

A nonmuscle caldesmon (CaD) is highly expressed in premigratory and migrating Xenopus cranial neural crest cells. A loss-of-function approach shows that CaD is critical for neural crest migration. The results further suggest that CaD influences cell morphology and motility by modulating actin dynamics in neural crest cells.
Abstract
Caldesmon (CaD) is an important actin modulator that associates with actin filaments to regulate cell morphology and motility. Although extensively studied in cultured cells, there is little functional information regarding the role of CaD in migrating cells in vivo. Here we show that nonmuscle CaD is highly expressed in both premigratory and migrating cranial neural crest cells of Xenopus embryos. Depletion of CaD with antisense morpholino oligonucleotides causes cranial neural crest cells to migrate a significantly shorter distance, prevents their segregation into distinct migratory streams, and later results in severe defects in cartilage formation. Demonstrating specificity, these effects are rescued by adding back exogenous CaD. Interestingly, CaD proteins with mutations in the Ca2+-calmodulin–binding sites or ErK/Cdk1 phosphorylation sites fail to rescue the knockdown phenotypes, whereas mutation of the PAK phosphorylation site is able to rescue them. Analysis of neural crest explants reveals that CaD is required for the dynamic arrangements of actin and, thus, for cell shape changes and process formation. Taken together, these results suggest that the actin-modulating activity of CaD may underlie its critical function and is regulated by distinct signaling pathways during normal neural crest migration.
INTRODUCTION
Caldesmon (CaD) is a multimodular protein that regulates contractility and actin cytoskeleton dynamics in smooth muscle and nonmuscle cells. In vertebrate, a single CaD gene is alternatively spliced to generate a high–molecular mass isoform in smooth muscle cells and a low–molecular mass nonmuscle isoform (Paul et al., 1995). In nonmuscle cells, CaD is associated with actin in a variety of structures, including stress fibers, membrane ruffles, the actin core of podosomes, division furrows, and the leading edge of migrating cells (Tanaka et al., 1993; Yamashiro et al., 2001; Eves et al., 2006). Generally, CaD binds to actin in a staple-like manner with its two actin-binding domains to stabilize the actin cytoskeleton and facilitate actin assembly (Sobue and Sellers, 1991; Yamashiro et al., 1994; Foster et al., 2004). CaD can also compete with other actin-binding proteins, such as gelsolin, fascin, and filamin, thus modulating actin severing, reannealing, bundling, and cross-linking (Nomura et al., 1987; Ishikawa et al., 1989a, 1989b, 1998; Dabrowska et al., 1996). In addition to actin, CaD binds to myosin, tropomyosin, and Ca2+-calmodulin (CaM; Sobue and Sellers, 1991; Lin et al., 2009). CaD enhances the binding of myosin to actin and, by recruiting tropomyosin, increases its own affinity to actin filaments (Hemric et al., 1994; Chalovich et al., 1995; Goncharova et al., 2001). On the other hand, binding to CaM, as well as phosphorylation by several kinases, partially dissociates CaD from actin cytoskeleton, which leads to loss of stress fibers and cell motility (Yamashiro et al., 1990; Huang et al., 2003; Foster et al., 2004). Such a myriad of binding -partners makes CaD a potent and versatile regulator of cell morphology and motility. Indeed, interfering with CaD levels in cultured cells results in diverse effects, ranging from stabilization or inhibition of focal adhesion to shortening of actin bundles, thickening or decreasing of stress fibers, disruption of adherens junctions, promotion or suppression of podosome formation, inhibition of cell cycle progression, and enhancement or reduction in cell motility (Novy et al., 1991; Helfman et al., 1999; Numaguchi et al., 2003; Grosheva et al., 2006; Gu et al., 2007; Yoshio et al., 2007).
In contrast to the wealth of information regarding CaD's activity in cultured cells, relatively little is known about its in vivo role during embryonic development. In zebrafish, CaD knockdown has been shown to cause defective cardiovascular development, with abnormal heart looping, disrupted proliferation and migration of smooth muscle, and partial loss of axial and trunk vessels (Zheng et al., 2009a, 2009b). In rat brain, both induction of CaD by glucocorticoids and inhibition of CaD by microRNA impair the radial migration of neural progenitor cells (Fukumoto et al., 2009).
In this study, we explore the role of nonmuscle CaD in migrating neural crest cells. An excellent system for the study of cell motility, neural crest cells migrate extensively in vivo along defined routes throughout the vertebrate embryo to populate a wide variety of tissues. In the cranial region, they contribute to cartilage, bone, and connective tissue, as well as to the peripheral nervous system of the head. We report that a nonmuscle CaD is highly expressed in premigratory and migrating Xenopus cranial neural crest cells and demonstrate that it is critical for neural crest migration in vivo and in vitro. Moreover, combined use of loss-of-function and rescue with wild-type and mutant forms of CaD suggests that CaD plays an important role in regulating cell morphology and motility by modulating actin organization in neural crest cells.
RESULTS
Isolation of Xenopus caldesmon
The partial sequence of Xenopus CaD was isolated from a cDNA microarray screen for genes up-regulated in naive animal caps treated with both Wnt and BMP antagonists, which mimics neural crest induction and induces neural crest markers, as previously described (Nie et al., 2009). To obtain the full-length cDNA of Xenopus laevis CaD (GenBank Accession No. HQ880575), we performed 5′ rapid amplification of cDNA ends (RACE). Figure 1 shows the full-length amino acid sequences of Xenopus CaD compared with chick, mouse, and human nonmuscle CaD homologues. This analysis reveals highly conserved domains common to nonmuscle CaD proteins across species: myosin-binding domain, tropomyosin-binding domain, actin-binding domain, and CaM-binding domain with amino acid identity of 82, 91, 93, and 100%, respectively.
FIGURE 1:

Sequence alignment of the Xenopus CaD and other CaD isoforms. Amino acid sequences of CaD isoforms from Xenopus CaD, chicken nonmuscle CaD (M59762), mouse CaD 1 (NM_145575), and human CaD (M83216) were aligned using the Clustal W program in DNAstar. Major binding domains for myosin, tropomyosin, actin, and Ca2+-calmodulin are highlighted in the green, yellow, red, and blue boxes, respectively. Xenopus CaD is highly conserved relative to those from other species in the binding domains, with 82, 91, 93, and 100% identity at the amino acid level, respectively.
CaD is expressed in cranial neural crest cells during Xenopus embryogenesis
Because CaD was identified in an in vitro screen for genes up-regulated by neural crest inducing signals, it was important to confirm that CaD is present at the right time and place to be functionally important for neural crest development. To this end, in situ hybridization analysis was performed to examine the expression pattern of CaD in Xenopus embryos from gastrula to tailbud stages (Figure 2). The results show that CaD expression was first detected at late gastrula stages (stages 12.5–13) at the lateral margin of the neural plate where prospective neural crest cells arise. At neurula stages when the neural plate folds to form the neural tube (stage 15), the expression level of CaD increased in the cranial neural crest (CNC) territory. CaD transcripts continued to mark CNC cells as they initiated migration into the future branchial arches (from stage 20 onward). By tailbud stages when these CNC cells differentiate, CaD expression decreased and was observed in portions of cranial ganglia and somites at low levels.
FIGURE 2:

CaD is expressed in neural crest tissue during early Xenopus development. Expression of CaD begins at late gastrula stage (stage 13) at the borders of the forming neural plate, the site of initial neural crest induction. Its expression increases in the CNC region by neurula stages (stage 15) and continues in the migrating neural crest cells as they populate the branchial arches (stage 20 onward). By tailbud stages, the expression decreases and only is present in part of the ganglia and somite at low levels (stage 26). All embryos are oriented with anterior to the left.
CaD is not required for neural crest induction
The expression of CaD in premigratory and migrating CNC cells is consistent with a possible role in neural crest development. To examine its loss-of-function effects, antisense morpholino oligonucleotides (MOs) against 5′ upstream sequences of the translation start site of Xenopus laevis CaD gene were generated to inhibit the translation of CaD. The efficiency of CaD-MO in inhibiting CaD protein translation was confirmed by Western blot (Supplemental Figure S1). CaD-MO (10 ng) together with a lineage tracer, nuclear β-galactosidase (nβGal) RNA, was injected into one cell of two-cell-stage embryos, and the morphants were compared with similar embryos injected with control-MO plus lineage tracer. To test whether CaD gene knockdown influenced early events in neural crest formation, embryos injected with MOs were collected at neurula stages, and in situ hybridization was performed against different neural crest marker genes. Gene expression was compared between the injected side and uninjected side, which served as an internal control, in the same embryo. The expressions of neural crest genes twist and sox10 were not affected during formation of the premigratory neural crest in control-MO– or CaD-MO–injected embryos (Figure 3). These results suggest that CaD is not essential for neural crest induction.
FIGURE 3:
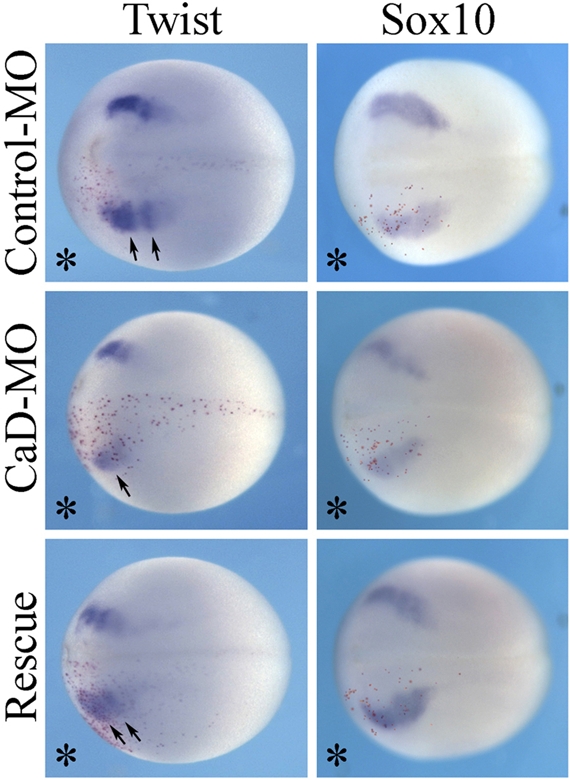
CaD is not required for neural crest induction. Embryos were injected with control-MO or CaD-MO (10 ng) in one cell at the two-cell stage, together with a lineage tracer (nβGal, stained with Red-Gal, bottom). No change in expression of neural crest genes twist and sox10 was observed by in situ hybridization at neurula stages, suggesting that they were not affected by CaD-MO. All embryos were in dorsal view, with anterior to the left.
CaD is required for CNC migration
We next examined whether CaD is required at subsequent stages for the process of neural crest migration. The embryos were injected with control-MO or CaD-MO into one cell at the two-cell stage and collected at tailbud stages. The morphants were subjected to in situ hybridization with markers of migrating cranial neural crest cells—twist, sox10, and EphA4, which marks the third branchial arch (Figure 4). Whereas twist, sox10, and EphA4 were expressed in distally migrating neural crest cells on the control side, these markers were confined to much more dorsal regions of the embryo in the CaD-MO—injected half (77% of 70 embryos examined). This likely reflects a general decrease in the distance that neural crest cells have migrated. In addition, the different migratory streams, particularly the third and fourth streams, marked by twist and sox10, were not well separated on the injected half. The third and fourth branchial arches separate relatively late, starting at stage 23 and becoming evident by stage 26, whereas the other arches are separate from the onset of CNC migration (Sadaghiani and Thiebaud, 1987). Thus it may not be surprising that the former exhibits the most dramatic effects after CaD knockdown. It is significant that this migration defect was rescued (in 74% of 46 embryos) by adding back CaD RNA, demonstrating that the effect is specific to knockdown of CaD.
FIGURE 4:

CaD-MO disrupted the expression of neural crest genes at tailbud stages. Embryos receiving control-MO or CaD-MO (10 ng) in one cell at the two-cell stage were collected at tailbud stages and analyzed for neural crest gene expression by in situ hybridization. The injected side for each embryo is marked by an asterisk and the control side of the same embryo is placed below serving as internal control. Whereas control-MO did not affect the expression of general neural crest markers twist and sox10 or the third branchial arch marker EphA4, the expression of all three genes was disrupted by CaD-MO (arrows). Marker analysis suggests that neural crest cells failed to extend into the lateral portion of the branchial arches and to separate into clear streams in the cases of twist and sox10. Simultaneous injection of CaD (100–200 pg) rescued the expression of neural crest genes. All embryos were oriented with anterior to the left and dorsal up.
In addition to the neural crest migration defect, CaD-MO also affected otic placode formation, as shown by sox10 expression. By in situ hybridization, expression of marker genes for other placodes was also examined, and development of both trigeminal placode and epibranchial placode was impaired to some extent, whereas lens formation was not affected (unpublished data). Because CaD is expressed in the preplacodal domain at early-neurula stages (Supplemental Figure S2), it could play either a direct role in placode specification or patterning or an indirect role in regulating the movement of surrounding neural crest cells. As shown in Figure 3, the premigratory crest cells starts to segregate according to their future migratory paths in control conditions, but such segregation was not apparent on the CaD-MO–injected side (arrows). As the otic placode forms at the dorsal junction of the second and the third arches, failure to separate the migration routes of cells could secondarily interrupt the organization of otic placode.
CaD is required for cartilage formation
Because CNC cells give rise to cartilage and bone in the head, we asked whether the migration defects caused by CaD-MO had later consequences on cartilage formation. To this end, embryos were collected at stage 45+, by which time craniofacial skeletal structures had formed, and Alcian blue staining was performed to look at cartilage elements. The results show that neural crest–derived cartilage was often malformed, smaller, or even missing on the CaD-MO–injected side (Figure 5). Coexpression of CaD RNA efficiently rescued the cartilage formation. This is consistent with the aforementioned finding that CNC cells fail to migrate to their proper destination and therefore do not properly differentiate into cartilage.
FIGURE 5:

CaD-MO inhibited the formation of cranial cartilage. Ventral view of cartilage from stage 45+ tadpoles stained with Alcian blue. Embryo halves received 10 ng of control-MO or CaD-MO on the left (marked by an asterisk). Cartilage on the CaD-MO–injected side was often reduced in size, distorted, or missing completely, whereas coexpression of CaD rescued the cartilage formation effectively. CB, ceratobranchial cartilage (from third and fourth branchial arch streams); CH, ceratohyal cartilage (from hyoid stream); M, Meckel's cartilage (from mandibular stream).
CaD controls CNC cell spreading and segregation on fibronectin
To examine the effect of CaD knockdown on cranial neural crest cells directly in the absence of neighboring tissues, CNC explants were dissected from early neurula stage embryos (stages 13–14) and cultured on fibronectin (FN)-coated dishes. After a few hours, control explants spread on FN, and by 10 h, cells migrated extensively and segregated into distinct lobes (Figure 6A), resembling the migratory streams into different branchial arches in vivo. In contrast, CaD-MO–expressing explants spread less efficiently and failed to segregate. We measured the areas covered by the explants at the start and end time points of the experiment and calculated the relative changes in the area as a measure of the degree of spreading. Whereas control-MO–expressing explants spread 3.6-fold on average (n = 33), CaD-MO–expressing explants spread only ∼2.2-fold (n = 28). The number of distinct cell streams was also counted for each explant, and CaD-MO decreased the average number of streams from 2.3 to 1.5. Adding back CaD successfully rescued both the spreading and segregation phenotypes (3.4-fold spreading and 2.0 streams; statistically comparable to control and significantly different from CaD-MO alone; n = 29).
FIGURE 6:

CaD is required for CNC explants to spread and segregate on FN matrix. (A) CNC explants were dissected from early neurula embryos injected with control-MO or CaD-MO (10 ng) and plated on FN. By 10 h, control-MO–expressing cells spread and migrated extensively and segregated into three streams. However, cells from CaD-MO–expressing explants failed to spread efficiently or separate into streams. Coinjection of 100–200 pg of CaD largely rescued the spread and segregation of the explants. Arrows with numbers marked the segregated lobes. High-magnification differential interference contrast imaging of the explants (B, 40×) and fluorescence imaging of cells with membrane labeling (C, 20×) show that whereas control-MO–expressing cells extended multiple membrane protrusions, CaD-MO–expressing cells remained unpolarized and were often rounded. (D) The cells were tracked for 2 h and trajectories plotted. Although the speed of migration was largely unaffected, the directionality of migration was significantly impaired by CaD-MO. Black traces indicate that the ratio of the final distance to the entire route covered is >0.3, and red indicates that it was <0.3.
Higher-resolution images revealed that CNC cells often adhered well to FN and formed multiple filopodia and lamellipodia types of protrusions (Figure 6, B and C; n = 24). When CaD was knocked down, however, neural crest cells failed to adhere tightly to FN and tended to round up (n = 19). They formed few membrane protrusions and displayed little interaction with each other. In time-lapse movies that visualized cell behavior in the explants, control-MO–expressing cells formed protrusions dynamically and migrated very actively. In contrast, CaD-MO–expressing cells tended to remain round and appeared disoriented (Supplemental Movies S1 and S2). These observations suggest that CaD is important for cell–cell and cell–matrix adhesion and also for cell process formation, thus facilitating spread, migration, and segregation of CNC cells.
To test the adhesive properties of these cells directly, two experiments were performed. To examine cell–cell adhesion, CNC cells were briefly dissociated for 20 min in calcium magnesium–free buffer and then allowed to reaggregate in high-salt buffer (high-salt modified Barth's saline [MBSH]) on an agarose bed. In 6 h, control cells formed a single large clump, reflecting high adherence. In contrast, CaD-MO–expressing cells only formed medium-sized clusters (Supplemental Figure S3), indicating reduced adhesiveness compared with controls. To examine adhesion between cells and the FN matrix, neural crest cells were dissociated in the same manner and plated on FN-coated dishes in high-salt buffer (0.5× Marc's modified Ringer's [MMR]). After 1 h, the plate was flipped to remove loosely bound cells. The cells were counted before and after the wash, and the percentage of tightly attached cells was determined. The result showed that 47% of control-MO–expressing cells remained attached, whereas only 15% of CaD-MO–expressing cells adhered to FN. Coinjection of CaD RNA increased the percentage of adhesion between cells and FN to 32%. This result shows that CaD plays a role in controlling cell–matrix adhesion.
The results from both the spreading assay and time-lapse movies indicate that CaD-MO–expressing cells migrate a shorter distance on FN. This could result from reduced migratory speed or diminished persistence during migration. To distinguish between these possibilities, we tracked H2B–enhanced green fluorescent protein (EGFP)–labeled cells of CNC explants for 2 h and calculated their velocity and directionality (Figure 6D). Because a large number of CaD-MO–injected cells failed to migrate, only those having a speed greater than 4 μm/min were considered. Directionality was calculated by dividing the final distance of translocation by the total length of the route covered. Whereas the control-MO and CaD-MO cells traveled at very similar speeds (20 and 19 μm/min, respectively, on average), their abilities to maintain direction were quite different. Control-MO–treated cells had a directionality of 0.45, whereas the directionality of CaD-MO–expressing cells was significantly lower at 0.20.
CaD regulates cranial crest cell migration in vivo
The in vitro explant assay indicated that CaD is important for spreading and segregation of CNC cells. To test whether this effect was cell autonomous, we performed in vivo grafting experiments. Cranial neural crest was dissected from donor embryos expressing membrane-tethered EGFP with control-MO or CaD-MO, grafted isotopically and homotopically into uninjected host embryo, from which CNC cells has been removed and raised to tailbud stages.
Whereas control-MO–expressing cells migrated nicely into all four branchial arches, CaD-MO–expressing cells migrated in a disorganized manner and traversed a much shorter distance, such that no clear boundary was formed between different streams (Figure 7A). High-resolution images revealed that cells migrated as individuals in CaD-MO–expressing grafts rather than together as a group as in control grafts. The behavior of different grafts was categorized into four classes (Figure 7B). Seventy-nine percent of control-MO–expressing grafts migrated normally (n = 61). In contrast, 18% of CaD-MO–expressing grafts failed to migrate, and 67% migrated either very short distances or without forming distinct streams (n = 45). Adding back CaD mRNA resulted in partial rescue, with more extensive migration and better separation of the migratory streams, such that 83% of the explants were able to segregate normally, of which 44% migrated indistinguishably from controls (n = 18).
FIGURE 7:

CaD-MO disrupted CNC migration in vivo. (A) GFP-labeled CNC grafts receiving control-MO or CaD-MO (10 ng) were transplanted into unlabeled host embryos at neurula stages and imaged at tailbud stages. Fluorescent and merged images are shown side by side with anterior to the left. Whereas control-MO–receiving grafts migrated laterally into separate branchial arches, CaD-MO–expressing grafts migrated less far and failed to segregate into streams. Coexpression of a low-dose CaD restored the migration of CNC transplants. (B) The embryos were binned into four categories on the basis of the migratory behaviors. Whereas 79% of control grafts migrated and segregated normally (n = 61), 18% of CaD-MO–expressing grafts failed to migrate at all. Another 67% of the grafts migrated shorter distances, over half of which did not segregate (n = 45). Coexpression of CaD (100–200 pg) rescued the segregation (83%) and migration (44%, n = 18) remarkably.
Time-lapse sequences were generated to further characterize the loss-of-function phenotype. In contrast to control grafts, cells in CaD-MO grafts lost their directionality and cohesiveness and moved in a rather random manner, failing to form distinct streams in the branchial arches (Supplemental Movies S3–S5). Taken together, these results indicate that CaD is required by CNC cells for proper migration into branchial arches.
CaD regulates actin organization in CNC cells
Because CaD is an actin-binding protein believed to control actin assembly and stabilization (Sobue and Sellers, 1991; Yamashiro et al., 1994; Kordowska et al., 2006), we tested whether inhibiting CaD function in CNC cells affects actin organization. GFP was injected at 16- to 32-cell stages together with a low dose (2–3 ng per embryo) of control-MO or CaD-MO to avoid strong effects on cell attachment and spreading. Because injections at this stage result in mosaic expression, cells receiving both GFP and MO can be compared with neighboring cells lacking GFP expression. Rhodamine-conjugated phalloidin was applied to fixed CNC explants (Figure 8A). In control-MO–expressing cells, actin filaments associated with lamellipodia, filopodia, and stress fibers (arrows) were similar to those in uninjected cells (arrowheads). Cells located at the periphery of the explant extended more membrane processes than internal cells, and the cell processes tended to orient toward the outside. Dissociated cells appeared to lose their orientation and instead spread extensively and formed multiple cell processes in every direction (Figure 8A, right). In contrast, actin was distributed relatively evenly throughout the cells receiving CaD-MO (arrows) compared with neighboring uninjected cells (arrowheads). Many cells located at the border rounded up, whereas dissociated cells spread moderately and formed membrane blebs, suggesting a failure to stabilize the actin cortex. Quantification of membrane protrusions from these cells confirmed that CaD-MO significantly decreased the formation of protrusions to 31% of control (n = 54/95).
FIGURE 8:

CaD-MO disrupts the organization of actin filament. (A) CNC explants receiving membrane-tethered EGFP together with control-MO or CaD-MO at 16- to 32-cell stages were fixed on FN and stained with rhodamine-conjugated phalloidin. Whereas actin associated with protrusions and stress fibers in control-MO–expressing cells, actin distributed in a relatively ubiquitous manner in CaD-MO–expressing cells. Arrows indicate injected cells, and arrowheads mark uninjected cells in the same explant. Right, actin staining alone in dissociated cells. (B) Phalloidin staining in cells expressing 0.1 ng of EGFP-CaD revealed colocalization of CaD with actin filaments in membrane protrusions and stress fibers (arrows). Images were taken at 40×. Scale bars, 20 μm.
To visualize how CaD might interact with actin filament to modulate its organization, a low dose of EGFP-CaD fusion protein (0.1 ng of RNA) was introduced into CNC explants prior to phalloidin staining. As shown in Figure 8B, CaD colocalized with actin filaments on membrane ruffles, at stress fibers, and in protrusions (arrows), confirming that CaD associates with actin during multiple events of actin arrangements. When a higher dose of the fusion construct (0.2 ng of RNA) was expressed, a dramatic enrichment of short actin bundles was observed (Figure 9). These were in the form of either thick membrane ruffles or fragmented stress fibers. This may indicate that excessive CaD disrupts actin polymerization but stabilizes actin filaments at ectopic locations.
FIGURE 9:

Overexpression of various CaD constructs affects actin-associated structures. Phalloidin staining was performed on CNC explants receiving 0.2 ng of EGFP-CaD, CaD39-AB, CaD39-6F, and CaD39-PAKA. To distinguish cells receiving the RNA, H2B-EGFP was coinjected with the mutant constructs. Whereas overexpression of EGFP-CaD produced abundant ectopic actin-rich protrusions, CaD39-AB increased stress fiber formation. CaD39-6F and CaD39-PAKA caused enrichment of short actin bundles around the cell periphery to different extents. Arrows indicate actin-rich structures. Images were taken using 40× objectives. Scale bars, 20 μm.
CaD activity is regulated by different signaling pathways
CaD's actin-binding activity can be modulated by CaM binding or by phosphorylation via multiple kinases, such as Cdc2 kinase (Cdk1), extracellular signal–regulated kinase (Erk), and/or p21 activated kinase (PAK; Mak et al., 1991; Yamashiro et al., 1991; Childs et al., 1992; Van Eyk et al., 1998; Foster et al., 2000). To gain a better understanding of how CaD might regulate actin dynamics, mutant versions of a truncated human CaD with substitutions in the CaM-binding sites (CaD39-AB; with two W-to-A substitutions at amino acids 461 and 494), ErK/Cdk1 phosphorylation sites (CaD39-6F; with T383A, S469A, T475A, T498A, S504A, and S534A), or PAK phosphorylation sites (CaD39-PAKA; with two S-to-A substitutions at amino acids 459 and 489) were used to rescue neural crest migration defects by CaD knockdown. The level of rescue was measured by the expression of neural crest markers Twist and Sox10. Injections of 0.1 ng of wild-type truncated CaD (CaD39; C-terminal fragment of CaD consists of amino acids 244–538, which contain the entire binding domains for tropomyosin, actin, and CaM) RNA with CaD-MO rescued the migration defect such that 74% of embryos were normal, compared with 37% with morpholino alone (n = 86). Coinjection of CaD39-PAKA mutant form with CaD-MO also successfully rescued up to 63% of embryos (n = 62). In contrast, the same concentration of CaD39-AB or of CaD39-6F failed to bring significant improvement, with only 44 and 33% of embryos remaining normal (n = 48 and 42), respectively. These results suggest that Ca2+-calmodulin and Erk/Cdk1 phosphorylation may modulate CaD function during neural crest cell migration, whereas PAK phosphorylation is not critical for this function.
Because we found that increased CaD activity leads to massive actin accumulation, we next overexpressed CaD mutants and visualized phalloidin staining to examine how they affected actin structure (Figure 9). H2B-EGFP was coinjected to trace cells receiving the CaD variants. When 0.2 ng of CaD39-AB was expressed in CNC explants, formation of actin bundles in protrusions and at cell–cell boundaries was largely unaffected, but the number and thickness of stress fibers increased significantly (arrows). In contrast, overexpression of CaD39-6F led to increased membrane ruffles and clusters of short actin bundles (arrows), resulting from recruitment of actin subunits to ectopic positions. Similar but more severe phenotypes were observed after overexpression of CaD39-PAKA, suggesting a partially redundant activity for Cdk1/Erk and PAK phosphorylation. These results support a critical role of Ca2+-calmodulin in regulating CaD activity in neural crest development. Because the CaM binding would be expected to partially dissociate CaD from actin and allow actin reorganization and cell shape changes during migration, mutations in CaD39-AB appear to prevent such dynamic regulation, thus suppressing cell motility due to the formation of extra stress fibers.
DISCUSSION
Nonmuscle CaD is present in all cell types except skeletal muscle and plays a role in controlling actin dynamics. Here we examined whether CaD plays a role in regulating cell morphology and motility during neural crest development. We showed that CaD is selectively expressed in premigratory and migrating neural crest cells in Xenopus embryos. We further provided the first evidence that CaD is essential for CNC migration. After CaD knockdown, CNC cells not only migrate a shorter distance, but they also fail to migrate in organized streams. The results suggest that CaD modulates neural crest cell adhesion, spread, and motility by regulating actin dynamics. In addition, rescue experiments with various mutated forms of CaD suggest that calmodulin is the major regulator of CaD activity during neural crest migration.
CaD in neural crest migration
Loss of CaD leads to fusion of third and fourth arch streams of migrating neural crest cells. One possible explanation is that cell adhesion, in which Eph/ephrin signaling may play a role, was disrupted. In the chick, adhesion of early migratory cells to an ephrinB substratum results in cell rounding and disruption of the actin cytoskeleton, whereas plating of melanoblasts on an ephrinB substratum induces the formation of microspikes (Santiago and Erickson, 2002). In mice, formation of gap junctions is inhibited at ectopic ephrin boundaries, and ephrinB1 regulates the distribution of connexin-43 (Mellitzer et al., 1999). In Xenopus, repulsive signals by the Eph/ephrin family are important for confining neural crest cells to appropriate paths (Robinson et al., 1997; Smith et al., 1997; Helbling et al., 1998). The EphA4 and EphB1 receptors are expressed in migrating crest cells and mesoderm of the third arch and the third plus fourth arches respectively, whereas the ligand ephrinB2 is expressed in the adjacent second arch neural crest and mesoderm. Such complementary arrangement inhibits the intermingling of second and third arch neural crest cells and targets third arch neural crest cells to their correct destination. However, it is unclear what restricts the mixing of third and fourth arch crest. In our study, CaD knockdown affects the posterior arches more severely than the anterior arches. The third arch gene EphA4 was expressed in a truncated pattern, whereas the expression of second arch genes EphA2 and ephrinB2 was not significantly altered (unpublished data). This may due to the larger population of cells contained in the more anterior arches. Thus CaD may not regulate Eph signaling itself, but instead may converge with Eph to control cell adhesion.
Another intriguing possibility is that CaD may interact with the Wnt pathway to influence the distribution of actin cytoskeleton and thus control the migration of neural crest cells. The directionality of Xenopus neural crest migration is primarily guided by noncanonical Wnt signaling (De Calisto et al., 2005; Carmona-Fontaine et al., 2008; Matthews et al., 2008a). Inhibition of Wnt11, Wnt11r, Frizzled-7, or Dishevelled completely abolishes neural crest migration. Furthermore, contact inhibition of locomotion plays a major role in controlling CNC migration by defining the leading edge and trailing edge of the cells. During CNC migration, Frizzled-7 and Dishevelled accumulate at cell–cell contacts and mediate the localized activation of RhoA, which antagonizes Rac and decreases protrusive activity between cells (Carmona-Fontaine et al., 2008). In contrast, Rac at the ventral free edge of CNC promotes actin polymerization and lamellipodia extension and thus drives directional migration.
CaD and cell adhesion
Our in vitro study and adhesion assay show that CaD-MO affected both cell–cell and cell–matrix adhesion. Consistent with our cell tracking results, coherent groups of cells migrate faster, maintain directionality better, and are more sensitive to chemoattractant than individual cells (Theveneau et al., 2010; Winklbauer et al., 1992). Cell–cell adhesion is important for cell migration. In Xenopus, cadherin-11 is expressed in premigratory and migrating CNC cells, and a precise level of cadherin-11 is required for normal CNC migration (Vallin et al., 1998; Borchers et al., 2001). In addition, cadherin-11 localizes to the filopodia of CNC cells cultured on FN and is critical for the activation of Rho GTPases in those cells (Kashef et al., 2009). Other adhesion proteins, such as the protocadherin PCNS, also have been implicated in regulating CNC spread and migration (Rangarajan et al., 2006). Failure to sort into distinguishable migrating streams, as observed in our experiment, is likely due to altered expression of different cadherins. In kidney epithelial cells, CaD is involved in the formation of adherens junctions (Grosheva et al., 2006). Thus it is possible that CaD associates in a similar manner with cell–cell adhesion complex in neural crest cells.
Cell–matrix adhesion is also essential for neural crest migration. Fibronectin is a major extracellular matrix component along the paths of neural crest migration. Interaction between integrin α5β1 and FN is required for CNC cell migration and segmentation (Alfandari et al., 2003). A coreceptor for Wnt signaling, syndecan-4, can also interact with FN and mediates contact inhibition of locomotion (Matthews et al., 2008b). Consistent with the findings that disrupting CaD activity leads to dysregulated focal adhesion formation (Warren et al., 1996; Helfman et al., 1999; Numaguchi et al., 2003; Mayanagi et al., 2008), our experiment shows that CaD-MO disturbs CNC cell adhesion on FN matrix.
CaD in cell morphology and dynamics
Our in vitro study reveals for the first time that CaD is important in neural crest cells for dynamic actin organization, cell shape changes, and protrusion formation. In CNC cells, CaD is highly associated with actin at protrusions, membrane ruffles, and stress fibers. When CaD was depleted by CaD-MO, the formation of protrusions and stress fibers was disrupted. In contrast, when CaD was overexpressed, large quantities of thick membrane ruffles and short stress fibers were produced, suggesting enhanced stabilization of actin filaments. However, the orientation of these cells was lost due to the random distribution of actin structures. Consistent with this possibility, lamellipodia protrusions were reduced in cells expressing a low level of EGFP-CaD, probably due to the inhibition of Arp2/3 complex–mediated actin polymerization and branching processes by actin-bound CaD (Yamakita et al., 2003).
When associated with Ca2+-calmodulin or phosphorylated by kinases, CaD partially dissociates from actin filament and leads to disassembly of stress fibers, formation of focal adhesions, extension of lamellipodia, and persistence of cell movement (McFawn et al., 2003; Li et al., 2004; Eppinga et al., 2006; Kordowska et al., 2006). In this study, we found that overexpression of different CaD mutants led to differential effects on actin organization. Overexpression of CaD mutant in Ca2+-calmodulin binding enhanced the formation of stress fibers, which is different from its reported disruption of stress fibers in CHO cells (Li et al., 2004). This may be due to the different cell types examined and/or the different levels of overexpression, since exogenous CaD was introduced at more than 80-fold that of the endogenous levels in the previous study. This excess CaD would compete with endogenous CaD and lock the actin filament in an inhibitory conformation for actin bundling and thus disintegrate stress fibers. In our case, the moderate increase of CaD (0.2 ng of RNA injected) may serve to stabilize actin stress fibers. However, similar levels of CaD39-6F and CaD39-PAKA disrupted stress fiber formation. It was reported that phosphorylated, not unphosphorylated, CaD colocalizes with vinculin at focal adhesions (Kordowska et al., 2006). In our experiments, one intriguing possibility is that CaD39-AB, not CaD39-6F or CaD39-PAKA, may recruit essential partners to form focal adhesions required for the contraction and extension of stress fibers.
In summary, our results suggest that CaD is critical for cranial neural crest migration and appears to play an important role in dynamic rearrangement of the actin cytoskeleton during their motility. This in turn is critical for the proper cell–cell and cell–substrate adhesion that allows neural crest cells to properly navigate and interact during their migration.
MATERIAL AND METHODS
Embryo manipulations, morpholino oligonucleotides, and RNA preparation
Xenopus laevis embryos, both pigmented and albino, were microinjected with capped RNAs or MOs as described (Chang et al., 1997), using a standard control MO (Gene Tools, Philomath, OR) and CaD-MO (5′-AATGAAAGGGTGTTTCAACAACTG-3′) that hybridizes to −28 to −5 position relative to the translational start site of Xenopus CaD (GenBank Accession No. HQ880575). Capped RNAs of nβGal, nuclear EGFP (H2B-EGFP), membrane-tethered EGFP (kindly provided by Chenbei Chang), CaD without the 5′ untranslated region, and EGFP-CaD were synthesized with an Ambion (Austin, TX) mMessage mMachine Kit. Mutant constructs of CaD were kindly provided by Jim J. Lin and subcloned into pCS2+ vector. To selectively target the neural crest, MOs and mRNAs were injected into dorsal animal region of 2- to 32-cell-stage embryos. Five nanoliters of MO at 2 μg/μl was injected into one blastomere of 2- to 8-cell-stage embryos (10 ng per embryo) in most experiments, and 100–200 pg of CaD RNA was coinjected in rescue experiment. For mosaic labeling and a milder effect on cell adhesion, 2–3 nl of CaD-MO at 1 μg/μl (2–3 ng of MO per injection) was injected into one blastomere of 16- to 32-cell-stage embryos. For tracing purposes, nβGal or EGFP was injected at 0.1 ng per embryo.
Red-Gal staining, in situ hybridization, and cartilage staining
For lineage tracing, embryos coinjected with nβGal were fixed for 1/2 h in the fixative MOPS-EGTA-MgSO4-formaldehyde (MEMFA), rinsed twice with phosphate-buffered saline (PBS), and stained with the Red-Gal substrate (Research Organics, Cleveland, OH) until they turned red. The embryos were refixed for 2 h in MEMFA, and in situ hybridization was performed as previously described (Chang et al., 1997). Cartilage staining was performed according to Richard Harland's lab protocol (http://tropicalis.berkeley.edu/home/gene_expression/cartilage-stain/alcian.html).
Reverse transcription and rapid amplification of cDNA ends
Total RNA was extracted from CNC explants using an RNAqueous-Micro Kit (Ambion), and RACE was performed to generate the full-length sequence of CaD using a GeneRacer Kit (Invitrogen, Carlsbad, CA). The following primers were used: 5′ RACE, 5′- CATTTCCTCCCGCTTCTGTCTCCTG-3′; 3′ RACE, 5′-TGAAGCCACTCTCTTGGATAGACT-3′.
Western blot analysis
Protein from control and CaD-MO–expressing embryos were extracted at stage 16 and resolved on SDS-PAGE gel, transferred to nitrocellulose (Pierce, Thermo Fisher Scientific, Rockford, IL), and detected in Western blots with CaD antibody (sc-15374; Santa Cruz Biotechnology, Santa Cruz, CA), coupled with horseradish peroxidase–conjugated secondary antibody by chemiluminescence. α-Tubulin antibody was used for loading control.
CNC explant cultures, grafting, and microscopy
CNC explants were dissected from stage 13–15 embryos as previously described (Borchers et al., 2000; Alfandari et al., 2003; DeSimone et al., 2005). Explants were plated onto FN (20 μg/ml in PBS)–coated dishes in MBSH media, and their behavior was photographically documented every 2–3 h for up to 10 h. The relative surface area and number of segments in explants were counted to provide a measure of their spread and segregation. For observation of cell migration and membrane protrusions, CNC explants labeled with membrane-EGFP were plated on FN-coated coverslips, and cell behaviors were recorded for 5 h with 3-min frame intervals with a 20× objective lens by time-lapse cinematography using a Zeiss (Jena, Germany) LSM5 PASCAL confocal microscope. To examine the translocation of cells, CNC explant labeled with nuclear EGFP (H2b-EGFP) was recorded for 2 h by time-lapse cinematography. Cells were traced by the ImageJ (National Institutes of Health, Bethesda, MD) Manual Tracking plug-in, and their migration speed or directionality was measured by the ImageJ Chemotaxis Tool plug-in. For grafting experiments, CNC explants were dissected similarly from EGFP-labeled donor embryos and inserted isotopically and isochronically into unlabeled host embryos from which CNC tissue was removed. The grafted embryos were allowed to heal in MBSH media for 3 h and then transferred to 0.1× MMR before imaging at late tailbud stages. In addition, embryos were immobilized by embedding in 3% methylcellulose with 0.1% tricaine, and time-lapse movies were recorded for 6–10 h using a 5× objective lens to follow the migratory behavior of CNC cells in vivo. To directly visualize actin filament, CNC explants were fixed in formaldehyde after plating on FN for 2–3 h. They were then rinsed in PBS and PBS with Tween-20 (PBT) and stained with rhodamine phalloidin (Invitrogen) at 1:40 dilution in PBT for 20 min. Cells were rinsed again and mounted in Fluoromount-G (SouthernBiotech, Birmingham, AL) before being imaged using a 40× objective lens.
Cell–cell and cell–matrix adhesion assay
To analyze cell–cell adhesion, CNC explants were dissected from stage 13–15 embryos and dissociated for 20 min in calcium- and magnesium-free buffer. Loose cells were transferred to 0.5× MMR in agarose-coated dishes and allowed to aggregate by orbital horizontal shaking for 6 h. Pictures were taken before and after aggregation. To examine cell–FN matrix adhesion, CNC cells were dissociated as described earlier and plated onto FN-coated dishes in high-salt buffer MBSH. Cells were settled on FN for 1 h before the dish was flipped over in MBSH buffer to wash off unattached cells. The cells on FN were imaged before and after the wash.
Supplementary Material
Acknowledgments
This work was supported by DE017911 and HD037105 from the National Institutes of Health to M.B.F. and by a 2011 Research Grant from Kangwon National University to Y.K.
Abbreviations used:
- CaD
caldesmon
- CNC
cranial neural crest
- FN
fibronectin
- MO
morpholino oligonucleotide
- nβGal
nuclear β-galactosidase
- RACE
rapid amplification of cDNA ends
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-02-0165) on July 27, 2011.
REFERENCES
- Alfandari D, Cousin H, Gaultier A, Hoffstrom BG, DeSimone DW. Integrin alpha5beta1 supports the migration of Xenopus cranial neural crest on fibronectin. Dev Biol. 2003;260:449–464. doi: 10.1016/s0012-1606(03)00277-x. [DOI] [PubMed] [Google Scholar]
- Borchers A, David R, Wedlich D. Xenopus cadherin-11 restrains cranial neural crest migration and influences neural crest specification. Development. 2001;128:3049–3060. doi: 10.1242/dev.128.16.3049. [DOI] [PubMed] [Google Scholar]
- Borchers A, Epperlein HH, Wedlich D. An assay system to study migratory behavior of cranial neural crest cells in Xenopus. Dev Genes Evol. 2000;210:217–222. doi: 10.1007/s004270050307. [DOI] [PubMed] [Google Scholar]
- Carmona-Fontaine C, Matthews HK, Kuriyama S, Moreno M, Dunn GA, Parsons M, Stern CD, Mayor R. Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature. 2008;456:957–961. doi: 10.1038/nature07441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalovich JM, Chen YD, Dudek R, Luo H. Kinetics of binding of caldesmon to actin. J Biol Chem. 1995;270:9911–9916. doi: 10.1074/jbc.270.17.9911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Wilson PA, Mathews LS, Hemmati-Brivanlou A. A Xenopus type I activin receptor mediates mesodermal but not neural specification during embryogenesis. Development. 1997;124:827–837. doi: 10.1242/dev.124.4.827. [DOI] [PubMed] [Google Scholar]
- Childs TJ, Watson MH, Sanghera JS, Campbell DL, Pelech SL, Mak AS. Phosphorylation of smooth muscle caldesmon by mitogen-activated protein (MAP) kinase and expression of MAP kinase in differentiated smooth muscle cells. J Biol Chem. 1992;267:22853–22859. [PubMed] [Google Scholar]
- Dabrowska R, Hinssen H, Galazkiewicz B, Nowak E. Modulation of gelsolin-induced actin-filament severing by caldesmon and tropomyosin and the effect of these proteins on the actin activation of myosin Mg(2+)-ATPase activity. Biochem J. 1996;315:753–759. doi: 10.1042/bj3150753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Calisto J, Araya C, Marchant L, Riaz CF, Mayor R. Essential role of non-canonical Wnt signalling in neural crest migration. Development. 2005;132:2587–2597. doi: 10.1242/dev.01857. [DOI] [PubMed] [Google Scholar]
- DeSimone DW, Davidson L, Marsden M, Alfandari D. The Xenopus embryo as a model system for studies of cell migration. Methods Mol Biol. 2005;294:235–245. doi: 10.1385/1-59259-860-9:235. [DOI] [PubMed] [Google Scholar]
- Eppinga RD, Li Y, Lin JL, Mak AS, Lin JJ. Requirement of reversible caldesmon phosphorylation at P21-activated kinase-responsive sites for lamellipodia extensions during cell migration. Cell Motil Cytoskeleton. 2006;63:543–562. doi: 10.1002/cm.20144. [DOI] [PubMed] [Google Scholar]
- Eves R, Webb BA, Zhou S, Mak AS. Caldesmon is an integral component of podosomes in smooth muscle cells. J Cell Sci. 2006;119:1691–1702. doi: 10.1242/jcs.02881. [DOI] [PubMed] [Google Scholar]
- Foster DB, Huang R, Hatch V, Craig R, Graceffa P, Lehman W, Wang CL. Modes of caldesmon binding to actin: sites of caldesmon contact and modulation of interactions by phosphorylation. J Biol Chem. 2004;279:53387–53394. doi: 10.1074/jbc.M410109200. [DOI] [PubMed] [Google Scholar]
- Foster DB, Shen LH, Kelly J, Thibault P, Van Eyk JE, Mak AS. Phosphorylation of caldesmon by p21-activated kinaseImplications for the Ca(2+) sensitivity of smooth muscle contraction. J Biol Chem. 2000;275:1959–1965. doi: 10.1074/jbc.275.3.1959. [DOI] [PubMed] [Google Scholar]
- Fukumoto K, Morita T, Mayanagi T, Tanokashira D, Yoshida T, Sakai A, Sobue K. Detrimental effects of glucocorticoids on neuronal migration during brain development. Mol Psychiatry. 2009;14:1119–1131. doi: 10.1038/mp.2009.60. [DOI] [PubMed] [Google Scholar]
- Goncharova EA, Shirinsky VP, Shevelev AY, Marston SB, Vorotnikov AV. Actomyosin cross-linking by caldesmon in non-muscle cells. FEBS Lett. 2001;497:113–117. doi: 10.1016/s0014-5793(01)02445-0. [DOI] [PubMed] [Google Scholar]
- Grosheva I, Vittitow JL, Goichberg P, Gabelt BT, Kaufman PL, Borras T, Geiger B, Bershadsky AD. Caldesmon effects on the actin cytoskeleton and cell adhesion in cultured HTM cells. Exp Eye Res. 2006;82:945–958. doi: 10.1016/j.exer.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Gu Z, Kordowska J, Williams GL, Wang CL, Hai CM. Erk1/2 MAPK and caldesmon differentially regulate podosome dynamics in A7r5 vascular smooth muscle cells. Exp Cell Res. 2007;313:849–866. doi: 10.1016/j.yexcr.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbling PM, Tran CT, Brandli AW. Requirement for EphA receptor signaling in the segregation of Xenopus third and fourth arch neural crest cells. Mech Dev. 1998;78:63–79. doi: 10.1016/s0925-4773(98)00148-8. [DOI] [PubMed] [Google Scholar]
- Helfman DM, Levy ET, Berthier C, Shtutman M, Riveline D, Grosheva I, Lachish-Zalait A, Elbaum M, Bershadsky AD. Caldesmon inhibits nonmuscle cell contractility and interferes with the formation of focal adhesions. Mol Biol Cell. 1999;10:3097–3112. doi: 10.1091/mbc.10.10.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemric ME, Tracy PB, Haeberle JR. Caldesmon enhances the binding of myosin to the cytoskeleton during platelet activation. J Biol Chem. 1994;269:4125–4128. [PubMed] [Google Scholar]
- Huang R, Li L, Guo H, Wang CL. Caldesmon binding to actin is regulated by calmodulin and phosphorylation via different mechanisms. Biochemistry. 2003;42:2513–2523. doi: 10.1021/bi0268605. [DOI] [PubMed] [Google Scholar]
- Ishikawa R, Yamashiro S, Kohama K, Matsumura F. Regulation of actin binding and actin bundling activities of fascin by caldesmon coupled with tropomyosin. J Biol Chem. 1998;273:26991–26997. doi: 10.1074/jbc.273.41.26991. [DOI] [PubMed] [Google Scholar]
- Ishikawa R, Yamashiro S, Matsumura F. Annealing of gelsolin-severed actin fragments by tropomyosin in the presence of Ca2+. Potentiation of the annealing process by caldesmon. J Biol Chem. 1989a;264:16764–16770. [PubMed] [Google Scholar]
- Ishikawa R, Yamashiro S, Matsumura F. Differential modulation of actin-severing activity of gelsolin by multiple isoforms of cultured rat cell tropomyosin. Potentiation of protective ability of tropomyosins by 83-kDa nonmuscle caldesmon. J Biol Chem. 1989b;264:7490–7497. [PubMed] [Google Scholar]
- Kashef J, Kohler A, Kuriyama S, Alfandari D, Mayor R, Wedlich D. Cadherin-11 regulates protrusive activity in Xenopus cranial neural crest cells upstream of Trio and the small GTPases. Genes Dev. 2009;23:1393–1398. doi: 10.1101/gad.519409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordowska J, Hetrick T, Adam LP, Wang CL. Phosphorylated l-caldesmon is involved in disassembly of actin stress fibers and postmitotic spreading. Exp Cell Res. 2006;312:95–110. doi: 10.1016/j.yexcr.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Li Y, Lin JL, Reiter RS, Daniels K, Soll DR, Lin JJ. Caldesmon mutant defective in Ca(2+)-calmodulin binding interferes with assembly of stress fibers and affects cell morphology, growth and motility. J Cell Sci. 2004;117:3593–3604. doi: 10.1242/jcs.01216. [DOI] [PubMed] [Google Scholar]
- Lin JJ, Li Y, Eppinga RD, Wang Q, Jin JP. Chapter 1: roles of caldesmon in cell motility and actin cytoskeleton remodeling. Int Rev Cell Mol Biol. 2009;274:1–68. doi: 10.1016/S1937-6448(08)02001-7. [DOI] [PubMed] [Google Scholar]
- Mak AS, Carpenter M, Smillie LB, Wang JH. Phosphorylation of caldesmon by p34cdc2 kinase. Identification of phosphorylation sites. J Biol Chem. 1991;266:19971–19975. [PubMed] [Google Scholar]
- Matthews HK, Broders-Bondon F, Thiery JP, Mayor R. Wnt11r is required for cranial neural crest migration. Dev Dyn. 2008a;237:3404–3409. doi: 10.1002/dvdy.21758. [DOI] [PubMed] [Google Scholar]
- Matthews HK, Marchant L, Carmona-Fontaine C, Kuriyama S, Larrain J, Holt MR, Parsons M, Mayor R. Directional migration of neural crest cells in vivo is regulated by Syndecan-4/Rac1 and non-canonical Wnt signaling/RhoA. Development. 2008b;135:1771–1780. doi: 10.1242/dev.017350. [DOI] [PubMed] [Google Scholar]
- Mayanagi T, Morita T, Hayashi K, Fukumoto K, Sobue K. Glucocorticoid receptor-mediated expression of caldesmon regulates cell migration via the reorganization of the actin cytoskeleton. J Biol Chem. 2008;283:31183–31196. doi: 10.1074/jbc.M801606200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFawn PK, Shen L, Vincent SG, Mak A, Van Eyk JE, Fisher JT. Calcium-independent contraction and sensitization of airway smooth muscle by p21-activated protein kinase. Am J Physiol Lung Cell Mol Physiol. 2003;284:L863–870. doi: 10.1152/ajplung.00068.2002. [DOI] [PubMed] [Google Scholar]
- Mellitzer G, Xu Q, Wilkinson DG. Eph receptors and ephrins restrict cell intermingling and communication. Nature. 1999;400:77–81. doi: 10.1038/21907. [DOI] [PubMed] [Google Scholar]
- Nie S, Kee Y, Bronner-Fraser M. Myosin-X is critical for migratory ability of Xenopus cranial neural crest cells. Dev Biol. 2009;335:132–142. doi: 10.1016/j.ydbio.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura M, Yoshikawa K, Tanaka T, Sobue K, Maruyama K. The role of tropomyosin in the interactions of F-actin with caldesmon and actin-binding protein (or filamin) Eur J Biochem. 1987;163:467–471. doi: 10.1111/j.1432-1033.1987.tb10892.x. [DOI] [PubMed] [Google Scholar]
- Novy RE, Lin JL, Lin JJ. Characterization of cDNA clones encoding a human fibroblast caldesmon isoform and analysis of caldesmon expression in normal and transformed cells. J Biol Chem. 1991;266:16917–16924. [PubMed] [Google Scholar]
- Numaguchi Y, Huang S, Polte TR, Eichler GS, Wang N, Ingber DE. Caldesmon-dependent switching between capillary endothelial cell growth and apoptosis through modulation of cell shape and contractility. Angiogenesis. 2003;6:55–64. doi: 10.1023/a:1025821517679. [DOI] [PubMed] [Google Scholar]
- Paul ER, Ngai PK, Walsh MP, Groschel-Stewart U. Embryonic chicken gizzard: expression of the smooth muscle regulatory proteins caldesmon and myosin light chain kinase. Cell Tissue Res. 1995;279:331–337. doi: 10.1007/BF00318489. [DOI] [PubMed] [Google Scholar]
- Rangarajan J, Luo T, Sargent TD. PCNS: a novel protocadherin required for cranial neural crest migration and somite morphogenesis in Xenopus. Dev Biol. 2006;295:206–218. doi: 10.1016/j.ydbio.2006.03.025. [DOI] [PubMed] [Google Scholar]
- Robinson V, Smith A, Flenniken AM, Wilkinson DG. Roles of Eph receptors and ephrins in neural crest pathfinding. Cell Tissue Res. 1997;290:265–274. doi: 10.1007/s004410050931. [DOI] [PubMed] [Google Scholar]
- Sadaghiani B, Thiebaud CH. Neural crest development in the Xenopus laevis embryo, studied by interspecific transplantation and scanning electron microscopy. Dev Biol. 1987;124:91–110. doi: 10.1016/0012-1606(87)90463-5. [DOI] [PubMed] [Google Scholar]
- Santiago A, Erickson CA. Ephrin-B ligands play a dual role in the control of neural crest cell migration. Development. 2002;129:3621–3632. doi: 10.1242/dev.129.15.3621. [DOI] [PubMed] [Google Scholar]
- Smith A, Robinson V, Patel K, Wilkinson DG. The EphA4 and EphB1 receptor tyrosine kinases and ephrin-B2 ligand regulate targeted migration of branchial neural crest cells. Curr Biol. 1997;7:561–570. doi: 10.1016/s0960-9822(06)00255-7. [DOI] [PubMed] [Google Scholar]
- Sobue K, Sellers JR. Caldesmon, a novel regulatory protein in smooth muscle and nonmuscle actomyosin systems. J Biol Chem. 1991;266:12115–12118. [PubMed] [Google Scholar]
- Tanaka J, Watanabe T, Nakamura N, Sobue K. Morphological and biochemical analyses of contractile proteins (actin, myosin, caldesmon and tropomyosin) in normal and transformed cells. J Cell Sci. 1993;104:595–606. doi: 10.1242/jcs.104.2.595. [DOI] [PubMed] [Google Scholar]
- Theveneau E, Marchant L, Kuriyama S, Gull M, Moepps B, Parsons M, Mayor R. Collective chemotaxis requires contact-dependent cell polarity. Dev Cell. 2010;19:39–53. doi: 10.1016/j.devcel.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallin J, Girault JM, Thiery JP, Broders F. Xenopus cadherin-11 is expressed in different populations of migrating neural crest cells. Mech Dev. 1998;75:171–174. doi: 10.1016/s0925-4773(98)00099-9. [DOI] [PubMed] [Google Scholar]
- Van Eyk JE, Arrell DK, Foster DB, Strauss JD, Heinonen TY, Furmaniak-Kazmierczak E, Cote GP, Mak AS. Different molecular mechanisms for Rho family GTPase-dependent, Ca2+-independent contraction of smooth muscle. J Biol Chem. 1998;273:23433–23439. doi: 10.1074/jbc.273.36.23433. [DOI] [PubMed] [Google Scholar]
- Warren KS, Shutt DC, McDermott JP, Lin JL, Soll DR, Lin JJ. Overexpression of microfilament-stabilizing human caldesmon fragment, CaD39, affects cell attachment, spreading, and cytokinesis. Cell Motil Cytoskeleton. 1996;34:215–229. doi: 10.1002/(SICI)1097-0169(1996)34:3<215::AID-CM5>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Winklbauer R, Selchow A, Nagel M, Angres B. Cell interaction and its role in mesoderm cell migration during Xenopus gastrulation. Dev Dyn. 1992;195:290–302. doi: 10.1002/aja.1001950407. [DOI] [PubMed] [Google Scholar]
- Yamakita Y, Oosawa F, Yamashiro S, Matsumura F. Caldesmon inhibits Arp2/3-mediated actin nucleation. J Biol Chem. 2003;278:17937–17944. doi: 10.1074/jbc.M208739200. [DOI] [PubMed] [Google Scholar]
- Yamashiro S, Chern H, Yamakita Y, Matsumura F. Mutant caldesmon lacking cdc2 phosphorylation sites delays M-phase entry and inhibits cytokinesis. Mol Biol Cell. 2001;12:239–250. doi: 10.1091/mbc.12.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro S, Yamakita Y, Hosoya H, Matsumura F. Phosphorylation of non-muscle caldesmon by p34cdc2 kinase during mitosis. Nature. 1991;349:169–172. doi: 10.1038/349169a0. [DOI] [PubMed] [Google Scholar]
- Yamashiro S, Yamakita Y, Ishikawa R, Matsumura F. Mitosis-specific phosphorylation causes 83K non-muscle caldesmon to dissociate from microfilaments. Nature. 1990;344:675–678. doi: 10.1038/344675a0. [DOI] [PubMed] [Google Scholar]
- Yamashiro S, Yoshida K, Yamakita Y, Matsumura F. Caldesmon: possible functions in microfilament reorganization during mitosis and cell transformation. Adv Exp Med Biol. 1994;358:113–122. doi: 10.1007/978-1-4615-2578-3_11. [DOI] [PubMed] [Google Scholar]
- Yoshio T, Morita T, Kimura Y, Tsujii M, Hayashi N, Sobue K. Caldesmon suppresses cancer cell invasion by regulating podosome/invadopodium formation. FEBS Lett. 2007;581:3777–3782. doi: 10.1016/j.febslet.2007.06.073. [DOI] [PubMed] [Google Scholar]
- Zheng PP, Severijnen LA, van der Weiden M, Willemsen R, Kros JM. A crucial role of caldesmon in vascular development in vivo. Cardiovasc Res. 2009a;81:362–369. doi: 10.1093/cvr/cvn294. [DOI] [PubMed] [Google Scholar]
- Zheng PP, Severijnen LA, Willemsen R, Kros JM. Caldesmon is essential for cardiac morphogenesis and function: in vivo study using a zebrafish model. Biochem Biophys Res Commun. 2009b;378:37–40. doi: 10.1016/j.bbrc.2008.10.165. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.