

Abstract
Heritable neurodevelopmental disorders are multifaceted disease conditions encompassing a wide range of symptoms including intellectual disability, cognitive dysfunction, autism and myriad other behavioral impairments. In cases where single, causative genetic defects have been identified, such as Angelman syndrome, Rett syndrome, Neurofibromatosis Type 1 and Fragile X syndrome, the classical Drosophila genetic system has provided fruitful disease models. Recent Drosophila studies have advanced our understanding of UBE3A, MECP2, NF1 and FMR1 function, respectively, in genetic, biochemical, anatomical, physiological and behavioral contexts. Investigations in Drosophila continue to provide the essential mechanistic understanding required to facilitate the conception of rational therapeutic treatments.
Keywords: Fragile X syndrome, FMRP, Angelman syndrome, UBE3A, Rett syndrome, MeCP2, Neurofibromatosis Type 1, NF1, Drosophila
Introduction
Neurodevelopmental disorders (NDDs) are often characterized by defects in synaptogenesis, synaptic refinement and activity-dependent modulation, resulting in pathologically imbalanced excitatory versus inhibitory neural circuit connectivity [1]. The root of these disorders is often multifactorial, involving atypical genetic susceptibilities coupled with galvanizing environmental influences, as widely hypothesized for autism spectrum disorders (ASDs). However, a smaller number of NDDs are linked to specific, causative single gene disruptions, as in the case of fragile x mental retardation 1 (FMR1) loss of function yielding Fragile X syndrome (FXS). Such monogenic disorders are readily modeled for investigations of disease etiology leading to informed, methodical development of therapeutic intervention strategies. One powerful genetic disease model is Drosophila, whose genome encodes 75% of human disease-associated genes [2]. The rapid generation time, comparatively low cost, and vast arsenal of genetic and transgenic capabilities available in Drosophila provide a proven, fruitful avenue for NDD mechanistic studies. This review will discuss recent Drosophila modeling advances for Angelman syndrome, Rett syndrome, Neurofibromatosis Type 1 and FXS.
Angelman Syndrome
With a prevalence of 1:12–20,000, Angelman syndrome (AS) is clinically characterized by severe developmental delay including marked speech and cognitive impairments, gait ataxia/limb tremulousness, altered electroencephalographic measures often with seizures, and disrupted sleep. AS patients also typically exhibit a uniquely happy demeanor and ready excitability. AS manifests with delayed, progressive onset often symptomatically noted by 3–6 months, with clinical description after 1 year [3]. The genetic disease culprit is ubiquitin ligase 3A (UBE3A), a HECT domain, E6-associated protein that drives ubiquitylation to mediate substrate degradation by the 26S proteasome. In 65–75% of AS cases, UBE3A loss of function in the maternally inherited allele, preferentially expressed in the brain [4], is cytogenetically caused by deletion events spanning 5–7Mb of chromosome 15q11–q13. The remainder of cases are linked to paternal uniparental disomy, imprinting defects and UBE3A mutations [3,5,6].
Drosophila UBE3A (Dube3a), containing a C-terminal HECT domain of 350 amino acids sharing 62% identity with human UBE3A, is ubiquitously expressed during early embryogenesis and readily detected in the developing nervous system [7]. In adult brain, Dube3a remains broadly distributed, including discernible concentration within the mushroom body (MB) learning and memory center [8]. An established model of Dube3a disruption demonstrates robust behavioral defects, including deficient locomotor climbing activity, circadian rhythms and long-term associative olfactory memory (Table 1) [8]. Dube3a variants containing human AS patient missense mutations (R626C, I925K, C55Y, T447P) mirror the behavioral loss of function phenotypes of the Drosophila Dube3a null, indicating strong conservation of function. The single published cellular study in peripheral dendritic arborization (DA) mechanosensory neurons shows cell autonomous neuronal architecture defects, including reduced terminal branching and incomplete receptive target field coverage (Table 1) [9].
Table 1. Comparison of Phenotypic Characterization in Drosophila NDD Models.
The neural circuit and behavioral defects often examined in Drosophila NDD models are illustrated. Neural circuits with both structural and functional outputs are color-coded (locomotor (red), circadian (green), learning/memory (blue)). Model-associated defects are indicated (√), as is one stated lack of discernible defect though data was not shown (—). Absence of published studies is also duly noted (not determined; N.D.). Abbreviations: AS – Angelman syndrome, RTT – Rett syndrome, NF1 – Neurofibromatosis Type 1, FXS – Fragile X syndrome, DA – dendritic arborization neurons, NMJ – neuromuscular junction, sLNv – small ventrolateral circadian neurons, MB – mushroom body neurons, L+M – learning and memory.
| Drosophila-Modeled Disorder | AS | RTT | NF1 | FXS | |
|---|---|---|---|---|---|
| Human Gene | UBE3A | MECP2 | NF1 | FMR1 | |
| Neuronal Architecture Defects | DA | √ | N.D. | N.D. | √ |
| NMJ | N.D. | N.D. | — | √ | |
| sLNv | N.D. | N.D. | N.D. | √ | |
| MB | N.D. | N.D. | N.D. | √ | |
| Behavioral Defects | Sociabilty | N.D. | N.D. | N.D. | √ |
| Locomotion | √ | √ | √ | √ | |
| Sleep | N.D. | N.D. | N.D. | √ | |
| Circadian Rhythms | √ | N.D. | √ | √ | |
| Conditioned Courtship L+M | N.D. | N.D. | N.D. | √ | |
| Pavlovian Olfactory L+M | √ | N.D. | √ | √ | |
Earlier proteomic screens of Drosophila brain with transgenically elevated Dube3a/hUBE3A identified proteins differentially driven toward polyubiquitination and subsequent degradation, including the Rho-GEF Pebble (Figure 1) [7]. Recently, the same approach demonstrated UBE3A-mediated modulation of the GTP cyclohydrolase Punch (Figure 1), an enzyme that produces the rate-limiting co-factor in monoamine biosynthesis, tetrahydrobiopterin (THB) [10]. Punch was elevated by Dube3a overexpression and depressed by loss of function. Accordingly, brain THB, neopterin and dopamine levels were elevated by Dube3a overexpression and decreased by Dube3a RNAi or mutation [10]. This study therefore suggests that altered dopaminergic function may contribute to AS etiology, and may shed light on symptoms associated with UBE3A copy number variants linked to ASD [11,12]. Clinically, two AS patients presenting with Parkinsonism responded positively to levodopa/carbidopa treatment [13], suggesting dopaminergic function involvement in AS. Further evidence for dopaminergic involvement in AS has been uncovered in a murine maternal loss of Ube3a model, in which loss of tyrosine hydroxylase-reactive neurons in the substantia nigra was linked to motor deficits [14]. Interestingly, a much earlier study in the Drosophila FXS disease model similarly showed Punch misregulation and altered brain dopamine synthesis, providing an intriguing FXS and AS molecular link (Figure 1) [15].
Figure 1. Molecular Intersections in Drosophila NDD Models.
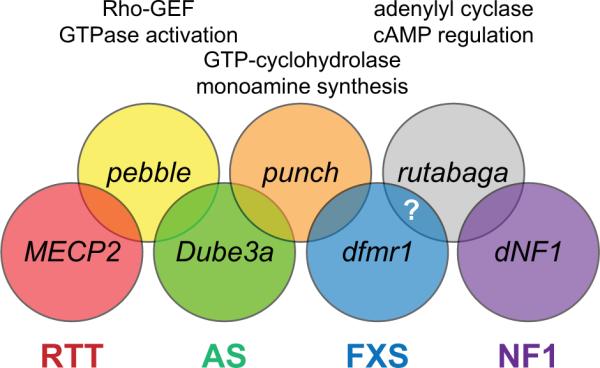
The diagram bottom row illustrates the primary genetic players in Drosophila models of Rett syndrome (RTT; MECP2, red), Angelman syndrome (AS; Dube3a, green), Fragile X syndrome (FXS; dfmr1, blue) and Neurofibromatosis Type 1 (NF1; dNF1, purple). The top row illustrates overlapping molecular interactors between the disease models, including the Rho-GEF pebble (yellow), the GTP cyclohydrolase punch (orange) and the adenylyl cyclase rutabaga (gray). Pebble is a target of the E3-ligase Dube3a, and Pebble attenuation also serves to suppress MeCP2 overexpression phenotypes. Punch is elevated by UBE3A overexpression and dfmr1 loss of function mutations. Rutabaga has long been tied to dNF1 and is likely to interact with dfmr1, as cAMP levels are altered in the Drosophila FXS model and FXS disease state [76].
Rett Syndrome
The X-linked NDD Rett syndrome (RTT) is a leading cause of female intellectual disability, with a prevalence of 1:10,000. In RTT patients, development usually progresses normally until 6–18 months, with subsequent, regressive loss of acquired proficiencies in expressivity and motor skills coupled with ongoing cognitive impairment, autistic behaviors and seizures [16–18]. In 90% of RTT cases, the genetic culprit is disruption of the transcriptional regulator methyl-CpG-binding protein 2 (MECP2; Xq28). The remainder of cases are associated with MECP2 duplication events [19,20] or mutations in 1) cyclin-dependent kinase-like 5 (CDKL5) [21–23] or 2) the transcriptional repressor Forkhead box protein G1 (FOXG1) [24,25]. These two latter molecular players colocalize with MeCP2 in nuclei of postnatal cortical neurons during maturation and synaptogenesis [21,22]. MeCP2 and CDKL5 interact directly, both in vitro and in vivo via GST-pull down and co-immunoprecipitation assays, with CDKL5 mediating MeCP2 phosphorylation [22].
Drosophila lacks an identifiable MECP2 homolog, and thus deletion modeling cannot be pursued [26]. Instead, modeling has been based on overexpression of human MECP2 and three RTT patient mutant alleles (R106W, R294X, and Δ166) [27–29]. The resultant eye, wing vein and locomotor phenotypes (Table 1) have formed the bases for enhancer/suppressor genetic screens [30]. In such screens, mutagenized animals are examined for second-site genetic hits that either enhance or suppress the base phenotypes, identifying genetic effectors and/or molecular interactors impinging upon the disease pathway. A candidate gene approach examining MeCP2-associating proteins linked to mammalian transcriptional repression revealed loss of function in histone deacetylase complex component Sin3 homolog A (Sin3A) acts as an enhancer, whereas loss of function in the co-repressive SMRT-related ecdysone receptor-interacting factor (Smr) and the multiple zinc finger-containing transcription factor crooked legs (crol) both act as suppressors. Suppression of wildtype MECP2 overexpression phenotypes also occured with concomitant partial loss of function in chromatin remodeling genes (additional sex combs (Asx), sex combs on midleg (Scm), corto and osa), overexpression of the kinase tricornered (trc), and loss of the Dube3a target pebble (pbl) [30]. These findings suggest potential RTT treatment strategies targeting these MeCP2 interactors, as well as a possible molecular link between AS and RTT (Figure 1).
Neurofibromatosis Type 1
Neurofibromatosis type 1 (NF1) is a more common NDD (1:2,500–5,000) characterized by hallmark benign tumors on peripheral nerves (neurofibromas) and specific cognitive impairments, including difficulties with attention, executive function, language, visual perception and learning [31–33]. The genetic disease culprit is loss of NF1 at cytological location 17q11.2, which encodes the Ras GTPase activating protein (RasGAP)/adenylate cyclase (AC) activator, neurofibromin. Disruption of NF1 function alters the ability to restrain cellular proliferation and inhibit protein translation via the mammalian target of rapamycin (mTOR) pathway [34]. In addition, increased Ras signaling in inhibitory interneurons increases activity-dependent GABA release in the hippocampus, likely causing a number of the NF1 behavioral symptoms [35].
A Drosophila model based on the dNF1 homolog (60% identity to human NF1) was established 14 years ago [36]. dNF1 is involved in both Ras- and cAMP-dependent pathways interacting with the rutabaga (rut) gene encoding AC (Figure 1) [36,37]. NF1 activation of the Rut/AC pathway is an essential component of Drosophila learning and memory (Table 1) [38]. In these behavioral studies, a Pavlovian olfactory assay trains animals by associating an odor with an electric shock punishment, paired with exposure to another odorant without shock, thus promoting learning and memory consolidation of conditioned-avoidance [39,40]. The immediate association assay after training (~15 minutes) measures learning, whereas short-term, middle-term, anesthesia-resistant and long-term memory (STM, MTM, ARM and LTM, respectively) are tracked through training paradigm variations (e.g. massed vs. spaced) coupled with time-dependent examination of retained avoidance behavior (e.g. STM decays within one hour of training, and LTM is detectable at 24 hours) [41]. In addition to learning and STM, NF1 also regulates the behavioral escape flight response [36] and circadian rhythmicity [42].
More recently, introduction of human NF1 into the Drosophila disease model has proved conservation of gene function. Human NF1 deletion constructs and NF1 patient point mutations (R1391S, K1423E – reducing Ras affinity; R1276P – reducing GAP activity) yield normal learning and ARM but defective LTM in the Drosophila disease model, whereas the NF1 C-terminal region differentially mediates immediate memory [43]. Most recently, dNF1 was shown to act during the operational phase of memory acquisition, and not during stabilization and maintenance [44]. RNA in situ studies have revealed previously elusive dNF1 expression within the Drosophila central brain, including the MB learning and memory center. Exploitation of the spatially and temporally inducible transgenic Gene-Switch system has shown that adult dNF1 expression is sufficient to rescue 3-hour memory defects in the Drosophila disease model, with a selective requirement in MB α/β neurons, as α′/β′ or γ expression does not rescue behavioral defects [44]. These precise cellular delineations should aid in elucidating NF1 roles within key populations of neurons differentially involved in learning and memory.
Fragile X Syndrome
Fragile X syndrome (FXS) is the most common heritable cause of intellectual disability and ASD, conservatively affecting 1:4,000 males and 1:8,000 females, with recent estimates suggesting full mutation frequency as high as 1:2,500 [45]. The genetic culprit is most commonly an unstable CGG-trinucleotide repeat expansion in the 5' regulatory region of the fragile x mental retardation 1 gene (FMR1; Xq27.3). This expansion causes hypermethylation and transcriptional silencing, resulting in loss of the mRNA-binding FMRP involved in transcript stability, trafficking and translation control. FXS is clinically characterized by delayed and depressed developmental trajectories, working memory deficits, disordered sleep, hypersensitivity to sensory stimuli, seizures, elevated anxiety, hyperactivity and 30% autism comorbidity [46,47].
FMRP loss has long been studied in both murine [48] and Drosophila models [49] with well-characterized defects in neuronal architecture, inappropriate activity-dependent pruning, altered neurotransmission and compromised behavioral output including disrupted circadian rhythms and defects in learning and memory (Table 1) [50–52]. Drosophila has a single FMR1 gene (dfmr1), as opposed to the murine tripartite FMR1/FXR1/FXR2 gene family, but unique conservation of FMR1 function was recently established [53]. In the Drosophila disease model (dfmr1 null), each human gene family member was expressed both in the germline and nervous system. Neither hFXR1 nor hFXR2 could restore any aspect of neuronal dysfunction, whereas hFMR1 fully rescued all defects, including elevated translation and overelaborated architectural complexity, as effectively as native dFMR1 [53]. In contrast, all three human gene family members were equally competent in rescuing non-neuronal dfmr1 phenotypes in the male testes. These results show that FMR1 maintains a unique function within neurons required for the translational control governing synaptogenesis and synaptic refinement.
Outside of these well-established functions, dFMRP was recently shown to play roles in cellular proliferation. This requirement was first evident in the germ line and during early embryonic development [54–58]. Importantly, dFMRP later also regulates the exit from quiescence and proliferative capacity of neural stem cells [59]. In dfmr1 null neuroblasts, elevated cyclin E during cell cycle progression drives the G1/S transition and increases cell numbers in G2/M, enhancing 5-bromo-2-deoxyuridine incorporation (an S-phase indicator). Clonal analyses using the mosaic analysis with a repressible cell marker (MARCM) technique demonstrated that dfmr1 null neuroblasts generate more neurons than controls, which were retained into adulthood [59]. These findings suggest that FXS neurological dysfunction may be due to aberrant connectivity resulting from supernumerary neuron incorporation, in addition to known defects in synaptogenesis and synaptic pruning.
FMRP has long been suggested to act as a translational repressor, first demonstrated in vivo in the Drosophila disease model by dFMRP acting as a negative regulator of the microtubule-binding MAP1B homolog Futsch [49]. More recently, a genetic suppressor screen based on the retinal disorganization caused by dfmr1 overexpression sought to uncover interactors in this translation mechanism and identified poly-A binding protein (pabp), discs overgrown/doubletime (dco/dbt), oo18 RNA-binding protein 2 (orb2), DEAD-box, ATP-dependent RNA helicase p62/68 (rm62/Dmp68) and small ribonucleoprotein smD3 (smD3) [60]. These players are dFMRP-RNA granule components, and their overexpression inhibited dendritic branching and complexity in class IV-type sensory DA neurons, phenocopying overexpression of dfmr1 (Table 1). With transport of such FMRP-RNA complexes achieved via microtubule motors, it is important to note that dFMRP was shown to complex with the dynein-binding Bicaudal-D (BicD) [61]. Interestingly, BicD mutants depress neuronal dFMRP::GFP particle abundance and motility, suggesting BicD serves to positively regulate dFMRP distribution in neuronal processes. Recently, dfmr1 has also been shown to genetically interact with dspastin, encoding a microtubule-severing protein whose loss is causally linked to neurodegenerative Hereditary Spastic Paraplegia (HSP), also modeled in Drosophila [62]. Genetic cooperativity between dfmr1 and dspastin was evident in microtubule network organization, neuromuscular junction (NMJ) synapse formation, and locomotor function (Table 1) [63]. There was an inverse correlation between dFMRP levels and mitochondrial abundance in axons and NMJ synaptic terminals, with dfmr1 also negatively regulating the flux and processivity of mitochondrial transport. Thus, dFMRP seems to bidirectionally interact with the neuronal microtubule cytoskeleton to mediate neuronal architecture and trafficking mechanisms.
The Drosophila FXS model has focused particularly on neural circuits driving two disease-relevant behaviors: 1) circadian activity and 2) learning/memory [50]. In dfmr1 mutants, the small ventrolateral clock neurons (sLNvs) exhibit synaptic overgrowth and overelaboration in the dorsal brain protocerebrum (Table 1) [53,64–67]. Conditional reintroduction of wildtype dFMRP using the Gene-Switch system showed that transient expression during late pupal brain development (P3/4) results in striking rescue of synaptic defects. In contrast, dFMRP expression either earlier in development (larval, P1/2) or in the adult provided absolutely no benefit [64]. This study indicates dFMRP plays a temporally restricted role during late brain development, when synaptogenesis and use-dependent pruning occurs, suggesting that FXS therapeutic interventions may be most beneficially targeted towards children. As predicted by clock circuit defects, dfmr1 nulls exhibit profound disruption of circadian rhythmicity and sleep regulation (Table 1) [68]. Reportedly, dfmr1 null mutants display elevated sleep, including increased daytime sleep, prolonged sleep through the anticipatory period at the termination of dark phase, and increased initiation of sleep episodes, with additional impairments in waking, improper sleep rebound after deprivation and increased locomotor activity. Importantly, dFMRP expression in the MB circuit was sufficient to rescue these defects [68]. The MB is more typically studied based on its central role in olfactory learning and memory. Null dfmr1 MB defects include axon lobe malformations, inappropriate axon midline crossing, and excessive dendritic/axonal structural elaboration (Table 1) [50,52]. Recently, MB calcium signaling has been studied using a transgenic Ca2+ reporter (UAS-GCaMP) [69]. Null dfmr1 MB neurons display development stage-specific increases in depolarization-induced Ca2+ transients as well as excessive Ca2+ mobilization from internal stores. Mutants also show decreased brain transcript levels for several Ca2+-binding proteins, including frequenin1/2, calmodulin and calbindin [69]. These findings suggest that calcium signaling defects likely contribute to FXS pathophysiology underlying learning and memory impairments.
Null dfmr1 mutants show striking defects in two commonly employed learning/memory assays: 1) Pavlovian olfactory association [70] and 2) courtship conditioning [71] (Table 1). Recent studies of the former show dfmr1 genetically interacts with cheerio, the actin remodeling Filamin A homolog associated with Periventricular Nodular Heterotopia (PNH), with double heterozygotes defective in acquisition of protein synthesis-dependent LTM [72]. Moreover, cheerio expression is decreased in dfmr1 MB after LTM-inducing spaced training, suggesting that disrupted actin organization may underlie defects in memory formation and retention. In courtship studies, recent work suggests differential requirements for dfmr1 splice isoforms [73]. An alternatively spliced C-terminal glutamine/asparagine (Q/N)-rich domain is essential for socialization enabling normal naïve courtship levels, influences STM, and is required, but not sufficient, for LTM [73]. A separate study illustrated age-dependent cognitive impairment in dfmr1 nulls with learning loss during training [74]. Importantly, treatment with metabotropic glutamate receptor (mGluR) antagonists or lithium rescued both the novel learning and previously identified STM defects when introduced either during development or adulthood [74]. The fact that early developmental treatment remains effective in adults suggests age-dependent phenotypes are largely predetermined by developmental defects. Thus, early FXS interventions may be curative for otherwise maintained behavioral impairments. Conversely, it is encouraging that adult-onset intervention also successfully alleviated these behavioral phenotypes, as it suggests a retained, accessible plasticity sufficient for correction at maturity.
Finally, the Drosophila FXS model has even taken a step towards assaying social interaction relevant to autism (Table 1) [75]. A recent study examining exploratory behavior and interfly distances reported that dfmr1 mutants were surprisingly hypoactive and interact less with one another than with controls, showing a decreased sociability index. Interestingly, dfmr1 nulls interact more with a control wildtype fly than another dfmr1 mutant [75], suggesting mutants may exhibit a form of motor dyspraxia, often described in ASD, wherein a normal receptive response to interaction is intact but internally compromised due to failure of appropriate motor output display required to maintain engagement with a partner. Such assays may aid in the detection and study of autistic phenotypes in other Drosophila NDD models.
Conclusions
The development of Drosophila NDD models has advanced considerably in recent years, including AS, RTT, NF1 and FXS models. Owing to powerful advantages in forward genetic and candidate-based screens, these models have identified key interactors of UBE3A, MeCP2, neurofibromin and FMRP (Figure 1). These discoveries shed light on molecular pathways that are viable targets for the design of pharmaceutical intervention strategies. Moreover, study of defined neural circuits and disease-related behaviors is rapidly advancing in Drosophila NDD models, including the circuitry and output for locomotor capacity, circadian rhythmicity and learning/memory (Table 1). These advances are now allowing for coupled developmental and cell autonomous dissection of molecular requirements. We are confident that the continuing evolution of Drosophila NDD models will enable understanding of the mechanistic bases underlying, and likely intersecting amongst, these devastating neurological disorders.
Research Highlights
75% of identified human disease-associated genes are present in Drosophila.
Angelman, Rett, Neurofibromatosis and Fragile X syndromes have been modeled.
Genetics, biochemistry, anatomy, physiology and behavior have been examined.
Drosophila has advanced understanding of UBE3A, MECP2, NF1 and FMR1 biology.
Acknowledgements
We would like to thank members of the Broadie lab, especially Saul Siller, for helpful discussions during manuscript preparation. This work was supported by National Institutes of Mental Health R01 MH084989 to K.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest The authors declare that this body of work was completed in the absence of any commercial or financial relationships that could be possibly construed as a potential conflict of interest.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Gatto CL, Broadie K. Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models. Front Syn Neurosci. 2010;2 doi: 10.3389/fnsyn.2010.00004. doi: 10.3389/fnsyn.2010.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This recent review synthesizes our current understanding of excitatory vs. inhibitory synaptic ratio imbalance in murine and Drosophila NDD models. Molecular alterations observed in ASD, epilepsy, RTT and FXS are discussed with disease commonalities highlighted.
- 2.Reiter LT, Potocki L, Chien S, Gribskov M, Bier E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001;11:1114–1125. doi: 10.1101/gr.169101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williams CA, Driscoll DJ, Dagli AI. Clinical and genetic aspects of Angelman syndrome. Genet Med. 2010;12:385–395. doi: 10.1097/GIM.0b013e3181def138. [DOI] [PubMed] [Google Scholar]
- 4.Yamasaki K, Joh K, Ohta T, Masuzaki H, Ishimaru T, Mukai T, Niikawa N, Ogawa M, Wagstaff J, Kishino T. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum Mol Genet. 2003;12:837–847. doi: 10.1093/hmg/ddg106. [DOI] [PubMed] [Google Scholar]
- 5.Chamberlain SJ, Lalande M. Neurodevelopmental disorders involving genomic imprinting at human chromosome 15q11–q13. Neurobiol Dis. 2010;39:13–20. doi: 10.1016/j.nbd.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 6.Dan B. Angelman syndrome: current understanding and research prospects. Epilepsia. 2009;50:2331–2339. doi: 10.1111/j.1528-1167.2009.02311.x. [DOI] [PubMed] [Google Scholar]
- 7.Reiter LT, Seagroves TN, Bowers M, Bier E. Expression of the Rho-GEF Pbl/ECT2 is regulated by the UBE3A E3 ubiquitin ligase. Hum Mol Genet. 2006;15:2825–2835. doi: 10.1093/hmg/ddl225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu Y, Bolduc FV, Bell K, Tully T, Fang Y, Sehgal A, Fischer JA. A Drosophila model for Angelman syndrome. Proc Natl Acad Sci U S A. 2008;105:12399–12404. doi: 10.1073/pnas.0805291105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu Y, Wang F, Li Y, Ferris J, Lee JA, Gao FB. The Drosophila homologue of the Angelman syndrome ubiquitin ligase regulates the formation of terminal dendritic branches. Hum Mol Genet. 2009;18:454–462. doi: 10.1093/hmg/ddn373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferdousy F, Bodeen W, Summers K, Doherty O, Wright O, Elsisi N, Hilliard G, O'Donnell JM, Reiter LT. Drosophila Ube3a regulates monoamine synthesis by increasing GTP cyclohydrolase I activity via a non-ubiquitin ligase mechanism. Neurobiol Dis. 2010 doi: 10.1016/j.nbd.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; • A proteomic screen of Dube3a/hUBE3A overexpression in Drosophila identified the GTP cyclohydrolase Punch as a target of UBE3A regulation controlling brain dopamine levels in this AS model. A strikingly similar result was previously shown for the Drosophila FXS model.
- 11.Bucan M, Abrahams BS, Wang K, Glessner JT, Herman EI, Sonnenblick LI, Alvarez Retuerto AI, Imielinski M, Hadley D, Bradfield JP, et al. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 2009;5:e1000536. doi: 10.1371/journal.pgen.1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harbord M. Levodopa responsive Parkinsonism in adults with Angelman Syndrome. J Clin Neurosci. 2001;8:421–422. doi: 10.1054/jocn.2000.0753. [DOI] [PubMed] [Google Scholar]
- 14.Mulherkar SA, Jana NR. Loss of dopaminergic neurons and resulting behavioural deficits in mouse model of Angelman syndrome. Neurobiol Dis. 2010;40:586–592. doi: 10.1016/j.nbd.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 15.Zhang YQ, Friedman DB, Wang Z, Woodruff E, 3rd, Pan L, O'Donnell J, Broadie K. Protein expression profiling of the drosophila fragile X mutant brain reveals up-regulation of monoamine synthesis. Mol Cell Proteomics. 2005;4:278–290. doi: 10.1074/mcp.M400174-MCP200. [DOI] [PubMed] [Google Scholar]
- 16.Gonzales ML, LaSalle JM. The role of MeCP2 in brain development and neurodevelopmental disorders. Curr Psychiatry Rep. 2010;12:127–134. doi: 10.1007/s11920-010-0097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Na ES, Monteggia LM. The role of MeCP2 in CNS development and function. Horm Behav. 2010 doi: 10.1016/j.yhbeh.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuishi T, Yamashita Y, Takahashi T, Nagamitsu S. Rett syndrome: The state of clinical and basic research, and future perspectives. Brain Dev. 2011 doi: 10.1016/j.braindev.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 19.Ramocki MB, Peters SU, Tavyev YJ, Zhang F, Carvalho CM, Schaaf CP, Richman R, Fang P, Glaze DG, Lupski JR, et al. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann Neurol. 2009;66:771–782. doi: 10.1002/ana.21715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramocki MB, Tavyev YJ, Peters SU. The MECP2 duplication syndrome. Am J Med Genet A. 2010;152A:1079–1088. doi: 10.1002/ajmg.a.33184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ariani F, Hayek G, Rondinella D, Artuso R, Mencarelli MA, Spanhol-Rosseto A, Pollazzon M, Buoni S, Spiga O, Ricciardi S, et al. FOXG1 is responsible for the congenital variant of Rett syndrome. Am J Hum Genet. 2008;83:89–93. doi: 10.1016/j.ajhg.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mari F, Azimonti S, Bertani I, Bolognese F, Colombo E, Caselli R, Scala E, Longo I, Grosso S, Pescucci C, et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum Mol Genet. 2005;14:1935–1946. doi: 10.1093/hmg/ddi198. [DOI] [PubMed] [Google Scholar]
- 23.Sprovieri T, Conforti FL, Fiumara A, Mazzei R, Ungaro C, Citrigno L, Muglia M, Arena A, Quattrone A. A novel mutation in the X-linked cyclin-dependent kinase-like 5 (CDKL5) gene associated with a severe Rett phenotype. Am J Med Genet A. 2009;149A:722–725. doi: 10.1002/ajmg.a.32711. [DOI] [PubMed] [Google Scholar]
- 24.Jacob FD, Ramaswamy V, Andersen J, Bolduc FV. Atypical Rett syndrome with selective FOXG1 deletion detected by comparative genomic hybridization: case report and review of literature. Eur J Hum Genet. 2009;17:1577–1581. doi: 10.1038/ejhg.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mencarelli MA, Spanhol-Rosseto A, Artuso R, Rondinella D, De Filippis R, Bahi-Buisson N, Nectoux J, Rubinsztajn R, Bienvenu T, Moncla A, et al. Novel FOXG1 mutations associated with the congenital variant of Rett syndrome. J Med Genet. 2010;47:49–53. doi: 10.1136/jmg.2009.067884. [DOI] [PubMed] [Google Scholar]
- 26.Hendrich B, Tweedie S. The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet. 2003;19:269–277. doi: 10.1016/S0168-9525(03)00080-5. [DOI] [PubMed] [Google Scholar]
- 27.Meins M, Lehmann J, Gerresheim F, Herchenbach J, Hagedorn M, Hameister K, Epplen JT. Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J Med Genet. 2005;42:e12. doi: 10.1136/jmg.2004.023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Esch H, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gecz J, et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.del Gaudio D, Fang P, Scaglia F, Ward PA, Craigen WJ, Glaze DG, Neul JL, Patel A, Lee JA, Irons M, et al. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet Med. 2006;8:784–792. doi: 10.1097/01.gim.0000250502.28516.3c. [DOI] [PubMed] [Google Scholar]
- 30.Cukier HN, Perez AM, Collins AL, Zhou Z, Zoghbi HY, Botas J. Genetic modifiers of MeCP2 function in Drosophila. PLoS Genet. 2008;4:e1000179. doi: 10.1371/journal.pgen.1000179. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This report details the establishment of a RTT MECP2 duplication model in Drosophila and provides evidence of its utility in the identification of MeCP2 interactors.
- 31.Payne JM, Moharir MD, Webster R, North KN. Brain structure and function in neurofibromatosis type 1: current concepts and future directions. J Neurol Neurosurg Psychiatry. 2010;81:304–309. doi: 10.1136/jnnp.2009.179630. [DOI] [PubMed] [Google Scholar]
- 32.Shilyansky C, Lee YS, Silva AJ. Molecular and cellular mechanisms of learning disabilities: a focus on NF1. Annu Rev Neurosci. 2010;33:221–243. doi: 10.1146/annurev-neuro-060909-153215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferner RE. The neurofibromatoses. Pract Neurol. 2010;10:82–93. doi: 10.1136/jnnp.2010.206532. [DOI] [PubMed] [Google Scholar]
- 34.Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci U S A. 2005;102:8573–8578. doi: 10.1073/pnas.0503224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cui Y, Costa RM, Murphy GG, Elgersma Y, Zhu Y, Gutmann DH, Parada LF, Mody I, Silva AJ. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell. 2008;135:549–560. doi: 10.1016/j.cell.2008.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.The I, Hannigan GE, Cowley GS, Reginald S, Zhong Y, Gusella JF, Hariharan IK, Bernards A. Rescue of a Drosophila NF1 mutant phenotype by protein kinase A. Science. 1997;276:791–794. doi: 10.1126/science.276.5313.791. [DOI] [PubMed] [Google Scholar]
- 37.Guo HF, The I, Hannan F, Bernards A, Zhong Y. Requirement of Drosophila NF1 for activation of adenylyl cyclase by PACAP38-like neuropeptides. Science. 1997;276:795–798. doi: 10.1126/science.276.5313.795. [DOI] [PubMed] [Google Scholar]
- 38.Guo HF, Tong J, Hannan F, Luo L, Zhong Y. A neurofibromatosis-1-regulated pathway is required for learning in Drosophila. Nature. 2000;403:895–898. doi: 10.1038/35002593. [DOI] [PubMed] [Google Scholar]
- 39.Tully T, Quinn WG. Classical conditioning and retention in normal and mutant Drosophila melanogaster. J Comp Physiol [A] 1985;157:263–277. doi: 10.1007/BF01350033. [DOI] [PubMed] [Google Scholar]
- 40.Connolly JB, Tully T. Behaviour, learning, and memory. In: Roberts DB, editor. Drosophila, a practical approach. 2nd edn. Oxford University Press; 1998. pp. 265–318. [Google Scholar]
- 41.Margulies C, Tully T, Dubnau J. Deconstructing memory in Drosophila. Curr Biol. 2005;15:R700–713. doi: 10.1016/j.cub.2005.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams JA, Su HS, Bernards A, Field J, Sehgal A. A circadian output in Drosophila mediated by neurofibromatosis-1 and Ras/MAPK. Science. 2001;293:2251–2256. doi: 10.1126/science.1063097. [DOI] [PubMed] [Google Scholar]
- 43.Ho IS, Hannan F, Guo HF, Hakker I, Zhong Y. Distinct functional domains of neurofibromatosis type 1 regulate immediate versus long-term memory formation. J Neurosci. 2007;27:6852–6857. doi: 10.1523/JNEUROSCI.0933-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buchanan ME, Davis RL. A distinct set of Drosophila brain neurons required for neurofibromatosis type 1-dependent learning and memory. J Neurosci. 2010;30:10135–10143. doi: 10.1523/JNEUROSCI.0283-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study critically demonstrates dNF1 RNA distribution in the MB learning and memory center of the Drosophila brain, with delineation of both developmental and Kenyon cell type-specific dNF1 requirements for appropriate functions in behavioral output.
- 45.Hagerman PJ. The fragile X prevalence paradox. J Med Genet. 2008;45:498–499. doi: 10.1136/jmg.2008.059055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harris SW, Hessl D, Goodlin-Jones B, Ferranti J, Bacalman S, Barbato I, Tassone F, Hagerman PJ, Herman H, Hagerman RJ. Autism profiles of males with fragile X syndrome. Am J Ment Retard. 2008;113:427–438. doi: 10.1352/2008.113:427-438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hagerman R. Commonalities in the neurobiology between autism and fragile X. J Intellect Disabil Res. 2008;52:817. [Google Scholar]
- 48.Bakker C, Verheij C, Willemsen R, van der Helm R. Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- 49.Zhang YQ, Bailey AM, Matthies HJ, Renden RB, Smith MA, Speese SD, Rubin GM, Broadie K. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell. 2001;107:591–603. doi: 10.1016/s0092-8674(01)00589-x. [DOI] [PubMed] [Google Scholar]
- 50.Gatto CL, Broadie K. The fragile x mental retardation protein in circadian rhythmicity and memory consolidation. Mol Neurobiol. 2009;39:107–129. doi: 10.1007/s12035-009-8057-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tessier CR, Broadie K. Activity-dependent modulation of neural circuit synaptic connectivity. Front Mol Neurosci. 2009;2:8. doi: 10.3389/neuro.02.008.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bhogal B, Jongens TA. Fragile X syndrome and model organisms: identifying potential routes of therapeutic intervention. Dis Model Mech. 2010;3:693–700. doi: 10.1242/dmm.002006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coffee RL, Jr., Tessier CR, Woodruff EA, 3rd, Broadie K. Fragile X mental retardation protein has a unique, evolutionarily conserved neuronal function not shared with FXR1P or FXR2P. Dis Model Mech. 2010;3:471–485. doi: 10.1242/dmm.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study proved the functional conservation of FMRP between human and Drosophila, illustrating that FMRP has a unique neuronal requirement not shared by either FXR1P or FXR2P.
- 54.Epstein AM, Bauer CR, Ho A, Bosco G, Zarnescu DC. Drosophila Fragile X protein controls cellular proliferation by regulating cbl levels in the ovary. Dev Biol. 2009;330:83–92. doi: 10.1016/j.ydbio.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 55.Yang Y, Xu S, Xia L, Wang J, Wen S, Jin P, Chen D. The bantam microRNA is associated with drosophila fragile X mental retardation protein and regulates the fate of germline stem cells. PLoS Genet. 2009;5:e1000444. doi: 10.1371/journal.pgen.1000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Monzo K, Dowd SR, Minden JS, Sisson JC. Proteomic analysis reveals CCT is a target of Fragile X mental retardation protein regulation in Drosophila. Dev Biol. 2010;340:408–418. doi: 10.1016/j.ydbio.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Papoulas O, Monzo KF, Cantin GT, Ruse C, Yates JR, 3rd, Ryu YH, Sisson JC. dFMRP and Caprin, translational regulators of synaptic plasticity, control the cell cycle at the Drosophila mid-blastula transition. Development. 2010;137:4201–4209. doi: 10.1242/dev.055046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pepper AS, Beerman RW, Bhogal B, Jongens TA. Argonaute2 suppresses Drosophila fragile X expression preventing neurogenesis and oogenesis defects. PLoS One. 2009;4:e7618. doi: 10.1371/journal.pone.0007618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Callan MA, Cabernard C, Heck J, Luois S, Doe CQ, Zarnescu DC. Fragile X protein controls neural stem cell proliferation in the Drosophila brain. Hum Mol Genet. 2010;19:3068–3079. doi: 10.1093/hmg/ddq213. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• The authors examine neural stem cell hyperproliferation caused by loss of dfmr1, resulting in elevated neuronal cell numbers maintained into adulthood. This defect suggests that aberrant cell numbers in neural circuits may contribute to FXS dysfunction.
- 60.Cziko AM, McCann CT, Howlett IC, Barbee SA, Duncan RP, Luedemann R, Zarnescu D, Zinsmaier KE, Parker RR, Ramaswami M. Genetic modifiers of dFMR1 encode RNA granule components in Drosophila. Genetics. 2009;182:1051–1060. doi: 10.1534/genetics.109.103234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bianco A, Dienstbier M, Salter HK, Gatto G, Bullock SL. Bicaudal-D regulates fragile X mental retardation protein levels, motility, and function during neuronal morphogenesis. Curr Biol. 2010;20:1487–1492. doi: 10.1016/j.cub.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Trotta N, Orso G, Rossetto MG, Daga A, Broadie K. The hereditary spastic paraplegia gene, spastin, regulates microtubule stability to modulate synaptic structure and function. Curr Biol. 2004;14:1135–1147. doi: 10.1016/j.cub.2004.06.058. [DOI] [PubMed] [Google Scholar]
- 63.Yao A, Jin S, Li X, Liu Z, Ma X, Tang J, Zhang YQ. Drosophila FMRP regulates microtubule network formation and axonal transport of mitochondria. Hum Mol Genet. 2010;20:51–63. doi: 10.1093/hmg/ddq431. [DOI] [PubMed] [Google Scholar]; •• This study details a genetic interaction between dfmr1 and dspastin initially identified via a genetic screen. Novel implications for FXS pathophysiology are uncovered with the observation of dFMRP-mediated regulation of microtubule network organization and changes in axonal mitochondrial abundance and transport.
- 64.Gatto CL, Broadie K. Temporal requirements of the fragile x mental retardation protein in modulating circadian clock circuit synaptic architecture. Front Neural Circuits. 2009;3:8. doi: 10.3389/neuro.04.008.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study shows that dFMRP is required during a brief period of late brain development and early use refinement to establish correct circadian neural circuit architecture. Earlier or later replacement of dFMRP was insufficient to provide any detectable restoration of circuit connectivity. This conclusion has important implications for the timing of FXS interventions.
- 65.Dockendorff TC, Su HS, McBride SM, Yang Z, Choi CH, Siwicki KK, Sehgal A, Jongens TA. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron. 2002;34:973–984. doi: 10.1016/s0896-6273(02)00724-9. [DOI] [PubMed] [Google Scholar]
- 66.Reeve SP, Bassetto L, Genova GK, Kleyner Y, Leyssen M, Jackson FR, Hassan BA. The Drosophila fragile X mental retardation protein controls actin dynamics by directly regulating profilin in the brain. Curr Biol. 2005;15:1156–1163. doi: 10.1016/j.cub.2005.05.050. [DOI] [PubMed] [Google Scholar]
- 67.Reeve SP, Lin X, Sahin BH, Jiang F, Yao A, Liu Z, Zhi H, Broadie K, Li W, Giangrande A, et al. Mutational analysis establishes a critical role for the N terminus of fragile X mental retardation protein FMRP. J Neurosci. 2008;28:3221–3226. doi: 10.1523/JNEUROSCI.5528-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bushey D, Tononi G, Cirelli C. The Drosophila fragile X mental retardation gene regulates sleep need. J Neurosci. 2009;29:1948–1961. doi: 10.1523/JNEUROSCI.4830-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tessier CR, Broadie K. The fragile X mental retardation protein developmentally regulates the strength and fidelity of calcium signaling in Drosophila mushroom body neurons. Neurobiol Dis. 2010 doi: 10.1016/j.nbd.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This is the first use of a genetically encoded calcium sensor in the Drosophila FXS model. The authors examine both developmentally regulated and acute depolarization-induced calcium dynamics in the MB learning/memory circuit, changes that are likely linked to misexpressed calcium-binding proteins in the brain.
- 70.Bolduc FV, Bell K, Cox H, Broadie KS, Tully T. Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat Neurosci. 2008;11:1143–1145. doi: 10.1038/nn.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McBride SM, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D, Sehgal A, Siwicki KK, Dockendorff TC, Nguyen HT, et al. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron. 2005;45:753–764. doi: 10.1016/j.neuron.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 72.Bolduc FV, Bell K, Rosenfelt C, Cox H, Tully T. Fragile x mental retardation 1 and filamin a interact genetically in Drosophila long-term memory. Front Neural Circuits. 2010;3:22. doi: 10.3389/neuro.04.022.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Using the oflactory learning/memory paradigm, this report describes the interaction between the Filamin A homolog cheerio and dfmr1 in protein synthesis-dependent LTM providing further evidence that cytoskeletal disruptions are linked to a host of FXS phenotypes.
- 73.Banerjee P, Schoenfeld BP, Bell AJ, Choi CH, Bradley MP, Hinchey P, Kollaros M, Park JH, McBride SM, Dockendorff TC. Short- and long-term memory are modulated by multiple isoforms of the fragile X mental retardation protein. J Neurosci. 2010;30:6782–6792. doi: 10.1523/JNEUROSCI.6369-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study dissects dFMRP isoform-specific function in conditioned courtship learning and memory, critically demonstrating the necessity of a Q/N-rich C-terminal domain for socialization, STM and LTM.
- 74.Choi CH, McBride SM, Schoenfeld BP, Liebelt DA, Ferreiro D, Ferrick NJ, Hinchey P, Kollaros M, Rudominer RL, Terlizzi AM, et al. Age-dependent cognitive impairment in a Drosophila fragile X model and its pharmacological rescue. Biogerontology. 2010;11:347–362. doi: 10.1007/s10522-009-9259-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bolduc FV, Valente D, Nguyen AT, Mitra PP, Tully T. An assay for social interaction in Drosophila fragile X mutants. Fly (Austin) 2010;4 doi: 10.4161/fly.4.3.12280. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This report details the technical development of a novel social interaction assay applied to the FXS model. Such a paradigm indicates the ability to identify and study of ASD-like features in a range of Drosophila NDD models.
- 76.Kelley DJ, Bhattacharyya A, Lahvis GP, Yin JC, Malter J, Davidson RJ. The cyclic AMP phenotype of fragile X and autism. Neurosci Biobehav Rev. 2008;32:1533–1543. doi: 10.1016/j.neubiorev.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]