

Abstract
Plectin, a major linker and scaffolding protein of the cytoskeleton, has been shown to be essential for the mechanical integrity of skin, skeletal muscle, and heart. Studying fibroblast and astroglial cell cultures derived from plectin (−/−) mice, we found that their actin cytoskeleton, including focal adhesion contacts, was developed more extensively than in wild-type cells. Also it failed to show characteristic short-term rearrangments in response to extracellular stimuli activating the Rho/Rac/Cdc42 signaling cascades. As a consequence, cell motility, adherence, and shear stress resistance were altered, and morphogenic processes were delayed. Furthermore, we show that plectin interacts with G-actin in vitro in a phosphatidylinositol-4,5-biphosphate-dependent manner and associates with actin stress fibers in living cells. The actin stress fiber phenotype of plectin-deficient fibroblasts could be reversed to a large degree by transient transfection of full-length plectin or plectin fragments containing the amino-terminal actin-binding domain (ABD). These results reveal a novel role of plectin as regulator of cellular processes involving actin filament dynamics that goes beyond its proposed role in scaffolding and mechanical stabilization of cells.
Keywords: Cytoskeleton, plectin, Rho/Rac/Cdc42, actin dynamics, fibroblasts
Plectin is a multidomain protein of exceptionally large size and versatile binding properties. It has been shown to interact in vitro with intermediate filament (IF) proteins of various types (Foisner et al. 1988) and to physically link IFs with microfilaments and microtubules, as demonstrated by whole mount electron microscopy of cultured cells (Foisner et al. 1995; Svitkina et al. 1996). The interaction with vimentin and cytokeratin filaments involves a specific binding domain located near to the carboxyl terminus of the protein (Nikolic et al. 1996), whereas the amino-terminal domain of the molecule harbors an actin-binding domain (ABD) of the spectrin type, as deduced from its cDNA sequence (Liu et al. 1996; McLean et al. 1996; Elliott et al. 1997). Moreover, in various cell types and tissues plectin has been localized at specific cytoskeleton-plasma membrane junctional complexes, including epidermal hemidesmosomes, dense plaques of smooth muscle, and focal adhesion contacts of cultured cells (Wiche et al. 1983, 1984; Seifert et al. 1992; Rezniczek et al. 1998). On the molecular level, we have demonstrated interactions of plectin with membrane skeleton proteins, such as fodrin and α-spectrin (Herrmann and Wiche 1987), the hemidesmosome integrin receptor subunit protein β4 (Rezniczek et al. 1998), and desmosome-associated desmoplakin (Eger et al. 1997). Other prominent locations of plectin are Z-lines, intracellular junctional structures of striated muscle, and intercalated discs of cardiac muscle (Wiche et al. 1983). Consistent with the versatile binding properties and strategic cellular localization of plectin, the phenotypic analyses of plectin-deficient epidermolysis bullosa simplex–muscular dystrophy (EBS–MD) patients (Chavanas et al. 1996; Gache et al. 1996; McLean et al. 1996; Pulkkinnen et al. 1996; Smith et al. 1996; Mellerio et al. 1997) and plectin gene knockout mice (Andrä et al. 1997) revealed that plectin is essential for the mechanical integrity of skin and muscle cells.
To investigate the function of plectin in more detail on the cellular level, we characterized the cytoskeleton of dermal fibroblasts and astroglial cells derived from plectin-deficient mice. No or only minor changes in microtubule and IF network organization were detectable in such cells; however, contrary to expectations, actin stress fibers and focal adhesion contacts were substantially increased in numbers. In addition, the actin stress fiber system of plectin-deficient cells failed to show characteristic short-term rearrangments in response to extracellular stimuli, and as a consequence the adhesion properties, migration abilities, and shear stress resistance of plectin-deficient cells were influenced and/or compromised. Furthermore, we found that the actin binding domain of plectin is functional, can be modulated by phosphatidylinositol-4,5-biphosphate (PIP2), and is able to partially restore the wild-type phenotype upon expression in plectin-deficient cells. These results support the idea that plectin not only provides cells with mechanical strength but also plays an important role as regulator of cellular processes linked to actin filament dynamics.
Results
Plectin deficiency leads to an increased number of actin stress fibers and focal adhesion contacts
To study the effects of plectin deficiency on cytoarchitecture, cultivated dermal mouse fibroblasts expressing (plectin+/+) or lacking (plectin−/−) plectin were used at passages 10–15. At this stage both types of cells appeared to be of similar morphology, as judged by phase-contrast microscopy (not shown), and exhibited similar growth properties (Fig. 1G). Immunofluorescence microscopy of plectin+/+ cells using antiserum to plectin (Fig. 1A) revealed a filamentous staining pattern throughout the cytoplasm that was undetectable in plectin−/− fibroblasts (Fig. 1B). Unexpectedly, when plectin-deficient fibroblasts were plated out for a short time (2 hr), the cells displayed a dramatic increase in actin stress fibers (Fig. 1D) compared to control cells (Fig. 1C). Their stress fibers were also more resistant toward treatment with cytochalasin B (data not shown). The higher abundance of actin stress fibers in plectin−/− cells was paralleled by an increase in number and size of vinculin-positive focal adhesion contacts (FACs). In control fibroblasts these structures were predominantly observed at the periphery of cells, displaying typical dash-like shapes (Fig. 1E), whereas in plectin−/− fibroblasts they appeared to be abnormally enlarged and scattered all over the cell (Fig. 1F). A statistical analysis of 50 cells randomly chosen from confocal microscopy images demonstrated that the increased numbers of actin stress fibers and FACs in plectin−/− cells were cell size-independent (Fig. 1H,I). This analysis also revealed a slight increase in the overall size of plectin-deficient cells compared to controls (Fig. 1H,I). Interestingly, after a prolonged time in culture (24 hr), the abundance of stress fibers in plectin-deficient fibroblasts was found to be comparable to control cells (data not shown), suggesting that other regulatory mechanisms could compensate for the absence of plectin upon long-term cultivation.
Figure 1.
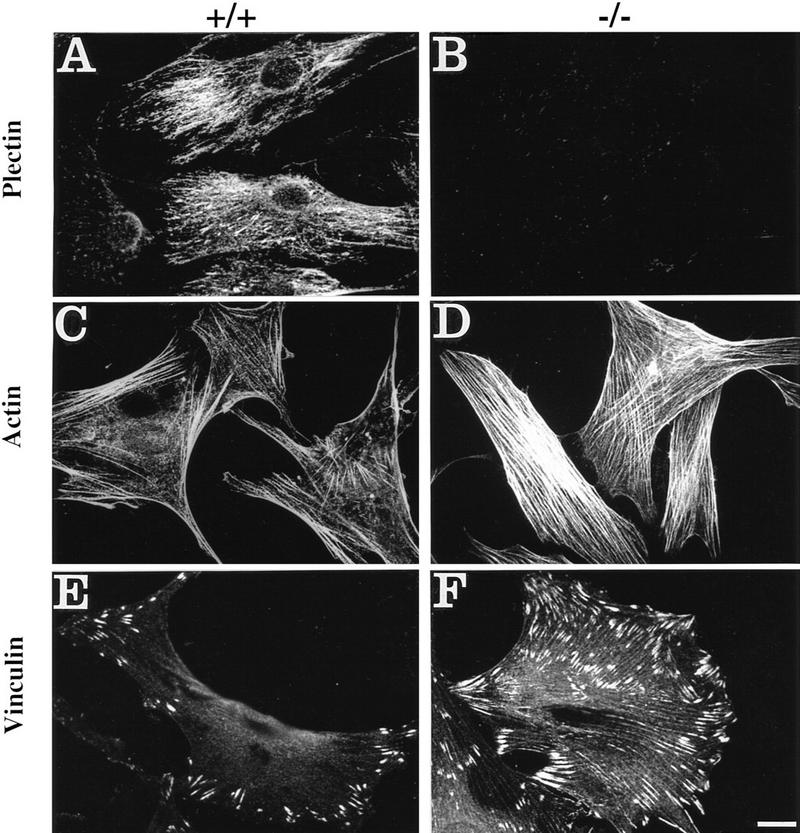

Augmentation of actin stress fibers and focal adhesion contacts in plectin-deficient fibroblasts during early spreading. Plectin+/+ and plectin−/− fibroblasts were processed for immunofluorescence microscopy after an attachment period of 2 hr. Anti-plectin (A,B), anti-actin (C,D), and anti-vinculin (E,F) were used as primary antibodies. Note higher numbers of stress fibers (C,D) and more numerous and enlarged focal adhesion contacts (E,F) in plectin-deficient cells (D,F), compared to control cells; also, in plectin−/− cells FACs are evenly, rather than peripherally, distributed over the cell (F). (G) Growth curves of plectin-deficient (• ± s.d., n = 6) and control fibroblasts (⋄ ± s.d., n = 6). (H,I) Numerical analysis of actin stress fibers (H) and FACs (I) in control (□) and plectin-deficient fibroblasts (•). Data points represent individual cells. Note higher number of stress fibers and FACs in plectin-deficient cells vs. control cells of similar sizes. Bar, 30 μm.
The actin cytoskeleton of plectin-deficient fibroblasts shows delayed responses to stimuli of the Rho/Rac/Cdc42 pathways
The formation of stress fibers in fibroblasts, as well as other short-term responses involving the reorganization of the actin cytoskeleton of motile cells, specifically the formation of lamellipodia, microspikes, and filopodia, are regulated by small GTPases named Rho, Rac, and Cdc42. These GTPases can be activated via receptor-linked processes by soluble ligands, such as lysophosphatidic acid (LPA), PDGF, or bradykinin (for review, see Hall 1998). Omission of LPA by starving cells in serum-free medium for 24 hr led to a marked reduction of stress fibers and partial redistribution of actin to the cell periphery in control cells (Fig. 2A), but not in the majority of plectin−/− cells (Fig. 2B). Normal, serum-starved fibroblasts formed surface membrane ruffles and lamellipodia upon treatment with PDGF for 1 hr (Fig. 2C, arrowheads), and after a 10-min incubation with bradykinin they developed numerous microspikes and a few filopodia, which were visualized by immunofluorescence microscopy as small actin-positive cell protrusions (Fig. 2E, arrowheads). Small cellular extensions resembling ruffles were also observed in some serum-starved plectin−/− cells after a similar treatment with PDGF (Fig. 2D, arrowheads). However, the majority of cells (∼90%) exhibited unaltered prominent arrays of stress fibers (Fig. 2D), which was true also for bradykinin-treated plectin−/− cells (Fig. 2F). This clearly indicated that the capability of cells to rearrange their actin cytoskeleton upon short-term stimulation with soluble ligands was compromised in the absence of plectin.
Figure 2.
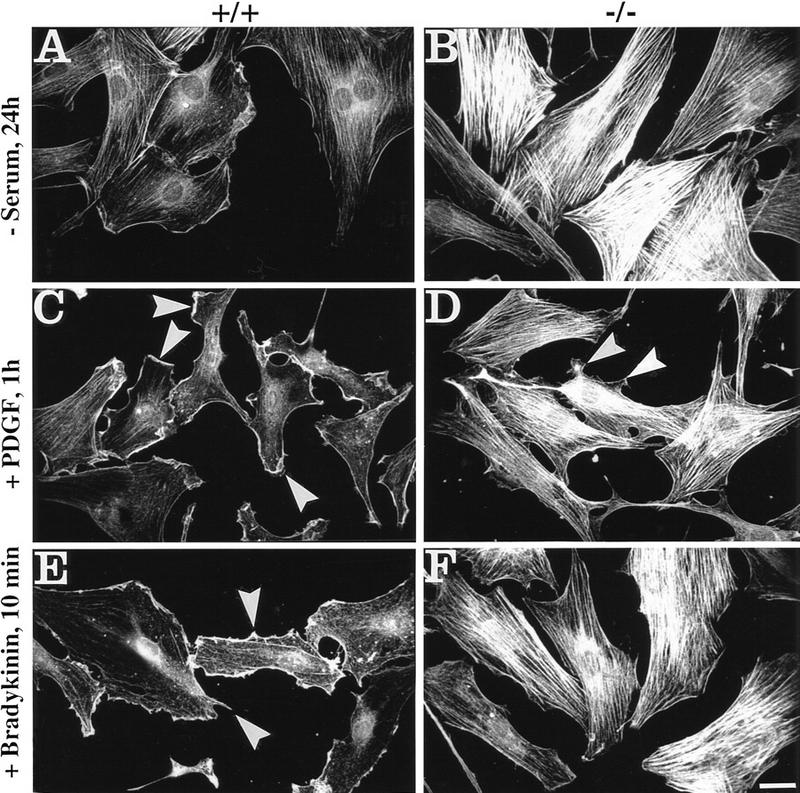
Impairment of actin cytoskeleton rearrangement in response to Rho, Rac, and Cdc42 activation in plectin-deficient vs. control cells. Cells exposed to serum starvation (−serum), or treatment with PDGF or bradykinin, as indicated, were analyzed by immunofluorescence microscopy using antibodies to actin. Note contrast between serum-starved plectin+/+ (A) and plectin−/− (B) mouse fibroblasts in number and density of actin stress fibers. In normal cells formation of membrane ruffles and lamellipodia (C, arrowheads) and microspikes (E, arrowheads) were induced upon treatment with PDGF and bradykinin, respectively, but not in plectin−/− cells (D,F), except for very few cell protrusions resembling ruffles (D, arrowheads). Bar, 30 μm.
To assess whether the actin cytoskeleton of serum-starved plectin−/− cells would eventually become responsive to the same stimuli at later times, 24 hr-starved cells were left for another 24 hr without serum or were incubated with PDGF, or bradykinin, over the same period. Under these conditions, both types of cells showed similar organization of their actin cytoskeleton. The actin stress fiber system, still prominent in most plectin−/− cells after 24 hr of serum starvation (Fig. 2B), was hardly visible (Fig. 3B). The appearances of actin structures in PDGF- and bradykinin-treated plectin−/− cells (Fig. 3D,F) overall were very similar to those in control cells (Fig. 3C,E), except for a slight tendency of fewer ruffles and a more prominent localization of actin at the cell periphery in PDGF-treated mutant cells compared to wild-type cells (Fig. 3, cf. C and D). These results indicated that plectin deficiency does not prevent actin cytoskeleton rearrangements but causes delays in their execution.
Figure 3.
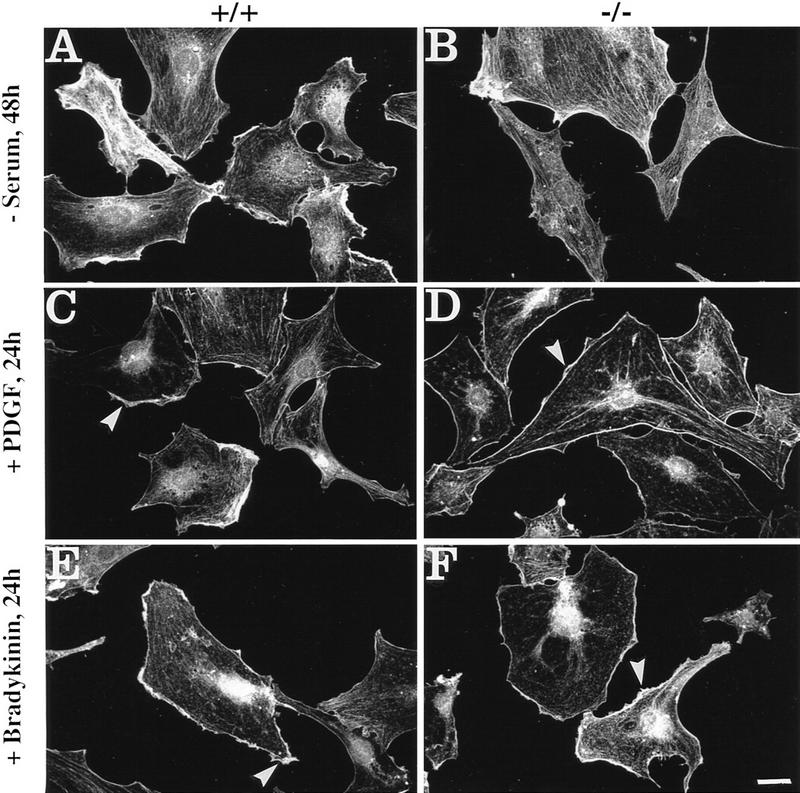
Impairment of actin cytoskeleton rearrangement in response to Rho, Rac, and Cdc42 activation is abolished in plectin-deficient cells upon 48 hr serum starvation and prolonged treatment (24 hr) with PDGF or bradykinin. Cells were analyzed by immunofluorescence microscopy using antibodies to actin. Note loss of actin stress fibers in serum-starved, PDGF-treated, or bradykinin-treated normal fibroblasts (A,C,E), similar to plectin−/− mouse fibroblasts (B,D,F), and the visualization of similar membrane-associated structures in both cell types after incubation with PDGF (C,D, arrowheads), and short microspikes after treatment with bradykinin (E,F, arrowheads).
Plectin-deficient fibroblasts exhibit reduced motility and increased adherence
Cell motility depends to a large degree on a precisely balanced regulation of the formation and rearrangement of the actin–myosin-based cytoskeleton. We therefore investigated whether the impaired capability of cells to rearrange actin structures, as observed in plectin−/− fibroblasts, influences their migratory abilities. In a wound-healing assay, plectin−/− fibroblasts turned out to migrate into the denuded monolayer area more slowly than normal fibroblasts, with significant differences observed particularly at later time points (Fig. 4A). In another migration assay, in which cells, after an adhesion time of 2 hr, could migrate through a porous membrane in response to a gradient of the chemoattractant PDGF, only about half the number of plectin−/− cells had passed through the membrane as compared to control cells (Fig. 4B). A regulated cooperation between FACs and the actin stress fibers is believed to be essential for tight cellular adhesion and stabilization of cells against external mechanical stress. When plectin−/− fibroblasts and control cells were plated out for only 2 hr before being exposed to a defined fluid shear stress (15 dyn/cm2), plectin−/− cells remained attached to the glass plate more frequently than control cells, indicating greater resistence to shear stress (Fig. 4C). However, when cells were allowed to attach for 24 hr, hardly any difference was seen between controls and plectin-deficient cells (Fig. 4D). These results correlated well with a decrease in both the number of stress fibers and FACs observed in plectin−/− cells after 24 hr versus 2 hr plating. This suggested that the reduced motility of plectin-deficient fibroblasts was a consequence of their tighter adherence.
Figure 4.

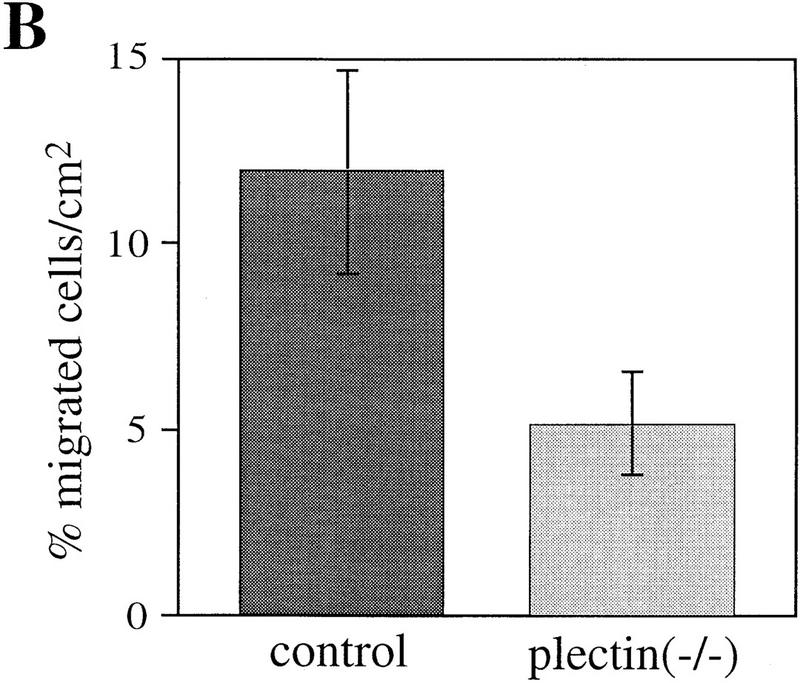
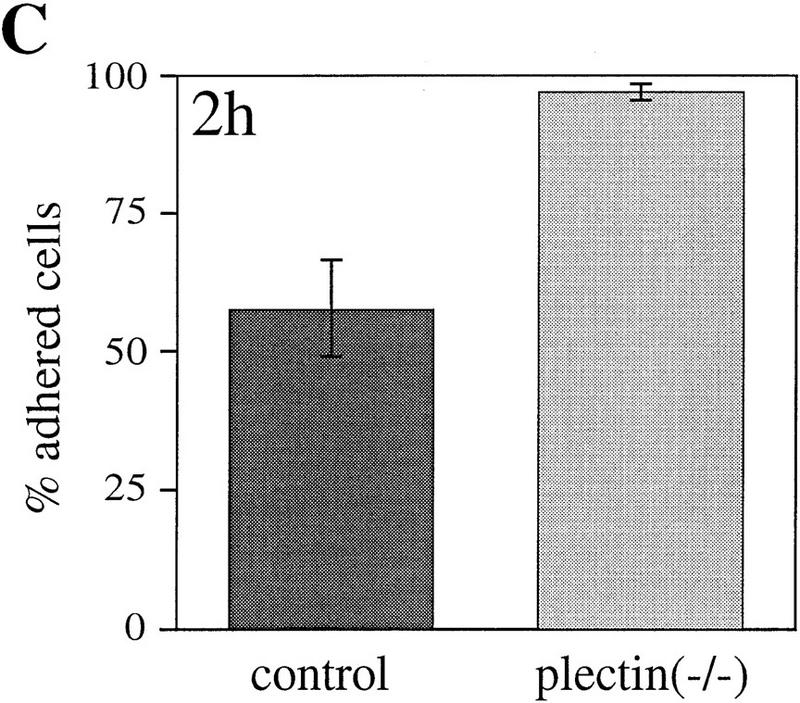
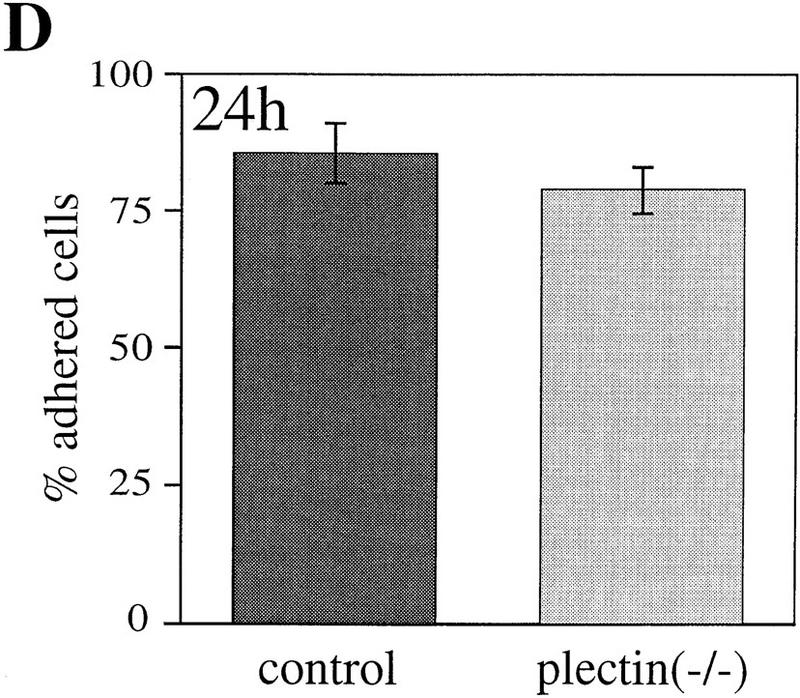
Reduced motility and enhanced adhesion of plectin-deficient fibroblasts. (A) Wound-healing assay. Note significantly reduced migration rate of plectin-deficient fibroblasts (•) compared to control cells (□). Each data point represents the mean ± s.d. (n = 10); significant differences are observed after 6–7 hr. (B) Migration through filter membrane in response to a gradient of PDGF (mean ± s.d., n = 6). (C,D) Shear stress resistance measurements (mean ± s.d., n = 6). Assays were carried out 2 hr (B,C) or 24 hr (D) after plating of cells.
Astroglial cells lacking plectin show delayed morphological differentiation upon cAMP stimulation
The effects of plectin deficiency on cytoarchitecture and actin cytoskeleton dynamics were further investigated in primary cultures of astroglial cells derived from brains of normal and plectin-deficient mice. Astroglia cells are known to express plectin at high level (Wiche and Baker 1982; Errante et al. 1994) and to undergo dramatic morphological changes involving actin-based mechanisms mediated by the GTPase Rho in response to dibutyryl–cAMP (db-cAMP) treatment (Koyama et al. 1996; Yoshimura et al. 1997). As identified by immunostaining using antibodies to the glia-specific IF subunit protein GFAP, no differences in the morphological appearance of GFAP IF networks in plectin−/− and control astroglial cells were detectable by immunofluorescence microscopy (Fig. 5A,B). After a 6-hr incubation period with db-cAMP, the majority of control astroglial cells had undergone the expected conversion from a flat and well-spread shape (Fig. 5A) to a stellate appearance with numerous long processes extending from the cell body (Fig. 5C, arrowheads). In contrast, the majority of plectin-deficient astroglial cells retained their flattened cell shape during this time and displayed only a few protrusions (Fig. 5B,D). Quantitative measurements indicated that the percentage of plectin-deficient astroglial cells, having undergone process formation after 6 hr of incubation with db-cAMP (∼30%), was only about half of the value (∼60%) found in normal astroglial cells (Fig. 5E), whereas after a 24-hr exposure to the drug nearly 100% of control cells and 80%–90% of plectin−/− cells were found to have arborized, indicating a delay in the morphological transformation of the cells due to the lack of plectin.
Figure 5.
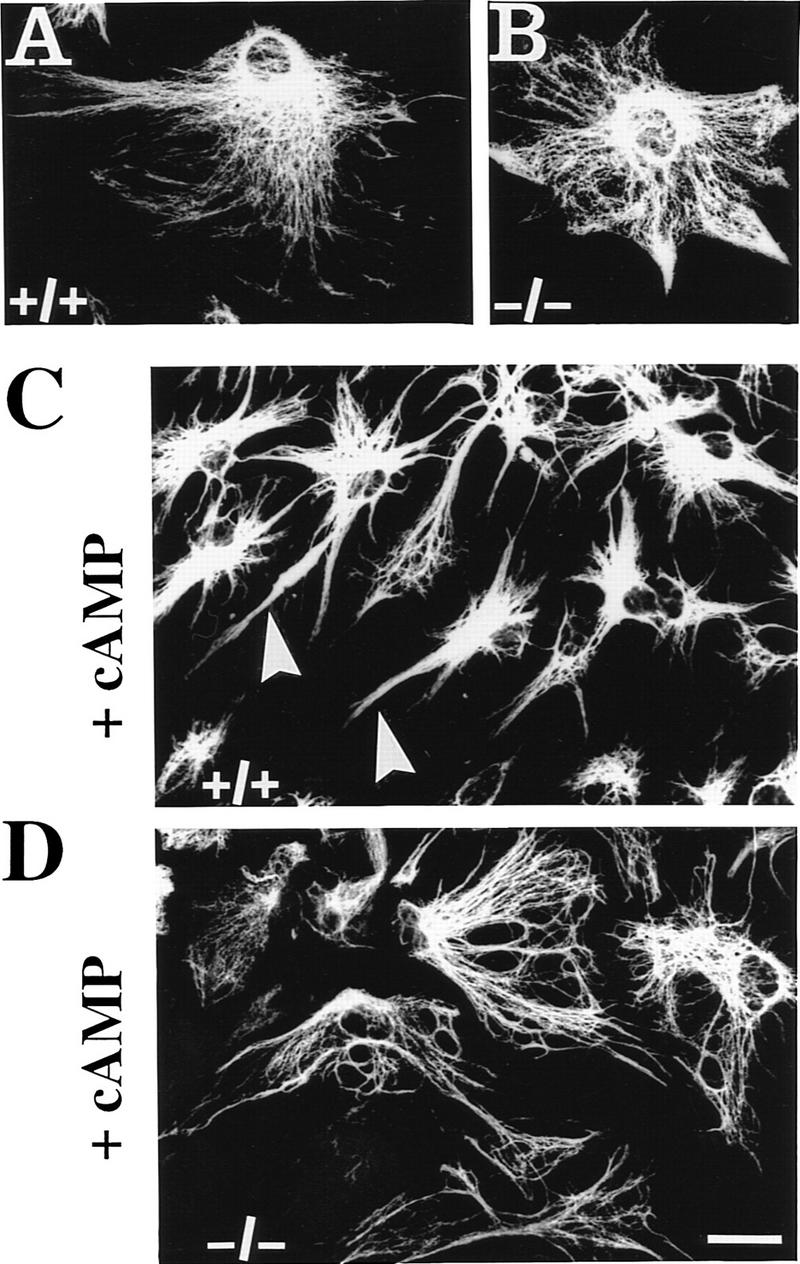

Impairment of db-cAMP-induced morphological differentiation of plectin-deficient astroglial cells. (A–D) Immunofluorescence microscopy of GFAP IF networks in 6-day-old primary astroglial cells derived from plectin+/+ and plectin−/− mouse brain, after incubation with (C,D) or without (A,B) 1 mm db-cAMP for 6 hr. Note stellate morphology with long protrusions in db-cAMP-treated normal astroglial cells (C, arrowheads), compared to more regular and flat appearance of plectin-deficient astroglia with hardly any processes (D). Bar, 20 μm. (E) Quantification (mean ± s.d., n = 10) of differentiation after 6 hr treatment with 1 mm db-cAMP. (Shaded bar) Plectin+/+; (open bar) plectin−/−.
The actin-binding domain of plectin is functional and can be modulated by PIP2 in vitro
The above results convincingly demonstrated an important role of plectin in regulating motility and cellular morphogenesis, especially involving short-term responses of the actin cytoskeleton. To investigate whether the highly conserved ABD of plectin, identified at the amino terminus of the molecule by sequence homology (Elliott et al. 1997), was important for these effects, a series of in vitro experiments were carried out. First, recombinant plectin protein fragments containing (ABD/2–6, ABD/1–24, and ABD/1b–24) or not containing [Ple–R4, Ple–R5, and Ple–R5(ANAA)] the putative ABD were purified from transfected bacteria (Fig. 6A; see also Fig. 5A in Nikolic et al. 1996), and subjected to a microtiter plate overlay assay (Nikolic et al. 1996) using Eu3+-labeled G-actin. Binding of G-actin was observed to all fragments containing the ABD but to none of the plectin mutant proteins corresponding to molecular domains excluding this domain (Fig. 6B,C). Preincubation of ABD/2–6, ABD/1–24, and ABD/1b–24 with PIP2, a signaling molecule known to alter the actin-binding properties of many proteins that regulate actin assembly (for review, see Janmey 1994), led to a significant (concentration-dependent) reduction in their actin-binding abilities (Fig. 6C). From Lineweaver–Burk analysis of binding data obtained with fragment ABD/1–24, a dissociation constant of Kd = 3.2 ± 0.6 × 10−7 m was estimated (Fig. 6D), which was within the range of that of other actin binding proteins, such as BPAG1n and tensin (Lo et al. 1994; Yang et al. 1996). These experiments indicated that plectin’s ABD was fully functional under in vitro assay conditions and could be modulated by PIP2.
Figure 6.


Binding of plectin to actin is influenced by PIP2. (A) SDS-PAGE of purified plectin fragments containing the ABD. ABD/2–6, ABD/1b–24, and ABD/1–24, fragments corresponding to plectin exons 2–6 (human), 1b–24 (rat), and 1–24 (rat), respectively. Positions of molecular mass markers are indicated. (B–D) Purified His-tagged recombinant plectin fragments were coated onto microtiter plates at concentrations of 50–500 nm (B) and 500 nm (C,D), and overlaid with Eu3+-labeled G-actin at concentrations of 100 nm (B,C) and 0–800 nm (D). Data are presented as the mean ± s.d. of triplicate determinations. (B) Overlay assay of ABD/2–6 and recombinant plectin fragments lacking the ABD [PleR–4, Ple–R5, Ple–R5(ANAA)]. Data have been corrected for nonspecific binding to BSA. (C) Recombinant proteins ABD/1–24, ABD/1b–24, ABD/2–6, and BSA (negative control) immobilized on microtiter plates were preincubated with 0, 8.5, and 42 μg/mL PIP2 for 1 hr before being overlaid with Eu3+-labeled G-actin. Note strong reduction of plectin–actin binding in the presence of PIP2. (D) Concentration-dependent binding of actin to ABD/1–24.
Plectin−/− actin stress fiber phenotype is partially restored by forced ABD expression
To assess functionality of plectin’s ABD under conditions closer to the in vivo situation and to examine its potential to restore the phenotype of plectin+/+ cells, plectin-deficient fibroblasts were transiently transfected using Myc-tagged expression plasmids encoding full-length rat plectin (pBN60) or an amino-terminal fragment (pBK23). Transfection of cells with pBN60 led to the collapse of vimentin IF networks into perinuclear aggregates (Fig. 7A, arrowheads), which were immunoreactive with anti-vimentin (not shown) and anti-plectin (Myc) antibodies. A similar IF phenotype has been observed previously in various cell types overexpressing full-length plectin or carboxy-terminal fragments of the protein (Wiche et al. 1993; Nikolic et al. 1996). Interestingly, in pBN60-transfected cells (Fig. 7B, arrowheads) the actin stress fibers appeared considerably reduced in number and complexity, compared to nontransfected cells (Fig. 7B, arrow).
Figure 7.
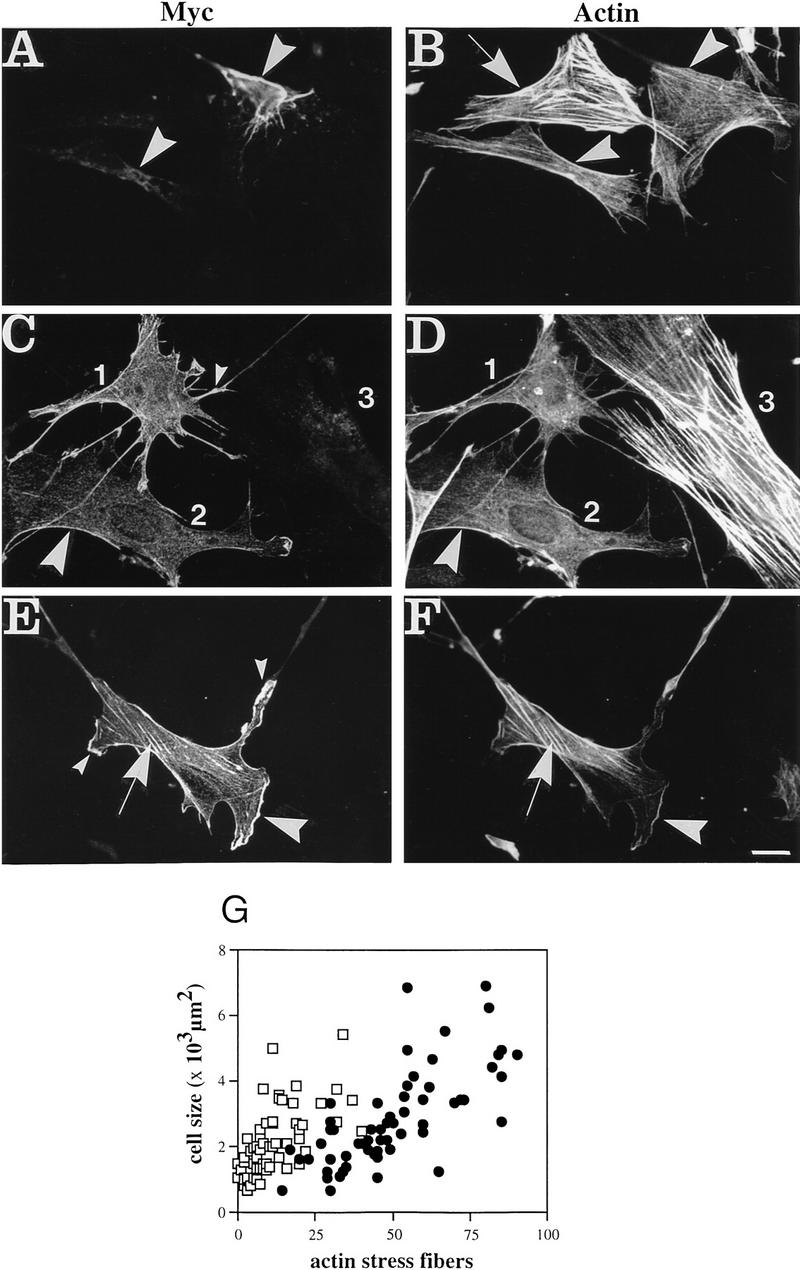
Transfection of cDNAs encoding full-length plectin (A,B) or the amino-terminal plectin fragment ABD/1a–14 (C–G) into plectin−/− fibroblast cells. (A–F) Double immunofluorescence microscopy was carried out 6 hr post-transfection using primary antibodies as indicated; expression plasmids used were pBN60 (A,B) and pBK23 (C–F). Note that the staining pattern of full-length plectin in the two transfected cells visualized in A (arrowheads) would hardly be distinguishable from that of the collapsed IF network (not shown) in these cells. Furthermore, expression of both full-length plectin (A,B) and ABD/1a–14 (C–F) clearly correlates with a decrease of actin stress fibers in transfected cells (B, arrowheads; C,D, cells 1 and 2) compared to untransfected cells (B, arrow; D, cell 3). Numerous plectin+/+ cell protrusions in cells transfected with ABD/1a–14 (C,E, small arrowheads) are missing in full-length plectin-transfected cells (cf. A with C and D). Note also, codistribution of ABD/1a–14 and actin at peripheral structures and protrusions (C–F, large arrowheads) and remaining stress fibers (E,F, arrows). (G) Numerical analysis of actin stress fibers in plectin−/− (•) and ABD/1a–14-transfected cells (□). Data points represent individual cells. Note higher number of actin stress fibers in plectin-deficient cells compared to ABD/1a–14-expressing cells of similar size. Bar, 30 μm.
To study the effects of ABD expression independently of vimentin network rearrangment, plectin-deficient cells were transfected using plasmid pBK23, which encodes an amino-terminal fragment of the molecule (ABD/1a–14), containing the ABD but not the IF-binding domain. The actin phenotype of (−/−) fibroblasts transfected with this plasmid was similar to that of cells transfected with full-length plectin. In this case too, the actin stress fiber system appeared to be largely, and in some cells completely, abolished upon transfection of cells (Fig. 7C,D, cells 1 and 2), whereas it was fully retained in nontransfected cells (Fig. 7C,D, cell 3). In most of the transfected cells, the expressed plectin mutant protein showed colocalization with actin primarily at the cell periphery (Fig. 7C–F, large arrowheads). Furthermore, >70% of transfected cells exhibited cellular protrusions, in which plectin fragments, identified by their Myc tag (Myc), were found in relatively high concentration (Fig. 7C,E, small arrowheads). Such protrusions were hardly found in cells transfected with expression plasmids encoding full-length plectin, suggesting that different domains of the molecule may have distinct effects on cell morphology. Apart from codistribution in peripheral areas of cells, anti-Myc staining was also observed along the few stress fibers that were retained in some of the transfected cells (Fig. 7E,F, arrows).
The substantial reduction of actin stress fibers in pBK23-transfected (ABD/1a–14) cells compared to untransfected cells could be documented clearly by statistical analysis of 60 randomly chosen cells of both types from the same culture dishes (Fig. 7G). This analysis also revealed a slightly larger size of nontransfected plectin-deficient cells compared to their transfected counterparts. Thus, the phenotypic differences revealed between pBK23-transfected and untransfected plectin−/− cells closely resembled those observed in a direct comparison of plectin−/− and plectin+/+ control cells (cf. Figs. 7G and 1H). This suggested that an amino-terminal fragment of plectin containing a functional ABD was sufficient to reverse important phenotypic aspects of plectin-deficient fibroblasts. The formation of long cellular protrusions as observed in some of the pBK23-transfected cells (see Fig. 7C) was, however, rarely noticed in plectin+/+ control cells.
Association with actin structures could be demonstrated even in the case of full-length plectin in the IF (vimentin)-free environment of SW-13 cells (Sarria et al. 1994). Upon transfection of cDNA encoding full-length plectin into these cells, codistribution of Myc-tagged overexpressed plectin with actin structures was predominantly observed at the periphery of cells (data not shown). These data, in conjunction with those obtained from in vitro-binding assays (see Fig. 6) and cDNA transfection into plectin-deficient fibroblasts (Fig. 7), provided strong evidence for plectin being a genuine actin-binding protein, presumably regulated by PIP2.
Discussion
Previous studies indicated a major role of plectin as scaffolding and a cytoskeletal linker protein. On these grounds a destabilizing effect on cytoskeletal filament systems may have been expected to occur in cells devoid of plectin. In contrast, our results demonstrated that the absence of plectin in cultured mouse fibroblasts resulted in a short-term stabilization of their actin filament cytoskeleton. It might be argued that the formation of stress fibers and FACs could be a secondary response of the cell to mechanical stress resulting from a destabilization of other cytoskeletal systems due to the lack of plectin. However, the microtubule system, known to have an influence on stress fibers and FACs (Bershadsky et al. 1996), was found to be intact in plectin−/− fibroblasts, and so was the vimentin filament system (data not shown). Moreover, plectin-deficient fibroblast and astroglial cells failed to promptly show typical morphological changes in response to activation of the Rho/Rac/Cdc42 pathways by extracellular factors. Such a phenotype would not be expected if the increase in stress fibers were just a stress response of the cell.
It is known that as primary cells grow in culture they often appear to become larger and more spread out on solid substrates. Therefore, the increased amount of actin stress fibers in plectin−/− cells could be a secondary response of a more rapid aging process in plectin−/− versus plectin+/+ control cells. However, we consider this not very likely in view of the very similar growth behavior of both cell types (see Fig. 1G). Furthermore, the increase in the number of stress fibers in plectin-deficient cells was independent of cell size, as clearly revealed by statistical analysis.
Our report is the first showing that the amino-terminal ABD of plectin is fully functional and presumably subject to PIP2 regulation. With a value of Kd = 3.2 ± 0.6 × 10−7 m, plectin’s dissociation constant turned out to be very similar to that of other actin-binding proteins containing binding domains of the same type as the one found in plectin. Thus, the dissociation constant of plectin–actin binding was considerably higher than that determined for plectin–vimentin interaction [Kd = 0.5 − 0.9 × 10−7 m (Nikolic et al. 1996)]. The lower affinity of plectin binding to actin compared to vimentin is in agreement with transfection experiments (Fig. 7; see also Nikolic et al. 1996), showing preferential association of overexpressed full-length plectin with IFs.
Regarding molecular mechanisms operating in plectin-mediated regulation of the actin cytoskeleton, the following scenarios may apply. First, as a component of FACs (Seifert et al. 1992) plectin might interfere with the assembly of mature FACs and the subsequent anchorage of stress fibers, which requires tyrosine phosphorylation of specific FAC proteins (Yamada and Geiger 1997) as well as costimulation of the Rho/Rac/Cdc42 pathways (Hall 1998). Formation of FACs per se is likely independent of plectin, because vinculin-positive FACs were found to be present in plectin-deficient fibroblasts. However, FACs appeared to be increased in number and size and not properly localized, similar to the phenotype of fibroblasts derived from mice deficient in focal adhesion kinase (FAK; Ilic et al. 1995), a main regulatory component of FACs. Not only the formation of FACs but also the turnover of these structures and their dynamic assembly and disassembly might ensure the motility of cells. Thus, similar to FAK, plectin may either directly take part in regulating the dynamics of FAC assembly or provide a structural matrix for other regulatory proteins contributing to this process. In both cases, the absence of plectin may impair FAC and stress fiber dynamics, keeping plectin-deficient fibroblasts firmly attached to their underlying substratum, thereby counteracting mechanisms driving cellular movement.
Second, in addition to its linker capacity, plectin may act as a sequestering or capping protein controlling polymerization/depolymerization of actin or the assembly of preexisting filaments to actin–myosin stress fibers. The latter possibility would agree with several reports indicating a correlation between the overexpression of different actin binding proteins and increased motility of cells (Cunningham et al. 1991, 1992; Hug et al. 1995; Sun et al. 1995), whereas the underexpression or absence of these actin-binding proteins resulted in a decrease of motility (Hug et al. 1995; Witke et al. 1995) and, at least in one case, in increased numbers of actin stress fibers (Witke et al. 1995). This concept is also supported by the finding that PIP2, a signaling molecule known to interact with numerous actin regulating proteins (Janmey 1994), interfered with actin-plectin binding. Actin polymerization leading to actin stress fibers, actin meshworks, or actin bundles can be controlled in a number of ways. First, the inhibition of actin filament capping leads to increased numbers of free barbed actin filament ends available for polymerization; second, the dissociation of a complex consisting of G-actin-binding proteins and actin leads to increased levels of polymerizable monomeric actin; and third, F-actin becomes stabilized. At least two of these mechanisms (filament capping and G-actin complex formation) have been demonstrated to be PIP2 controlled, involving capping proteins such as gelsolin, cap 32/34, and cap100 (Haus et al. 1991; Hofmann et al. 1992; Hartwig et al. 1995) or the G-actin-binding protein profilin (Lassing and Lindberg 1985, 1988). Thus, both of these mechanisms may be relevant also for plectin, which can bind to G-actin, as shown by in vitro assays (Fig. 6), as well as to F-actin, as demonstrated by colocalization of plectin and actin stress fibers in living cells (Fig. 7).
Taking into account plectin’s abundance and PIP2-binding ability, the absence of plectin may result in an alteration of the steady-state level of PIP2. This in turn could lead to increased actin polymerization and could be the reason for the longer persistence of actin stress fibers in plectin-deficient fibroblasts after stimulation with extracellular ligands. The finding that stress fibers in plectin-deficient cells failed to disassemble after removal of Rho-activating LPA would be consistent with such a mechanism. Alternatively, plectin could act downstream of the Rho-signaling cascade as a negative regulatory element causing cells to form fewer stress fibers. Increases in PIP2 levels have been demonstrated to influence FAC formation (Gilmore and Burridge 1996), and consequently they might be responsible for the increased number of FACs observed in plectin-deficient cells. At this point our experiments do not allow us to distinguish whether plectin acts as a direct or indirect regulator of actin cytoskeleton dynamics. Further studies are necessary to fully understand the molecular mechanisms that underlie the observed phenomena. As the abnormalities in actin filament dynamics observed in plectin-deficient cells seem to affect primarily short-term responses of the cell, it will be of particular interest to investigate the molecular mechanisms that can compensate for the lack of plectin in the long run.
In conclusion, the results reported extend the previous view of plectin as a major cytoskeletal linker and scaffolding protein to an important regulator of cellular processes linked to actin cytoskeleton dynamics. Future studies aimed at elucidating the molecular mechanism(s) involved in this previously undetected regulatory function of plectin may provide important new insights into the role of this protein, and the cytoskeleton in general, in cell motility and differentiation.
Materials and methods
cDNA constructs
Rat plectin cDNA constructs were generated by PCR and/or other cloning techniques on the basis of the complete rat and human cDNA sequences (GenBank/EMBL/DDBJ accession nos. X59601 and Z54367). Rat exons 1a and 1b sequences are published in Elliott et al. (1997). Briefly, the plasmid pBN60 used for transfection studies encoding full-length plectin was generated by releasing full-length rat plectin cDNA fragment (14049 bp) from pAD29 vector (Nikolic et al. 1996) by HindIII and XbaI digestion and directional cloning into expression vector pRC/CMV (Invitrogen). For the plasmid pBK23 corresponding to exons 1a–14a, PCR product was generated by using the primers rPE1a/U (5′-GATAAGCTTGAATTCATGTCTCAGCAACGGCTCCG) and AS7-8 1237 (5′-CGAAGCTTATTGGCCCTCACTCCATC) and rat cDNA as template. After digestion of the PCR product with HindIII and BglII and directional cloning into the vector AD43, the insert was again released by digestion with HindIII and XbaI and cloned into a modified version of pRC/CMV (Invitrogen).
All constructs used for in vitro experiments were subcloned into the bacterial expression vector pBN120 (Nikolic et al. 1996) which enabled expression of amino-terminally His-tagged proteins, except for the ones encoding ABD/1–24 and ABD/1b–24, which were inserted into the unique EcoRI site of pFS23, a pET23a (Novagen) derivative driving the expression of carboxy-terminally His-tagged proteins. Plectin plasmids encoded the following segments of the protein: ABD/1–24 (amino acids M1–L1129), ABD/1b–24 (amino acids corresponding to exon 1b and D181–L1129), ABD/2–6 (amino acids E176–K338), Ple–R4 (amino acids Y3780–T4024), Ple–R5 (amino acids F4025–L4367), Ple–R5ANAA (amino acids F4025–L4367-mutated A4277NAA).
Cell culture and transfection
Plectin+/+ and plectin−/− fibroblasts were cultivated from mouse skin explants, as described (Jones 1996), and analyzed at passage number 10–15. Growth properties were analyzed by plating out cells in triplicate wells in the appropriate medium. At each time point, cells were trypsinized and counted in a hemocytometer. No difference was found in the rate of cell division between plectin+/+ and plectin−/− cells. For drug treatment, cells were incubated for 30 min with 0.2 mm cytochalasin B (Sigma). For other treatments, cells were incubated with serum-free DMEM overnight and then with 10 ng/ml PDGF (Sigma) for 1 or 24 hr, or with 100 ng/ml bradykinin (Sigma) for 10 min or 24 hr. Astroglial cells were isolated from neopallium of brains of 1-day old plectin−/− and normal control mice and cultivated as described (Hao et al. 1992). For differentiation assays, 6-day old subconfluent cultures, containing 80%–90% GFAP-positive cells, were treated with 1 mm db-cAMP for 6 or 24 hr. For quantification, differentiated cells were counted in randomly chosen microscopic fields. Mouse fibroblast cells, maintained in 10% FCS/DMEM, were transfected with Myc-tagged CMV promoter-driven expression plasmids (Invitrogen) encoding full-length plectin starting with exon 1 (pBN60), or an amino-terminal fragment of plectin corresponding to exons 1a–14 (pBK23), using lipofectamin (GIBCO) according to the manufacturer’s instructions.
Immunfluorescence microscopy
Cells were grown on glass coverslips and processed for immunofluorescence microscopy as described (Wiche et al. 1993). Microscopy and photography were performed using a Zeiss Axiophot microscope and Ilford FP4 film. The following primary antibodies were used: anti-plectin and anti-vimentin antisera (Wiche and Baker 1982), both diluted 1:500; anti-tubulin mAb B5-1-2 (Sigma), diluted 1:1000; anti-actin mAb AC40 (Sigma), diluted 1:100; anti-vinculin mAb (Genosys), diluted 1:30; and antiserum to GFAP (Dako), diluted 1:300. We used the following as secondary antibodies: Texas Red-conjugated goat anti-rabbit or anti-mouse IgG (Jackson Immunoresearch) and Bodipy-conjugated goat anti-rabbit or anti-mouse IgG (Molecular Probes), all diluted 1:80.
The size of individual cells, the number of actin stress fibers, and FACs were calculated from confocal microscopic images of 50–60 randomly chosen cells, generated by the software COMOS (Bio-Rad).
Cell motility and adhesion assays
Fibroblast motility measurements were made by wounding a subconfluent culture dish with a sterile 18-gauge needle (Cunningham et al. 1991). Migration of cells was measured over time with the scratch serving as reference point. Only cells migrating completely into the wound were scored. Filter migration assays were performed by growing 3 × 104 cells/cm2 on 12-mm Transwell filter devices with 12 μm pore size (Costar) in 12-well tissue culture plates. Cells were allowed to attach for 2 hr, starved in serum-free DMEM for 1 hr, and incubated with 3% FCS–DMEM containing 50 ng/ml PDGF (Sigma) for 24 hr in the lower compartment of the 12-well plate. Cells were washed, fixed with 3% formaldehyde in PBS for 10 min at room temperature, stained with 10% methanol, 10% acetic acid, and 0.05% Coomassie blue for 10 min and destained with 10% methanol and 10% acetic acid. All cells on the upper side of the filter were removed with a cotton swab, and cells that had migrated through the filter were photographed and counted. Shear stress assays were performed by growing plectin+/+ and plectin−/− cells for 2 or 24 hr in 10% FCS–DMEM in three separate reaction fields of the same glass plate. Glass plates were mounted into a flow chamber (Koslow et al. 1986), and cells exposed to a defined shear stress level of 15 dynes/cm2 by continuous flow of DMEM for 1 hr at 37°C. Cells were then fixed with 3% paraformaldehyde, photographed, and counted.
Recombinant protein expression in bacteria, actin overlay, and PIP2-binding assays
After expression of His-tagged versions of human and rat plectin cDNA (Liu et al. 1996; Elliot et al. 1997) corresponding to exons 2–6 (ABD/2–6), exons 1–24 (ABD/1–24), and exons 1b–24 (ABD/1b–24) comprising the ABD of plectin (Elliott et al. 1997), the recombinant proteins were purified by affinity chromatography as described (Nikolic et al. 1996). For overlay assays on microtiter plates, actin purified from rabbit skeletal muscle was labeled with Eu3+ essentially following manufacturers’ instructions (Wallac, Turku, Finland) and the protocol given for vimentin (Nikolic et al. 1996); this previous report also contains details of the binding assay. Briefly, 100 μl of 50–500 nm actin solutions was used for overnight coating of plates at 4°C, followed by blocking with 4% BSA in TBS for 2–4 hr and incubation with Eu3+-labeled actin in 100 μl overlay buffer (TBS at pH 7.5, containing 1 mm EGTA, 2 mm MgCl2, 1 mm DTT, and 0.1% Tween 20) for 90 min at room temperature. After extensive washing with overlay buffer, the amount of bound actin was determined by releasing complexed Eu3+ with enhancement solution and measuring the fluorescence with a Delfia time-resolved fluorometer (Wallac). The fluorescence values were converted to concentrations by comparison with an Eu3+ standard. PIP2 (Sigma) was dissolved in H2O to a final concentration of 1 mg/ml and sonicated three times for 10 sec using a sonicator; suspensions were frozen in small aliquots at −80°C. Prior to use, solutions were thawed quickly and sonicated again. For use in actin-binding assays, 100 μl of PIP2 diluted to 8.5 or 42 μg/ml in overlay buffer (pH 8.5) was overlaid onto plate-immobilized recombinant plectin mutant proteins for 1 hr at room temperature. After extensive washing with overlay buffer, the assay was continued using Eu3+-labeled actin as described.
Acknowledgments
We thank Winfried Ness for his help with shear-stress experiments, Birgit Kopecky for expression plasmid pBK23, and Irmgard Fischer for providing purified recombinant plectin protein fragments. We are also very grateful to Michael Schleicher and Mario Gimona for their so prompt reaction in supplying us with urgently needed materials, and to Anthony Infante for critical comments on the manuscript. The major part of this work was supported by grant SFB 006/611 from the Austrian Science Research Fund.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL wiche@abc.univie.ac.at; FAX 43(1)4277-52854.
References
- Andrä K, Lassmann H, Bittner R, Shorny S, Fässler R, Propst F, Wiche G. Targeted inactivation of plectin reveals essential function in maintaining integrity of skin, muscle, and heart cytoarchitecture. Genes & Dev. 1997;11:3143–3156. doi: 10.1101/gad.11.23.3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bershadsky A, Chausovsky A, Becker E, Lyubimova A, Geiger B. Involvement of microtubules in the control of adhesion-dependent signal transduction. Curr Biol. 1996;6:1279–1289. doi: 10.1016/s0960-9822(02)70714-8. [DOI] [PubMed] [Google Scholar]
- Chavanas S, Pulkkinen L, Gache Y, Smith FJD, McLean WHI, Uitto J, Ortonne JP, Meneguzzi G. A homozygous nonsense mutation in the PLEC1 gene in patients with epidermolysis bullosa simplex with muscular dystrophy. J Clin Invest. 1996;98:2196–2200. doi: 10.1172/JCI119028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CC, Stossel TP, Kwiatkowski DJ. Enhanced motility in NIH 3T3 fibroblasts that overexpress gelsolin. Science. 1991;251:1233–1236. doi: 10.1126/science.1848726. [DOI] [PubMed] [Google Scholar]
- Cunningham CC, Gorlin JB, Kwiatkowski DJ, Hartwig JH, Janmey PA, Byers HR, Stossel TP. Actin-binding protein requirement for cortical stability and efficient locomotion. Science. 1992;255:325–327. doi: 10.1126/science.1549777. [DOI] [PubMed] [Google Scholar]
- Eger A, Stockinger A, Wiche G, Foisner R. Polarization dependent association of plectin with desmoplakin at the lateral submembrane skeleton in MDCK cells. J Cell Sci. 1997;110:1307–1316. doi: 10.1242/jcs.110.11.1307. [DOI] [PubMed] [Google Scholar]
- Elliott CE, Becker B, Oehler S, Castañón MJ, Hauptmann R, Wiche G. Plectin gene transcription diversity: Multiple alternative first exons, rod-less and other isoforms. Genomics. 1997;42:115–125. doi: 10.1006/geno.1997.4724. [DOI] [PubMed] [Google Scholar]
- Errante LD, Wiche G, Shaw G. Distribution of plectin, an intermediate filament-associated protein, in the adult rat central nervous system. J Neurosci Res. 1994;37:515–528. doi: 10.1002/jnr.490370411. [DOI] [PubMed] [Google Scholar]
- Foisner R, Leichtfried FE, Herrmann H, Small JV, Lawson D, Wiche G. Cytoskeleton-associated plectin: In situ localization, in vitro reconstitution, and binding to immobilized filament proteins. J Cell Biol. 1988;106:723–733. doi: 10.1083/jcb.106.3.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foisner R, Bohn W, Mannweiler K, Wiche G. Distribution and ultrastructure of plectin arrays in subclones of rat glioma C6 cells differing in intermediate filament protein (vimentin) expression. J Struct Biol. 1995;115:304–317. doi: 10.1006/jsbi.1995.1055. [DOI] [PubMed] [Google Scholar]
- Gache Y, Chavanas S, Lacour JP, Wiche G, Owaribe K, Meneguzzi G, Ortonne JP. Defective expression of plectin/HD1 in epidermolysis bullosa simplex with muscular dystrophy. J Clin Invest. 1996;97:2289–2298. doi: 10.1172/JCI118671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore AP, Burridge K. Regulation of vinculin binding to talin and actin by phosphatidyl-inositol-4-5-biphosphate. Nature. 1996;381:531–535. doi: 10.1038/381531a0. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hao C, Richardson A, Fedoroff S. Isolation of microglia from mouse astroglia cultures. In: Fedoroff S, Richardson A, editors. Protocols for neural cell culture. Totowa, NJ: Humana Press; 1992. pp. 97–104. [Google Scholar]
- Hartwig JH, Bokoch GM, Carpenter CL, Janmey PA, Taylor LA, Toker A, Stossel TP. Thrombin receptor ligation and activated Rac uncap actin filament barbeolenol through phosphoinositide synthesis in permeabilized human platelets. Cell. 1995;82:643–653. doi: 10.1016/0092-8674(95)90036-5. [DOI] [PubMed] [Google Scholar]
- Haus U, Hartmann H, Trommler P, Noegel AA, Schleicher M. F-actin capping by cap32/34 requires heterodimeric conformation and can be inhibited with PIP2. Biochem Biophys Res Commun. 1991;181:833–839. doi: 10.1016/0006-291x(91)91265-e. [DOI] [PubMed] [Google Scholar]
- Herrmann H, Wiche G. Plectin and IFAP-300k are homologous proteins binding to microtubules-associated proteins 1 and 2 and to the 240-kilodalton subunit of spectrin. J Biol Chem. 1987;262:1320–1325. [PubMed] [Google Scholar]
- Hoffmann A, Eichinger L, Andre E, Rieger D, Schleicher M. Cap100, a novel phosphatidylinositol 4,5-biphosphate-regulated protein that caps actin filaments but does not nucleate actin assembly. Cell Motil Cytoskelet. 1992;23:133–144. doi: 10.1002/cm.970230206. [DOI] [PubMed] [Google Scholar]
- Hug C, Jay PY, Reddy I, McNally JG, Bridgman PC, Elson EL, Cooper JA. Capping protein levels influence actin assembly and cell motility in dictyostelium. Cell. 1995;81:591–600. doi: 10.1016/0092-8674(95)90080-2. [DOI] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T, Aizawa S. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Science. 1995;377:539–543. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Janmey PA. Phosphoinositides and calcium as regulators of cellular actin assembly and disassembly. Annu Rev Physiol. 1994;56:169–191. doi: 10.1146/annurev.ph.56.030194.001125. [DOI] [PubMed] [Google Scholar]
- Jones GE. Establishment, maintenance, and cloning of human primary cell strains. In: Jones GE, editor. Methods in molecular medicine. Totowa, NJ: Humana Press; 1996. pp. 15–18. [DOI] [PubMed] [Google Scholar]
- Koslow AR, Stromberg RR, Friedman LI, Lutz RJ, Hilbert SL, Schuster P. A flow system for the study of shear forces upon cultured endothelial cells. J Biomech Eng. 1986;108:338–341. doi: 10.1115/1.3138625. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Fukuda T, Baba A. Inhibition of vanadate-induced astrocytic stress fiber formation by C3 ADP-ribosyltransferase. Biochem Biophys Res Commun. 1996;218:331–336. doi: 10.1006/bbrc.1996.0058. [DOI] [PubMed] [Google Scholar]
- Lassing I, Lindberg U. Specific interaction between phosphatidyl-inositol 4,5-biphosphate and profilactin. Nature. 1985;314:472–474. doi: 10.1038/314472a0. [DOI] [PubMed] [Google Scholar]
- ————— Specificity of the interaction between phosphatidylinositol 4,5-biphosphate and the profilin: Actin complex. J Cell Biochem. 1988;37:255–267. doi: 10.1002/jcb.240370302. [DOI] [PubMed] [Google Scholar]
- Liu C-G, Maercker C, Castañón MJ, Hauptmann R, Wiche G. Human plectin: Organization of the gene, sequence analysis, and chromosome localization (8q24) Proc Natl Acad Sci. 1996;93:4278–4283. doi: 10.1073/pnas.93.9.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SH, Janmey PA, Hartwig JH, Chen LB. Interactions of tensin with actin and identification of its three distinct actin-binding domains. J Cell Biol. 1994;125:1067–1075. doi: 10.1083/jcb.125.5.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean WHI, Pulkkinen L, Smith FDJ, Rugg EL, Lane EB, Bullrich F, Burgeson RE, Armano S, Hudson DL, Owaribe K, McGrath JA, McMillan JR, Eady RAJ, Leigh IM, Christiano AM, Uitto J. Loss of plectin causes epidermolysis bullosa with muscle dystrophy: cDNA cloning and genomic organization. Genes & Dev. 1996;10:1724–1735. doi: 10.1101/gad.10.14.1724. [DOI] [PubMed] [Google Scholar]
- Mellerio JE, Smith FJD, McMillan JR, McLean WHI, McGrath JA, Morrison GAJ, Tierny P, Albert DM, Wiche G, Leigh IM, Geddes JF, Lane EB, Uitto J, Eady RAJ. Recessive epidermolysis bullosa simplex associated with plectin mutations: Infantile respiratory complications in two unrelated cases. Br J Dermatol. 1997;137:989–996. [PubMed] [Google Scholar]
- Nikolic B, Mac Nulty E, Mir B, Wiche G. Basic amino acid residue cluster within nuclear targeting sequence motif is essential for cytoplasmic plectin-vimentin network junctions. J Cell Biol. 1996;134:1455–1467. doi: 10.1083/jcb.134.6.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulkkinen L, Smith FJD, Shimizu H, Murata S, Yaoita H, Hachisuka H, Nishikawa T, McLean WHI, Uitto J. Homozygous deletion mutations in the plectin gene (PLEC1) in patients with epidermolysis bullosa simplex associated with late-onset muscular dystrophy. Hum Mol Genet. 1996;5:1539–1546. doi: 10.1093/hmg/5.10.1539. [DOI] [PubMed] [Google Scholar]
- Rezniczek GA, de Pereda JM, Reipert S, Wiche G. Linking integrin α6/β4-based cell adhesion to the intermediate filament cytoskeleton: Direct interaction between the β4 subunit and plectin at multiple molecular sites. J Cell Biol. 1998;141:209–225. doi: 10.1083/jcb.141.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarria AJ, Lieber JG, Nordeen SK, Evans RM. The presence or absence of a vimentin-type intermediate filament network affects the shape of the nucleus in human SW-13 cells. J Cell Sci. 1994;107:1593–1607. doi: 10.1242/jcs.107.6.1593. [DOI] [PubMed] [Google Scholar]
- Seifert GJ, Lawson D, Wiche G. Immunolocalization of the intermediate filament-associated protein plectin at focal contacts and actin stress fibers. Eur J Cell Biol. 1992;59:138–147. [PubMed] [Google Scholar]
- Smith FJD, Eady RAJ, Leigh IM, McMillan JR, Rugg EL, Kelsell DP, Bryant SP, Spurr NK, Geddes JF, Kirtschig G, Milana G, de Bono AG, Owaribe K, Wiche G, Pulkkinen L, Uitto J, McLean WHI, Lane EB. Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nat Genet. 1996;13:450–457. doi: 10.1038/ng0896-450. [DOI] [PubMed] [Google Scholar]
- Sun HQ, Kwiatkowska K, Wooten DC, Yin HL. Effects of CapG overexpression on agonist-induced motility and second messenger generation. J Cell Biol. 1995;129:147–156. doi: 10.1083/jcb.129.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Verkhovsky AB, Borisy GG. Plectin sidearms mediate interactions of intermediate filaments with microtubules and other components of the cytoskeleton. J Cell Biol. 1996;135:991–1007. doi: 10.1083/jcb.135.4.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiche G, Baker MA. Cytoplasmic network arrays demonstrated by immunolocalization using antibodies to a high molecular weight protein present in cytoskeletal preparations from cultured cells. Exp Cell Res. 1982;138:15–29. doi: 10.1016/0014-4827(82)90086-6. [DOI] [PubMed] [Google Scholar]
- Wiche G, Krepler R, Artlieb U, Pytela R, Denk H. Occurrence and immunolocalization of plectin in tissues. J Cell Biol. 1983;97:887–901. doi: 10.1083/jcb.97.3.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiche G, Krepler R, Artlieb U, Pytela R, Aberer W. Identification of plectin in different human cell types and immunolocalization at epithelial basal cell surface membranes. Exp Cell Res. 1984;155:43–49. doi: 10.1016/0014-4827(84)90766-3. [DOI] [PubMed] [Google Scholar]
- Wiche G, Gromov D, Donovan A, Castañón MJ, Fuchs E. Expression of plectin mutant cDNA in cultured cells indicates a role of C-terminal domain in intermediate filament association. J Cell Biol. 1993;121:607–619. doi: 10.1083/jcb.121.3.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witke W, Sharpe AH, Hartwig JH, Azuma T, Stossel TP, Kwiatkowski DJ. Hemostatic, inflammatory, and fibroblast response are blunted in mice lacking gelsolin. Cell. 1995;81:41–51. doi: 10.1016/0092-8674(95)90369-0. [DOI] [PubMed] [Google Scholar]
- Yamada KM, Geiger B. Molecular interactions in cell adhesion complexes. Curr Opin Cell Biol. 1997;9:76–85. doi: 10.1016/s0955-0674(97)80155-x. [DOI] [PubMed] [Google Scholar]
- Yang Y, Dowling J, Yu QC, Kouklis P, Cleveland DW, Fuchs E. An essential cytoskeletal linker protein connecting actin microfilaments to intermediate filaments. Cell. 1996;86:655–665. doi: 10.1016/s0092-8674(00)80138-5. [DOI] [PubMed] [Google Scholar]
- Yoshimura S, Sakai H, Nakashima S, Nozawa Y, Shinoda J, Sakai N, Yamada H. Differential expression of Rho family GTP-binding proteins and protein kinase C isozymes during C6 glial cell differentiation. Brain Res Mol Brain Res. 1997;45:90–98. doi: 10.1016/s0169-328x(96)00239-2. [DOI] [PubMed] [Google Scholar]