

In contrast to the classical model, in which unfolded proteins accumulated in the endoplasmic reticulum trigger the unfolded-protein response (UPR), we show that membrane aberrancy also evokes this protective cellular event. This finding may explain UPR activation under various physiological conditions.
Abstract
Eukaryotic cells activate the unfolded-protein response (UPR) upon endoplasmic reticulum (ER) stress, where the stress is assumed to be the accumulation of unfolded proteins in the ER. Consistent with previous in vitro studies of the ER-luminal domain of the mutant UPR initiator Ire1, our study show its association with a model unfolded protein in yeast cells. An Ire1 luminal domain mutation that compromises Ire1's unfolded-protein–associating ability weakens its ability to respond to stress stimuli, likely resulting in the accumulation of unfolded proteins in the ER. In contrast, this mutant was activated like wild-type Ire1 by depletion of the membrane lipid component inositol or by deletion of genes involved in lipid homeostasis. Another Ire1 mutant lacking the authentic luminal domain was up-regulated by inositol depletion as strongly as wild-type Ire1. We therefore conclude that the cytosolic (or transmembrane) domain of Ire1 senses membrane aberrancy, while, as proposed previously, unfolded proteins accumulating in the ER interact with and activate Ire1.
INTRODUCTION
The unfolded-protein response (UPR), the basic concept of which was initially proposed by Kozutsumi et al. (1988), has been generally considered as transcriptional induction of endoplasmic reticulum (ER) chaperone genes in response to accumulation of unfolded secretory proteins in the ER. Of the ER membrane proteins that mediate the intracellular signal for the UPR, only Ire1 is known to be evolutionarily conserved throughout eukaryotes (Mori, 2009). Ire1 is a type 1 transmembrane protein carrying endoribonuclease activity in its cytosolic region. The best-documented function of Ire1 is cytoplasmic splicing of the yeast HAC1 and the metazoan XBP1 mRNAs that produce RNAs that are translated into transcription factor proteins (Ron and Walter, 2007).
Cellular stress conditions evoking the UPR are cumulatively called ER stress; this has been generally believed to mean accumulation of unfolded proteins in the ER. We and others previously reported that the ER chaperone BiP associates with and deactivates Ire1 under nonstress conditions (Bertolotti et al., 2000; Okamura et al., 2000; Kimata et al., 2003). ER stress causes dissociation of BiP from Ire1, which then leads to clustering of Ire1 (Kimata et al., 2007). The formation of clusters of Ire1 contributes to gathering and efficient splicing of its target RNA (Aragón et al., 2009; Korennykh et al., 2009). The molecular mechanism by which BiP dissociates from Ire1 is still obscure, since some observations (reviewed in Kimata and Kohno, 2011) argue against a simple model in which unfolded proteins accumulating in the ER deprive Ire1 of BiP. Thus it is unclear whether unfolded proteins are directly involved in the dissociation of BiP from Ire1.
On the other hand, further studies argue for direct involvement of unfolded proteins in activation of yeast Ire1. As illustrated in Figure 1A, the BiP-binding site is located on subregion V of the luminal domain of Ire1 (Kimata et al., 2004). Since Ire1 mutants carrying deletions in subregion V are still regulated by ER stress almost as well as wild-type Ire1, BiP is unlikely to be the principal determinant of Ire1's activity (Kimata et al., 2004; Pincus et al., 2010). Credle et al. (2005) reported the x-ray crystal structure of the core stress-sensing region (CSSR; Figure 1B) of Ire1, which, when dimerized, forms a cavity that may capture unfolded proteins. Interaction of unfolded proteins with Ire1 has also been supported by our study showing a recombinant CSSR protein inhibits aggregation of denatured model proteins in vitro (Kimata et al., 2007). A truncation mutation, ΔIII (see Figure 1C for mutation location) abolished this ability of the CSSR. The direct interaction of unfolded proteins with Ire1 is likely to contribute to activation of Ire1, since ΔIII Ire1 was poorly activated by ER stress induced by treatment of cells with the thiol-reducing agent dithiothreitol (DTT) or N-glycosylation inhibitor tunicamycin (Kimata et al., 2007). We therefore propose that Ire1 is activated upon ER stress conditions via two sequential steps, namely, its clustering, followed by the dissociation of BiP and direct interaction of unfolded proteins with the Ire1 cluster.
FIGURE 1:

Current model for structure and function of the luminal domain of S. cerevisiae Ire1. The luminal domain of yeast Ire1 can be divided into five subregions. Subregions I (aa 32–111), III (aa 243–272), and V (aa 455–524) are loosely folded, while subregions II (aa 112–242) and IV (aa 273–454) form the tightly folded CSSR (Kimata et al., 2004; Oikawa et al., 2005; Credle et al., 2005). (A) Under nonstress conditions, BiP associates with subregion V (Kimata et al., 2004). Dissociation of BiP from Ire1 leads to cluster formation of Ire1, the structural basis of which is two different modes of CSSR homoassociation (homoassociation via interfaces I and II; Credle et al., 2005; Kimata et al., 2007). Unfolded proteins then directly interact with the Ire1 cluster, causing full activation of Ire1 (Kimata et al., 2007). (B) The CSSR dimer associated via interface I forms a cavity-like structure, by which unfolded proteins may be captured (Credle et al., 2005). (C) The dashed lines indicate the positions of amino acid residues deleted in the ΔI (aa 32–91), ΔIII (aa 253–272), and ΔV (aa 463–524) mutations. The bZIP mutant of Ire1 carries the bZIP domain of Gcn4 instead of subregions I to V.
Nevertheless, it is unlikely that the UPR is activated only in order to cope with unfolded proteins accumulated in the ER. Comprehensive gene expression studies showed that UPR transcriptionally induces not only ER chaperones and protein degradation factors, but also various proteins, including ER translocon components, COPII coat components, and enzymes for ER membrane biogenesis via the Ire1-HAC1 pathway (Travers et al., 2000; Kimata et al., 2006). Deletion of either the IRE1 or the HAC1 gene confers auxotrophy for inositol, an important component of phospholipids, on yeast cells. It should be noted that expression of Ire1 is induced by depletion of inositol from yeast culture (Cox et al., 1997). As reviewed in Rutkowski and Hegde (2010) and argued in the Discussion, the UPR signal pathway is activated and required under various physiological and pathological situations in mammals, some of which do not seem to be tightly related to protein load in the ER.
In this study, we have examined activation steps of yeast Ire1 upon various stress conditions. Our findings indicate that unfolded proteins actually associate with Ire1 for its activation, while other stress stimuli that are strongly related to aberrancy of membrane homeostasis activate Ire1 in a different manner. This observation emphasizes roles of the UPR not restricted to response to unfolded proteins accumulated in the ER.
RESULTS
In vivo association of a model unfolded protein with Ire1
The ability of Ire1 to associate with unfolded proteins has been proposed by structural and biochemical analyses of recombinant CSSR proteins (Credle et al., 2005; Kimata et al., 2007). To further support the idea of a physical interaction between unfolded proteins and Ire1, we investigated whether such complexes are formed in yeast cells. The G255R mutant of Prc1 carboxypeptidase Y (CPY), known as CPY*, fails to be correctly folded and transported to the vacuole (Finger et al., 1993). Green fluorescent protein (GFP)-tagged, wild-type CPY (CPY-GFP) or CPY* (CPY*-GFP) was constitutively produced from the strong TEF1 promoter (Figure 2). Activation of the UPR by these proteins was checked by induction of a lacZ reporter controlled by the UPR promoter element (UPRE), which the Hac1 protein directly activates (Kawahara et al., 1997; Figure 2A). As expected, the reporter was induced by expression of CPY*-GFP, and less strongly induced by CPY-GFP. This observation was reproduced by an assay for Ire1-dependent HAC1i-mRNA production, in which cellular RNA samples were used for reverse transcriptase PCR (RT-PCR) amplification of the HAC1 mRNAs (Figure 2B). We therefore think that CPY-GFP may be somewhat unfolded, while CPY*-GFP acts as a more obviously unfolded-protein model. This insight is supported by immunofluorescence images showing an ER localization pattern that coincides with the BiP-staining pattern for CPY*-GFP, while CPY-GFP seems to be further partially transported (Figure 2C).
FIGURE 2:

UPR inducibility and cellular localization of CPY-GFP and CPY*-GFP. (A) An ire1Δ strain KMY1015 carrying both the wild-type IRE1 (WT Ire1) plasmid pRS315-IRE1-HA and the UPRE-lacZ reporter plasmid pCZY1 was further transformed with the CPY-GFP or the CPY*-GFP expression plasmid (pRS313-TEF1pr-CPY-GFP or pRS313-TEF1pr-CPY*-GFP) or empty vector pRS313 (Vector). The transformant strains were then assayed for cellular β-galactosidase activity, the values of which are normalized against that of vector control cells (set at 1.00). In the “no ire1” sample, cells carried vector plasmids pRS315 and pRS313. Error bars represent the SDs from three independent transformants. According to Student's t test, all values are statistically different from each other (p < 0.05). (B) Total RNA samples from the duplicate transformants used in (A) were subjected to RT-PCR to amplify the HAC1 products, HAC1u (u) and HAC1i (i), which were then fractionated by 13% acrylamide-gel electrophoresis and visualized by fluorescence signal from a fluorescently labeled PCR primer. (C) The transformant strains used in (A) were double-stained with chicken anti-GFP antibody/fluorescein isothiocyanate (FITC)-labeled secondary antibody and rabbit anti–yeast BiP antibody/Cy3-labeled secondary antibody.
Cells were treated with the chemical cross-linker dithiobis(succinimydylpropionate) (DSP) before cell lysis and anti-GFP immunoprecipitation (IP; Figure 3A). In agreement with the ER retention of CPY*-GFP, this protein appeared as a single protein band in an anti-GFP Western blot of the lysate and the anti-GFP IP samples. Also consistent with the above result, CPY-GFP partially converted to the fast-mobility vacuolar form. Significantly, coexpressed hemagglutinin (HA) epitope–tagged Ire1 (Ire1-HA) was coimmunoprecipitated with CPY*-GFP, but less abundantly with CPY-GFP.
FIGURE 3:

In vivo association of CPY*-GFP with Ire1. (A) The ire1Δ strain KMY1015 carrying both HA-tagged Ire1 plasmid pRS426-IRE1-HA and pRS313-TEF1pr-CPY-GFP or pRS313-TEF1pr-CPY*-GFP (or empty vector pRS313; Vector) was incubated with protein cross-linker DSP before cell lysis and anti-GFP IP. Subsequently, the lysate and the anti-GFP IP samples were analyzed by anti-HA or anti-GFP Western blotting. (B) The ire1Δ strain transformed with both pRS426-IRE1-HA (or empty vector pRS426 for lanes 1 and 7) and a GAL1 promoter-inducible CPY-GFP or CPY*-GFP plasmid, pRS313-GAL1pr-CPY-GFP or pRS313-GAL1pr-CPY*-GFP, was cultured in galactose-containing medium (see Materials and Methods for detail). After incubation with DSP, cells were lysed and analyzed by anti-HA IP, followed by anti-HA or anti-GFP Western blotting. In lanes 2, 3, and 4 and 8, 9, and 10, samples from three independent clones were analyzed. Cells for lane 6 carried an empty vector pRS313 instead of the CPY-GFP or CPY*-GFP plasmid. A molecular mass marker (M) was loaded in lane 5.
In Figure 3B, we present the results of a reverse immunoprecipitation experiment, in which CPY*-GFP was expressed from the inducible GAL1 promoter, since we failed to transform ire1-null strains with the CPY*-GFP constitutive expression plasmid. Consistent with the results shown in Figure 3A, Ire1-HA coimmunoprecipitates CPY*-GFP and, less abundantly, the ER-retained form of CPY-GFP. Since the vacuolar form of CPY-GFP, which migrates faster on PAGE, was not coimmunoprecipitated with Ire1-HA, we think the results of our immnoprecipitation experiments represent in vivo interaction between Ire1-HA and the ER-located model protein.
Effect of Ire1 mutations on Ire1 association with a model unfolded protein
Structural requirements of Ire1 for its association with CPY*-GFP were explored by using luminal domain mutants of IRE1. In an M229A/F285A/Y301A triple-point (MFY) mutation, all three mutation points are located on the inner surface of the CSSR cavity (Figure 1, B and C; Credle et al., 2005). The T226W/F246A double-point mutation and the W426 mutation are deduced to impair two different modes of CSSR homoassociation (Figure 1, A and C; Credle et al., 2005; Kimata et al., 2007; Aragón et al., 2009).
Cells were treated with DSP were subjected to anti-GFP immunoprecipitation in order to check association between CPY*-GFP and Ire1 mutants (Figure 4A). The cellular level of CPY*-GFP was highest in ire1-null cells and lowest in cells carrying wild-type Ire1-HA (Figure 4A, top panel). This observation correlates with the profile of UPR activation by the IRE1 mutants shown in Figure 4C. Potent UPR in the wild-type Ire1-HA cells is likely to accelerate ER-associated protein degradation of CPY*-GFP, which is compromised in the ire1-null cells (Travers et al., 2000).
FIGURE 4:

Effect of the CSSR mutations on relationship between Ire1 and CPY*-GFP. (A) The ire1Δ strain KMY1015 transformed with both pRS313-GAL1pr-CPY*-GFP and pRS426-IRE1-HA or its mutants (or empty vector pRS426; no ire1) was cultured in galactose-containing medium and analyzed as per Figure 3A. The asterisk denotes nonspecific bands. (B) The ratios of Ire1-HA to CPY*-GFP signal in anti-GFP IP in triplicate experiments demonstrated in (A) were normalized to that of wild-type Ire1 and are presented as “Binding efficiency to CPY*-GFP.” (C) The ire1Δ strain triply transformed with UPRE-lacZ plasmid pCZY1, plasmid pRS313-GAL1pr-CPY*-GFP (or empty vector pRS313; Vector), and pRS315-IRE1-HA (WT) or its mutants (or empty vector pRS315; no ire1) was cultured in galactose-containing medium before measuring cellular β-galactosidase activity, the values of which are normalized against that of WT IRE1 Vector cells (set at 1.00). Error bars represent the SDs from three independent transformants. According to Student's t test, the values of all CPY*-GFP mutant Ire1 samples are statistically different from those of the CPY*-GFP WT IRE1 sample (p < 0.01). Also, when not carrying the CPY*-GFP plasmid, the WT IRE1 sample exhibited statistical difference from the W426A, T226W/F247A, and MFY mutant samples (p < 0.05). (D) Cells used in (C) were analyzed by RT-PCR to evaluate splicing efficiency of the HAC1 mRNA.
The results shown in Figure 4A are quantitatively expressed in Figure 4B. The T226W/F247A mutation partially compromised the association of Ire1 with CPY*-GFP, while the W426A mutation showed only a slight effect. This finding seems to agree with the idea that homoassociation via interface I but not via interface II is required to form the cavity (see Figure 1B). Importantly, the MFY cavity mutation and the ΔIII mutation impaired the association of Ire1 with CPY*-GFP. This observation is consistent with our previous report that these mutations abolish in vitro ability of the CSSR to interact with model unfolded proteins (Kimata et al., 2007).
As shown in Figure 4C, all of the Ire1 mutations used here compromised the ability of Ire1 to evoke the UPR upon cellular accumulation of CPY*-GFP. This observation was reproduced by checking cellular HAC1 mRNA splicing (Figure 4D). We think that W426A Ire1 exhibited weak UPR activity, despite having unfolded-protein–interacting ability, due to its inability to form clusters. Meanwhile, the compromising of activity of the other Ire1 mutants seems to be explained in part by their impaired ability to interact with unfolded proteins.
Since ΔIII Ire1 clusters as well as wild-type Ire1 upon potent ER stress (Kimata et al., 2007), we think that the ΔIII mutation specifically impairs the association between Ire1 and unfolded proteins. In contrast, the MFY mutation has been reported to somehow confer more diverse damage on the CSSR structure and function than the ΔIII mutation (Kimata et al., 2007). It should be noted that ΔIII Ire1 showed wild-type-like activity under the nonstress conditions, while the MFY mutation, as well as the homoassociation mutations T226W/F247A and W426A, compromised Ire1's activity (Figure 4C). This observation strongly suggests that the unfolded-protein association with the CSSR is not required for basal-level activity of Ire1 under nonstress conditions, and confirms an unexpected side effect of the MFY mutation. We therefore used the ΔIII mutation to address the requirement for an unfolded protein–CSSR interaction for activation of Ire1 by various stress stimuli.
ΔIII Ire1 is activated as well as wild-type Ire1 upon inositol depletion
We monitored time-course changes in cellular HAC1 mRNA-splicing efficiency and expressed them as quantified data (HAC1 mRNA splicing%; Figure 5, A–C) obtained from raw gel images (examples shown in Supplemental Figure S1). Because ire1Δ cells showed no HAC1 mRNA splicing under various conditions used here (Figure S2), this assay actually enabled us to estimate Ire1's activity.
FIGURE 5:

Effect of the ΔIII mutation on activation of Ire1 by external stress stimuli. An ire1Δ strain KMY1516 transformed with the wild-type IRE1 plasmid (pRS313-IRE1; WT) or its ΔIII mutant version was stressed by adding DTT (A and D) or tunicamycin (B) or by shifting to inositol depletion medium (C and D) at time 0. The total RNA samples were then analyzed by RT-PCR to evaluate splicing efficiency of the HAC1 mRNA. Error bars represent the SDs from three independent transformants.
As shown in Figure 5A, upon addition of 3 mM final concentration of DTT into the culture, wild-type Ire1 exhibited rapid and potent activation, which reached a peak level ∼30 min after the stimulus onset and then gradually attenuated. In the case of ΔIII Ire1, such a rapid activation was not observed. Instead, and unexpectedly, ΔIII Ire1 gradually enhanced its HAC1 mRNA splicing activity, which 5 h after the stimulus onset was comparable to that of wild-type Ire1. Similar results were obtained when cells were treated with 0.6 μg/ml tunicamycin (Figure 5B). In contrast, we also noticed that inositol depletion from culture media conferred similar activation profiles, which reached a peak ∼5 h after the stress onset, in wild-type Ire1 and ΔIII Ire1 (Figure 5C). Figure 5D shows that the ΔIII mutation impaired the acute activation (30 min after stress onset) at any concentration of DTT.
Activation steps of Ire1 upon inositol depletion
We have previously proposed that upon conventional ER stress by treatment with DTT or tunicamycin, BiP dissociates from Ire1, which then clusters (Kimata et al., 2007; Aragon et al., 2009; Li et al., 2010). In this study, we explored whether activation of Ire1 by inositol depletion also accompanies these molecular events. In vivo association of BiP with wild-type Ire1 was checked by anti-HA co-IP of BiP with Ire1-HA (Figure 6A). In a concentration-dependent manner, cellular exposure to DTT for 30 min caused BiP dissociation from Ire1. Moreover, inositol depletion for 4 h also caused BiP dissociation from Ire1. It should be noted that the 3 mM DTT sample showed a level of BiP dissociation similar to the inositol-depleted sample, in agreement with similar HAC1 splicing efficiencies under these two experimental conditions (Figure 5, A and C). A similar tendency was seen when we checked cluster formation of Ire1 (Figure 6B). As performed in our previous study (Kimata et al., 2007), cellular localization of Ire1-HA was visualized by anti-HA immunofluorescence staining, which showed that not only DTT treatment but also inositol depletion changes the cellular localization of Ire1-HA from an ER-like double-ring pattern to a dot-like pattern. We think the dot-like staining pattern actually represents clustering of Ire1, since it was not observed when the homoassociation mutation W426A was introduced into Ire1.
FIGURE 6:

Activation steps of Ire1 by DTT imposition and inositol depletion. (A) The ire1Δ strain KMY1516 transformed with an HA-tagged Ire1 plasmid pRS423-IRE1-HA (or empty vector pRS423; no ire1) was cultured in the indicated conditions, and the lysate and anti-HA IP samples were analyzed by anti-HA or anti-BiP Western blotting. (B) Cells used in A (or carrying the W426A mutant version of pRS423-IRE1-HA) were stressed by the indicated conditions and stained by mouse anti-HA antibody/FITC-labeled secondary antibody. (C) The ire1Δ strains transformed with the indicated mutant versions of pRS313-IRE1 were stressed by the indicated conditions, and the total RNA samples were then analyzed by RT-PCR to evaluate splicing efficiencies of the HAC1 mRNA. Error bars represent the SDs from three independent transformants. (D) Cell lysates from the ire1Δ strain KMY1015 transformed with pRS315-IRE1-HA or its mutant versions were analyzed by anti-HA Western blotting.
On the other hand, the cluster formation is not sufficient for full HAC1 mRNA-splicing activity of Ire1. According to our previous report (Kimata et al., 2007), the ΔIΔV double-deletion mutation (see Figure 1C for mutation location) causes nonregulated cluster formation of Ire1, probably because in addition to the binding of BiP to subregion V, subregion I somehow suppresses the cluster formation of Ire1. Significantly, ΔIΔV Ire1 is strongly up-regulated by tunicamycin or DTT treatment, while the ΔIΔIIIΔV triple-deletion mutant is not (Oikawa et al., 2007; Kimata et al., 2007). These observations have led us to propose that the physical interaction between unfolded proteins and the CSSR causes full activation of clustered Ire1. In this study, we investigated inositol depletion (Figure 6C). Confirming our previous report (Kimata et al., 2007), acute (30 min after stress onset) activation of ΔIΔV Ire1 by DTT treatment was markedly compromised by introduction of the ΔIII mutation. At the same time, we noticed that inositol depletion also resulted in considerable activation of ΔIΔV Ire1, which, however, was only moderately compromised by the ΔIII mutation. Ire1-HA and its mutants were detected by anti-HA Western blot analysis of cell lysates, indicating that, for unknown reason(s), introduction of the ΔIII mutation markedly reduces cellular expression level of ΔIΔV Ire1 but not of wild-type Ire1 (Figure 6D). This finding explains why the ΔIII mutation compromises, albeit moderately, the activation of ΔIΔV Ire1, but not of wild-type Ire1, upon inositol depletion.
The results shown in Figure 7 support our idea that ER stress induced by inositol depletion is essentially different from that induced by DTT. We broke cells in the presence of the nonionic detergent Triton X-100 and centrifuged them to obtain protein-aggregate fractions (Figure 7A). Via subsequent anti-BiP Western-blot analysis, a considerable amount of BiP was detected in the pellet fraction from cells stressed by DTT, suggesting formation of unfolded-protein aggregates in the ER. However, inositol depletion did not result in such an effect, although it activated Ire1 to a similar level as DTT exposure under the conditions used here (3 mM DTT for 30 min; see Figure 5).
FIGURE 7:
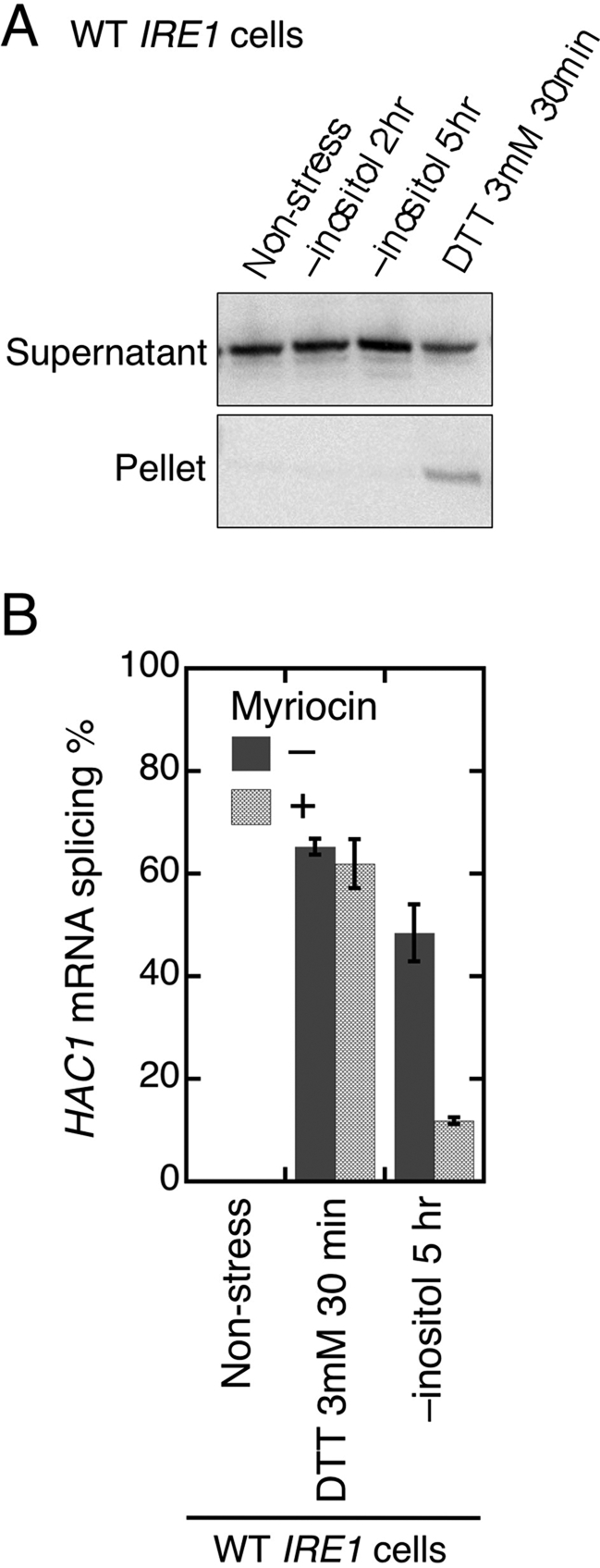
Different properties of stress stimuli by DTT imposition and inositol depletion. (A) The ire1Δ strain KMY1516 carrying an IRE1 plasmid pRS316−IPE1 was stressed by the indicated conditions and lysed by vortexing with glass beads in Triton X-100-containing buffer as described in Kimata et al. (2003). After removal of debris by centrifugation at 700 × g for 3 min, the lysates were further fractionated by centrifugation at 8000 × g for 20 min. The pellet (equivalent to 0.5 OD600 cells) and supernatant (equivalent to 0.1 OD600 cells) fractions were then analyzed by anti-BiP Western blotting. (B) After being cultured with (+) or without (–) 0.5 μg/ml myriocin for 5 h, KMY1516 cells carrying pRS316-IRE1 were stressed by the indicated stimuli. In the myriocin (+) samples, 0.5 μg/ml myriocin was also added into the stressing media. The total RNA samples were then analyzed by RT-PCR to evaluate splicing efficiency of the HAC1 mRNA. Error bars represent the SDs from three independent cultures.
Cells were cultured with the sphingolipid synthesis inhibitor, myriocin, 5 h before and during ER stress imposition (Figure 7B). Notably, myriocin compromised cellular HAC1 mRNA splicing not via DTT exposure but through inositol depletion.
Activation of wild-type and ΔIII Ire1 by gene deletion
We next asked what stress stimuli other than inositol depletion activates ΔIII Ire1 to an intensity similar to wild-type Ire1. Instead of external stimuli, we used here deletion of nonessential genes. Cellular UPR activity was monitored by induction of a genome-integrated UPRE-lacZ reporter (Figure 8A), which allowed us to obtain low-deviation data in comparison with a 2-μ plasmid-borne UPRE-lacZ reporter used in Figures 2A and 4C. Activation of Ire1 was also checked through cellular HAC1 mRNA splicing (Figure S3). To identify gene deletions that evoke the UPR, we referred to Jonikas et al. (2009), in which a UPRE-GFP reporter was utilized to comprehensively screen the yeast deletion strain stock. As detailed in Materials and Methods, we checked the gene deletions reported to evoke the UPR by Jonikas et al. (2009), reproduced their results with some exceptions, and finally chose the 18 genes listed in Table 1.
FIGURE 8:

Gene deletion-induced activation profile of wild-type and ΔIII Ire1. (A) As described in Materials and Methods, the ire1Δ strain KMY1516 (chromosomally carrying the 5X UPRE-lacZ reporter gene) was modified to carry the wild-type (WT) or the ΔIII IRE1 gene (pRS313-IRE1 or its mutant) and a deletion of the indicated genes. Subsequently, β-galactosidase activity of cells cultured without external stress stimuli was assayed and normalized against that of wild-type IRE1 wild-type cells (set at 1.00). Error bars represent the SDs from three independent transformants. According to Student's t test, the ΔIII mutation significantly compromised cellular β-galactosidase activity of cells carrying deletion of SCJ1, SPC2, STE24, ALG3, EOS1, PMT2, ERV14, ERV25, or ERD1, but not of WT cells or cells carrying deletion of the other genes (p < 0.05). (B) The ratio of the value for ΔIII Ire1 cells against that for WT Ire1 cells shown in (A) is calculated for each deletion and presented.
TABLE 1:
S. cerevisiae genes deleted for UPR induction in this study
| Processing or folding of secretory proteins in the ER | |
| SCJ1 | Co-chaperone of BiP |
| SPC2 | Subunit of signal peptidase |
| Glycosylation | |
| ALG3 | Synthesis of oligosaccharide donor for N-linked glycosylation of proteins |
| EOS1 | N-linked glycosylation |
| PMT2 | Protein O-mannosyltransferase |
| Vesicle transport and related events | |
| ERV14 | Cargo receptor in ER-to-Golgi transport |
| ERV25 | Cargo receptor in ER-to-Golgi transport |
| SEC28 | Subunit of COPI vesicle coat |
| GPI-anchor production | |
| BST1 | GPI inositol deacylase |
| LAS21 | Synthesis of the GPI core structure |
| Lipid metabolism and homeostasis | |
| ARV1 | Intracellular transport of lipidic components |
| OPI3 | Phosphatidylcholine biosynthesis |
| SCS3 | FIT family protein (triglyceride droplet biosynthesis) |
| ISC1 | Phosphosphingolipid phospholipase C |
| MGA2 | Transcriptional regulation of OLE1 encoding δ9 fatty acid desaturase |
| Other function related to ER membrane | |
| GET1 | Insertion of tail-anchored proteins into the ER membrane from cytosol |
| Unknown function | |
| STE24 | Zinc metalloprotease |
| ERD1 | Retention of BiP in the ER |
Gene functions described here are based on SGD.
Figures 8A and S3 show activation of wild-type Ire1 and ΔIII Ire1 by deletion of these 18 genes. The values shown in Figure 8A were then used to obtain the values in Figure 8B, which clearly demonstrates whether activation of Ire1 by a gene deletion was compromised by the ΔIII mutation. Deletion of any of SCJ1, SPC2, STE24, ERV14, ERV25, ALG3, EOS1, PMT2, or ERD1 activated wild-type Ire1 more strongly than ΔIII Ire1. At the same time, wild-type Ire1 and ΔIII Ire1 were similarly activated by deletion of any of SEC28, BST1, LAS21, ARV1, GET1, OPI3, SCS3, ISC1, or MGA2. This finding indicates that Ire1 is activated by certain stress stimuli, which are likely to cause membrane- or lipid-related abnormalities (see Discussion for detailed explanations), without the interaction between Ire1 and unfolded proteins.
Luminal domain-independent mode of stress sensing by Ire1
Liu et al. (2000) reported that Ire1 is somehow activated by tunicamycin treatment, even when its luminal domain is replaced by a dimer-forming “basic-region leucine zipper” (bZIP) motif obtained from nuclear transcription factor proteins. To support the idea that Ire1 can be activated via a manner other than interaction between the CSSR and unfolded proteins, we used bZIP-Ire1, in which the bZIP domain of yeast Gcn4 was substituted into subregions I–V (see Figure 1C). Unexpectedly, anti-HA immunofluorescence staining of bZIP-Ire1-HA (Figure 9A) showed that bZIP-Ire1 is clustered under both nonstress and inositol-depleted conditions. As well as the clusters of wild-type Ire1 (Kimata et al., 2007; Aragón et al., 2009), the bZIP-Ire1 clusters were formed on the ER, since their location could be merged with an anti-BiP antibody-staining image (Figure 9B). We speculate that the oligomer-forming ability of the cytosolic domain of Ire1 (Korennykh et al., 2009) contributes to the cluster formation of bZIP-Ire1.
FIGURE 9:

Cellular localization and activity of bZIP-Ire1. (A) The ire1Δ strain KMY1516 transformed with wild-type (WT) pRS423-IRE1-HA or its bZIP mutant version (or empty vector pRS423 [Vector]) was cultured under the indicated conditions and stained by mouse anti-HA antibody/FITC-labeled secondary antibody. (B) bZIP-Ire1-HA cells used in (A) were double-stained with rabbit anti-HA antibody/Cy5-labeled secondary antibody and rabbit anti–yeast BiP antibody/FITC-labeled secondary antibody. (C and D) The ire1Δ strain transformed with the bZIP mutant version of pRS313-IRE1 was stressed by the indicated stimuli. Total RNA samples were then analyzed by RT-PCR to evaluate splicing efficiency of the HAC1 mRNA. Error bars represent the SDs from three independent transformants.
As anticipated from Liu et al. (2000), treatment of cells with DTT or tunicamycin activated bZIP-Ire1 (Figure 9C) but more slowly than wild-type Ire1 (compare to Figure 5, A and B). Meanwhile, bZIP-Ire1 was activated by inositol depletion with a time course and a level comparable to wild-type Ire1 (compare Figures 9C and 5C). Unlike wild-type Ire1, bZIP-Ire1 failed to show acute activation (30 min after stress onset) with any concentration of DTT (Figure 9D).
The stress-dependent activation of bZIP-Ire1 strongly suggests that Ire1 is capable of stress recognition using its cytosolic or transmembrane domain. Wiseman et al. (2009) indicated that Ire1 is artifactually activated by its interaction with small molecules such as quercetin. Nevertheless, we do not think that this is the cause of the luminal domain-independent activation of Ire1 shown here, since activation of bZIP-Ire1 by inositol depletion, as well as activation of wild-type Ire1 by DTT treatment, was observed even when point mutations abolishing the small-molecule interaction (Wiseman et al., 2009) were introduced into the cytosolic domain (Figure S4).
DISCUSSION
On the basis of structural and biochemical analyses of recombinant CSSR proteins (Credle et al., 2005; Kimata et al., 2007), we and others previously proposed that the luminal domain of Ire1 has the ability to interact with unfolded proteins. The in vivo association between Ire1 and CPY*-GFP shown in this study (Figure 3) provides further supporting evidence for this insight. Binding of CPY-GFP to Ire1 was less obvious than that of CPY*-GFP, probably because CPY-GFP was partially transported out of the ER and/or because folding status is not the same in CPY-GFP and CPY*-GFP. As described in Results, the ability of the Ire1 mutants to associate with CPY*-GFP (shown in Figure 4, A and B) is consistent with the notion that unfolded proteins are captured by the CSSR cavity (Figure 1). Taking into consideration observations from previous studies (Credle et al., 2005; Kimata et al., 2007), the impaired activation of each Ire1 mutant (Figure 4C) has to be considered on a case-by-case basis. For instance, the W426A mutation abolishes the cluster formation of Ire1 without considerably compromising its ability to associate with unfolded proteins. In contrast, ΔIII Ire1 seems to be impaired with regard to its association with unfolded proteins, while having cluster-forming ability (Kimata et al., 2007).
This property of ΔIII Ire1 allowed us to explore involvement of the unfolded-protein association in activation of Ire1 under various stress stimuli. We noticed that, unlike DTT or tunicamycin treatment, inositol depletion activated ΔIII Ire1 at almost the same level and time course as wild-type Ire1 (Figure 5C). On the basis of the results from gene deletions (Figures 8), we propose that two different types of stress stimuli activate the UPR in distinct manners.
Initially, stress stimuli causing accumulation of unfolded proteins in the ER activate Ire1 via its interaction with the aberrant proteins. Cellular expression of aberrant proteins (CPY*-GFP in this study); DTT or tunicamycin treatment; and deletion of any of SCJ1, SPC2, STE24, ALG3, EOS1, PMT2, ERD1, ERV14, and ERV25 seem to fall into this category, since these stress stimuli activate wild-type Ire1 more strongly than ΔIII Ire1. Given that Scj1, Spc2, Alg3, and Eos1 can each function as a BiP cochaperone, a subunit of the signal peptidase complex or factors in N-glycosylation, the loss of these proteins is likely to produce aberrant proteins in the ER. Also, Pmt2 was recently reported to be an O-mannosyltransferase that participates in protein quality control in the ER (Goder and Melero, 2011). Evocation of the UPR by ERD1 deletion may be explained by mislocalization of BiP (Hardwick et al., 1990), which is also likely to impair protein folding in the ER. Erv14 and Erv25, although not required for COPII-coated vesicle formation per se (Matsuoka et al., 1998), contribute to ER-to-Golgi transport through their physical interaction with cargo proteins (Muñiz et al., 2000; Powers and Barlowe, 2002). We therefore think that loss of Erv14 or Erv25 causes stacking of cargo proteins in the ER that is sensed by Ire1 in a manner similar to sensing of unfolded-protein accumulation. However, we can offer no explanation as to why the STE24 deletion exhibits such a UPR activation profile.
In contrast, deletion of certain other genes, as well as inositol depletion, activates wild-type and ΔIII Ire1 almost equally. We propose that these stress stimuli lead to membrane- or lipid-related aberrations, which activate Ire1 even without its interaction with unfolded proteins. OPI3 and ISC1 encode enzymes that metabolize phospholipids. Scs3 is a member of the ER-located FIT family of proteins, which are involved in fat storage (Kaderei et al., 2008). Although the function of Arv1 and its metazoan orthologues is still obscure, their loss is reported to perturb intracellular distribution of lipidic components (Kajiwara et al., 2008; Tong et al., 2010). Since Get1 is a member of the GET complex, which mediates insertion of tail-anchored proteins from cytosol to the ER membrane (Schuldiner et al., 2008), its loss is likely to damage primarily the membrane, rather than the lumen of the ER. It also seems reasonable that membrane homeostasis is perturbed when genes involved in glycosylphosphatidylinositol (GPI) anchor biogenesis, such as LAS21 or BST1, are deleted. Although it is possible that such mutations also adversely affect the integrity of GPI-anchored proteins, this is not directly sensed by Ire1, because the mutations activate wild-type Ire1 and ΔIII Ire1 equally. Interestingly, SEC28, but not ERV14 or ERV25, falls into this category, although all three genes are involved in intracellular vesicle transport. While a possible role of Erv14 and Erv25 is to function as cargo-protein receptors in the ER-to-Golgi transport (Muñiz et al., 2000; Powers and Barlowe, 2002), Sec28 is a component of the coatomer (Duden et al., 1998; Kimata et al., 1999), which per se is responsible for formation of transport vesicles in Golgi-to-ER retrieval transport. We therefore speculate that loss of Sec28 primarily impairs membrane composition, but not protein flux, in the ER.
Inositol is one of the main components of phospholipids, and we propose that UPR evocation by inositol depletion is also due to a membrane-related abnormality. In other words, although inositol depletion is reported to lead to altered conditions in the ER lumen (Merksamer et al., 2008), this is not the main factor that activates Ire1 upon this stress stimulus. This is because, as mentioned in the next paragraph, inositol depletion causes up-regulation of bZiP-Ire1, which lacks the authentic Ire1 luminal domain, as well as wild-type Ire1 (Figure 9C and D). Moreover, while DTT exposure produced BiP-containing protein aggregates, inositol depletion failed to exhibit such an effect (Figure 7A). This observation implies that inositol depletion does not damage protein folding in the ER lumen as potently as DTT. It also should be noted that myriocin, an inhibitor of sphingolipid biosynthesis, compromises activation of Ire1 by inositol depletion but not by DTT exposure (Figure 7B). Although the link between sphingolipids or biosynthetic intermediates of sphingolipids and the UPR is obscure, this finding strongly suggests a tight relationship between cellular membrane conditions and Ire1 activation by inositol depletion.
The activation steps of Ire1 upon DTT or tunicamycin treatment have been documented in our previous report (Kimata et al., 2007), which proposed that dissociation of BiP from Ire1 causes cluster formation of Ire1, while unfolded proteins interact with the Ire1 clusters for full activation. To explore the activation steps of Ire1 that are independent of unfolded proteins, we used inositol depletion as a model stress condition. BiP dissociation and cluster formation of Ire1 were also observed upon inositol depletion (Figure 6, A and B). The ΔIΔV mutation abolishes BiP binding and causes constitutive clustering of Ire1 (Oikawa et al., 2007; Kimata et al., 2007). In the present study, we noticed that inositol depletion up-regulates ΔIΔV Ire1 and, albeit slightly weakly, ΔIΔIIIΔV Ire1. We therefore conclude that, instead of interaction between unfolded proteins and Ire1, an undisclosed molecular event occurs to give full activation of clustered Ire1 molecules upon inositol depletion. Since bZIP-Ire1 responded well to inositol depletion (Figure 9, C and D), the luminal domain of Ire1 is unlikely to contribute to sensing this stress stimulus.
Unlike the quick activation of wild-type Ire1 observed upon treatment with DTT or tunicamycin (peak activation within 30 min or 1 h after stimulus onset; Figure 5, A and B), activation of wild-type Ire1, and also of ΔIII Ire1 and bZIP-Ire1, by inositol depletion is rather slow (Figures 5C and 9C). Although this time lag may be due to residual cellular inositol stock, it is also possible that the cellular mechanism activating Ire1 upon inositol depletion per se is slow in responding. We therefore speculate that the cellular response to membrane-related ER stress need not be acute. Conversely, the response of Ire1 to unfolded-protein accumulation is so acute that cells can cope with such a stress condition quickly. It should be noted that DTT and tunicamycin somehow activated ΔIII Ire1 and bZIP-Ire1, albeit more slowly and weakly than observed for wild-type Ire1 (Figures 5, A and B, and 9C). We speculate that these conventional ER stressors also disturb membrane homeostasis and then slowly activate Ire1, even when it carries a mutation abolishing its ability to associate with unfolded proteins. Excess accumulation of aberrant proteins in the ER may concomitantly damage the ER membrane.
Mammals carry two Ire1 paralogues, of which IRE1α is the major version expressed ubiquitously. According to the x-ray crystal structure reported by Zhou et al. (2006), the luminal domain of IRE1α carries a cavity-like structure, which, however, is too narrow to capture unfolded proteins, unlike that of yeast Ire1. Moreover, we failed to demonstrate in vitro interaction between unfolded proteins and a recombinant luminal-domain fragment of IRE1α (Oikawa et al., 2009). While one explanation for these observations is that the size of the IRE1α cavity is somehow regulated and enlarged when it is required to capture unfolded proteins, it is also possible that IRE1α lacks the ability to associate with unfolded proteins. Meanwhile, BiP association/dissociation is not likely to be the sole determinant of IRE1α activity, since an IRE1α truncation mutant lacking the major BiP-binding region is still up-regulated by ER stress (Oikawa et al., 2009). We therefore think that the new mechanism of stress sensing presented here may also contribute to activation of IRE1α upon ER stress.
How does the cytosolic (or transmembrane) domain of Ire1 sense stress stimuli? It is an attractive idea, as proposed by Wiseman et al. (2009), that small molecules interact with the cytosolic domain of Ire1 to up-regulate Ire1. However, the newly found ligand-binding pocket (Wiseman et al., 2009) is unlikely to be involved in activation of Ire1 upon inositol depletion (Figure S4). In the case of mammalian IRE1α, its activity has been reported to be modulated by association with proteins other than BiP (Hetz et al., 2006; Luo et al., 2008; Lisbona et al., 2009; Qiu et al., 2010). This matter may have to be addressed in order to further elucidate the new mode of stress sensing by Ire1.
It should be noted that various reports have touched upon evocation of the UPR by lipid- or membrane-related stimuli. Pineau et al. (2009) and Deguil et al. (2011) proposed that an imbalanced fatty acid composition could activate yeast Ire1. In contrast to the concept we are now presenting, Pineau et al. (2009) argued that activation of Ire1 by the fatty acid imbalance involves accumulation of unfolded proteins, since it is attenuated by the chemical chaperone 4-phenyl butyrate. In mammals, obesity induces ER stress and may lead to type 2 diabetes (Özcan et al., 2004), the symptoms of which are alleviated by chemical chaperones (Özcan et al., 2006). Cunha et al. (2008) and Wei et al. (2006) described lipid-induced ER stress followed by cellular damage of pancreatic β cells and liver cells. Furthermore, as reviewed by Zheng et al. (2010), the UPR pathway is involved in lipogenesis of mammalian cells, as well as of yeast. It is widely accepted that IRE1α and its downstream target XBP1 are required for homeostasis maintenance of and/or differentiation to cells or tissues secreting high levels of proteins, such as antibody-producing plasma cells, pancreatic β cells, and placenta, in which the ER membrane is highly proliferated (Iwakoshi et al., 2003; Lipson et al., 2008; Iwawaki et al., 2009, 2010). One possible explanation for this is that excess influx of proteins into the ER leads to activation of ER stress sensors, including IRE1α. However, this idea is not supported by the observation that mutant B lymphocytes engineered to lack antibody production also activate XBP1 upon their differentiation to plasma cells (Hu et al., 2009). We therefore think that upon various lipid- or membrane-related stimuli, membrane stress per se and concomitant accumulation of unfolded proteins are recognized by ER stress sensors, including Ire1, in a complex manner.
In conclusion, ER stress that activates the UPR is not always accompanied by accumulation of unfolded proteins in the ER. While unfolded proteins are captured and quickly activate at least yeast Ire1, lipid- or membrane-related ER stress is likely to be sensed by ER stress sensors in a different manner. This new concept of ER stress and cellular responses to it may allow us to understand more about ER stress responses under various physiological and pathological conditions.
MATERIALS AND METHODS
Plasmids
As previously described (Kimata et al., 2003, 2004), Saccharomyces cerevisiae IRE1 plasmid pRS313-IRE1 and C-terminally HA-tagged IRE1 (Ire1-HA) plasmids pRS315-IRE1-HA and pRS423-IRE1-HA were respectively produced from centromeric vectors pRS313, pRS315, and 2 μ vector pRS423 (Sikorski and Hieter, 1989; Christianson et al., 1992). Plasmid pRS426-IRE1-HA is the URA3 marker variant (Christianson et al., 1992) of pRS423-IRE1-HA. Overlap PCR and in vivo homologous recombination (gap repair) techniques were used to introduce point or partial-deletion mutations into the IRE1 gene on these plasmids (Kimata et al., 2004, 2007). This methodology was also used to generate bZIP-Ire1 plasmids, where the bZIP domain sequence of S. cerevisiae GCN4 (corresponding to aa 249–281) was substituted into the luminal domain sequence (corresponding to aa 32–524) on the IRE1 gene. Because we assigned the initiation methionine according to the data from the Saccharomyces Genome Database (SGD; www.yeastgenome.org), the amino acid numbering of Ire1 in the present paper differs by 7 aa from that in two of our previous reports (Kimata et al., 2004; Oikawa et al., 2005). URA3 2 μ plasmid pCZY1, carrying a fusion of UPER-CYC1 core promoter-lacZ, was a gift of K. Mori (Kyoto University, Kyoto, Japan).
The PCR technique was also used to amplify the S. cerevisiae TEF1 and the GAL1 promoter sequences, the CPY or the CPY*-encoding sequences (S. cerevisiae PRC1 or prc1-1; Finger et al., 1993), and the codon-optimized GFP-coding sequence (y-enhanced GFP; Sheff and Thorn, 2004) from plasmid or yeast genomic DNA samples by using PCR primers listed in Table S1. The restriction sites attached to ends of the PCR products (see Table S1) were then digested with the corresponding restriction enzymes and ligated with the corresponding sites of pRS313, in order to obtain plasmids pRS313-TEF1pr-CPY-GFP, pRS313-TEF1pr-CPY*-GFP, pRS313-GAL1pr-CPY-GFP, and pRS313-GAL1pr-CPY*-GFP.
Yeast strains
S. cerevisiae ire1Δ strains KMY1015 (MATα ura3-52 leu2-3112 his3-Δ200 trp1-Δ901 lys2-801 ire1Δ::TRP1) and KMY1516 (MATα ura3-52 LEU2::UPRE-CYC1 core promoter-GFP::leu2-3112 his3-Δ200 trp1-Δ901 LYS2::(UPRE)5-CYC1 core promoter-lacZ::lys2-801 ire1Δ::TRP1) have been described previously (Kimata et al., 2004).
To select genes for deletion (see Figure 8), we referred to Jonikas et al. (2009), and picked genes for which deletions score more than 2.0 for the ratio values of UPRE-GFP fluorescence against TEF2pr-red fluorescent protein fluorescence, with exceptions for genes of unknown function (CSF1, YBL083C, YML013C-A, FYV6, ILM1, YDL096C, and YHR039C-B); genes for which deletion exhibited no significant UPR induction on our strategy described in the next paragraph (OTS3, SEL1, GUP1, LHS1, PMT1 and VAN1); genes for which deletion could not be introduced by us (probably because the deletions cause severe growth defects, as noted in SGD [www.yeastgenome.org]; SPF1, PER1, and GLO1); and genes belonging to the same functional pathway as ALG3 (ALG6, ALG9, and ALG12). We also checked lipid-related gene deletions that are reported to evoke weaker but detectable UPR in Jonikas et al. (2009), and picked up OPI3, SCS3, and ISC1. SEC28 was additionally checked, since our previous study showed UPR evocation by deletion of this gene (Kimata et al., 2000).
Gene deletion strains used in Figures 8 and S3 were generated from the ire1Δ strain KMY1516, which had been modified to carry the pRS316 (a URA3 marker centromeric vector; Sikorski and Hieter, 1989)-based variant of pRS313-IRE1 (pRS316-IRE1). To introduce a deletion mutation, we transformed this strain with a KanMX4-based gene disruption module that was PCR-amplified from a EUROSCARF deletion strain by using PCR primers hybridizing ∼100 base pairs upstream (sense) and downstream (antisense) of the deleted coding region. The resulting strain was transformed again with pRS313-IRE1 or its ΔIII variant (the HIS3 marker), and grown on an synthetic dextrose (SD) agar plate containing 0.1% 5-fluoroorotic acid and 0.01% uracil for counterselection against pRS316-IRE1.
Yeast media and culturing conditions
Unless otherwise noted, yeast cells were cultured at 30°C in SD medium (2% dextrose and 0.67% Difco yeast nitrogen base without amino acids [YNB w/o aa]) supplemented with appropriate auxotrophic requirements. Based on the Difco & BBL Manual (www.bd.com), pure chemicals were mixed to obtain inositol-depleted SD medium (inositol depletion medium). When changing medium to inositol depletion medium, we washed cells six times by centrifugation and resuspension in inositol depletion medium. For GAL1 promoter-dependent induction of CPY*-GFP, cells carrying pRS313-GAL1pr-CPY*-GFP were grown in synthetic raffinose medium (2% raffinose and 0.67% YNB w/o aa) containing auxotrophic requirements, into which 1/10 volume of 20% galactose water was then added before a further incubation for 14 h, which was followed by cell harvesting.
β-Galactosidase assay and data presentation
Two types of the UPRE-lacZ reporter were used in this study. One is borne on the 2 μ plasmid pCZY1 (Figures 2A and 4C). The other carries five tandem copies of the UPRE and is integrated into the genome of the ire1Δ strain KMY1516 (Figure 8). In both the cases, cellular β-galactosidase activity (see Kimata et al. [2003] for the assay method) of three independent clones was used to obtain the average and SD values presented in the figures. The value and error bar shown in Figure 8B are respectively calculated from the formula a/c and , where a and b, respectively, represent the value and the SD for ΔIII Ire1 cells shown in Figure 8A, while c and d, respectively, represent the value and the SD for WT Ire1 cells.
RNA analysis
Total RNA samples (2 μg) obtained as described in Kimata et al. (2003) were used for 10 μl-scale reverse transcription reaction with the SuperScript II Reverse Transcriptase kit (Invitrogen, Carlsbad, CA) and a dT18 oligonucleotide primer. The reaction products (2 μl) were then mixed with 1 μl each of 10 μM HAC1 primers (forward: TACAGGGATTTCCAGAGCACG; reverse: TGAAGTGATGAAGAAATCATTCAATTC), 2 μl of 2.5 mM each dNTP mix, 2.5 μl of the supplied 10X PCR buffer, 16.37 μl of water, and 0.13 μl of TAKARA Taq DNA polymerase (5 U/μl), and subjected to a 25-cycle thermal cycle reaction of 94°C for 30 s, 54°C 30 s, and 72°C for 60 s. In, the PCR products were then run on 2% agarose gels (0.5X TBE buffer), and the ethidium bromide–stained fluorescent images were captured by a LAS-4000 Cooled CCD camera system (Fujifilm, Tokyo Japan; Figures 5, A–D, 6C, 7B, 9, C and D, S1, S2, S3A, and S4). As shown in Figures 2B, 4D, and S3B, we used a modified method in which a 6-carboxyfluorescein-labeled oligonucleotide (Invitrogen Custom Primers) was used as the RT-PCR primer, and the PCR products were run on 13% acrylamide gels (40 mM Tris, 20 mM acetic acid, and 1 mM ethylene diamine tetraacetic acid buffer). This allowed us to detect and quantify even faint HAC1i bands, since background gel fluorescence was almost undetectable. The image data were then analyzed by Fujifilm ImageGauge software in order to quantify fluorescence intensity of the HAC1u and the HAC1i bands. The “HAC1 mRNA splicing%” values (100X(HAC1i band signal)/[(HAC1u band signal) + (HAC1i band signal)]) calculated from the data from three independent clones are averaged and presented with the SDs.
Antibodies
Antibodies used in this study are listed in Table S2.
Protein analysis
Cell lysis and immunoprecipitation were performed basically as described previously (Kimata et al., 2003). To obtain the data shown in Figures 3, A and B, and 4, A and B, cells (25 OD600 equivalent) were suspended in 800 μl of PBS and incubated with 2 mM DSP at room temperature for 1 h. The cross-linking reaction was then quenched by addition of Tris-HCl (pH 7.5) to 100 mM and further incubated at room temperature for 30 min before cell lysis. The lysate (equivalent to 1 OD600 cells) or IP (equivalent to 10 OD600 cells) samples were fractionated by using the standard SDS/DTT denaturing PAGE (8% acrylamide) protocol (Kimata et al., 2003). After Western blot analysis of the gel performed as per Kimata et al. (2003), horseradish peroxidase–enhanced chemiluminescence signal was detected on Fujifilm x-ray films (Figures 3A and 4A) or a LAS-4000 system, and quantified with Fujifilm ImageGauge software.
Immunofluorescence microscopy
Cell fixation and immunofluorescence staining were performed as described previously (Kimata et al., 2007). Cells were observed on a Carl Zeiss Axiophoto fluorescence microscope (100×/1.30 Plan-Neofluor objective) carrying an Olympus DP70 CCD camera system (Figures 2C, 6B, and 9A) or a Carl Zeiss Axiovert 200M fluorescence microscope (100×/1.40 Plan-Apochromat objective) equipped with an Apotome deconvolution system (Figure 9B). Adobe Photoshop software (San Jose, CA) was used for image overlapping.
Supplementary Material
Acknowledgments
We thank Kazutoshi Mori (Kyoto University, Kyoto, Japan) for yeast strains and Junko Hashimoto for technical assistance. This work is supported by KAKENHI grants 22657030 and 21112516 to Y.K. and 19058010 and 20380062 to K.K. from the Ministry of Education, Culture, Sports, Science & Technology or Japan Society for the Promotion of Science. This work is also partly supported by a grant from the Noda Institute for Scientific Research to K.K.
Abbreviations used:
- bZIP
basic-region leucine zipper
- CPY
carboxypeptidase Y
- CSSR
core stress-sensing region
- DSP
dithiobis(succinimydylpropionate)
- DTT
dithiothreitol
- ER
endoplasmic reticulum
- FITC
fluorescein isothiocyanate
- GFP
green fluorescent protein
- GPI
glycosylphosphatidylinositol
- HA
hemagglutinin
- IP
immunoprecipitation
- MFY mutation
M229A/F285A/Y301A triple-point mutation
- RT-PCR
reverse transcriptase PCR
- SD
synthetic dextrose
- SGD
Saccharomyces Genome Database
- UPR
unfolded-protein response
- UPRE
UPR promoter element
- YNB w/o aa
yeast nitrogen base without amino acids
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-04-0295) on July 20, 2011.
*These authors contributed equally to this work.
REFERENCES
- Aragon T, van Anken E, Pincus D, Serafimova IM, Korennykh AV, Rubio CA, Walter P. Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature. 2009;457:736–740. doi: 10.1038/nature07641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P. Multifunctional yeast high-copy-number shuttle vectors. Gene. 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- Cox JS, Chapman RE, Walter P. The unfolded protein response coordinates the production of endoplasmic reticulum protein and endoplasmic reticulum membrane. Mol Biol Cell. 1997;8:1805–1814. doi: 10.1091/mbc.8.9.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci USA. 2005;102:18773–18784. doi: 10.1073/pnas.0509487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha DA, et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J Cell Sci. 2008;121:2308–2318. doi: 10.1242/jcs.026062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguil J, Pineau L, Rowland Snyder EC, Dupont S, Beney L, Gil A, Frapper G, Ferreira T. Modulation of lipid-induced ER stress by fatty acid shape. Traffic. 2011;12:349–362. doi: 10.1111/j.1600-0854.2010.01150.x. [DOI] [PubMed] [Google Scholar]
- Duden R, Kajikawa L, Wuestehube L, Schekman R. ε-COP is a structural component of coatomer that functions to stabilize α-COP. EMBO J. 1998;17:985–995. doi: 10.1093/emboj/17.4.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger A, Knop M, Wolf DH. Analysis of two mutated vacuolar proteins reveals a degradation pathway in the endoplasmic reticulum or a related compartment of yeast. Eur J Biochem. 1993;218:565–574. doi: 10.1111/j.1432-1033.1993.tb18410.x. [DOI] [PubMed] [Google Scholar]
- Goder V, Melero A. Protein O-mannosyltransferases participate in ER protein quality control. J Cell Sci. 2011;124:144–153. doi: 10.1242/jcs.072181. [DOI] [PubMed] [Google Scholar]
- Hardwick KG, Lewis MJ, Semenza J, Dean N, Pelham HR. ERD1, a yeast gene required for the retention of luminal endoplasmic reticulum proteins, affects glycoprotein processing in the Golgi apparatus. EMBO J. 1990;9:623–630. doi: 10.1002/j.1460-2075.1990.tb08154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312:572–576. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- Hu CC, Dougan SK, McGehee AM, Love JC, Ploegh HL. XBP-1 regulates signal transduction, transcription factors and bone marrow colonization in B cells. EMBO J. 2009;28:1624–1636. doi: 10.1038/emboj.2009.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- Iwawaki T, Akai R, Kohno K. IRE1α disruption causes histological abnormality of exocrine tissues, increase of blood glucose level, and decrease of serum immunoglobulin level. PLoS One. 2010;5:e13052. doi: 10.1371/journal.pone.0013052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwawaki T, Akai R, Yamanaka S, Kohno K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc Natl Acad Sci USA. 2009;106:16657–16662. doi: 10.1073/pnas.0903775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonikas MC, et al. Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science. 2009;323:1693–1697. doi: 10.1126/science.1167983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaderei B, Kumar P, Wang WJ, Miranda D, Snapp EL, Severina N, Torregroza I, Evans T, Silver DL. Evolutionarily conserved gene family important for fat storage. Proc Natl Acad Sci USA. 2008;105:94–99. doi: 10.1073/pnas.0708579105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiwara K, Watanabe R, Pichler H, Ihara K, Murakami S, Riezman H, Funato K. Yeast ARV1 is required for efficient delivery of an early GPI intermediate to the first mannosyltransferase during GPI assembly and controls lipid flow from the endoplasmic reticulum. Mol Biol Cell. 2008;19:2069–2082. doi: 10.1091/mbc.E07-08-0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara T, Yanagi H, Yura T, Mori K. Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol Biol Cell. 1997;8:1845–1862. doi: 10.1091/mbc.8.10.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata Y, Higashio H, Kohno K. Impaired proteasome function rescues thermosensitivity of yeast cells lacking the coatomer subunit ε-COP. J Biol Chem. 2000;275:10655–10660. doi: 10.1074/jbc.275.14.10655. [DOI] [PubMed] [Google Scholar]
- Kimata Y, Ishiwata-Kimata Y, Ito T, Hirata A, Suzuki T, Oikawa D, Takeuchi M, Kohno K. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J Cell Biol. 2007;179:75–86. doi: 10.1083/jcb.200704166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata Y, Ishiwata-Kimata Y, Yamada S, Kohno K. Yeast unfolded protein response pathway regulates expression of genes for anti-oxidative stress and for cell surface proteins. Genes Cells. 2006;11:59–69. doi: 10.1111/j.1365-2443.2005.00921.x. [DOI] [PubMed] [Google Scholar]
- Kimata Y, Kimata YI, Shimizu Y, Abe H, Farcasanu IC, Takeuchi M, Rose MD, Kohno K. Genetic evidence for a role of BiP/Kar2 that regulates Ire1 in response to accumulation of unfolded proteins. Mol Biol Cell. 2003;14:2559–2569. doi: 10.1091/mbc.E02-11-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata Y, Kohno K. Endoplasmic reticulum stress-sensing mechanisms in yeast and mammalian cells. Curr Opin Cell Biol. 2011;23:135–142. doi: 10.1016/j.ceb.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Kimata Y, Lim CR, Kiriyama T, Nara A, Hirata A, Kohno K. Mutation of the yeast ε-COP gene ANU2 causes abnormal nuclear morphology and defects in intracellular vesicular transport. Cell Struct Funct. 1999;24:197–208. doi: 10.1247/csf.24.197. [DOI] [PubMed] [Google Scholar]
- Kimata Y, Oikawa D, Shimizu Y, Ishiwata-Kimata Y, Kohno K. A role for BiP as an adjustor for the endoplasmic reticulum stress-sensing protein Ire1. J Cell Biol. 2004;167:445–456. doi: 10.1083/jcb.200405153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, Walter P. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457:687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–464. doi: 10.1038/332462a0. [DOI] [PubMed] [Google Scholar]
- Li H, Korennykh AV, Behrman SL, Walter P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc Natl Acad Sci USA. 2010;107:16113–16118. doi: 10.1073/pnas.1010580107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson KL, Ghosh R, Urano F. The role of IRE1alpha in the degradation of insulin mRNA in pancreatic β-cells. PLoS One. 2008;3:e1648. doi: 10.1371/journal.pone.0001648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisbona F, et al. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1α. Mol Cell. 2009;33:679–691. doi: 10.1016/j.molcel.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CY, Schroder M, Kaufman RJ. Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J Biol Chem. 2000;275:24881–24885. doi: 10.1074/jbc.M004454200. [DOI] [PubMed] [Google Scholar]
- Luo D, He Y, Zhang H, Yu L, Chen H, Xu Z, Tang S, Urano F, Min W. AIP1 is critical in transducing IRE1-mediated endoplasmic reticulum stress response. J Biol Chem. 2008;283:11905–11912. doi: 10.1074/jbc.M710557200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka K, Orci L, Amherdt M, Bednarek SY, Hamamoto S, Schekman R, Yeung T. COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell. 1998;93:263–275. doi: 10.1016/s0092-8674(00)81577-9. [DOI] [PubMed] [Google Scholar]
- Merksamer PI, Trusina A, Papa FR. Real-time redox measurements during endoplasmic reticulum stress reveal interlinked protein folding functions. Cell. 2008;28:933–947. doi: 10.1016/j.cell.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K. Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem. 2009;146:743–750. doi: 10.1093/jb/mvp166. [DOI] [PubMed] [Google Scholar]
- Muñiz M, Nuoffer C, Hauri HP, Riezman H. The Emp24 complex recruits a specific cargo molecule into endoplasmic reticulum-derived vesicles. J Cell Biol. 2000;148:925–930. doi: 10.1083/jcb.148.5.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa D, Kimata Y, Kohno K. Self-association and BiP dissociation are not sufficient for activation of the ER stress sensor Ire1. J Cell Sci. 2007;120:1681–1688. doi: 10.1242/jcs.002808. [DOI] [PubMed] [Google Scholar]
- Oikawa D, Kimata Y, Kohno K, Iwawaki T. Activation of mammalian IRE1α upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp Cell Res. 2009;315:2496–2504. doi: 10.1016/j.yexcr.2009.06.009. [DOI] [PubMed] [Google Scholar]
- Oikawa D, Kimata Y, Takeuchi M, Kohno K. An essential dimer-forming subregion of the endoplasmic reticulum stress sensor Ire1. Biochem J. 2005;391:135–142. doi: 10.1042/BJ20050640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Kimata Y, Higashio H, Tsuru A, Kohno K. Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem Biophys Res Commun. 2000;279:445–450. doi: 10.1006/bbrc.2000.3987. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincus D, Chevalier MW, Aragon T, van Anken E, Vidal SE, El-Samad H, Walter P. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol. 2010;8:e1000415. doi: 10.1371/journal.pbio.1000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineau L, Colas J, Dupont S, Beney L, Fleurat-Lessard P, Berjeaud JM, Berges T, Ferreira T. Lipid-induced ER stress: synergistic effects of sterols and saturated fatty acids. Traffic. 2009;10:673–690. doi: 10.1111/j.1600-0854.2009.00903.x. [DOI] [PubMed] [Google Scholar]
- Powers J, Barlowe C. Erv14p directs a transmembrane secretory protein into COPII-coated transport vesicles. Mol Biol Cell. 2002;13:880–891. doi: 10.1091/mbc.01-10-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y, et al. A crucial role for RACK1 in the regulation of glucose-stimulated IRE1α activation in pancreatic β cells. Sci Signal. 2010;3:ra7. doi: 10.1126/scisignal.2000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Hegde RS. Regulation of basal cellular physiology by the homeostatic unfolded protein response. J Cell Biol. 2010;189:783–794. doi: 10.1083/jcb.201003138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner M, Metz J, Schmid V, Denic V, Rakwalska M, Schmitt HD, Schwappach B, Weissman JS. The GET complex mediates insertion of tail-anchored proteins into the ER membrane. Cell. 2008;134:634–645. doi: 10.1016/j.cell.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheff MA, Thorn KS. Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast. 2004;21:661–670. doi: 10.1002/yea.1130. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong F, Billheimer J, Shechtman CF, Liu Y, Crooke R, Graham M, Cohen DE, Sturley SL, Rader DJ. Decreased expression of ARV1 results in cholesterol retention in the endoplasmic reticulum and abnormal bile acid metabolism. J Biol Chem. 2010;285:33632–33641. doi: 10.1074/jbc.M110.165761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006;291:E275–E281. doi: 10.1152/ajpendo.00644.2005. [DOI] [PubMed] [Google Scholar]
- Wiseman RL, Zhang Y, Lee KP, Harding HP, Haynes CM, Price J, Sicheri F, Ron D. Flavonol activation defines an unanticipated ligand-binding site in the kinase-RNase domain of IRE1. Mol Cell. 2009;38:291–304. doi: 10.1016/j.molcel.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Zhang C, Zhang K. Role of unfolded protein response in lipogenesis. World J Hepatol. 2010;2:203–207. doi: 10.4254/wjh.v2.i6.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Liu CY, Back SH, Clark RL, Peisach D, Xu Z, Kaufman RJ. The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface required for activation of the unfolded protein response. Proc Natl Acad Sci USA. 2006;103:14343–14348. doi: 10.1073/pnas.0606480103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.