

Voa1 and Voa2 cooperatively regulate the acidification and transmitter uptake/storage of dense-core vesicles, although they might not be as critical for exocytosis as recently proposed.
Abstract
The Vo sector of the vacuolar H+-ATPase is a multisubunit complex that forms a proteolipid pore. Among the four isoforms (a1–a4) of subunit Voa, the isoform(s) critical for secretory vesicle acidification have yet to be identified. An independent function of Voa1 in exocytosis has been suggested. Here we investigate the function of Voa isoforms in secretory vesicle acidification and exocytosis by using neurosecretory PC12 cells. Fluorescence-tagged and endogenous Voa1 are primarily localized on secretory vesicles, whereas fluorescence-tagged Voa2 and Voa3 are enriched on the Golgi and early endosomes, respectively. To elucidate the functional roles of Voa1 and Voa2, we engineered PC12 cells in which Voa1, Voa2, or both are stably down-regulated. Our results reveal significant reductions in the acidification and transmitter uptake/storage of dense-core vesicles by knockdown of Voa1 and more dramatically of Voa1/Voa2 but not of Voa2. Overexpressing knockdown-resistant Voa1 suppresses the acidification defect caused by the Voa1/Voa2 knockdown. Unexpectedly, Ca2+-dependent peptide secretion is largely unaffected in Voa1 or Voa1/Voa2 knockdown cells. Our data demonstrate that Voa1 and Voa2 cooperatively regulate the acidification and transmitter uptake/storage of dense-core vesicles, whereas they might not be as critical for exocytosis as recently proposed.
INTRODUCTION
Acidification of intracellular compartments, including secretory vesicles, is established and maintained primarily by the vacuolar H+-ATPase (V-ATPase). V-ATPases are ATP-driven proton pumps that function to both acidify intracellular compartments and transport protons across the plasma membrane. The V-ATPase is a multimeric protein complex that consists of two sectors: the cytoplasmic V1 sector, which is responsible for ATP hydrolysis, and the transmembrane Vo sector, which forms a proteolipid pore responsible for proton translocation (Forgac, 2007; Jefferies et al., 2008; Marshansky and Futai, 2008; Hinton et al., 2009; Saroussi and Nelson, 2009).
The largest (∼110 kDa) subunit of the Vo sector, Voa, is an integral membrane protein with four isoform genes (a1–a4) identified in human, mouse, Caenorhabditis elegans, and Drosophila. In C. elegans, these four genes display highly tissue-specific distributions, with Voa1 (Unc-32) being predominantly expressed in neurons (Oka et al., 2001b; Pujol et al., 2001). Similarly, the function of Voa1 (Vha100-1) in Drosophila seems to be restricted to neurons, since its null mutation phenotypes are rescued by neuron-specific expression of Vha100-1 (Hiesinger et al., 2005). In mammals, although Voa1, Voa2, and Voa3 are widely expressed, Voa1 is more strongly expressed in the brain, whereas Voa3 is primarily expressed in osteoclasts (Nishi and Forgac, 2000; Toyomura et al., 2000). The expression of Voa4 is restricted to several ion-transporting epithelia of the kidney (Oka et al., 2001a), the inner ear (Stover et al., 2002), and the ocular ciliary bodies (Kawamura et al., 2010)
The essential functions of the isoforms of mammalian Voa subunit have been revealed by the discovery of mutations associated with certain diseases in humans, as well as by the analyses of knockout mice. Mutations in Voa3 cause osteopetrosis in humans (Frattini et al., 2000; Kornak et al., 2000) and mice (Li et al., 1999), whereas mutations in Voa4 cause recessive distal renal tubular acidosis (Smith et al., 2000; Stover et al., 2002) and in some cases hearing loss in humans (Stover et al., 2002). These defects have been interpreted to result from the inability of the V-ATPase containing mutated Voa3 or Voa4 to expel protons to the outside of the cell, as Voa3 is localized at the plasma membrane of osteoclasts and Voa4 is found at the plasma membrane of kidney intercalated cells. In addition, it was recently discovered that mutations in Voa2 cause autosomal recessive cutis laxa type 2, or wrinkly skin syndrome, in humans due to functional defects at the Golgi, including glycosylation (Kornak et al., 2008), as well as perturbations in general vesicular trafficking and tropoelastin secretion (Hucthagowder et al., 2009). However, it is unclear whether these phenotypes caused by mutations in Voa2 are due to acidification defects in intracellular compartments such as the Golgi. The phenotypes associated with mutations in mammalian Voa1 are unknown.
Independent of its role as a cellular proton pump, the Vo sector has also been suggested to function in membrane fusion (Wada et al., 2008). The process of membrane fusion depends on soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs; Söllner et al., 1993; Jahn et al., 2003; Jahn, 2004; Sudhof, 2004). In addition, SNAREs have been shown to be sufficient in driving fusion in liposome fusion assays (Weber et al., 1998; Hu et al., 2003). However, the reaction of SNARE-mediated liposome fusion is much slower than exocytotic membrane fusion in vivo. A recent study using yeast vacuolar fusion assay demonstrated that the proteolipid pore-forming Vo sector is required for membrane fusion downstream of SNARE (Peters et al., 2001). This study suggests that following the tethering of donor and acceptor membranes via the SNARE complex formation, the Vo sector directly catalyzes the mixing of the two lipid bilayers by virtue of its highly hydrophobic subunit composition. The same authors further provided genetic evidence that a mutation in VPH1, a yeast homologue of Voa, resulted in vacuolar fusion defects in yeasts (Bayer et al., 2003). Additional genetic evidence supporting a role for the Vo sector, particularly the Voa1 subunit, in membrane fusion has emerged from studies of synaptic vesicle release in Drosophila (Hiesinger et al., 2005; Williamson et al., 2010) and phagocytotic membrane fusion in zebrafish (Peri and Nüsslein-Volhard, 2008).
In neurons and neuroendocrine cells, the lumen of secretory vesicles (i.e., synaptic vesicles and dense-core vesicles) is kept acidic (∼pH 5.5) via V-ATPase activity, and the proton gradient maintained by a properly acidified vesicle allows the loading of neurotransmitters into the vesicle (Moriyama and Futai, 1990a, 1990b; Amara and Kuhar, 1993; Schuldiner et al., 1998; Masson et al., 1999; Gasnier, 2000; Fremeau et al., 2004). However, the specific Voa isoform responsible for secretory vesicle acidification has yet to be identified. Although Voa1 has long been known to be the major isoform of Voa in the brain (Perin et al., 1991; Peng et al., 1994), the roles of Voa1 in acidification and transmitter uptake of secretory vesicles have not been demonstrated. To address the roles of Voa1 and Voa2 in secretory vesicle acidification, transmitter loading/storage, and exocytotic membrane fusion, we engineered PC12 cells in which Voa1, Voa2, or both are strongly down-regulated. PC12 cells, derived from rat adrenal chromaffin cells, retain robust regulated catecholamine and peptide secretion through SNARE-mediated membrane fusion/exocytosis (Han et al., 2009). Our results indicate that Voa1 and, to a lesser degree, Voa2 play critical but overlapping roles in secretory vesicle acidification and transmitter uptake and storage. Surprisingly, our study suggests that they may not play a major role in exocytotic membrane fusion.
RESULTS
Differential localization of Voa1, Voa2, and Voa3 in PC12 cells
To examine the isoform-specific localization of the Voa subunit in neuroendocrine PC12 cells, we engineered PC12 cells that stably express Voa1, Voa2, or Voa3 as Emerald green fluorescent protein (GFP)– and/or mCherry-fusion proteins through lentivirus-mediated infection (Figure 1, A–C; Materials and Methods). A recent study of the membrane topology of the Voa homologue VPH1 in yeast supports an eight-transmembrane-helix model of subunit a in which the C-terminus is located on the cytoplasmic side of the membrane (Wang et al., 2008). Reverse transcription (RT)-PCR revealed that Voa4 is present in normal rat kidney (NRK) cells but is absent in PC12 cells (Figure 1D), confirming that the expression of this isoform is confined to the kidney, the inner ear, and the optical ciliary bodies (Oka et al., 2001a; Stover et al., 2002; Kawamura et al., 2010). We therefore excluded the analysis of Voa4 from this study. Although the expression constructs of the Voa isoforms clearly produced fluorescence signals in transfected HEK293-FT cells, fluorescence signals from the stable PC12 cells were very weak, suggesting that the expression of these fluorescent protein-fused Voa proteins is low. Nevertheless, the fusion proteins were detected using either anti-mCherry or anti-GFP antibodies in Western blot analyses (Figure 1, B and C). Hence we used anti-GFP antibody in our immunofluorescence studies to detect the localizations of Voa1-, Voa2-, and Voa3-EmGFP. Specifically, we examined colocalization of these fluorescence-tagged Voa isoforms with synaptotagmin-1, a marker protein for secretory vesicles (i.e., both synaptic-like clear microvesicles and dense-core vesicles), as well as with GM130, a marker protein of cis and medial Golgi (Figures 2 and 3; also see Supplemental Figure S1 for lower-magnification photographs). We found that Voa1-EmGFP showed punctate staining both at the soma and at the tip of neurites (Figure 2, A and B). At the tip of neurites, it was colocalized with synaptotagmin-1 (Figure 2A). In contrast, Voa2-EmGFP showed strong perinuclear staining (Figure 2, C and D) and was only weakly colocalized with synaptotagmin-1 (Figure 2C), whereas it was well colocalized with GM130 (Figure 2D), indicating that Voa2 is enriched at the Golgi. On the other hand, Voa1 was not colocalized with GM130 (Figure 2B). This enrichment of Voa2 at the Golgi is in good agreement with defects in Golgi function resulting from loss-of-function Voa2 mutations (Kornak et al., 2008). Unlike Voa1- or Voa2-EmGFP, Voa3-EmGFP stained with anti-GFP antibody did not colocalize with synaptotagmin-1 (Figure 3A) or GM130 (Figure 3B). However, it strongly colocalized with EEA1, a marker protein of early endosomes (Figure 3C), but not with LAMP1, a marker protein of lysosomes (Figure 3D). Our results suggest that Voa3 is primarily localized on early endosomes and is not likely to play a major role in secretory vesicle acidification in PC12 cells.
FIGURE 1:

Stable expression of fluorescence-tagged Voa1-3 in PC12 cells, as well as the absence of Voa4 isoform in PC12 cells. (A) A schematic representation of Voa protein fused with a fluorescent protein (mCherry or EmGFP). (B, C) Immunoblot analyses of the cells that stably express Voa1, Voa2, or Voa3 fused with mCherry or EmGFP. Total 20-μg homogenates from the PC12 cells overexpressing the various recombinant Voa proteins were analyzed by SDS–PAGE, and immunoblotting was performed by using antibodies against DsRED or GFP. (D) RT-PCR analyses of gene expression in wild-type PC12 and NRK cells.
FIGURE 2:

Confocal immunofluorescence microscopy reveals that stably expressed Voa1 better colocalizes with synaptotagmin-1, whereas Voa2 better colocalizes with GM130 in PC12 cells. NGF-differentiated PC12 cells that stably express Voa1-EmGFP (A, B) or Voa2-EmGFP (C, D) were costained with anti-GFP rabbit polyclonal antibody and either anti–synaptotagmin-1 mouse monoclonal antibody (A, C) or anti-GM130 mouse monoclonal antibody (B, D). For secondary staining Alexa 488–conjugated goat anti–rabbit antibody and Rhodamine Red-X–conjugated goat anti–mouse antibody were used against the appropriate primary antibodies. Right, merged pictures. Scale bar, 10 μm
FIGURE 3:

Stably expressed Voa3 is localized on early endosomes. NGF-differentiated PC12 cells that stably express Voa3-EmGFP were costained with anti-GFP rabbit polyclonal antibody and anti–synaptotagmin-1 mouse monoclonal antibody (A), anti-GM130 antibody (B), anti-EEA1 goat polyclonal antibody (C), or anti-LAMP1 antibody (D). For secondary staining Alexa 488–conjugated goat anti–rabbit antibody, Rhodamine Red-X–conjugated goat anti–mouse antibody, or Alexa 568–conjugated donkey anti–goat antibody were used against the appropriate primary antibodies. Right, merged pictures. Scale bar, 10 μm.
The data from fluorescence-tagged Voa isoforms suggest that Voa1 is the major isoform expressed on secretory vesicles. We examined whether endogenous Voa1 is also localized on secretory granules using rabbit anti-Voa1 antibody. In strong agreement with the Voa1-EmGFP data (Figure 2A), we found that endogenous Voa1 showed a strong enrichment at the tip of neurites, where it was colocalized with synaptotagmin-1 (Figure 4A; higher magnification in Figure 4B). We also attempted to examine the localization of endogenous Voa2 using rabbit anti-Voa2 antibody (Peng et al., 1999). Unfortunately, the staining by this antibody was less clear. Nonetheless, we found a partial enrichment of anti-Voa2 staining at the perinuclear region, where Voa2 appeared to partially colocalize with GM130 (Figure 4C), which is in agreement with the Voa2-EmGFP data (Figure 2D).
FIGURE 4:

Endogenous Voa1 colocalizes with syntaptotagmin-1. NGF-differentiated PC12 cells were costained with anti-Voa1 rabbit polyclonal antibody and anti–synaptotagmin-1 mouse monoclonal antibody (A, B) or with anti-Voa2 rabbit polyclonal antibody and GM130 mouse monoclonal antibody (C). For secondary staining Alexa 488–conjugated goat anti–rabbit antibody and Rhodamine Red-X–conjugated goat anti–mouse antibody were used against the appropriate primary antibodies. Right, merged pictures. Scale bar, 10 μm
Knockdown of Voa1 caused significant reductions in acidification of dense-core vesicles: a compensatory up-regulation of Voa2 and Ac45
To study the functional significance of Voa1 in neurosecretory cells, we generated stable Voa1 knockdown PC12 cells using lentivirus-mediated short hairpin RNA (shRNA). Lentiviruses produced from pLKO-puro-Voa1KD were highly efficient in silencing the Voa1 mRNA transcript, allowing us to successfully generate a heterogeneous pool of PC12 cells in which the Voa1 protein was strongly down-regulated after selection with puromycin (Figure 5A). Anti-Voa1 antibody from Synaptic Systems, which is specific to rodent Voa1, and that from Santa Cruz Biotechnology, which detects both rodent Voa1 and human Voa1 (see Figure 9 later in the paper), showed similar levels (∼90%) of knockdown. We next examined the expression levels of several proteins, which included other subunits of the Vo sector of the V-ATPase (Voa2 and Vod), an accessory protein of the Vo sector (Ac45; Supek et al., 1994), t-SNARE proteins (syntaxin-1 and SNAP-25; Söllner et al., 1993), a marker protein for cis and medial Golgi (GM130; Nakamura et al., 1995), a marker protein for the endoplasmic reticulum (calnexin; Wada et al., 1991), and a general membrane-trafficking protein (VCP/p97; Peters et al., 1990). We found significant increases in the expression of Voa2, which was detected by two different antibodies, and Ac45 (Figure 5B). No changes were detected in the other proteins examined. The up-regulation of Voa2 and perhaps of Ac45 is likely due to a compensatory reaction of the cell in response to the down-regulation of Voa1.
FIGURE 5:

Down-regulation of Voa1 results in significant reductions in acidification of dense-core vesicles. (A) Immunoblot analyses of a heterogeneous pool of stable Voa1 knockdown cells. Total 20-μg homogenates from control and stable Voa1 knockdown PC12 cells were analyzed by SDS–PAGE and immunoblotting using the antibodies indicated on the right. The signal was detected with an enhanced chemiluminescence detection system. The numbers on the left indicate the positions of the molecular weight markers. (B) Quantification of the changes in Voa2 and Ac45 expressions between control and Voa1 knockdown cells. Western blot images of these proteins were quantified using Image J software. The protein expression in the knockdown cells was normalized with that in control cells. The data are from four or five independent blots. (C) Summary data of FACS analysis of fluorescence signals from control and Voa1 knockdown cells that were transfected with NPY-epHluorin (n = 8 each). In each analysis, 104 PI-negative cells were quantified, and their mean value was used for the summary data. (D) Immunoblot analysis of the transfected NPY-epHluorin in control and Voa1 knockdown cells. Total 20-μg homogenates from control and stable Voa1 knockdown PC12 cells that were electroporated with 15 μg of pCMV-NPY-epHluorin were analyzed by SDS–PAGE and immunoblotting using anti-GFP antibody. The results of three independent electroporations are shown. (E) Quantification of the protein expression of transfected NPY-hPLAP.
FIGURE 9:

Expression of knockdown-resistant Voa1 suppresses the acidification defects caused by knockdown of Voa1 or Voa1/Voa2. (A) Immunoblot analyses of the PC12 cells that stably express mCherry (control) or human Voa1 (SNM) that is resistant to the Voa1KD shRNA. (B, C) Immunoblot analyses of the human Voa1 (SNM)–expressing cells in which endogenous Voa1 or Voa1/Voa2 was knocked down. (D, E) Summary data (n = 6 or 7) of FACS analysis of fluorescence signals from control and Voa1 knockdown or Voa1/Voa2 double-knockdown cells that expressed human Voa1 (SNM). The cells were incubated with the indicated concentrations of NH4Cl in substitution of equal concentrations of NaCl (buffered to pH 7.4).
To examine the effects of Voa1 knockdown on acidification inside dense-core vesicles, we generated a reporter construct, neuropeptide Y-fused super ecliptic pHluorin (NPY-epHluorin). Super ecliptic pHluorin is a pH-sensitive GFP protein (Miesenböck et al., 1998; Granseth et al., 2006). We also generated other reporter constructs, NPY-fused Emerald GFP (NPY-EmGFP) and NPY-fused ratiometric pHluorin (NPY-rpHluorin). Emerald GFP is less sensitive to pH than epHluorin (Shaner et al., 2005). NPY-rpHluorin would have been an ideal reporter protein to accurately estimate pH, since ratiometric analysis is possible for rpHluorin (Miesenböck et al., 1998). Unfortunately, the expression of NPY-rpHluorin was not confirmed by immunoblot analysis nor was its fluorescence signal detected. Therefore, we used NPY-epHluorin to detect changes in pH inside the dense-core vesicles.
We transfected control and Voa1 knockdown cells with NPY-epHluorin or NPY-EmGFP and observed their fluorescence signals using fluorescence microscopy. NPY-EmGFP exhibited bright, granule-like signals for both Voa1KD and control cells (see also Figure 12D later in the paper and Supplemental Figure S4), whereas NPY-epHluorin appeared dim in both control and knockdown cells. To quantify the fluorescence signals, we performed fluorescence activated cell sorting (FACS) analysis of the signals from NPY-epHluorin in transfected control and Voa1 knockdown cells. We found consistent increases in the fluorescence signals of NPY-epHluorin from Voa1KD cells compared with the control cells (Figure 5C), which suggests a disruption of dense-core vesicle acidification. This effect was statistically significant (Student's independent t test, n = 8 each, t14 = 3.20, p < 0.01). Because our observation is based on multiple, independent transfections by electroporation (n = 8 for each group), it is unlikely that the observed effects are due to transfection artifacts. However, from these data alone we cannot rule out the possibility that the Voa1KD cells are somehow more transfection prone and/or accumulate the transfected proteins due to defects in secretion. Therefore we performed immunoblot analyses of the expression levels of transfected NPY-epHluorin, which showed no increases in NPY-epHluorin in Voa1KD cells over control cells (Figure 5D). In addition, we transfected both control and Voa1KD cells with a construct that expresses NPY fused with the soluble domains of human placental alkaline phosphatase (NPY-hPLAP). The relative expression of this protein can be accurately quantified through its enzymatic activity (heat-stable alkaline phosphatase) using a commercially available kit (Materials and Methods). We found that the enzymatic activities of NPY-hPLAP extracted from the transfected cells and normalized by the total protein concentrations were comparable between control and knockdown cells (n = 6 each) (Figure 5E). Thus, we conclude that the observed increase in the fluorescence signals of NPY-epHluorin in Voa1 knockdown cells is not due to the increased expression and/or accumulation of these proteins, but instead is due to an increase in pH inside the dense-core vesicles of the Voa1 knockdown cells.
FIGURE 12:

Down-regulation of Voa1 or Voa1/Voa2 does not cause significant defects in Ca2+-dependent secretion of transfected peptide. (A) Secretion of transfected NPY-hPLAP was stimulated from control and Voa1KD cells with or without 70 mM KCl for 20 min. Each experiment was performed in triplicate. Error bars, SEM (n = 8). (B) Secretion of transfected NPY-hPLAP was stimulated from control and Voa1/Voa2 DKD cells with or without 70 mM KCl for 20 min. Each experiment was performed in triplicate. Error bars, SEM (n = 6). (C) Secretion of transfected NPY-hPLAP was stimulated from control and Munc18-1/2 double-knockdown cells with or without 70 mM KCl for 20 min. Each experiment was performed in triplicate. Error bars, SEM (n = 6). (D) NGF-differentiated control (top) and Voa1/Voa2 DKD (bottom) cells that were transfected with NPY-EmGFP with stained with anti–secretogranin II (SgII) rabbit polyclonal antibody. For secondary staining Rhodamine Red-X–conjugated goat anti–rabbit antibody was used. Right, merged pictures. Scale bar, 10 μm.
Knockdown of Voa2 alone showed no significant effects on vesicular acidification
The modest changes in vesicular acidification observed in Voa1 knockdown cells may be accounted for by the up-regulation of Voa2 (Figure 5A), which may be compensating for the function of Voa1 in vesicular acidification in Voa1 knockdown cells. To examine the functional significance of Voa2, we generated a heterogeneous pool of stable Voa2 knockdown cells (Voa2KD). We did not find consistent changes in the expression of the other proteins examined, including Voa1 and Ac45 (Figure 6A). FACS analysis of the signals from cells transfected with NPY-epHluorin revealed that the signal of NPY-epHluorin in Voa2KD is similar to that of control cells (Figure 6B). There was no statistical difference in the signal of NPY-epHluorin between control and Voa2KD cells (n = 9 each, t16 = 1.37, p = 0.19). Our results demonstrate that Voa2 is not the major determinant of vesicular pH in the presence of Voa1.
FIGURE 6:
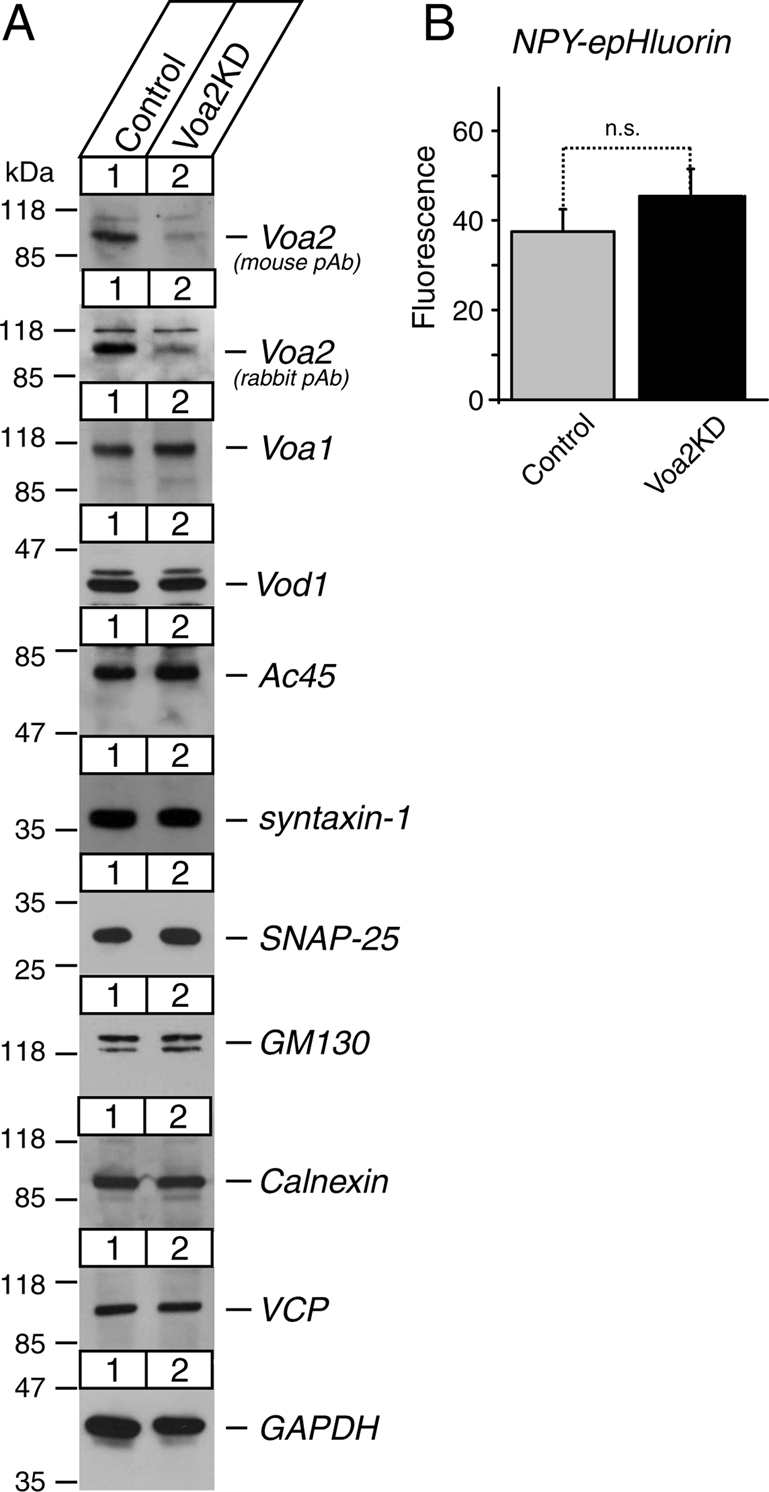
Down-regulation of Voa2 does not cause significant reductions in dense-core vesicle acidification. (A) Immunoblot analyses of a heterogeneous pool of stable Voa2 knockdown (Voa2KD) cells. Total 20-μg homogenates from control and stable Voa2 knockdown PC12 cells were analyzed by SDS–PAGE and immunoblotting using the antibodies indicated on the right. The signal was detected with enhanced chemiluminescence detection system. The numbers on the left indicate the positions of the molecular weight markers. (B) Summary data of FACS analysis of fluorescence signals from control and Voa2 knockdown cells that were transfected with NPY-epHluorin (n = 11 each). In each analysis, 104 PI negative cells were quantified, and their mean value was used for the summary data.
Double knockdown of Voa1 and Voa2 caused dramatic reductions in acidification of dense-core vesicles
Earlier we described an interesting up-regulation of Voa2 in response to the down-regulation of Voa1 (Figure 5). Although Voa2 knockdown alone did not induce significant changes in acidification of dense-core vesicles (Figure 6), Voa2 may still play a significant compensatory role in vesicular acidification in the absence of Voa1, which may explain the modest changes in vesicular acidification seen in Voa1 knockdown cells (Figure 5). To examine the functional significance of Voa1 and Voa2 as a whole, we generated Voa1/Voa2 double knockdown (DKD) cells by infecting PC12 cells with both Voa1KD and Voa2KD knockdown constructs (Materials and Methods) (Figure 7). In DKD cells, the expression levels of Ac45 were again up-regulated, whereas the expressions of the other proteins examined remained unchanged (Figure 7B).
FIGURE 7:

Down-regulation of Voa1 and Voa2 results in dramatic reductions in dense-core vesicle acidification, and application of NH4Cl reduces the difference in the signal of NPY-pHluorin between control and DKD cells in a dose-dependent manner. (A) Immunoblot analyses of a heterogeneous pool of stable Voa1/Voa2 double-knockdown (DKD) cells. Total 20-μg homogenates from control and DKD cells were analyzed by SDS–PAGE, and immunoblotting was performed using the antibodies indicated on the right. The signal was detected with an enhanced chemiluminescence detection system. The numbers on the left indicate the positions of the molecular weight markers. (B) Quantification of changes in protein expression of Ac45 between control and double-knockdown cells. Western blot images of this protein were quantified using Image J software. The protein expression in knockdown cells was normalized with that in control cells. The data are from four independent blots. (C) Examples of FACS analysis of fluorescence signals compare the fluorescence signal distribution from control and DKD cells that are transfected with NPY-epHluorin. For each sample, 104 PI negative cells were analyzed. (D) Summary data of the FACS analysis of fluorescence signals from control and Voa1/Voa2 double-knockdown cells that were transfected with NPY-epHluorin (n = 11 each). (E) Summary data (n = 10) of FACS analysis of fluorescence signals from control and Voa1/Voa2 double-knockdown cells that were transfected with NPY-epHluorin. The cells were incubated with the indicated concentrations of NH4Cl in substitution of equal concentrations of NaCl (buffered to pH 7.4). In each analysis, 104 PI-negative cells were quantified, and their mean value was used for the summary data. (F) The NPY-epHluorin signal in the absence of NH4Cl was normalized with the signal in the presence of 100 mM NH4Cl.
When the double-knockdown cells were transfected with NPY-epHluorin and then quantified with FACS, the distribution of the fluorescence signals in DKD cells was evidently shifted to the right as compared to the distribution of the signal in the control cells (Figure 7C). The averaged signals in DKD cells were two to seven times that of the control cells (Figure 7D). There was a strong statistical difference in the signal of NPY-epHluorin between control and DKD cells (n = 13 each, t24 = 5.16, p < 0.0001). Our results demonstrate that Voa1 and, to a lesser degree, Voa2 play overlapping roles in the acidification of dense-core vesicles.
We also examined whether this dramatic difference in acidification of dense-core vesicles between control and DKD cells is abolished upon application of NH4Cl. NH4Cl rapidly alkalinizes luminal pH due to the rapid influx of NH3 (dissociated from NH4+) into intracellular compartments and the subsequent combination of most of these NH3 molecules with luminal H+. If the difference in NPY-epHluorin fluorescence between control and DKD cells is due to the difference in their luminal pH, this difference should be reduced or abolished in the presence of NH4Cl. For these experiments, we used 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; 15 mM, pH 7.4) buffered saline (instead of phosphate-buffered saline [PBS] used in Figures 5C, 6B, and 7D) containing different concentrations of NH4Cl. The concentration of NaCl was also adjusted accordingly to maintain a constant osmolarity (Kim and Ryan, 2010; Materials and Methods). Using the HEPES-buffered saline, we again observed a clear difference in the FACS signal of NPY-epHluorin between control and DKD cells in the absence of NH4Cl (i.e., at 0 mM NH4Cl in Figure 7E, n = 10 each, t18 = 3.81, p < 0.0001). NH4Cl increased the signal of NPY-epHluorin of both control and DKD cells in a dose-dependent manner. However, the increase in the NPY-epHluorin signal was steeper in the control cells than in the DKD cells (Figure 7E). In the presence of 100 mM NH4Cl, there was no statistically significant difference in NPY-epHluorin signal between control and DKD cells (n = 10 each, t18 = 1.65, p = 0.12). When normalized by the signal of NPY-epHluorin in the presence of 100 mM NH4Cl—presumably the maximal possible fluorescence—the signal without NH4Cl was 13.6% in the control cells, whereas the signal without NH4Cl was 41.4% in the DKD cells (Figure 7F). These results confirm that the lumen of dense-core vesicles are significantly alkalized in DKD cells compared with control cells.
However, the foregoing results did not reveal exact changes in pH of the intracellular compartments (primarily dense-core vesicles) containing the reporter protein NPY-pHluorin. To accurately measure the changes in pH, we used a KCl-based solution containing a combination of ionophores—monensin (5 μM) and nigericin (5 μg/ml)—to control organelle luminal pH (calibration buffer, pH 5.5, 6, 6.5, buffered with 2-(N-morpholino)ethanesulfonic acid [MES], pH 7, 7.5, buffered with HEPES; see Materials and Methods; Kim et al., 1996; Demaurex et al., 1998) and calibrated the signal of our reporter construct NPY-pHluorin with respect to pH. Specifically, the FACS signal of NPY-epHluorin in control and DKD cells that was measured in the normal HEPES-buffered saline was calibrated with the FACS signal of NPY-epHluorin measured in the KCl calibration buffers (Figure 8). The results indicate that the pH inside dense-core vesicles of control cells is ∼6.0, whereas the pH of DKD cells is ∼6.6. This is a significant shift in pH of dense-core vesicles by the double knockdown of Voa1/Voa2, considering that cytosolic pH is between 7.0 and 7.2. We also observed that the signal of NPY-epHluorin was higher in the DKD cells than in the control cells at each respective pH (Figure 8), which indicates that the total amount of NPY-epHluorin expressed in the DKD cells was higher than that in the control cells. This also explains why differences in the signal of NPY-epHluorin between DKD and controls cells were not completely abolished in the presence of 100 mM NH4Cl (Figure 7F). One potential explanation for the higher expression (20–30%) of NPY-epHluorin in the DKD cells over the control cells is that the degradation of transfected DNA in lysosomes and endosomes of DKD cells may be compromised due to acidification defects in these organelles resulting from the down-regulation of Voa1/Voa2. Nevertheless, our results clearly indicate a significant shift in pH in dense-core vesicles by the knockdown of Voa1 and Voa2.
FIGURE 8:

Calibration of NPY-epHluorin signal with respect to pH reveals a substantial shift in pH in dense-core vesicles by knockdown of Voa1/Voa2. Summary data of the FACS analysis of the fluorescence signals from control and DKD cells that were transfected with NPY-epHluorin (n = 6 each). Cells that were used for the calibration were incubated with a combination of nigericin (5 μg/ml) and monensin (5 μM) in KCl-based solutions that were either MES buffered (pH 5.5, 6.0, and 6.5) or HEPES buffered (pH 7.0 and 7.5). Cells that were used to measure intact fluorescence were incubated in an NaCl-based solution that is HEPES buffered (pH 7.4).
The expression of knockdown-resistant human Voa1 suppressed the acidification defects of knockdown of Voa1 and Voa1/Voa2
We then examined whether the expression of knockdown-resistant Voa1 can rescue acidification defects observed by the knockdown of Voa1 or Voa1/Voa2. For this purpose, we first engineered PC12 cells in which knockdown-resistant human Voa1 is stably expressed (Figure 9A). The human Voa1–expressing lentivirus was applied to PC12 cells, and the stable infectants were isolated using IRES-blasticidin resistance gene as a marker of infection. Human Voa1 contains silent nucleotide mutations (SNMs), which makes it refractory to the shRNA knockdown construct of rat Voa1 (Materials and Methods). Anti-Voa1 antibody from Synaptic Systems, which is specific to rodent Voa1, did not detect an increase in Voa1 immunoreactivity, whereas anti-Voa1 antibody from Santa Cruz Biotechnology, which detects both rodent and human Voa1, showed a clear increase in Voa1 immunoreactivity in the PC12 cells that were infected with human Voa1–expressing lentivirus compared with those that were infected with mCherry-expressing lentivirus (Figure 9A). We then proceeded to knock down Voa1 or both Voa1 and Voa2 in the human Voa1–expressing PC12 cells (Figure 9, B and C) and examined whether the signal of transfected NPY-pHluorin is increased compared with control in the presence of the indicated concentrations of NH4Cl in HEPES buffer (pH 7.4). We found no increases in NPY-pHluorin signal upon down-regulation of Voa1 or Voa1/Voa2 in the presence of knockdown-resistant human Voa1 (Figure 9, D and E). Thus the expression of knockdown-resistant Voa1 prevented acidification defects caused by the knockdown of Voa1 or Voa1/Voa2 (Figures 5 and 7). These results clearly indicate that the acidification defects seen in Voa1 and Voa1/Voa2 knockdown are not due to off-target effects of the knockdown, but rather are due to the specific down-regulation of endogenous rat Voa1 or Voa1/Voa2.
Catecholamine uptake is significantly reduced in Voa1/Voa2 double-knockdown cells
What are the major consequences of the acidification defect in secretory vesicles? The uptake of catecholamine into dense-core vesicles is mediated by vesicular monoamine transporter, which requires a proton gradient across the vesicular membrane that is generated by the V-ATPase (Amara and Kuhar, 1993; Schuldiner et al., 1998; Masson et al., 1999; Gasnier, 2000; Fremeau et al., 2004). Thus, it is anticipated that reduced acidification of dense-core vesicles should lead to a reduction in catecholamine uptake into these vesicles. To quantify catecholamine uptake activity, we established uptake assays using [3H]noradrenaline (NA). We made a small modification to the published protocols of the catecholamine uptake assay (Ahnert-Hilger et al., 1998; Brunk et al., 2009) by using a ball homogenizer instead of streptolysin O to permeabilize the PC12 cells (Materials and Methods). We first confirmed that vesicular uptake of [3H]NA is greatly increased in the presence of ATP, demonstrating the ATP-dependent nature of the uptake of catecholamine into dense-core vesicles in native PC12 cells (Supplemental Figure S2).
Using this assay, we examined whether there are significant differences in [3H]NA uptake between control and Voa knockdown cells. We found a tendency that knockdown of Voa1 reduced the uptake of [3H]NA (control: 9354 ± 1419 dpm, n = 9; Voa1KD: 6559 ± 2603 dpm, n = 9). However, this tendency did not reach statistical significance (t16 = 1.68, p = 0.11) as analyzed by Student's independent t test (Figure 10A). With respect to Voa2KD, we found almost no change in the uptake of [3H]NA compared with the control cells (Figure 10B). However, the Voa1/Voa2 DKD cells showed a strong reduction (by 60%) in the uptake of [3H]NA compared with control, and this effect was highly significant (n = 6 each, t10 = 3.69, p < 0.01; Figure 10C). These results suggest that Voa1 and Voa2 play overlapping roles in the uptake of catecholamines into dense-core vesicles by generating a proton gradient across the membrane of dense-core vesicles, with Voa1 playing a more important role.
FIGURE 10:

Down-regulation of Voa1/Voa2 but not of Voa1 or Voa2 results in significant decreases in the uptake of [3H]NA. [3H]NA uptake assays in the presence or absence of 2 mM MgATP were performed using mechanically permeabilized Voa1 knockdown and its control (A, n = 9 each), Voa2 knockdown and its control (B, n = 5 each) or Voa1/Voa2 double-knockdown and its control (C, n = 6 each) cells. Error bars, SEM.
Endogenous dopamine concentrations are significantly reduced in Voa1 knockdown or Voa1/Voa2 double-knockdown cells
We next examined one physiological consequence of reduced catecholamine uptake in our Voa knockdown cells. Similar to parental adrenal chromaffin cells, PC12 cells store catecholamines inside dense-core vesicles. We hypothesized that endogenous catecholamine contents inside these knockdown cells are reduced. To test this hypothesis, we measured catecholamine contents of control and knockdown cells using high-performance liquid chromatography (HPLC), which effectively separated adrenaline, noradrenaline, and dopamine (Supplemental Figure 3A). In PC12 cells dopamine is the major catecholamine and noradrenaline is the minor one (Greene and Tischler, 1976; Fujita et al., 2007; Supplemental Figure S3B). Thus, we compared the concentration of dopamine stored in control and Voa knockdown cells. The value of dopamine concentration was normalized by the respective total protein concentration for each population of control and knockdown cells.
We found that dopamine concentrations inside the cells were reduced in Voa1 knockdown cells (0.78 ± 0.06 ng/μg protein, n = 8) compared with those in control cells (1.48 ± 0.13 ng/μg protein, n = 9) and that this difference was highly significant (t16 = 4.97, p < 0.001; Figure 11A). Although the effect was more modest, we also observed that dopamine concentrations of Voa2KD (0.64 ± 0.08 ng/μg protein, n = 25) were lower than their control counterpart (0.95 ± 0.11 ng/μg protein, n = 25), which was statistically significant (t48 = 2.13, p < 0.05; Figure 11B). Thus, in addition to Voa1 knockdown, Voa2 knockdown also has significant effects on dopamine concentrations stored inside the cells.
FIGURE 11:

Down-regulation of Voa1, Voa2, and Voa1/Voa2 causes significant reductions in the amount of endogenous dopamine stored in PC12 cells. Endogenous dopamine concentrations were measured using HPLC for Voa1 knockdown and its control (A, n = 9 each), Voa2 knockdown and its control (B, n = 25 each), or Voa1/Voa2 double knockdown and its control (D, n = 15 each). Values were normalized with the total protein concentrations of the samples. Error bars, SEM. (C) Sample picture of HPLC data showing differences in dopamine amounts between control and Voa1/Vao2 double-knockdown cells. Endogenous dopamine concentrations were measured using HPLC for Voa1/Voa2 double knockdown and its control (F, n = 6 each) from purified plasma membrane–associated DCV. Of this purified sample, 2.5 μg was immunoblotted with anti–secretogranin II rabbit polyclonal antibody (E).
We also found more dramatic (∼70%) reductions of dopamine concentrations in the DKD cells (0.29 ± 0.04 ng/μg protein, n = 15) compared with their control cells (0.91 ± 0.10 ng/μg protein, n = 15), which is highly significant (t28 = 5.53, p < 0.001; Figure 11, C and D). To ensure that the differences in dopamine concentrations between control and DKD cells using whole-cell lysates reflect the differences in dopamine concentrations inside dense-core vesicles, we measured dopamine concentrations from partially purified dense-core vesicles (Martin and Kowalchyk, 1997) of the control and DKD cells. We found similar reductions of dopamine concentrations in the partially purified dense-core vesicles of the DKD cells compared with those of the control cells (n = 6 each, t10 = 5.20, p < 0.001; Figure 11, E and F). Thus our results of HPLC measurements of dopamine concentrations (Figure 11) demonstrate that Voa1 is the critical isoform for the maintenance of endogenous catecholamine inside the cells, although Voa2 also appears to play a minor role.
Knockdown of Voa1 or double knockdown of Voa1/Voa2 has a very limited effect on Ca2+-dependent regulated secretion of transfected peptide
So far our results indicate that Voa1 and, to a lesser degree, Voa2 play critical and overlapping roles in acidification and transmitter uptake and storage in dense-core vesicles. Recently, an independent function was suggested for Voa1 in exocytotic and phagocytotic membrane fusion (Hiesinger et al., 2005; Peri and Nüsslein-Volhard, 2008; Williamson et al., 2010). Therefore, we examined the exocytotic abilities of Voa1 single-knockdown cells, as well as those of Voa1/Voa2 double-knockdown cells. To distinguish the effects of transmitter uptake defects from secretion defects in these knockdown cells, we examined the abilities of the knockdown cells to secrete a transfected peptide, NPY-fused human placental alkaline phosphatase (NPY-hPLAP). It should be noted that packaging of this peptide in dense-core vesicles does not steeply rely on proton gradient across the vesicular membrane produced by the V-ATPase. We found that both control and Voa1 knockdown cells exhibited robust secretion of transfected NPY-hPLAP upon high K+ stimulation (70 mM), reaching 25–35% secretion of the total amount of NPY (Figure 12A). There was no statistically significant difference in high K+–induced NPY-hPLAP secretion between control and Voa1KD cells (n = 8 each, t14 = 0.28, p = 0.79). There was also no statistical significant difference (n = 6 each, t10 = 1.49, p = 0.17; Figure 12B) in high K+–induced NPY-hPLAP secretion between control and DKD cells. Thus, Voa1 knockdown or Voa1/Voa2 double knockdown did not show significant effects on peptide secretion compared with their controls. In contrast, we found that double knockdown of Munc18-1/-2 (Han et al., 2009) resulted in a strong decrease in regulated secretion of the peptide (n = 6 each, t10 = 9.80, p < 0.001), indicating that secretion of the peptide is dependent on the Sec1/Munc18–family proteins (Figure 12C). Our data, which show that peptide release in Voa1 knockdown or Voa1/Voa2 double-knockdown cells is fairly normal compared with control cells, does not support the hypothesis that Voa1 functions critically for membrane fusion downstream of the SNARE proteins (Hiesinger et al., 2005). Our results suggest, rather, that the functions of Voa1 and Voa2 are limited to the acidification of secretory vesicles. This lack of effects on regulated secretion by Voa1/Voa2 double knockdown, however, might due to the presence of residual Voa1, Voa2, or Voa3.
We also verified whether NPY-fusion proteins are properly stored in dense-core vesicles in the absence of Voa1 and Voa2. For this purpose, we transfected DKD cells and their respective control with NPY-EmGFP expression plasmid and differentiated with NGF. We observed that NPY-EmGFP was enriched at the tip of the neurites and was clearly colocalized with secretogranin II in both control and DKD cells, suggesting that the sorting of NPY fusion protein remains largely unaffected by the knockdown of both Voa1 and Voa2 (Figure 12D; see Figure 4 for lower magnifications).
DISCUSSION
Although Voa proteins have been widely known to regulate the acidification of intracellular compartments, most previous studies, which involved down-regulating specific isoforms of Voa, failed to reveal defects in the acidification of intracellular organelles, particularly of secretory vesicles (Sun-Wada et al., 2006; Peri and Nüsslein-Volhard, 2008). For example, in Voa1 knockdown zebrafish, vesicular acidification in microglia, measured by LysoSensor, could take place to a similar degree as in the wild type (Peri and Nüsslein-Volhard, 2008). The failure to observe any acidification defects in secretory vesicles upon the down-regulation of Voa isoforms in these previous studies may be explained by two reasons. The first reason to account for the lack of observation of acidification defects is the overlapping function of the Voa isoforms, which is supported by our observations that the knockdown of Voa1 resulted in compensatory up-regulation of Voa2 (Figure 5A). As a consequence, the effects of knocking down Voa1 on vesicular acidification as measured using NPY-epHluorin were relatively mild (Figure 5C). However, once both Voa1 and Voa2 were removed, we found a more dramatic increase in the signal of NPY-epHluorin (Figure 7D). To our knowledge, our work is the first to examine the effects of double knockdown of Voa1 and Voa2 on the acidification of intracellular compartments. We also suspected that the acidification function of our Voa1 and Voa2 double-knockdown cells is partially compensated for by the up-regulation of Voa3 and/or Voa4. Data from a recently generated knock-in mouse of Voa3-GFP showed that Voa3 is enriched on late endosomes, lysosomes, and phagosomes (Sun-Wada et al., 2009). Our results using Voa3-EmGFP did not show this protein to be localized on lysosomes but instead on early endosomes in PC12 cells (Figure 3). In any case, both the results from Sun-Wada et al. (2009) and from the present study exclude the Voa3 isoform as having a major role in secretory vesicle acidification. Finally, our RT-PCR analyses showed that Voa4 signal was not present in PC12 cells, although it was clearly expressed in NRK cells (Figure 1D), supporting previous findings that this protein is specifically expressed in the kidney, inner ears, and ocular ciliary bodies (Oka et al., 2001a; Stover et al., 2002; Kawamura et al., 2010).
The second possible reason as to why acidification defects were not observed in previous studies involving the knockdown of the Voa subunit may be due to the choice of pH reporter used in those studies. Previous studies examined the acidification of intracellular compartments using LysoSensor or LysoTracker (Sun-Wada et al., 2006; Peri and Nüsslein-Volhard, 2008). When we stained acidic components of the cells using LysoSensor, we found that this dye stained a majority of the areas in PC12 cells. We suspect that relatively nonspecific staining of LysoSensor made it difficult to detect changes in acidification in these previous studies. In contrast to LysoSensor, the localization of NPY-epHluorin is likely restricted to secretory vesicles and the trans-Golgi network, similar to NPY-EmGFP, which showed good colocalization with secretogranin II (Figure 12D and Supplemental Figure S4). Thus, by using NPY-epHluorin as a sensor for secretory vesicle acidification together with FACS analysis, which objectively measures the signal at a single-cell level from a large number of cells, we were able to detect reliable increases in pH inside the secretory vesicles of Voa1 knockdown or Voa1/Voa2 double-knockdown cells but not of Voa2 knockdown cells (Figures 5C, 6B, 7D, and 8). Furthermore, the presence of knockdown-resistant Voa1 completely abolished the acidification defects induced by Voa1 or Voa1/Voa2 knockdown (Figure 9, D and E), excluding the possibility of off-target effects of the knockdown.
We also examined the effects of Voa knockdown on catecholamine uptake and storage and found that double knockdown of Voa1 and Voa2 caused significant reductions in acute uptake of [3H]NA (Figure 10C). We also found significant reductions in the concentrations of an endogenous catecholamine, dopamine, in Voa1 single-, Voa2 single- and Voa1/Voa2 double-knockdown cells (Figure 11). Thus the uptake and storage of catecholamines inside dense-core vesicles are indeed dependent on the proton gradient across the membrane of dense-core vesicles generated by proton pumps containing Voa1 and, to a lesser degree, Voa2.
Finally, we tested a recently proposed and highly controversial hypothesis that the Vo sector, in particular the Voa1 subunit, is involved in exocytotic fusion reactions without having a significant role in vesicular acidification (Peters et al., 2001; Hiesinger et al., 2005; Williamson et al., 2010). Despite the striking effects on vesicular acidification and catecholamine uptake/storage in our Voa1 knockdown or Voa1/Voa2 double-knockdown cells, we did not see major defects in the secretion of the transfected peptide, NPY-hPLAP, from these cells (Figure 12, A and B). Thus our results suggest that Voa1 is not critical for membrane fusion during exocytosis per se. Rather, our data indicate that Voa1 mutation may affect neurotransmitter release indirectly through defects in transmitter uptake/storage (Figures 10 and 11), which may underlie the uncoordinated movement phenotype in unc-32, a mutant of Voa1 homologue in C. elegans (Pujol et al., 2001), as well as the abnormal photoreceptor response in vha100-1 mutant Drosophila. Thus, our view is in contrast to the perspective proposed from the study of vha100-1 mutant in Drosophila (Hiesinger et al., 2005). In that study the authors did not see changes in the size of miniature EPSP in vha100-1 mutants, indicating that the uptake and storage of transmitters (glutamate) in synaptic vesicles is normal and that vesicular acidification is also normal, leading the authors to suggest a direct role of vha100-1 in exocytosis (Hiesinger et al., 2005). However, our results do not completely exclude the potential role of Voa1 or other isoforms in regulated exocytosis because of the presence of residual Voa1, Voa2, or Voa3 in DKD cells.
Due to the fundamental importance of V-ATPases in various cellular processes, knockout studies of the proteins comprising V-ATPases have been hampered by embryonic lethality. Without a proper mammalian system to study the function of the V-ATPase, previous studies aiming to understand the structure/function relationship of the V-ATPases relied heavily on the use of yeast V-ATPase as a model system (Hirata et al., 2003; Kawasaki-Nishi et al., 2003; Shao and Forgac, 2004; Wang et al., 2004b, 2007; Inoue et al., 2005; Jefferies et al., 2008; Qi and Forgac, 2008). One major reason for using yeast as a model system in studying the function of V-ATPase is that the structure of the V-ATPase is highly conserved from yeast to human. Nevertheless, there are a few differences between mammalian V-ATPase and its yeast counterpart. For instance, the Vo sector of mammalian V-ATPase does not contain the Voc′ subunit, which is a critical component of the yeast V-ATPase. Instead, mammalian V-ATPase contains an accessory protein, Ac45, that has no homologue in yeast (Supek et al., 1994; Getlawi et al., 1996). Thus the yeast model may not be an ideal model in which to study the function of V-ATPase in higher organisms. The results from our work suggest that PC12 cells may serve this purpose. Our EmGFP-fusion constructs of Voa1 and Voa2 together with the Voa1, Voa2–stable knockdown cells will provide unique opportunities for further structure/function analyses of Voa isoforms, as well as of Ac45.
MATERIALS AND METHODS
General materials
Parental pLKO-puro plasmid for lentivirus-mediated knockdown was purchased from Sigma-Aldrich (Oakville, ON, Canada). pLKO-neo plasmid was generated by replacing the puromycin resistant gene with the neomycin resistant gene (Han et al., 2009). Parental pLVX-IRES-puro plasmid for the lentivirus-mediated expression of proteins was purchased from Clontech (Mountain View, CA). The plasmids pCMV8.74 and pMD2G used to generate recombinant lentivirus were kind gifts from Tomoyuki Mashimo (University of Texas Southwestern Medical Center at Dallas, Dallas, TX). We obtained rabbit polyclonal antibodies against Voa1 from Synaptic Systems (Göttingen, Germany) and Santa Cruz Biotechnology (Santa Cruz, CA), GFP from Invitrogen (Carlsbad, CA), and calnexin from Sigma-Aldrich; mouse polyclonal antibodies against Voa2 from Abnova (Taipei, Taiwan), goat polyclonal antibodies against EEA1 (and mouse monoclonal antibodies against Vod1 (clone 34-Z) from Santa Cruz Biotechnology, syntaxin-1A/1B (clone HPC-1; Barnstable et al., 1985) from Sigma-Aldrich, Ac45 (clone 3A2) from Abnova, SNAP-25 (clone SMI 81) from Covance (Princeton, NJ), GM130 (clone 35) from BD Biosciences (Mississauga, ON, Canada), LAMP1 (clone LY1C6) from StreeMarq Biosciences (Victoria, BC, Canada), and DsRed from Clontech. Mouse monoclonal anti–synaptotagmin-1 (Cl41.1), rabbit polyclonal anti-Voa2 antibody (Peng et al., 1999), and anti-VCP/p97 antibody (Sugita and Südhof, 2000) were kind gifts from Reinhard Jahn (Max Planck Institute for Biophysical Chemistry, Göttingen, Germany), Xiao-Song Xie (University of Texas Southwestern Medical Center at Dallas), and Thomas Südhof (Stanford University, Stanford, CA), respectively.
Lentivirus-mediated stable expression of fluorescence tagged Voa1, Voa2, and Voa3 in PC12 cells
We also generated the lentivirus-mediated expression constructs of Voa1, Voa2, or Voa3 fused with Emerald GFP (EmGFP) or mCherry for the purpose of stably expressing these recombinant proteins in PC12 cells. We first amplified the cDNAs of Emerald GFP and mCherry by PCR from pCMV-Emerald GFP and pcDNA3.1-myc/HisA-mCherry (a kind gift from Herbert Gaisano, University of Toronto, Toronto, ON, Canada), respectively. The PCR products were then digested with BglII and BamHI and ligated into the BamHI site of pLVX-IRES-blast to generate pLVX-EmGFP-IRES-blast and pLVX-mCherry-IRES-blast. Human cDNA of Voa1 (IMAGE clone ID 5195776), mouse cDNA of Voa2 (IMAGE clone ID 3670722), and human cDNA of Voa3 (IMAGE clone ID), obtained from the ATCC (Manassas, VA) or Open Biosystems (Huntsville, AL), were amplified by PCR and ligated to EcoRI/XbaI site (Voa1, Voa2) or XhoI/XbaI site (Voa3) of pLVX-EmGFP-IRES-blast and pLVX-mCherry-IRES-blast, respectively. These recombinant expression plasmids were cotransfected with pCMV8.74 and pMD2G into HEK-293FT cells to generate recombinant lentiviruses that express Voa1, Voa2, or Voa3 fused with EmGFP or mCherry. The infected cells were eventually selected with blasticidin (5 μg/ml) to obtain populations of cells that stably express the proteins of interest.
RT-PCR
Total RNA was extracted from wild-type or knockdown and their control PC12 cells, as well as from NRK cells (CRL-6509, ATCC) using RNeasy (Qiagen, Valencia, CA). RT-PCR was performed using a One Step RT-PCR Kit (Qiagen) in a reaction involving denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and elongation at 72°C for 1 min. The number of cycles was 30. The following primers were used: Voa1, sense, TCTCCACCCATTCAGAGGAC; antisense, CCTTCCATGATCAGCAGGAT; product size, 301 base pairs. Voa3, sense, GCTTCCACCTTGGAGAACAG; antisense, CCCAGAGACGCAAGTAGGAG; product size: 169 base pairs. Voa4, sense, CATGGGCATCTTCTCCATCT; antisense, TTGAAGCCAGGTTCCAAATC; product size, 230 base pairs. Glyceraldehyde-3-phosphate dehydrogenase, sense, CTCATGACCACAGTCCATGC; antisense: TTCAGCTCTGGGATGACCTT; product size: 155 base pairs. PCR products were electrophoresed on a 1.5% agarose gel containing ethidium bromide.
Cell preparation for confocal immunofluorescence microscopy
Confocal immunofluorescence microscopy was performed for wild-type PC12 cells, PC12 cells that stably expressed Voa1-EmGFP, Voa2-EmGFP, or Voa3-EmGFP, as well as for control and DKD cells that were transfected with NPY-EmGFP. Sterilized circular glass coverslips (0.25 mm width, 1.8 cm diameter) were placed in 2.2-cm wells within 12-well cell culture plates. The coverslips were then coated for 30 min with poly-d-lysine (0.1 mg/ml) at room temperature. Cells were allowed to adhere to the coverslips overnight and then differentiated on the coverslips for 3–4 d in DMEM containing 100 ng/ml nerve growth factor (NGF; Sigma-Aldrich), 1% horse serum, 1% calf serum, and penicillin/streptomycin. After the differentiation period cells were washed with PBS, fixed for 15 min with PBS containing 4% paraformaldehyde, and permeabilized with PBS containing 0.2% Triton X-100 for 5 min. Nonspecific sites were blocked for 1 h at room temperature in blocking buffer, PBS containing 0.3% bovine serum albumin (BSA). For PC12 cells stably expressing Voa1-EmGFP, Voa2-EmGFP, or Voa3-EmGFP primary antibodies against GFP (rabbit polyclonal, 1:1000 dilution) and synaptotagmin-1 (Cl41.1 mouse monoclonal, 1:1000 dilution), GM130 (mouse monoclonal, 1:500 dilution), EEA1 (goat polyclonal, 1:1000 dilution), or LAMP1 (mouse monoclonal, 1:1000) in blocking buffer were then applied for 1 h at room temperature. Following three washes in blocking buffer, Alexa 488–conjugated goat anti–rabbit antibody (1:1000 dilution) and Rhodamine Red-X–conjugated goat anti–mouse antibody (1:1000 dilution) or Alexa 568–conjugated donkey anti–goat antibody (1:1000 dilution) in blocking buffer were applied to the samples for 1 h in the dark at room temperature. Samples were washed again three times in blocking buffer before being mounted on microscope slides. Similar steps were taken in preparing control and DKD cells that were transfected with NPY-EmGFP. In these cells, however, rabbit anti–secretogranin II polyclonal antibody (SgII, 1:1000 dilution) and Alexa 568–conjugated goat anti–rabbit antibody (1:1000 dilution) were used for primary and secondary staining, respectively. For the localization of endogenous Voa1 and Voa2 in wild-type PC12 cells similar steps were taken in preparing these cells up to the cell-fixing stage with 4% paraformaldehyde. Subsequently, permeabilization of the cells was achieved by incubating for 15 min with 0.1% SDS, 0.4% saponin, 1% normal goat serum (NGS), and 1% BSA in PBS. Primary antibodies against Voa1 (rabbit polyclonal, 1:1000 dilution) or Voa2 (rabbit polyclonal, 1:1000 dilution) and synaptotagmin-1 or GM130 in PBS containing 0.4% saponin, 1% NGS, and 1% BSA were applied overnight to the permeabilized cells. The next day cells were washed three times (10 min each time) with PSB containing 0.4% saponin, 1% NGS, and 1% BSA. Secondary antibodies against Voa1 or Voa2 (Alexa 488–conjugated goat anti–rabbit antibody, 1:1000 dilution) and synaptotagmin-1 or GM130 (Rhodamine Red-X–conjugated goat anti–mouse antibody, 1:1000 dilution) in PBS containing 0.4% saponin, 1% NGS, and 1% BSA were applied for 1 h in the dark. Cells were then washed three times (10 min each time). All samples were mounted onto microscope slides using Fluoromount-G reagent (SouthernBiotech, Birmingham, AL). Immunofluorescence staining was recorded with a Zeiss (Jena, Germany) laser confocal scanning microscope (LSM 510) with an oil immersion objective lens (63×).
Construction of Voa1 and Voa2 knockdown plasmids
To knock down the Voa1 gene, we targeted the 21-nucleotide sequence GCTGCTTATTGTTGTGTCAGT (residues 61–81, Voa1KD) of rat Voa1. To knock down the Voa2 gene, we targeted the 21-nucleotide sequence GGTGGAGCTCAGAGAAGTCAC (residues 315–335, Voa2KD) of rat Voa2. We used CTCGAG as a linker sequence. Fifty-eight–base pair oligos containing sense and antisense of the target sequences were annealed and subcloned into the AgeI-EcoRI sites of pLKO-puro or pLKO-neo, generating the VOa1 knockdown plasmid (pLKO-puro-Voa1KD) and Voa2 knockdown plasmids (pLKO-puro-Voa2KD, pLKO-neo-Voa2KD). Inserted sequences were verified by sequencing.
Construction of plasmids that transiently express neuropeptide Y fused with super ecliptic pHluorin
The plasmids to express NPY fused with epHluorin, rpHluorin, and EmGFP were generated using pCMV5 as the parental plasmid. cDNAs of super ecliptic pHluorin and ratiometric pHluorin were amplified by PCR from pGM6 and pGM1 (kind gifts from Gero Miesenböck, University of Oxford, Oxford, United Kingdom), respectively. The PCR products containing epHluorin and rpHluorin were digested with ClaI and XbaI and ligated to the same site of pCMV5, generating pCMV-epHluorin and pCMV-rpHluorin, respectively. pCMV-EmGFP was a kind gift from Weiping Han (University of Texas Southwestern Medical Center at Dallas). cDNA of NPY was amplified by PCR on pVenus-N1-NPY (a kind gift from Atsushi Miyawaki, Riken, Japan) and digested with BglII and ClaI and ligated to the same site of pCMV-epHluorin, pCMV-rpHluorin, or pCMV-EmGFP, generating pCMV-NPY-epHluorin, pCMV-NPY-rpHluorin, and pCMV-NPY-EmGFP, respectively.
Isolation of stable Voa1 and Voa2 single- and Voa1/Voa2 double-knockdown PC12 cells
PC12 cells were maintained in DMEM (Invitrogen, or HyClone, Logan, UT) containing 5% calf serum, 5% horse serum (both from HyClone), penicillin (100 U/ml)/streptomycin (0.1 mg/ml; Sigma-Aldrich; Wang et al., 2004a, 2005; Fujita et al., 2007; Li et al., 2007; Arunachalam et al., 2008), and, in some cases, 250 ng/ml amphotericin B (Sigma-Aldrich) and 1.25 μg/ml plasmocin (InvivoGen, San Diego, CA). To knockdown Voa1, we generated recombinant lentiviruses by cotransfecting each of pLKO-puro (control, 9 μg) or pLKO-puro-Voa1KD (9 μg) with two other plasmids (pMD2G, 3 μg; pCMV-dR8.74, 4.8 μg) into HEK-293FT (Invitrogen) cells using 40 μl of polyethylenimine (1.2 mg/ml, pH 7.2). These recombinant lentiviruses were then applied to wild-type PC12 cells, and the infected host cells were selected using puromycin (2.5 μg/ml). For each recombinant virus, we isolated a pool of heterogeneous cells that had survived in puromycin-containing medium over a period of 2 wk. These surviving cells were then subjected to immunoblot analysis using anti-Voa1 antibody. We confirmed the strong knockdown of Voa1 in the surviving cells, which were then grown, frozen, and kept in a liquid nitrogen tank until future use. We found that the cells maintain their phenotypes for 2–3 mo.
The efficacy of the Voa2 knockdown was not as strong as that of Voa1 knockdown. To maximize the knockdown level, we incorporated the knockdown sequence of Voa2 into both pLKO-puro and pLKO-neo plasmids, respectively, generating pLKO-puro-Voa2KD and pLKO-neo-Voa2KD. We infected PC12 cells successively with the recombinant viruses generated from pLKO-puro-Voa2KD and pLKO-neo-Voa2KD, and the infected PC12 cells were successively selected by puromycin and G418 (0.7 mg/ml). The controls for these knockdown cells are the PC12 cells infected with lentiviruses generated from pLKO-puro and pLKO-neo and successively selected by puromycin and G418. Our rationale for using two plasmids to target the same sequence of Voa2 is our expectation that having at least two copies of the knockdown sequences incorporated into the host PC12 cells may have stronger and more stable knockdown effects than having just a single copy of the knockdown sequence.
To generate the stable Voa1 and Voa2 DKD cells, we sequentially infected PC12 cells with lentiviruses generated from pLKO-puro-Voa1KD and pLKO-neo-Voa2KD and selected them with puromycin and G418.
Transfection of the reporter constructs NPY-epHluorin and NPY-EmGFP and subsequent FACS analysis of their fluorescence signals
Control and Voa knockdown cells were collected from 10-cm dishes that were 70–90% confluent. These cells were then transfected with 15 μg of pCMV5, pCMV-NPY-epHluorin, or pCMV-NPY-EmGFP by electroporation. Immediately after transfection the cells were placed into new 10-cm dishes and kept in growth medium (DMEM containing 5%, calf serum, 5% equine serum, and penicillin [100 U/ml]/streptomycin [0.1 mg/ml]). The transfected cells were replated onto six-well plates 3 d after transfection. At 4–6 d after transfection, the cells from each well were harvested and resuspended in 300–500 μl of various buffered solutions. Experiments to test acidification defects were performed in PBS containing 1% calf serum, 1% equine serum, and 10 μg/ml of propidium iodide (PI). Experiments that tested the effects of different concentrations of NH4Cl on the NPY-epHluorin were performed in HEPES-buffered saline (pH 7.4) containing 15 mM HEPES (pH 7.4), 5.6 mM KCl, 140 mM NaCl, 2.2 mM CaCl2, 0.5 mM MgCl, 5.6 mM glucose, and 10 μg/ml PI. NH4Cl applications were performed with various (20, 50, 100 mM) concentrations of NH4Cl in substitution of equal concentrations of NaCl. Experiments to calibrate the pH dependence of NPY-epHluorin were performed in either MES-buffered saline (adjusted to pH 5.5, 6.0, and 6.5) or HEPES-buffered saline (adjusted to pH 7.0, and 7.5) containing 15 mM HEPES or MES, 140 mM KCl, 0.5 mM MgCl2, 0.2 mM ethylene glycol tetraacetic acid (EGTA), 5 μg/ml nigericin, 5 μM monensin, and 10 μg/ml PI. In all experiments samples were triturated 10 times with a 1-ml pipette and then passed through a 35-μm nylon mesh strainer (Falcon, 352235; BD Biosciences, San Diego, CA). For each sample, 104 PI negative cells (FL3 channel) were analyzed for GFP intensity (FL1 channel) by FACSCalibur (BD Biosciences). The average fluorescence value of the cells transfected with pCMV5 was subtracted from the average values of the cells transfected with pCMV-NPY-epHluorin.
Lentivirus-mediated expression of knockdown-resistant human Voa1 in PC12 cells
The puromycin-resistance gene of pLVX-IRES-puro (Clontech) was replaced with the blasticidin-resistance gene, generating pLVX-IRES-blast. The cDNA sequence of Voa1, to which the shRNA-mediated knockdown was targeted, is identical between rat and human. We introduced eight SNMs (GCCTACTGCTGCGTGTCG; underlines indicate SNMs) within the target sequence in the human Voa1 gene (IMAGE clone ID 5195776) to protect the mRNA transcripts transcribed from the VOa1 expression plasmid from being degraded by the shRNA-mediated knockdown of Voa1. The human Voa1 with SNM was subcloned into the EcoRI/XbaI site of pLVX-IRES-blast.
Measurements of [3H]NA uptake into PC12 cells
We used an established method to measure ATP-dependent uptake of [3H]NA into dense-core vesicles in PC12 cells (Ahnert-Hilger et al., 1998; Brunk et al., 2009) with a small modification; the cell's plasma membrane was mechanically permeabilized using a ball homogenizer instead of using the pharmacological agent streptolysin O. PC12 cells were grown on 10-cm dishes with 70–80% confluency and were washed with physiological saline solution, harvested in K-glutamate buffer (20 mM HEPES, pH 7.1, 140 mM potassium glutamate, 2 mM EGTA, 1 mM MgCl2), and mechanically permeabilized with a ball homogenizer (Wang et al., 2000, 2004a; Fujita et al., 2007; Li et al., 2007). The permeabilized PC12 cells were washed once with K-glutamate buffer and incubated with the same buffer containing 50 nM [3H]NA (GE Healthcare, Montreal, QC, Canada), 450 nM unlabeled NA (Sigma-Aldrich), and 0.5 mM ascorbic acid in the presence of 2 mM MgATP or 2 mM MgCl2 at 37°C for 60 min. Reaction was halted by adding 1 ml of ice-cold K-glutamate buffer. Samples were then centrifuged at 4°C for 3 min at 14,000 rpm and the supernatants removed. An extra wash with 500 μl of ice-cold K-glutamate buffer was performed. After centrifugation at 4°C for 3 min at 14,000 rpm the pelleted samples were treated with 200 μl of 0.5% Triton X-100 (in deionized water) and subsequently collected for liquid scintillation counting. The amount of [3H]NA taken up into the permeabilized cells was normalized using the respective protein concentrations of the cells. Each assay was performed in quadruplicate.
Measurements of endogenous catecholamines from PC12 cells by HPLC
Concentrations of catecholamines were measured using an HPLC system that consists of a delivery pump (model HP1100; Agilent Technologies, Santa Clara, CA), a reversed-phase analytical column (ZORBAX Eclipse XDB-C8, 150 × 4.6 mm inner diameter, 5 μm; Agilent Technologies), a degasser, and a fluorescence detector. We followed the method developed by Lakshmana and Raju (1997) that used isocratic assay without derivatization. The mobile phase consisted of sodium acetate (0.02 M), methanol (16%), heptane sulfonic acid (0.055%), EDTA (0.2 mM), and dibutylamine (0.01% vol/vol). The solution was adjusted to pH 3.92 with o-phosphoric acid. The flow rate was set to 0.9 ml/min. As standards, noradrenaline, adrenaline, and dopamine were dissolved in 0.1 M perchloric acid (PCA) at the concentration of 250 ng/ml each, and the volume of injection was set to 10 μl. Good detection and separation of these compounds were first established at the amount of 2.5 ng/10 μl sample volume (Supplemental Figure S3). For whole-cell lysate measurements of the concentrations of catecholamines, PC12 cells were harvested from 10-cm dishes in 1 ml of Hank's buffer with 1 mM EDTA and supplemented with 9 ml of the growth medium. The cells were pelleted by centrifugation, washed once with 1 ml of physiological saline solution, and homogenized in 200 μl of 0.1 M PCA. Insoluble materials were removed by centrifugation (three times), and 10 μl of the cleared solution was applied to HPLC. We detected the clear peak of dopamine (in the range of 20–160 ng/10 μl sample volume). This result confirms that the primary catecholamine in PC12 cells is dopamine (Fujita et al., 2007). The concentrations of these catecholamines were normalized by the total protein concentrations (in the range of 10–30 μg/10 μl sample volume) of each sample of PC12 cells. To measure dopamine concentrations from partially purified dense-core vesicles, we followed the purification protocol established by Martin and Kowalchyk (1997). Briefly, PC12 cells in KGlu buffer (Martin and Kowalchyk, 1997) were permeabilized by passing them 10 times through a ball homogenizer (diameter, 0.2527 inches). The cell homogenate was then centrifuged at 800 × g to pellet the nucleus and other large cell pieces. The supernatant was then centrifuged at 5000 × g to pellet plasma membrane-associated dense-core vesicles. After dissolution of this pellet in 0.1 M PCA and the insoluble materials cleared, 50 μl of this sample was loaded for HPLC analyses. In all cases, the total protein concentration was measured by the Bradford method using bovine serum albumin as a standard.
NPY-hPLAP secretion assay from PC12 cells
PC12 cells (Voa1KD, Voa1/a2DKD, Munc18-1/-2 double-knockdown cells, and their respective controls) at 70–80% confluency in 10-cm dishes were transfected with 15 μg of a reporter plasmid, pCMV-NPY-hPLAP (Fujita et al., 2007; Li et al., 2007; Arunachalam et al., 2008), using electroporation. After 4 d, the cells were harvested and replated in 24-well plates. At 8–9 d after electroporation, the plated cells were washed once with physiological saline solution (PSS), and NPY-hPLAP secretion was stimulated with 200 μl of PSS containing 145 mM NaCl, 5.6 mM KCl, 2.2 mM CaCl2, 0.5 mM MgCl2, 5.6 mM glucose, and 15 mM HEPES, pH 7.4, or high-K+ PSS containing 81 mM NaCl and 70 mM KCl. Secretion was terminated after a 20-min incubation period at 37°C by chilling to 0°C, and samples were centrifuged at 4°C for 3 min. Supernatants were removed, and the pellets were solubilized in 200 μl of PSS containing 0.1% Triton X-100. The amounts of NPY-hPLAP secreted into the medium and retained in the cells were measured by the Phospha-Light Reporter Gene Assay System (Applied Biosystems, Foster City, CA). We treated the samples at 65°C for 30 min to inactivate nonplacental alkaline phosphatases and assayed an aliquot (10 μl) for placental alkaline phosphatase activity with the kit. The total volume of the assay was 120 μl. After 5–10 min, chemiluminescence was quantified by an FB12 luminometer (Berthold Detection Systems, Zylux, Oak Ridge, TN).
Supplementary Material
Acknowledgments
We thank W. Han, H. Gaisano, R. Jahn, T. Mashimo A. Miyawaki, G. Miesenböck, X. Xie, T. Südhof, W. Trimble, S. Grinstein, and A. Volchuk for providing reagents for this study. We thank H. Gaisano, W. Trimble, and S. Grinstein for critically reading the manuscript, as well as for advice. We also thank Nan Chang (Toronto Western Research Institute, Toronto, ON, Canada) for instructions on flow cytometry. This research was supported by the Heart and Stroke Foundation (NA6217, T6700), the Canada Research Chair Program, and the Canadian Institute of Health Research (MOP-57825 and MOP-93665).
Abbreviations used:
- BSA
bovine serum albumin
- FACS
fluorescence activated cell sorting
- GFP
green fluorescent protein
- NGF
nerve growth factor
- NGS
normal goat serum
- PBS
phosphate-buffered saline
- PCA
perchloric acid
- PSS
physiological saline solution
- RT-PCR
reverse transcription-PCR
- shRNA
short hairpin RNA
- SNARE
soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- SNM
silent nucleotide mutation
- V-ATPase
vacuolar H+-ATPase
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-02-0155) on July 27, 2011. The authors declare no conflict of interest.
REFERENCES
- Ahnert-Hilger G, Nürnberg B, Exner T, Schäfer T, Jahn R. The heterotrimeric G protein Go2 regulates catecholamine uptake by secretory vesicles. EMBO J. 1998;17:406–413. doi: 10.1093/emboj/17.2.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amara S, Kuhar M. Neurotransmitter transporters: recent progress. Annu Rev Neurosci. 1993;16:73–93. doi: 10.1146/annurev.ne.16.030193.000445. [DOI] [PubMed] [Google Scholar]
- Arunachalam L, et al. Munc18-1 is critical for plasma membrane localization of syntaxin1 but not of SNAP-25 in PC12 cells. Mol Biol Cell. 2008;19:722–734. doi: 10.1091/mbc.E07-07-0662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnstable CJ, Hofstein R, Akagawa K. A marker of early amacrine cell development in rat retina. Dev Brain Res. 1985;20:286–290. doi: 10.1016/0165-3806(85)90116-6. [DOI] [PubMed] [Google Scholar]
- Bayer M, Reese C, Buhler S, Peters C, Mayer A. Vacuole membrane fusion: V0 functions after trans-SNARE pairing and is coupled to the Ca2+-releasing channel. J Cell Biol. 2003;162:211–222. doi: 10.1083/jcb.200212004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunk I, Blex C, Speidel D, Brose N, Ahnert-Hilger G. Ca2+-dependent activator proteins of secretion promote vesicular monoamine uptake. J Biol Chem. 2009;284:1050–1056. doi: 10.1074/jbc.M805328200. [DOI] [PubMed] [Google Scholar]
- Demaurex N, Furuya W, D'Souza S, Bonifacino JS, Grinstein S. Mechanism of acidification of the trans-Golgi network (TGN). In situ measurements of pH using retrieval of TGN38 and furin from the cell surface. J Biol Chem. 1998;273:2044–2051. doi: 10.1074/jbc.273.4.2044. [DOI] [PubMed] [Google Scholar]
- Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007;8:917–929. doi: 10.1038/nrm2272. [DOI] [PubMed] [Google Scholar]
- Frattini A, et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet. 2000;25:343–346. doi: 10.1038/77131. [DOI] [PubMed] [Google Scholar]
- Fremeau RJ, Voglmaier S, Seal R, Edwards R. VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends Neurosci. 2004;27:98–103. doi: 10.1016/j.tins.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Fujita Y, et al. Ca2+-dependent activator protein for secretion 1 is critical for constitutive and regulated exocytosis but not for loading of transmitters into dense core vesicles. J Biol Chem. 2007;282:21392–21403. doi: 10.1074/jbc.M703699200. [DOI] [PubMed] [Google Scholar]
- Gasnier B. The loading of neurotransmitters into synaptic vesicles. Biochimie. 2000;82:327–337. doi: 10.1016/s0300-9084(00)00221-2. [DOI] [PubMed] [Google Scholar]
- Getlawi F, Laslop A, Schägger H, Ludwig J, Haywood J, Apps D. Chromaffin granule membrane glycoprotein IV is identical with Ac45, a membrane-integral subunit of the granule's H(+)-ATPase. Neurosci Lett. 1996;219:13–16. doi: 10.1016/s0304-3940(96)13151-7. [DOI] [PubMed] [Google Scholar]
- Granseth B, Odermatt B, Royle S, Lagnado L. Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron. 2006;51:773–786. doi: 10.1016/j.neuron.2006.08.029. [DOI] [PubMed] [Google Scholar]
- Greene L, Tischler A. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, et al. Rescue of Munc18-1 and -2 double knockdown reveals the essential functions of interaction between Munc18 and closed syntaxin in PC12 cells. Mol Biol Cell. 2009;20:4962–4975. doi: 10.1091/mbc.E09-08-0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiesinger P, et al. The v-ATPase V0 subunit a1 is required for a late step in synaptic vesicle exocytosis in Drosophila. Cell. 2005;121:607–620. doi: 10.1016/j.cell.2005.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton A, Bond S, Forgac M. V-ATPase functions in normal and disease processes. Pflugers Arch. 2009;457:589–598. doi: 10.1007/s00424-007-0382-4. [DOI] [PubMed] [Google Scholar]
- Hirata T, Iwamoto-Kihara A, Sun-Wada G, Okajima T, Wada Y, Futai M. Subunit rotation of vacuolar-type proton pumping ATPase: relative rotation of the G and C subunits. J Biol Chem. 2003;278:23714–23719. doi: 10.1074/jbc.M302756200. [DOI] [PubMed] [Google Scholar]
- Hu C, Ahmed M, Melia T, Söllner T, Mayer T, Rothman J. Fusion of cells by flipped SNAREs. Science. 2003;300:1745–1749. doi: 10.1126/science.1084909. [DOI] [PubMed] [Google Scholar]
- Hucthagowder V, et al. Loss-of-function mutations in ATP6V0A2 impair vesicular trafficking, tropoelastin secretion and cell survival. Hum Mol Genet. 2009;18:2149–2165. doi: 10.1093/hmg/ddp148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Wang Y, Jefferies K, Qi J, Hinton A, Forgac M. Structure and regulation of the V-ATPases. J Bioenerg Biomembr. 2005;37:393–398. doi: 10.1007/s10863-005-9478-8. [DOI] [PubMed] [Google Scholar]
- Jahn R. Principles of exocytosis and membrane fusion. Ann NY Acad Sci. 2004;1014:170–178. doi: 10.1196/annals.1294.018. [DOI] [PubMed] [Google Scholar]
- Jahn R, Lang T, Südhof T. Membrane fusion. Cell. 2003;112:519–533. doi: 10.1016/s0092-8674(03)00112-0. [DOI] [PubMed] [Google Scholar]
- Jefferies K, Cipriano D, Forgac M. Function, structure and regulation of the vacuolar (H+)-ATPases. Arch Biochem Biophys. 2008;476:33–42. doi: 10.1016/j.abb.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura N, Tabata H, Sun-Wada GH, Wada Y. Optic nerve compression and retinal degeneration in Tcirg1 mutant mice lacking the vacuolar-type H-ATPase a3 subunit. PLoS One. 2010;5:e12086. doi: 10.1371/journal.pone.0012086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki-Nishi S, Nishi T, Forgac M. Interacting helical surfaces of the transmembrane segments of subunits a and c′ of the yeast V-ATPase defined by disulfide-mediated cross-linking. J Biol Chem. 2003;278:41908–41913. doi: 10.1074/jbc.M308026200. [DOI] [PubMed] [Google Scholar]
- Kim JH, Lingwood CA, Williams DB, Furuya W, Manolson MF, Grinstein S. Dynamic measurement of the pH of the Golgi complex in living cells using retrograde transport of the verotoxin receptor. J Cell Biol. 1996;134:1387–1399. doi: 10.1083/jcb.134.6.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Ryan TA. CDK5 serves as a major control point in neurotransmitter release. Neuron. 2010;67:797–809. doi: 10.1016/j.neuron.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornak U, et al. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat Genet. 2008;40:32–34. doi: 10.1038/ng.2007.45. [DOI] [PubMed] [Google Scholar]
- Kornak U, Schulz A, Friedrich W, Uhlhaas S, Kremens B, Voit T, Hasan C, Bode U, Jentsch T, Kubisch C. Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum Mol Genet. 2000;9:2059–2063. doi: 10.1093/hmg/9.13.2059. [DOI] [PubMed] [Google Scholar]
- Lakshmana MK, Raju TR. An isocratic assay for norepinephrine, dopamine, and 5-hydroxytryptamine using their native fluorescence by high-performance liquid chromatography with fluorescence detection in discrete brain areas of rat. Anal Biochem. 1997;246:166–170. doi: 10.1006/abio.1996.9997. [DOI] [PubMed] [Google Scholar]
- Li G, et al. RalA and RalB function as the critical GTP sensors for GTP-dependent exocytosis. J Neurosci. 2007;27:190–202. doi: 10.1523/JNEUROSCI.2537-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chen W, Liang Y, Li E, Stashenko P. Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat Genet. 1999;23:447–451. doi: 10.1038/70563. [DOI] [PubMed] [Google Scholar]
- Marshansky V, Futai M. The V-type H+-ATPase in vesicular trafficking: targeting, regulation and function. Curr Opin Cell Biol. 2008;20:415–426. doi: 10.1016/j.ceb.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TF, Kowalchyk JA. Docked secretory vesicles undergo Ca2+-activated exocytosis in a cell-free system. J Biol Chem. 1997;272:14447–14453. doi: 10.1074/jbc.272.22.14447. [DOI] [PubMed] [Google Scholar]
- Masson J, Sagné C, Hamon M, El Mestikawy S. Neurotransmitter transporters in the central nervous system. Pharmacol Rev. 1999;51:439–464. [PubMed] [Google Scholar]
- Miesenböck G, De Angelis D, Rothman J. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Moriyama Y, Futai M. H(+)-ATPase, a primary pump for accumulation of neurotransmitters, is a major constituent of brain synaptic vesicles. Biochem Biophys Res Commun. 1990a;173:443–448. doi: 10.1016/s0006-291x(05)81078-2. [DOI] [PubMed] [Google Scholar]
- Moriyama Y, Futai M. Presence of 5-hydroxytryptamine (serotonin) transport coupled with vacuolar-type H(+)-ATPase in neurosecretory granules from bovine posterior pituitary. J Biol Chem. 1990b;265:9165–9169. [PubMed] [Google Scholar]
- Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis T, Warren G. Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol. 1995;131:1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi T, Forgac M. Molecular cloning and expression of three isoforms of the 100-kDa a subunit of the mouse vacuolar proton-translocating ATPase. J Biol Chem. 2000;275:6824–6830. doi: 10.1074/jbc.275.10.6824. [DOI] [PubMed] [Google Scholar]
- Oka T, Murata Y, Namba M, Yoshimizu T, Toyomura T, Yamamoto A, Sun-Wada G, Hamasaki N, Wada Y, Futai M. a4, a unique kidney-specific isoform of mouse vacuolar H+-ATPase subunit a. J Biol Chem. 2001a;276:40050–40054. doi: 10.1074/jbc.M106488200. [DOI] [PubMed] [Google Scholar]
- Oka T, Toyomura T, Honjo K, Wada Y, Futai M. Four subunit a isoforms of Caenorhabditis elegans vacuolar H+-ATPaseCell-specific expression during development. J Biol Chem. 2001b;276:33079–33085. doi: 10.1074/jbc.M101652200. [DOI] [PubMed] [Google Scholar]
- Peng S, Crider B, Xie X, Stone D. Alternative mRNA splicing generates tissue-specific isoforms of 116-kDa polypeptide of vacuolar proton pump. J Biol Chem. 1994;269:17262–17266. [PubMed] [Google Scholar]
- Peng SB, Li X, Crider BP, Zhou Z, Andersen P, Tsai SJ, Xie XS, Stone DK. Identification and reconstitution of an isoform of the 116-kDa subunit of the vacuolar proton translocating ATPase. J Biol Chem. 1999;274:2549–2555. doi: 10.1074/jbc.274.4.2549. [DOI] [PubMed] [Google Scholar]
- Peri F, Nüsslein-Volhard C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. 2008;133:916–927. doi: 10.1016/j.cell.2008.04.037. [DOI] [PubMed] [Google Scholar]
- Perin M, Fried V, Stone D, Xie X, Südhof T. Structure of the 116-kDa polypeptide of the clathrin-coated vesicle/synaptic vesicle proton pump. J Biol Chem. 1991;266:3877–3881. [PubMed] [Google Scholar]
- Peters C, Bayer M, Bühler S, Andersen J, Mann M, Mayer A. Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature. 2001;409:581–588. doi: 10.1038/35054500. [DOI] [PubMed] [Google Scholar]
- Peters J, Walsh M, Franke W. An abundant and ubiquitous homo-oligomeric ring-shaped ATPase particle related to the putative vesicle fusion proteins Sec18p and NSF. EMBO J. 1990;9:1757–1767. doi: 10.1002/j.1460-2075.1990.tb08300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol N, Bonnerot C, Ewbank J, Kohara Y, Thierry-Mieg D. The Caenorhabditis elegans unc-32 gene encodes alternative forms of a vacuolar ATPase a subunit. J Biol Chem. 2001;276:11913–11921. doi: 10.1074/jbc.M009451200. [DOI] [PubMed] [Google Scholar]
- Qi J, Forgac M. Function and subunit interactions of the N-terminal domain of subunit a (Vph1p) of the yeast V-ATPase. J Biol Chem. 2008;283:19274–19282. doi: 10.1074/jbc.M802442200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saroussi S, Nelson N. Vacuolar H(+)-ATPase-an enzyme for all seasons. Pflugers Arch. 2009;457:581–587. doi: 10.1007/s00424-008-0458-9. [DOI] [PubMed] [Google Scholar]
- Schuldiner S, Steiner-Mordoch S, Yelin R. Molecular and biochemical studies of rat vesicular monoamine transporter. Adv Pharmacol. 1998;42:223–227. doi: 10.1016/s1054-3589(08)60733-4. [DOI] [PubMed] [Google Scholar]
- Shaner N, Steinbach P, Tsien R. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- Shao E, Forgac M. Involvement of the nonhomologous region of subunit A of the yeast V-ATPase in coupling and in vivo dissociation. J Biol Chem. 2004;279:48663–48670. doi: 10.1074/jbc.M408278200. [DOI] [PubMed] [Google Scholar]
- Smith A, et al. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet. 2000;26:71–75. doi: 10.1038/79208. [DOI] [PubMed] [Google Scholar]
- Söllner T, Whiteheart S, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman J. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- Stover E, et al. Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J Med Genet. 2002;39:796–803. doi: 10.1136/jmg.39.11.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof T. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Sugita S, Südhof T. Specificity of Ca2+-dependent protein interactions mediated by the C2A domains of synaptotagmins. Biochemistry. 2000;39:2940–2949. doi: 10.1021/bi9920984. [DOI] [PubMed] [Google Scholar]
- Sun-Wada G, Tabata H, Kawamura N, Aoyama M, Wada Y. Direct recruitment of H+-ATPase from lysosomes for phagosomal acidification. J Cell Sci. 2009;122:2504–2513. doi: 10.1242/jcs.050443. [DOI] [PubMed] [Google Scholar]
- Sun-Wada G, Toyomura T, Murata Y, Yamamoto A, Futai M, Wada Y. The a3 isoform of V-ATPase regulates insulin secretion from pancreatic beta-cells. J Cell Sci. 2006;119:4531–4540. doi: 10.1242/jcs.03234. [DOI] [PubMed] [Google Scholar]
- Supek F, Supekova L, Mandiyan S, Pan Y, Nelson H, Nelson N. A novel accessory subunit for vacuolar H(+)-ATPase from chromaffin granules. J Biol Chem. 1994;269:24102–24106. [PubMed] [Google Scholar]
- Toyomura T, Oka T, Yamaguchi C, Wada Y, Futai M. Three subunit a isoforms of mouse vacuolar H(+)-ATPasePreferential expression of the a3 isoform during osteoclast differentiation. J Biol Chem. 2000;275:8760–8765. doi: 10.1074/jbc.275.12.8760. [DOI] [PubMed] [Google Scholar]
- Wada I, Rindress D, Cameron P, Ou W, Doherty JJ 2nd, Louvard D, Bell A, Dignard D, Thomas D, Bergeron J. SSR alpha and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J Biol Chem. 1991;266:19599–19610. [PubMed] [Google Scholar]
- Wada Y, Sun-Wada GH, Tabata H, Kawamura N. Vacuolar-type proton ATPase as regulator of membrane dynamics in multicellular organisms. J Bioenerg Biomembr. 2008;40:53–57. doi: 10.1007/s10863-008-9128-z. [DOI] [PubMed] [Google Scholar]
- Wang L, Li G, Sugita S. RalA-exocyst interaction mediates GTP-dependent exocytosis. J Biol Chem. 2004a;279:19875–19881. doi: 10.1074/jbc.M400522200. [DOI] [PubMed] [Google Scholar]
- Wang L, Li G, Sugita S. A central kinase domain of type I phosphatidylinositol phosphate kinases is sufficient to prime exocytosis: isoform specificity and its underlying mechanism. J Biol Chem. 2005;280:16522–16527. doi: 10.1074/jbc.M413263200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Cipriano D, Forgac M. Arrangement of subunits in the proteolipid ring of the V-ATPase. J Biol Chem. 2007;282:34058–34065. doi: 10.1074/jbc.M704331200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Inoue T, Forgac M. TM2 but not TM4 of subunit c′′ interacts with TM7 of subunit a of the yeast V-ATPase as defined by disulfide-mediated cross-linking. J Biol Chem. 2004b;279:44628–44638. doi: 10.1074/jbc.M407345200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Sugita S, Sudhof T. The RIM/NIM family of neuronal C2 domain proteinsInteractions with Rab3 and a new class of Src homology 3 domain proteins. J Biol Chem. 2000;275:20033–20044. doi: 10.1074/jbc.M909008199. [DOI] [PubMed] [Google Scholar]
- Wang Y, Toei M, Forgac M. Analysis of the membrane topology of transmembrane segments in the C-terminal hydrophobic domain of the yeast vacuolar ATPase subunit a (Vph1p) by chemical modification. J Biol Chem. 2008;283:20696–20702. doi: 10.1074/jbc.M803258200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Zemelman B, McNew J, Westermann B, Gmachl M, Parlati F, Söllner T, Rothman J. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- Williamson W, Wang D, Haberman A, Hiesinger P. A dual function of V0-ATPase a1 provides an endolysosomal degradation mechanism in Drosophila melanogaster photoreceptors. J Cell Biol. 2010;189:885–899. doi: 10.1083/jcb.201003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.