inhibitors (1 mm PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin). Clarified lysates were incubated with glutathione–Sepharose beads at 4°C for 1 hr. Beads were washed in 20 mm HEPES (pH 7.5), 150 mm NaCl, 10% glycerol, and 0.1% Triton X-100, and bound protein was eluted in SDS-containing sample buffer and resolved by SDS-PAGE. Proteins were transferred to nitrocellulose and detected with DOCK180 antibodies (kindly provided by M. Matsuda, International Medical Center of Japan, Tokyo) or M2 anti-Flag antibody (Sigma) followed by horseradish peroxidase-labeled secondary antibodies and development by enhanced chemiluminescence (ECL, DuPontNEN). Preparation of recombinant GST fusion proteins is described previously (Vincent and Settleman 1997). GST fusion proteins bound to glutathione beads were loaded with guanine nucleotides by incubation for 15 min at 37°C in 50 μl of exchange buffer (50 mm HEPES at pH 7.5, 5 mm EDTA, 0.1 mm EGTA, 50 mm NaCl, 0.1 mm DTT) containing 0.5 mm of the appropriate nucleotide. The reaction was stopped by the addition of MgCl2 to a final concentration of 20 mm on ice. Binding to the recombinant GST fusion proteins was carried out as described above.
c-Jun phosphorylation
Cos cells were transfected with a c-Jun vector (provided by M. Classon, Massachusetts General Hospital Cancer Center), together with RacV12, DOCK180, or DOCK180 and RacN17 plasmids. Cells were lysed directly in SDS-containing sample buffer 36 hr after transfection and sonicated briefly before SDS-PAGE analysis. For anisomycin activation, cells were treated with 50 μg/ml anisomycin for 20 min before collection. Phosphorylated c-Jun was detected by immunoblotting with an antibody against Ser-63-phosphorylated c-Jun (New England Biolabs).
Acknowledgments
We thank Susan Abmayr, John Blenis, Marie Classon, Ernst Hafen, Dan Kiehart, Liqun Luo, Alan Michelson, and Stephane Noselli for reagents. Ed Seling provided expert assistance with SEMs. June Kim assisted with the GTPase-binding studies. We are grateful to Michiyuki Matsuda for the DOCK180 reagents and for communicating results prior to publication. We also thank members of our laboratory and that of Iswar Hariharan for generous advice throughout these studies. The work was supported by an award to J.S. from the American Cancer Society.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL settleman@helix.mgh.harvard.edu; FAX (617) 726-7808.
References
- Barrett K, Leptin M, Settleman J. The Rho GTPase and a putative RhoGEF mediate a signaling pathway for the cell shape changes in Drosophila gastrulation. Cell. 1997;91:905–915. doi: 10.1016/s0092-8674(00)80482-1. [DOI] [PubMed] [Google Scholar]
- Campos-Ortega JA, Hartenstein V. The embryonic development of Drosophila melanogaster. New York, NY: Springer-Verlag; 1989. [Google Scholar]
- Coso OA, Chiariello M, Yu J-C, Teramoto H, Crespo P, Xu N, Miki T, Gutkind J. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- Eaton S, Wepf R, Simons K. Roles for Rac1 and Cdc42 in planar polarization and hair outgrowth in the wing of Drosophila. J Cell Biol. 1996;135:1277–1289. doi: 10.1083/jcb.135.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson MR, Galleta BJ, Abmayr SM. Drosophila myoblast city encodes a conserved protein that is essential for myoblast fusion, dorsal closure, and cytoskeletal organization. J Cell Biol. 1997;138:589–603. doi: 10.1083/jcb.138.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glise B, Noselli S. Coupling of Jun amino-terminal kinase and Decapentaplegic signaling pathway in Drosophila morphogenesis. Genes & Dev. 1997;11:1738–1747. doi: 10.1101/gad.11.13.1738. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Harden N, Loh HY, Chia W, Lim L. A dominant inhibitory version of the small GTP-binding protein Rac disrupts the cytoskeletal structures and inhibits developmental cell shape changes in Drosophila. Development. 1995;121:903–914. doi: 10.1242/dev.121.3.903. [DOI] [PubMed] [Google Scholar]
- Hariharan IK, Hu K-Q, Asha H, Quintanilla A, Ezzell RM, Settleman J. Characterization of Rho GTPase homologues in Drosophila melanogaster: Overexpression Rho1 causes a late developmental defect. EMBO J. 1995;14:292–302. doi: 10.1002/j.1460-2075.1995.tb07003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart MJ, Sharma S, elMasry N, Qiu R-Q, McCabe P, Polakis P, Bollag G. Identification of a novel guanine nucleotide exchange factor for the rho GTPase. J Biol Chem. 1996;271:25452–25458. doi: 10.1074/jbc.271.41.25452. [DOI] [PubMed] [Google Scholar]
- Hasegawa H, Kiyokawa E, Tanaka S, Nagashima K, Gotoh N, Shibuya M, Kurata T, Matsuda M. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol Cell Biol. 1996;16:1770–1776. doi: 10.1128/mcb.16.4.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay BA, Wolff T, Rubin GM. Expression of baculovirus p35 prevents cell death in Drosophila. Development. 1994;120:2121–2129. doi: 10.1242/dev.120.8.2121. [DOI] [PubMed] [Google Scholar]
- Hou XS, Goldstein ES, Perrimon N. Drosophila Jun relays the Jun amino-terminal kinase signal transduction pathway to the Decapentaplegic signal transduction pathway in regulating epithelial cell sheet movement. Genes & Dev. 1997;11:1728–1737. doi: 10.1101/gad.11.13.1728. [DOI] [PubMed] [Google Scholar]
- Joneson T, White MA, Wigler MH, Bar-Sagi D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of Ras. Science. 1996;271:810–812. doi: 10.1126/science.271.5250.810. [DOI] [PubMed] [Google Scholar]
- Keely PJ, Westwick JK, Whitehead IP, Der CJ, Parise LV. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature. 1997;390:632–636. doi: 10.1038/37656. [DOI] [PubMed] [Google Scholar]
- Kimmel BE, Heberlein U, Rubin GM. The homeodomain protein rough is expressed in a subset of cells in the developing Drosophila eye where it can specify photoreceptor cell subtype. Genes & Dev. 1990;4:712–727. doi: 10.1101/gad.4.5.712. [DOI] [PubMed] [Google Scholar]
- Klemke RL, Leng J, Molander R, Brooks PC, Vuori K, Cheresh DA. CAS/Crk coupling serves as a ‘molecular switch’ for induction of cell migration. J Cell Biol. 1998;140:961–972. doi: 10.1083/jcb.140.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarche N, Tapon N, Burbelo PD, Aspendrom P, Bridges T, Chant J, Hall A. Rac and Cdc42 induce actin polymerization and G1 cell cycle progression independently of p65pak and the JNK/SAPK MAP kinase cascade. Cell. 1996;87:519–529. doi: 10.1016/s0092-8674(00)81371-9. [DOI] [PubMed] [Google Scholar]
- Luo L, Yiao YJ, Jan LY, Jan YN. Distinct morphogenetic functions of similar small GTPases: Drosophila Drac1 is involved in axonal outgrowth and myoblast fusion. Genes & Dev. 1994;8:1781–1802. doi: 10.1101/gad.8.15.1787. [DOI] [PubMed] [Google Scholar]
- Manser E, Leung T, Salihuddin H, Zhao ZS, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–46. doi: 10.1038/367040a0. [DOI] [PubMed] [Google Scholar]
- Matsuda M, Hashimoto Y, Muroyu K, Hasegawa H, Kurata T, Tanaka S, Nakamura S, Hattori S. CRK protein binds to two guanine nucleotide-releasing proteins for the Ras family and modulates nerve growth factor induced activation of ras in PC12 cells. Mol Cell Biol. 1994;14:5495–5500. doi: 10.1128/mcb.14.8.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel NH. Imaging neuronal subsets and other cell types in whole-mount Drosophila embryos and larvae using antibody probes. In: Goldstein LSB, Fyrberg EA, editors. Drosophila melanogaster: Practical uses in cell and molecular biology. San Diego, CA: Academic Press; 1994. pp. 445–487. [DOI] [PubMed] [Google Scholar]
- Riesgo-Escovar JR, Hafen E. Common and distinct roles DFos and DJun during Drosophila development. Science. 1997a;278:669–672. doi: 10.1126/science.278.5338.669. [DOI] [PubMed] [Google Scholar]
- ————— Drosophila Jun kinase regulates expression of decapentaplegic via the ETS-domain protein Aop and the AP-1 transcription factor DJun during dorsal closure. Genes & Dev. 1997b;11:1717–1727. doi: 10.1101/gad.11.13.1717. [DOI] [PubMed] [Google Scholar]
- Rushton E, Drysdale R, Abmayr SM, Michelson AM, Bate M. Mutations in a novel gene, myoblast city, provide evidence in support of the founder cell hypothesis for Drosophila muscle development. Development. 1995;121:1979–1988. doi: 10.1242/dev.121.7.1979. [DOI] [PubMed] [Google Scholar]
- Sanger JW, Holtzer S, Holtzer H. Effects of cytochalasin B on muscle cells in tissue culture. Nature. 1971;229:121–123. doi: 10.1038/newbio229121a0. [DOI] [PubMed] [Google Scholar]
- Sakai R, Iwamatsu A, Hirano N, Ogawa S, Tanaka T, Mano H, Yazaki Y, Hirai H. A novel signaling molecule, p130, forms with v-Crk and v-Src in a tyrosine phosphorylation-dependent manner. EMBO J. 1994;13:3748–3756. doi: 10.1002/j.1460-2075.1994.tb06684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit L, van der Horst G, Borst J. Sos, Vav, and C3G participate in B cell receptor-induced signaling pathways and differentially associate with Shc-Grb2, Crk, and Crk-L adaptors. J Biol Chem. 1996;271:8564–8569. doi: 10.1074/jbc.271.15.8564. [DOI] [PubMed] [Google Scholar]
- Strutt DI, Weber U, Mlodzik M. The role of RhoA in tissue polarity and Frizzled signalling. Nature. 1997;387:292–295. doi: 10.1038/387292a0. [DOI] [PubMed] [Google Scholar]
- Tomlinson A, Ready DF. Cell fate in the Drosophila ommatidium. Dev Biol. 1987;123:264–275. doi: 10.1016/0012-1606(87)90448-9. [DOI] [PubMed] [Google Scholar]
- Van Aelst L, D’Souza-Schorey C. Rho GTPases and signaling networks. Genes & Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- Van Vactor D, Kopczynski C. Anatomical techniques for analysis of nervous system development in the Drosophila embryo. In: Richter J, editor. A comparative methods approach to the study of oocytes and embryos. New York, NY: Oxford University Press; 1998. . (In press). [Google Scholar]
- Vincent S, Settleman J. The PRK2 kinase is a potential effector target of both Rho and Rac GTPases and regulates actin cytoskeletal organization. Mol Cell Biol. 1997;17:2247–2256. doi: 10.1128/mcb.17.4.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwick J, Lambert Q, Clark G, Symonds M, Van Aelst L, Pestell R, Der C. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Mol Cell Biol. 1997;17:1324–1335. doi: 10.1128/mcb.17.3.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead IP, Campbell S, Rossman KL, Der CJ. Dbl family proteins. Biochim Biophys Acta. 1997;1332:F1–23. doi: 10.1016/s0304-419x(96)00040-6. [DOI] [PubMed] [Google Scholar]
- Wu Y-C, Horvitz HR. C. elegans phagocytosis and cell-migration protein CED-5 is similar to human DOCK180. Nature. 1998;392:501–503. doi: 10.1038/33163. [DOI] [PubMed] [Google Scholar]
- Zipkin ID, Kindt RM, Kenyon CJ. Role of a new Rho family member in cell migration and axon guidance in C. elegans. Cell. 1997;90:883–894. doi: 10.1016/s0092-8674(00)80353-0. [DOI] [PubMed] [Google Scholar]