

Abstract
The neural adhesion/recognition protein L1 (L1CAM; CD171) has been shown or implicated to function in stimulation of cell motility in several cancer types, including high-grade gliomas. Our previous work demonstrated the expression and function of L1 protein in stimulation of cell motility in rat glioma cells. However, the mechanism of this stimulation is still unclear. This study further investigated the function of L1 and L1 proteolysis in human glioblastoma multiforme (GBM) cell migration and invasion, as well as the mechanism of this stimulation. L1 mRNA was found to be present in human T98G GBM cell line but not in U-118 MG grade III human glioma cell line. L1 protein expression, proteolysis, and release were found in T98G cells and human surgical GBM cells by Western blotting. Exosome-like vesicles released by T98G cells were purified and contained full-length L1. In a scratch assay, T98G cells that migrated into the denuded scratch area exhibited upregulation of ADAM10 protease expression coincident with loss of surface L1. GBM surgical specimen cells exhibited a similar loss of cell surface L1 when xenografted into the chick embryo brain. When lentivirally introduced shRNA was used to attenuate L1 expression, such T98G/shL1 cells exhibited significantly decreased cell motility by time lapse microscopy in our quantitative Super Scratch assay. These cells also showed a decrease in FAK activity and exhibited increased focal complexes. L1 binding integrins which activate FAK were found in T98G and U-118 MG cells. Addition of L1 ectodomain-containing media (1) rescued the decreased cell motility of T98G/shL1 cells and (2) increased cell motility of U-118 MG cells but (3) did not further increase T98G cell motility. Injection of L1-attenuated T98G/shL1 cells into embryonic chick brains resulted in the absence of detectable invasion compared to control cells which invaded brain tissue. These studies support a mechanism where glioma cells at the edge of a cell mass upregulate ADAM10 to proteolyze surface L1 and the resultant ectodomain increases human glioma cell migration and invasion by binding to integrin receptors, activating FAK, and increasing turnover of focal complexes.
Keywords: Glioma, Glioblastoma, Cell motility, L1CAM, Xenograft, Focal complex, FAK
Introduction
Glioma is the most common primary tumor that occurs in adult human brain. The most malignant type, glioblastoma multiforme (GBM, WHO grade IV), is practically incurable [1–6]. The average survival time with aggressive treatment is around 12 months, and the five year survival rate is about 4% [2, 4]. Glioma often grows back after resection and becomes more aggressive than the original neoplasm. The main reason for the therapeutic failure of GBM is due to the extensive and diffuse cell migration. The infiltration of GBM cells into normal brain tissue allows the cells to escape surgical resection and cause recurrence. The mechanism by which malignant glioma aggressively invades into brain tissue still is not elucidated fully. Our previous study indicated the neural cell adhesion/recognition protein L1 (L1CAM; CD171) stimulates rat glioma cell motility through integrins in an autocrine matter [7] and suggested that this might be true for human glioma cells as well.
L1 is a 200–220 kDa type I membrane glycoprotein that belongs to the immunoglobulin superfamily and contains six immunoglobulin-like domains (Ig domains), five fibronectin-like repeats (FN repeats), a transmembrane domain, and a highly conserved cytoplasmic domain [8, 9]. In the normal adult nervous system, L1 expression is restricted to post-mitotic neurons in the central nervous system and pre- and non-myelinating Schwann cells in the peripheral nervous system [10, 11]. The function of L1 in central nervous system development is well established, which includes facilitating neuronal migration, survival, as well as axon outgrowth, guidance, fasciculation and regeneration [12–15]. Mutations of L1 in human cause a myriad of nervous system birth defects [16] collectively known as L1 syndrome.
L1 can be proteolyzed and released from the cell membrane by ADAM10 and ADAM17, two members of the disintegrin and metalloprotease (ADAM) family [17–21]. Constitutive proteolysis occurs by ADAM10, whereas ADAM17 activity must be induced by phorbol esters or cholesterol depletion [20]. The resultant soluble L1 ecto-domain has been suggested to be generated normally and to interact with integrins to facilitate axon myelination during peripheral nerve development [22]. However, this proteolysis is undetectable in the normal adult brain [23], which indicates that L1 proteolysis is under strict regulation during normal brain development. Interestingly, the most severe forms of L1 syndrome result from mutations that cause truncated and secreted L1 ectodomain [24].
Our previous study demonstrated that L1 expression and proteolysis stimulated cell motility in rat glioma cells [7]. However, the mechanism of this stimulation still needs to be revealed. Focal adhesion kinase (FAK) is a member of non-receptor protein tyrosine kinases that has important roles in cell migration by mediating focal complex turnover [25]. FAK is activated by the ligand binding and clustering of cell surface integrin and other receptors [reviewed in 26]. There are several receptors that can activate FAK, including integrins αvβ3, αvβ5, α5β1 and epidermal growth factor receptor (EGFR), which are upregulated in malignant glioma, especially GBM. The L1 ectodomain, after ADAM-mediated proteolysis, has been suggested to interact with integrins including αvβ3, αvβ5 and α5β1 [27–30] to stimulate cell motility and migration [17–21]. However, the potential function of L1 ectodomain in activation of FAK signaling pathways through those integrin receptors, therefore regulating cell migration, has not been explored.
In this study, we not only investigated L1 expression and proteolysis in a human glioma cell line and human GBM surgical specimen cells, but also demonstrated L1 function in human glioma cell migration in vitro, and cell invasion in vivo using our embryonic chick brain xenograft model. Furthermore, we also demonstrated that L1 stimulates human glioma cell migration by regulating FAK activation, possibly through interaction with integrin receptors after ADAM10 shedding, thereby controlling turnover of focal complexes.
Materials and methods
Primary GBM cells and cell lines
High-grade human primary glioma surgical samples were obtained through the Tissue Procurement Center at the Helen F. Graham Cancer Center, Christiana Care (Newark, DE). The surgical samples were transported in Hibernate A medium (Brainbits LLC, Springfield, IL) on ice. GBM sample #21 was processed to obtain dissociated cells or lysed directly in RIPA lysis buffer for Western blotting analysis as described previously [7]. Briefly, the sample was washed in ice-cold neurobasal medium (Invitrogen, Carlsbad, CA), the visible necrotic sites of the tumor were trimmed, and the remaining tissue was cut into <1 mm3 pieces. Some pieces were lysed directly in RIPA lysis buffer with Complete Mini EDTA-Free Protease Inhibitor cocktail tablets (PI) (Roche Diagnostics, Indianapolis, IN) followed by sonication for Western blotting analysis. The rest of the pieces were dissociated using enzymatic digestion in 0.25% trypsin–EDTA at 37°C for one half hour before termination using 0.03% SBTI (Soybean Trypsin Inhibitor) and 0.003% DNase I on ice. Single cells were dissociated by trituration in 2 ml SBTI–DNase I. Dissociated cells (GBM21) were cultured in DMEM (Mediatech, Inc., Herndon, VA) with 10% fetal bovine serum (FBS; Hyclone, Waltham, MA), 2 mM L-glutamine (Mediatech Inc.), and penicillin–streptomycin (Mediatech Inc.). Cell culture has been maintained through 12 passages.
Human glioma cell lines used in this study were obtained from the American Type Culture Collection (ATCC, Manassa, VA). T98G is a human GBM cell line [31]. U-118 MG is a human Grade III brain glioblastoma astrocytoma cell line [32]. QT6 is a quail fibrosarcoma cell line [33]. CHO is a Chinese hamster ovary cell line [34]. MDA-MB-231 is a human breast cancer cell line. HEK293T/17 is a human embryonic kidney cell line. QT6 cells were cultured in Medium 199 (Mediatech, Inc. Herndon, VA,) with 5% FBS, 20% tryptose phosphate broth, 2 mM L-glutamine and penicillin–streptomycin. Other cell lines were cultured in DMEM, 10% BGS (bovine growth serum; Hyclone, Waltham, MA), 2 mM L-glutamine, and penicillin–streptomycin and incubated in a humidified incubator with 37°C and 5% CO2.
Vectors, transfection and infection
As previously described [7], QT6 cells were transfected with a full length human L1 expression plasmid using standard calcium phosphate transfection method [35]. Transfected cells served as a positive control for L1 expression for Western blotting and immunofluorescent staining. CHO cells were infected with a lentiviral vector (1879hL1ecto) containing a CMV promoter followed by the complete human L1 ectodomain sequence and served as positive control for detecting human L1 ectodomain and collection of L1 ectodomain-conditioned medium. This vector was made by inserting 3350 bp nucleotides from the human L1 ectodomain into the SpeI and XhoI (NEB, Ipswich, MA) restriction sites of the 1879 lentiviral vector plasmid [36]. L1 ectodomain expression was confirmed by Western blotting of infected MDA-MB-468 breast cancer cells and culture supernatant. The human L1 cDNA was obtained from Dr. Vance Lemmon in a pcDNA3 vector. The lentiviral vector 1879 was derived from the # 72 vector (described below) by replacing the GFP gene with a multicloning site. Briefly, the BamHI/XhoI-containing GFP fragment of vector 72 was excised and replaced with the GGATCCACTAGTTGATCAGCTAGCACGCGTTCTAG ACTCGAG nucleotide fragment, comprising BamHI-SpeI-BclI-NheI-MluI-XbaI-XhoI. CHO cells were also infected with empty 1879 lentiviral vector and served as a negative control for infection. The viruses used for infection were packaged in HEK293T/17 cell line. The expressing vector (20 μg), with the VSV-G helper plasmid pMD.G (5 μg) and gag-pol helper plasmid pCMVΔR8.2 (15 μg) were transfected into HEK293T/17 cells using standard calcium phosphate method. The viruses were collected 48 and 72 h after transfection and filtered through 0.45 μm filters. The positively infected cells were selected using puromycin.
Five different unconfirmed L1 targeting shRNA (short hairpin RNA) expressing lentiviral vectors were purchased from Open Biosystems (Accession #: NM-000425; Cat. #: RHS3979-97052300; Thermo Fisher Scientific). The backbone vector is pLKO.1 which contains a human U6 promoter and a puromycin selective marker into which a hairpin RNA targeting sequence was inserted. After assessing the set of the vectors for ability to attenuate L1 expression in human T98G cells, one vector (Clone ID: TRCN0000063917) with the L1 target sequence GCCAATGCCTACATCTACGTT (sense sequence) CTCGAG (hairpin) AACGTAGATG TAGGCATTGG (antisense sequence) successfully attenuated L1 expression. The sequence targets the 21 bp at the end of the 4th Ig domain of L1. The control vector was the pLKO.1 vector (Plasmid 10878:pLKO.1-TRC cloning vector, Addgene Inc., Cambridge, MA) without the hairpin RNA insert. The shL1 viruses and control viruses were packaged in HEK293T/17 cell line as described above. The positively infected T98G and surgical GBM 21 cells were selected in 2 μg/ml puromycin.
The lentiviral vector (designated #72) constitutively expresses GFP under control of the CMV early promoter as described elsewhere [37, 38] and pLEGFP-C1 is a commercially available GFP-encoding retroviral vector (Clontech, Mountain View, CA; cat. # 6058-1) used by us previously [7]. The vector #72 GFP expressing viruses were packaged in HEK293T/17 cells as described above. Vector #72 GFP virus was used to label surgical GBM21 cells. The pLEGFP-C1 virus was produced as described previously [7]. pLEGFP-virus was used to label T98G/shL1 and T98G/control cells, as well as surgical GBM#21/shL1 and GBM#21/control cells for double selection, as pLEGFP vector contains the neor marker. The positive double-infected cells were selected using 2 μg/ml puromycin and 400 μg/ml G418.
Antibodies for western blotting, immunofluorescent staining and FACS analysis
NCAM-L1 C-20 (cat. sc-1508; Santa Cruz Biotechnology, Santa Cruz, CA) is a goat polyclonal antibody raised against a peptide mapping within a C-terminal cytoplasmic domain of L1CAM of human origin. UJ127 (cat. GTX72362, Gene Tex, Irvine, CA; sc-53386, Santa Cruz Biotechnology) is a mouse monoclonal antibody raised against the human ectodomain of L1 (within the fibronectin repeats and close to the membrane region). Anti-ADAM10 (cat. 28071; AnaSpec, Inc., Fremont, CA) is a rabbit polyclonal antibody used for Western blotting. Anti-ADAM10 (cat. AB936; R&D Systems Inc., Minneapolis, MN) is a goat polyclonal antibody used for immunofluorescent staining of live cells. Anti-human integrin αVβ5 (cat. # MAB2019Z) and anti-integrin αVβ3 (cat. # MAB1976Z) are mouse monoclonal antibodies and α5β1 is a goat polyclonal antibody (cat. # AB1950) and are all from Chemicon International (Billerica, MA). Anti-GAPDH is a rabbit polyclonal antibody (cat. # 2118; Cell Signaling, Boston, MA) and anti-β-tubulin (E7) is a mouse monoclonal antibody (Developmental Studies Hybridoma Bank, Iowa, IA) used as loading controls for Western blots. The monoclonal mouse anti-TSG101 antibody (4A10; cat. # ab83; Abcam, Cambridge, MA) was used as an exosome marker. Anti-FAK (pY397) (cat. # 611806; BD Biosciences, San Jose, CA) is a mouse monoclonal antibody. Anti-total FAK (cat. # 603801; Bio-Legend, San Diego, CA) is a rabbit polyclonal antibody.
Secondary antibodies used were HRP-conjugated donkey anti-goat IgG (Jackson Immunoresearch, West Grove, PA), HRP-conjugated goat anti-rabbit IgG (Jackson Immunoresearch), HRP-conjugated goat anti-mouse IgG (Jackson Immunoresearch), Alexa Fluor-488 or -594 donkey anti goat IgG (Molecular Probes, Invitrogen, Carlsbad, CA), Alexa Fluor-488 or -594 goat anti mouse IgG (Molecular Probes, Invitrogen). A biotin-conjugated goat anti-mouse or anti-rabbit IgG (Jackson Immunoresearch) followed by Alexa Fluor-488 streptavidin conjugate (Molecular Probes, Invitrogen) were also used for immunofluorescent staining.
RT-PCR
An RT-PCR protocol was used as previously detailed [7]. Briefly, the total RNA was extracted from the cell lines using the RNeasy Mini Kit (Qiagen, Vanlencia, CA). The first-strand cDNA synthesis was carried out using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA). The primer sequences used were designed against the transmembrane domain of human L1. The forward primer sequence was 5′-TACC GCTTCCAGCTTCAG and the reverse sequence was 5′-TGATGAAGCAGAGGATGAGC. PCR Master Mix (Invitrogen, Carlsbad, CA) was used and the reaction was carried out in a thermal cycler (Techne Touchgene Gradient, Albany, NY) using an initial denaturation step for 15 min at 95°C, after which the reactions were subjected to 40 cycles of amplification. Each cycle consisted of denaturation for 30 s at 94°C, annealing for 30 s at 55°C and extension for 1 min at 72°C. The final extension was for 10 min at 72°C and the final hold was at 4°C.
Western blotting
The Western blotting analysis was performed as described in our previous study [7]. Cell culture dishes were kept on ice for 10 min and rinsed with cold PBS/PI (Phosphate buffered saline with protease inhibitors) before solubilizing them in RIPA lysis buffer with PI for 2–3 min on ice. The cells were then scraped and the lysates were clarified by sonication. The primary human glioma surgical samples were minced and put into RIPA lysis buffer as described previously; lysates were sonicated as well. Protein quantification was performed using the BCA Assay (Pierce Biotechnology, Pittsburgh, PA). The samples were prepared by adding the NuPage 4× LDS sample buffer (Invitrogen) and the NuPage 10× reducing agent (Invitrogen), heating at 70°C for 10 min and centrifuging. Equal amounts of the proteins (8–30 μg, depending on experiment) were loaded on NuPage 4–12% gradient polyacrylamide gels (Invitrogen) along with protein ladders (See Blue Plus 2 prestained standard or Magic Mark XP, Invitrogen). A small volume (500 μl) of the NuPage anti-oxidant (Invitrogen) was also added to the top chamber of the running buffer tank prior to the electrophoresis to prevent the proteins from reoxidizing during electrophoresis. NuPage MOPS running buffer (Invitrogen) was used.
For Western transfer, polyvinylidene fluoride (PVDF) membranes (0.45 μm, Invitrogen) were used and carried out at 4°C at 30 VDC overnight. The membrane was blocked in 5% nonfat dry milk solution in Phosphate Buffered Saline with 0.01% Tween 20. The blots were then incubated in the primary antibody, for 1–2 h at room temperature or overnight at 4°C. After rinsing three times with PBS/Tween 20, HRP-conjugated secondary antibody was then added for 1 h at room temperature. The membranes were developed for 1–2 min using the enhanced chemiluminescence detection system (Amersham, Piscat-away, NJ or Pierce Biotechnology) and protein bands were visualized using Blue Basic Autorad Film (ISC Bioexpress, Kensville, UT).
Immunofluorescent staining
The immunofluorescent staining procedure was described in our previous study [7]. Fixed cell staining was carried out by growing cells on coverslips coated with 200 μg/ml poly-L-ornithine (Sigma-Aldrich, St. Louis, MO), followed by fixing cells in 1% paraformaldehyde in PBS for 30 min at room temperature. The cells were incubated in the primary antibody diluted in PBS + 5% FBS or 5% normal goat serum + 0.03% Triton X-100 for an hour at room temperature. Coverslips were incubated in Alexa Flour 488 or 594 secondary antibodies (Molecular Probes, Invitrogen) in the same diluents as the primary antibodies for 45 min −1 h at room temperature. Coverslips were incubated in 10 μg/ml bisbenzimide (Sigma-Aldrich) in PBS for 5–10 min to stain nuclei and mounted in buffered glycerol/paraphenylenediamine (pH 8.0) on glass slides. For live cell staining for L1 or ADAM10, cells were incubated in primary antibody in DMEM + 5% heat-inactivated FBS on ice, rinsed, fixed with 1% paraformaldehyde at room temperature for 30 min, rinsed, incubated in biotin-conjugated secondary antibody in PBS + 5% FBS, rinsed, incubated in Alexa 488-streptavidin, and rinsed before the nuclear staining and mounting. The staining was visualized using a Nikon Microphot-FX microscope with Fluor objectives or Zeiss LSM 510 confocal microscope.
FACS analysis
The FACS protocol used in this study was described in the previous study [7]. Briefly, the cells were trypsinized lightly in 0.05% Trypsin/0.02% EDTA for several minutes, resuspended in DMEM + 10% H-I FBS. For live cell staining for L1-binding integrins, the cells were incubated with primary antibody in the H-I FBS containing diluents on ice (as described above), rinsed with diluent twice, and incubated in a biotin-conjugated secondary antibody followed by Alexa Fluor-488 streptavidin conjugate (Molecular Probes, Invitrogen). The cells then were analyzed using a Becton-Dickinson FACSCalibur flow cytometer using Cell Quest software.
Exosome isolation and TEM analysis
Ultracentrifugation was performed to isolate exosomes from the culture media of T98G cells. Briefly, the T98G cells were seeded on 10 cm dishes with serum-containing media until 70–80% confluent. The media was then removed and replaced with serum-free media with PI and incubated for 18–24 h in order for L1-containing exosomes to accumulate in the media. The media was collected, filtered through a 0.2 μm filter to remove cell debris, and subjected to 3 rounds of centrifugation: 1,200×g for 10 min, 10,000×g for 20 min and 100,000×g for 22 h. The first two rounds of centrifugation were performed using a Sorvall RC-5B centrifuge with a fixed angle SS-34 rotor to further remove cell debris. Final centrifugation was performed at 4°C using a Beckman L8-55M Ultracentrifuge and SW41 Ti swinging bucket rotor. Following ultracentrifugation, the supernatant was removed and the glassy pellet was resuspended in 200 μl sterile PBS with PI for TEM (transmission electron microscopy) analysis or in 200 μl RIPA/PI lysis buffer for Western blotting. TEM analysis was performed using a Zeiss CEM 902 with soft imaging system Mega View II. Ammonium molybdate staining was used to visualize exosomal vesicles under TEM.
In vitro cell motility assay
Cell motility of U-118 MG, T98G/shL1, and T98G/control cells was measured by using the time-lapse microscopy Super Scratch assay as previously described [7, 39, 40]. Cells were grown to confluence on plastic tissue culture dishes and then “wounded” with a sterile plastic 1 ml pipettor tip in serum-free media. For the L1 ectodomain-containing medium “rescue” assay or stimulation experiments, the serum-free culture medium from infected CHO cells were filtered through 0.2 μm filters and added to U-118 MG cells, T98G/control cells and T98G/shL1cells. For some experiments, 40 μg/ml of a peptide (QPSITWRGDGRDLQE) were added to cells. Cells were then placed into a custom culture chamber mounted on a ProScan II automated stage (Prior Scientific, Rockland, MA) on a Nikon TE-2000E automated microscope. Temperature was maintained at 37°C by a combination of a warm air temperature controller (Air Therm, World Precision Instruments, Sarasota, FL) and thermoelectric warming with an optically clear temperature-controlled stage insert (Tokai Hit, Shizuokaken, Japan). The atmosphere within the chamber was kept at 5% CO2/95% air using a gas injection controller (Forma Scientific, Marietta, OH). A CoolSnap ES CCD camera (Photometrics, Tucson, AZ) was used to capture images over the course of the experiment using a Nikon Plan Fluor 20X ELWD objective at areas of interest on each plate for approximately 20 h. The experiments entailed collection of phase contrast images at 5 min intervals. The system was controlled using MetaMorph Premier Software (Molecular Devices Corporation, Downingtown, PA). Quantitative analysis of cell motility was performed on acquired sequential phase contrast images using the MetaMorph software “Track Points” feature with nucleoli serving as imaging targets. Data were evaluated statistically using Student’s two-tailed t-test and graphed using Microsoft Excel.
Focal complex analysis
T98G/shL1 and T98G/control cells were grown to confluence and a wound was introduced as above. The cells were then allowed to migrate in serum-free medium or the conditioned medium overnight. Then the cells were fixed and stained for FAK pY397. Ten cells on the edge of the scratch with similar morphology were chosen for analysis. Confocal images were then obtained using a Zeiss LSM510 confocal microscope and the images were analyzed using the MetaMorph Integrated Morphometry Analysis program. The stained areas in the selected region were analyzed for area (μm2). Statistics used were student’s t-test and standard error of the mean.
In vivo chick embryonic brain microinjection
T98G/shL1 and T98G/control cells were infected with pLEGFP retrovirus for microinjection. GBM21 cells were infected with vector #72-GFP lentivirus for microinjection. Virus production, infection and selection details were described above. Cells were trypsinized and resuspended in cell medium with (T98G cells) or without (GBM21) 30% Matrigel. The cell density for injection was 2 × 104/μl.
Fertile White Leghorn chicken embryos were obtained from the University of Delaware Department of Animal and Food Sciences. A humidified forced-draft incubator at 37.5°C was used to incubate the eggs until embryonic day 5 (E5). Chick embryonic OT (optic tecta, midbrains) were injected with the human GBM cells. The experimental procedure was detailed previously [39]. Briefly, a small window was cut over the air space at the top end of the eggs and approximately 10 μl of cell suspension (~105 cells) was microinjected into the OT using a PV830 pneumatic picopump (World Precision Instruments; Sarasota, FL). After injection, sterile ampicillin was added over the embryo, the window in the egg shell was sealed with transparent tape, and the embryo was placed back into the egg incubator until E9.
At E9, the embryos were sacrificed and brains were dissected. E9 chick embryo tecta were fixed in 2% paraformaldehyde in PBS for 2 h, rinsed in PBS and cryoprotected in 30% sucrose overnight. The next day, tecta were embedded in Tissue Freezing Media (cat. # H-TFM; Triangle Biomedical Sciences, Durham, NC) before being sectioned at 10 μm. The cryosections were used for immunostaining for L1. The dissected E9 chick brains also were fixed in 2% paraformaldehyde in PBS overnight and the next day were embedded in 3.5% agar and 8% sucrose in PBS and sectioned at 200 μm using a Vibratome Series 1000 Sectioning System (Ted Pella, Redding, CA) for observation using a Nikon SMZ-1500 zoom stereomicroscope equipped with epifluorescence attachment and Tucsen color CCD camera (Tucsen Image Technology Inc., Fuzhou, FuJian, China) and for confocal microscopy observation.
Results
L1 is expressed and proteolyzed in human glioma cells
L1 expression and proteolysis in human glioma cell lines were tested utilizing reverse transcription-polymerase chain reaction (RT-PCR), Western blotting and immunofluorescent staining. The primer used in RT-PCR targeted the transmembrane region of human L1 and was used previously for this purpose [7]. RT-PCR detected L1 mRNA in human GBM cell line T98G and to a lesser extent in U-118 MG (Fig. 1a). Human L1 cDNA served as a positive control for L1 mRNA expression and no-RT reactions served as negative controls.
Fig. 1.

L1 was expressed and proteolyzed in a human GBM cell line and surgical cells. a RT-PCR to detect L1 mRNA. T98G cell line exhibited the expected 461 bp band and U-118 MG showed the 461 bp band to a much less extent. The human L1 cDNA (hL1cDNA) was used as positive control. No-reverse transcriptase was used as a negative control. b Western blot analysis of L1 expression and proteolysis. NCAML1 antibody recognized the ~220 kDa full length and the ~32 kDa L1 bands in T98G cells. L1 protein expression was undetectable in U-118 MG cells. UJ127 antibody detected the full length L1 in surgical GBM21 cells. QT6 cells transfected with human full length L1 (QT6/hFL1) was used as positive control (PC) and mock transfected cells (QT6/mock) served as negative control (NC). c Western blot analysis demonstrated ADAM10 expression in T98G and U-118 MG cells, with unprocessed premature ADAM10 (~80 kDa) and the processed mature ADAM10 (~60 kDa band) in both cell lines. Jurkat cell extract was used as positive control. d Western blot analysis of cell culture medium versus cell lysate. T98G cell culture medium (under “Culture Medium” bar) showed a UJ127-positive band (~180 kDa) smaller than 220 kDa full-length band expressed by QT6/hFL1 cell lysate (PC). Mock transfected QT6 cell medium (NC) and medium from U-118 MG cells were negative controls. NCAML1 did not detect the full length L1 in the T98G cell culture medium as expected. e T98G cells immunostained for L1 and viewed by widefield fluorescence microscopy. Live cell staining using UJ127 antibody (upper panel) showed cell surface L1. Fixed cell staining using NCAML1 antibody (lower panel) detected intracellular as well as cell surface L1. QT6/hFL1 and QT6/mock cells were used as positive and negative controls. Nuclei were stained with bisbenzimide (lower row in each panel). Bar = 50 μm
Western blotting was used to investigate L1 protein expression and proteolysis in T98G, U-118 MG and human surgical GBM cells (Fig. 1b). T98G cells exhibited the 220 kDa full length as well as approximate 32 kDa transmembrane fragment by anti-L1 cytoplasmic domain antibody NCAML1, most likely resulting from L1 ectodomain shedding by ADAM10 proteolysis (see “Discussion” section). L1 protein expression was undetectable in U-118 MG cells (Fig. 1b). Analysis of a primary surgical human glioma specimen (GBM 21) also showed full length L1 expression using anti-L1 ectodomain antibody UJ127 (Fig. 1b). QT6 cells transfected with full length human L1 cDNA (QT6/hFL1) served as a positive control for L1 expression, and QT6 cells after mock transfection (QT6/mock) served as an L1 negative control.
To confirm the presence of ADAM10 protease in the glioma cell lines, Western blotting was used (Fig. 1c). Both forms of ADAM10 (unprocessed: ~80 kDa and processed: ~50 kDa) were found in T98G and U-118 MG cells. Although L1 protein expression in U-118 MG cells was not detectable, the ADAM10 expression might indicate other ADAM10 target proteins in these cells. Jurkat cells were used as a positive control for ADAM10 expression according to manufacturer’s suggestion.
To determine if the L1 ectodomain generated by proteolysis was released by T98G cells, cell culture medium was collected, concentrated, and also subjected to Western blotting (Fig. 1d). A slightly smaller than 220 kDa (~180 kDa) L1 fragment was detected by antibody UJ127, which represents the shed-L1 ectodomain resulting from membrane proximal proteolysis. U-118 MG cell culture medium was analyzed and was negative for released L1, which was predicted from the lack of L1 in cell extracts. To eliminate the possibility that the observed L1 that was easily detected in T98G cell medium was from exosomes released from the cells (details below), TSG101 (an exosome marker) and NCAML1 antibodies both were used to check for exosomes. There was no full length L1 or exosomes detected in the normal T98G cell culture medium. Exosomes can be concentrated from the media (see below), but this requires concentration by ultracentrifugation for them to be to be detected. QT6/hFL1 and QT6/mock cells served as positive and negative controls for L1 expression.
T98G cells were immunofluorescently stained for L1 to illustrate staining patterns on the cells using both NCAM-L1 and UJ127 antibodies (Fig. 1e). After live cell staining, T98G cells showed surface L1 using the UJ127 antibody. After fixed cell staining, T98G cells showed surface as well as prominent intracellular L1 staining (Fig. 1e). While intracellular L1 staining was fairly even in fixed T98G cells, surface staining was less even on live T98G cells. This may be a result of partial L1 surface proteolysis by low levels of ADAM10, which was explored further below in a scratch assay. QT6/hFL1 and QT6/mock cells were used as positive and negative controls for L1 immunostaining for both NCAML1 and UJ127 antibodies. Overall, our data show that L1 and its known protease ADAM10 were expressed in human GBM cells. L1 was proteolyzed, presumably by ADAM10, and the cleaved large L1 ectodomain fragment was released into the cell extracellular environment.
L1 is a component of exosomal vesicles released from T98G cells
Since we previously found that two rat and one human glioma cell lines released L1-containing exosomes into the culture medium, exosomes were concentrated from T98G cell culture media as described previously [7, 41]. The ultracentrifugation pellet was resuspended in a small volume for either transmission electron microscopy (TEM) analysis or Western blotting. TEM analysis after ammonium molybdate staining revealed ~24 nm diameter exosome-like vesicles (Fig. 2a, arrows show representative exosome vesicles). Antibodies UJ127 and NCAML1 detected full length L1 after Western blotting, but did not detect the 180 or 32 kDa ADAM10 cleavage products (Fig. 2b). The lower weight bands are unknown cleavage products that might have been generated by other known L1 proteases (e.g. plasmin and PC5A). The human breast cancer cell line MDA-MB-231 was used as positive control for exosome preparation, as they have been characterized before [7]. The TSG101 exosomal marker protein was detected, which confirmed the presence of exosomes in the preparations. ADAM10 was detected (Fig. 2b) in what appeared to be both the unprocessed and processed forms. This analysis showed that L1 was released from human T98G cells as a component of exosomes but was not proteolyzed in them. Because L1 shedding was not detected in exosomes from T98G cells, the RGD-containing L1 ecto-domain of membrane-bound exosomal L1 is available for binding to L1 receptors. Therefore, due to their minute size, exosomes might be able to serve as a second source of L1 ectodomain released from cells that could interact with glioma cell surface integrins to stimulate motility.
Fig. 2.
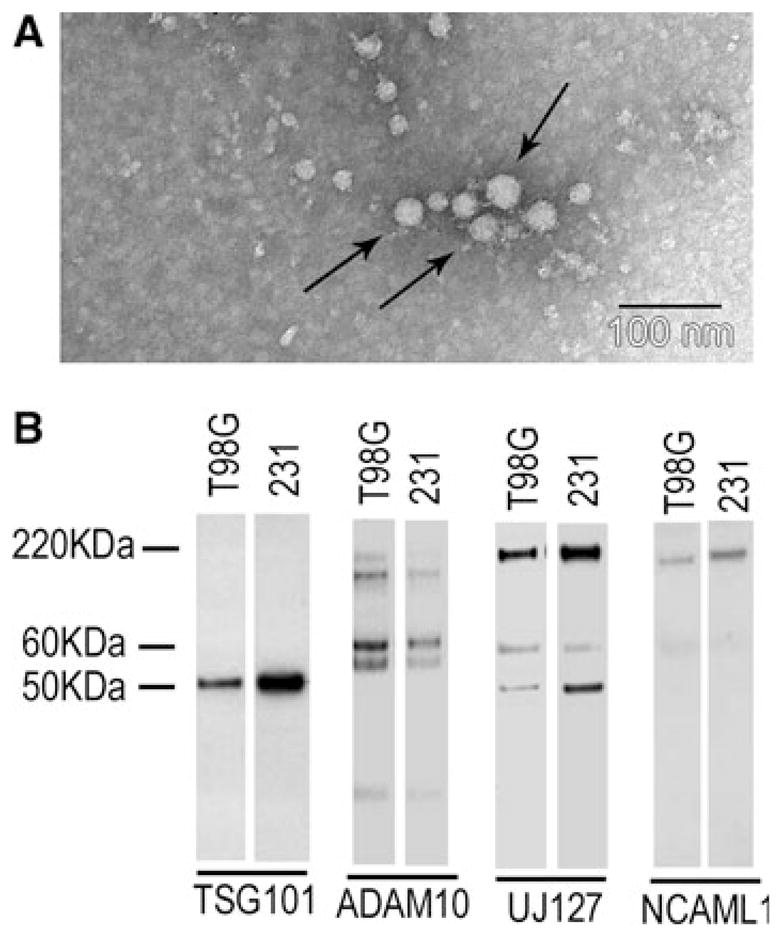
L1 was a component of exosome-like vesicles released from T98G cells. T98G cells and MDA-MB-231 human breast cancer cells were analyzed for secretion of exosomes as described in “Materials and methods” section. a Exosome vesicles in T98G cell culture using TEM showed an average of ~24 nm diameter vesicles (arrows show representative exosome vesicles). b Western blotting of exosome preparations. Mature ADAM10 (~60 kDa) was present in T98G and MDA-MB-231 exosomes. Full length L1 (220 kDa) was detected in T98G cells by UJ127 and NCAML1. UJ127 also detected two bands lower than 220 kDa, which are likely to be the L1 degradation products not generated by ADAM10, since a 32 kDa L1 cleavage product was not detected by NCAML1 antibody. MDA-MB-231 cells were used as a positive control for the exosome preparation. TSG101 is an exosome marker
L1 ectodomain shedding was observed in migrating glioma cells coincident with upregulation of surface ADAM10
As demonstrated above and previously [7], L1 proteolysis results in release of a large ectodomain fragment into the extracellular environment. To further investigate the regulation of this shedding, a wound healing assay followed by immunofluorescent staining was performed. A scratch was created in the T98G cell monolayer using a pipet tip and the cells were allowed to migrate into the denuded area overnight. The cells were either fixed, and stained for L1 using UJ127 antibody or stained live for ADAM10 using an antibody recognizing the ectodomain of ADAM10. Fixed cells were used for L1 staining so that both peripheral surface L1 and punctate intracellular L1 could be observed simultaneously. The cells behind the scratch edge exhibited bright peripheral L1 staining where it accumulated at areas of cell–cell contact (Fig. 3a, left panel) as well as dimmer punctate intracellular staining. Only the cells that migrated into the denuded area lost their bright peripheral L1 staining (Fig. 3a, middle panel). However, high magnification imaging clearly showed that these cells continued to exhibit punctate intracellular L1 staining (Fig. 3a, right panel) without peripheral staining. This indicates continued L1 expression in these cells and suggests a preferential loss of surface L1. Conversely, surface ADAM10 staining was not observed on cells behind the scratch edge (Fig. 3b, left panel) and was observed only on migrating cell surfaces as opposed to the non-migrating cells (Fig. 3b, middle and right panels). This is similar to cells observed in cultures of colon cancer [42], where the cleavage of L1 by ADAM10 was observed mainly in migrating cells. The findings here indicate that something relative to the existence of the scratch edge triggers L1 ectodomain shedding. This shedding is coincident with the upregulation of ADAM10 in those cells and, thus, is likely to be a direct result of cleavage by that protease.
Fig. 3.

L1 ectodomain shedding was observed in migrating glioma cells coincident with upregulation of surface ADAM10. a T98G cells were allowed to migrate into a denuded scratch area overnight. Cells behind the scratch edge (nonmigrating) showed surface L1 staining between cells (low mag., left upper panel). However, cells that migrated into the scratch showed loss of cell surface L1 staining (low mag., middle upper panel; scratch edge denoted by dotted line; bar = 100 μm). Arrows indicate the direction of cell migration into the scratch. A high magnification (right panels; bar = 10 μm) demonstrates the loss of peripheral cell surface L1 but not the punctate intracellular L1 in a cell that migrated into the denuded area. Cell nuclei are shown below the corresponding UJ127 image. b Corresponding ADAM10 expression. ADAM10 staining was visible only on the surface of migrating cells (at the scratch edge or in the scratch) (middle and right upper panels), but not on nonmigrating cells behind the scratch edge (left upper panel). Bar = 50 μm for all panels. a, b are widefield fluorescence images. c Surgical GBM21 cells labeled with GFP were microinjected into E5 chick embryonic OT and analyzed at E9 by confocal microscopy. GBM21 cells (shown in upper panel; green) clearly exhibited intercellular surface L1 (UJ127 staining;red) inside the tumor mass, but migrating cells (arrows) exhibited no surface L1 (UJ127) staining
A similar spatial observation was obtained from surgical GBM21 cells microinjected into embryonic chick brain (Fig. 3c). GFP-labeled GBM21 cells were microinjected into E5 embryonic OT and allowed to form a tumor for 4 days, and the embryo was sacrificed at E9. The brain was dissected, cryostat sectioned, immunofluorescently stained for L1 expression, and analyzed by confocal microscopy. Xenografted GBM21 cells expressed L1 that was clearly visible around cells inside the tumor, but cells that migrated away from the tumor mass exhibited no surface L1. Higher magnification inspection of these migrating cells revealed that they still were positive for intracellular L1 (not shown), so they did not stop expressing it. This in vivo data supports the hypothesis that L1 proteolysis occurs at the tumor margin as it does in peripheral T98G cells along a scratch edge, where cells initiate migration, thereby facilitating cell movement by soluble L1 ectodomain interaction with cell surface integrins.
Lentiviral shRNA attenuation of L1 decreased T98G cell motility
A short hairpin lentiviral vector targeting 21 bp within the end of the 4th L1 Ig domain was used to attenuate L1 expression in T98G and surgical specimen cells. T98G and GBM21 cells were infected either with shRNAL1 vector (T98G/shL1 and GBM21/shL1) or empty control vector (T98G/control and GBM21/control). Puromycin was used to select positively infected cells, Western blotting was used to evaluate attenuation of L1 expression in selected cells, and quantitation was performed. Compared to control vector-infected cells, T98G/shL1 cells were reduced in their L1 levels by more than 90% (Fig. 4a). GBM21/shL1 cells also showed reduction of L1 expression compared to controls. Quantitative Super Scratch assays [7, 39, 40] were performed using time-lapse microscopy, and T98G/shL1 cells migrated at a lower average velocity throughout the experiment (Fig. 4b) and had significantly reduced overall average velocity compared to T98G/control cells (Fig. 4c) (40% velocity of controls; P ≪ 0.001). The slower motility rate of L1-attenuated T98G cells showed that L1 is important in human GBM cell migration and suggests that released L1 ectodomain might be their stimulatory factor, since surface L1 proteolysis accompanies motility.
Fig. 4.

L1 attenuation decreased T98G cell motility. a L1 expression in T98G and surgical GBM21 cells infected either with a control lentiviral vector (T98G/control and GBM21/control) or L1-targeting shRNA lentiviral vector (T98G/shL1 and GBM21/shL1). L1 expression was dramatically down-regulated in both T98G and GBM21 cells infected with shL1 vector. b T98G/shL1 cells showed decreased motility (red lower trace on graph) compared to T98G/control cells (blue upper trace on graph). The graph shows the average velocities (thin lines) and 3rd order polynomial best-fit curves (thick lines) for cells analyzed above at 5 min intervals. c The overall average velocities were calculated using all individual cell velocities collected during the course of the experiment and graphed as single values. T98G/shL1 cell velocity was 0.070 μm/min ± 0.002 s.e.m. and T98G/control cell velocity was 0.191 μm/min ± 0.003 s.e.m. (*P < 0.001). The experiment was repeated three times with similar results
Exogenous L1 ectodomain stimulated human glioma cell migration
In order to demonstrate the stimulatory function of the L1 ectodomain that results from ADAM10 proteolysis of endogenous L1 in human glioma cell migration, this was mimicked by adding exogenous L1 ectodomain. To do this, CHO cells were infected with either a lentiviral vector encoding the entire human L1 ectodomain region (CHO/hL1ED) or a control vector (CHO/control). After infection, cells were selected using puromycin, and the serum-free culture medium was collected and examined by Western blotting, which demonstrated L1ED in the medium from CHO/hL1ED cells (Fig. 5a). Medium from CHO/control cells was also collected and used as a negative control medium. The L1ED medium or the control medium was added to T98G/control and T98G/shL1 cells, and the motility of the cells was quantitated by time-lapse microscopy using the Super Scratch assay.
Fig. 5.

Exogenous L1 ectodomain stimulated human glioma cell migration. a CHO cells either were infected with a lentiviral vector encoding the human L1 ectodomain (CHO/hL1ED) or the empty control vector (CHO/control). Cells and conditioned medium were probed for L1 with UJ127 antibody. L1 ectodomain was present in CHO/hL1ED cells and conditioned media but not in the CHO/control cells and conditioned media. QT6/hFL1 and QT6/mock cells were used as positive and negative controls for full length L1 expression. b The Super Scratch assay was used to quantitate cell motility at 5 min intervals for 20 h. T98G/shL1 cells showed increased cell motility (green upper trace on graph) when they were incubated in the CHO/hL1ED conditioned medium compared to when they were incubated in the CHO/control medium (red lower trace on graph). c T98G/shL1 cells incubated in the CHO/hL1ED conditioned medium (green slightly lower trace on graph) showed similar motility to T98G/control cells incubated with control medium (dark blue slightly higher trace on graph). d T98G/control cells incubated with the CHO/hL1ED conditioned medium showed no increase in motility compared to T98G/control cells incubated with control medium (traces almost identical on graph). e T98G/shL1 cell incubated with control medium (red lower trace on graph) did not show rescued velocity and had much reduced velocity compared to T98G/control cells incubated with control medium (dark blue upper trace on graph). f The overall average velocity of each cell population is shown. Overall average velocity of T98G/shL1 incubated with CHO/hL1ED conditioned medium (0.172 ± 0.004 s.e.m.) was increased significantly compared to T98G/shL1 cells incubated with control media (0.097 ± 0.003; *P < 0.001), which was nearly recovered to that of T98G/control cells in control medium (0.190 ± 0.004 s.e.m.). However, T98G/control cell velocity was not increased by the addition of L1ED conditioned medium (0.192 ± 0.004 s.e.m.). Error bars are s.e.m. g U-118 MG cells showed increased velocity when incubated with the CHO/hL1ED conditioned medium (red upper trace on graph) compared to control medium (blue lower trace on graph). h The overall average velocities of U-118 MG cells were significantly increased by 29% (0.179 ± 0.003 s.e.m. vs. 0.141 ± 0.003 s.e.m.; *P < 0.001). The experiments were repeated twice with similar results
The average velocity of T98G/shL1 incubated with L1ED conditioned medium was increased throughout the time course of the experiment compared to T98G/shL1 incubated with control medium (Fig. 5b), and was rescued almost completely to the level of the velocity of T98G/control cells incubated with control medium (Fig. 5c). Interestingly, T98G/control cells incubated with L1ED medium did not show increased cell velocity compared to T98G/control cells incubated with control medium (Fig. 5d). One possibility to explain this lack of further stimulation might be saturation of L1ED binding integrin receptors (see Fig. 6 below for integrin expression profiles) in those cells by their own released L1ED. T98G/shL1 cells incubated with control medium showed substantially decreased velocity compared to T98G/control cells incubated with control medium (Fig. 5e), which is consistent with the previous finding shown in Fig. 4b. The overall average velocity of T98G/shL1 cells incubated with L1ED medium was increased significantly compared to the T98G/shL1 cells incubated with control media (76% increase; Fig. 5f; *P < 0.001) and was rescued almost to the level of T98G/control cells (90% of control cells). The overall average velocities of all cell lines and conditions were calculated and are shown in Fig. 5f.
Fig. 6.

Focal complex size was increased in T98G/shL1 cells. a T98G and U-118 MG cells were analyzed by flow cytometry for integrins αvβ3, αvβ5 and α5β1. T98G cells expressed all three integrins (blue unfilled line graphs; controls are red filled graphs). U-118 MG cells expressed αvβ3 and αvβ5 integrins, but no α5β1 integrin staining was detected. b Western blot analysis of phosphorylated FAK in T98G cells. Antibodies against phosphorylated FAKY397 and total FAK were used. T98G/shL1 cells exhibited decreased pFAKY397 compared to T98G/control cells. GAPDH was used as loading control. c T98G/shL1 cells incubated in CHO/L1ED conditioned medium exhibited increased or rescued pFAKY397 levels back to control levels of T98G/control cells incubated in CHO/control medium. Total FAK (tFAK) also was substantially increased. GAPDH was used as loading control. d The total area of focal complexes in ten T98G/shL1 cells (indicated by pFAKY397 staining) was significantly larger (31.3 ± 1.5 s.e.m. vs. 23.1 ± 1.0 s.e.m.; *P = 0.001) than in ten T98G/control cells, as measured by MetaMorph Integrated Morphometry analysis. e After L1ED conditioned medium incubation, the total area of focal complexes in “rescued” T98G/shL1 cells was reduced to slightly less than the level of control cells incubated with control medium (24.7 ± 1.5 vs. 21.4 ± 1.1 s.e.m.; *P = 0.1). f Examples of a T98G/shL1 and a T98G/control cell stained for pFAK that was used for morphometry analysis in d. The arrows point to a subset of the focal complexes at the cell periphery that was included in the analysis
Similar experiments were performed with U-118 MG cells. As shown above, U-118 MG cells lack endogenous L1 protein expression, but express L1-binding integrin receptors (shown below). L1ED-conditioned medium and control medium were added to U-118 MG cells and subjected to the Super Scratch assay. U-118 MG cells incubated with L1ED containing medium showed increased cell velocity throughout the experiment (Fig. 5g) and significantly increased overall average velocity (29% increase; Fig. 5h; *P < 0.001). The overall collected data from these experiments demonstrates that the soluble L1 ectodomain, which results from ADAM10 proteolysis, facilitated cell migration in human glioma cells. This was true also for cells that did not express L1 protein themselves. This supports the proposed model of autocrine and paracrine stimulation of glioma cell motility by the cleaved L1 ectodomain.
To obtain evidence that the L1 ectodomain was binding to integrin receptors via the RGD sequence within the 6th Ig domain, we performed Super Scratch motility assays on T98G cells with addition of an RGD-containing peptide. The peptide sequence was that of L1 centered on the RGD sequence (QPSITWRGDGRDLQE). Two experiments were performed, and addition of the peptide decreased T98G cell motility by 11% (0.155–0.138 μm/min.; P < 0.001) and 31% (0.248–0.171 μm/min.; P < 0.001), which are very significant but only partial decreases compared to 60% decrease after attenuation of L1 expression above. Thus, we conclude that at least some stimulation of glioma cell motility by the L1 ectodomain was through binding to integrin receptors via the RGD domain.
Focal complex area was increased in T98G/shL1 cells
Our proposed mechanism of L1 ectodomain-stimulated glioma cell motility is that the cleaved L1 ectodomain binds to integrin receptors and subsequently activates FAK by phosphorylation at Y397, therefore controlling focal complex turnover in migrating glioma cells. To support this, first the expression of L1-binding integrins that associate with FAK activation was assessed. Flow cytometry analysis revealed that T98G cells express L1 binding integrins αvβ3, αvβ5, and α5β1 (Fig. 6a). However in U-118 MG cells, α5β1 was not detected (Fig. 6a).
Next, to investigate if FAK activation was associated with L1 expression and L1 ectodomain shedding, phosphorylated FAKY397 was examined and found to be dramatically down-regulated (by 93% normalized to GAPDH; by 75% normalized to total FAK) in T98G/shL1 cells compared to control cells (Fig. 6b). However, phosphorylated FAKY397 was substantially increased (about 30% normalized to GAPDH) in T98G/shL1 cells incubated with the L1 ectodomain conditioned medium compared to T98G/control cells incubated with control medium (Fig. 6c). Interestingly, total FAK also was increased about 5-fold (normalized to GAPDH) in T98G/shL1 cells incubated with L1 ectodomain containing medium compared to T98G/control cells incubated with control medium. Thus, it appears that L1 ectodomain might also regulate FAK gene expression levels, but the mechanism is unknown. Nonetheless, the most important findings are the changes in pFAK normalized to GAPDH, since it is pFAK levels (not total FAK) that modulates focal complex turnover [25, 26].
Because FAK activation modulates focal complex assembly and disassembly at the cell front during cell migration [25, 26] and focal complex size is inversely correlated with cell velocity [43], we analyzed the total area of focal complexes in migrating T98G cells. The scratch assay was used with T98G/shL1 and T98G/control cells, and after overnight migration, cells were fixed and stained for phosphorylated FAKY397 to indicate the location and size of focal complexes at the cell front. Antibodies against the focal complex protein vinculin were also used initially, but they did not allow visualization of focal complexes as distinctly as did anti-pFAK antibodies. The total areas of the focal complexes were then quantitated using MetaMorph Integrated Morphometry Analysis. Examples of pFAK stained cells that were used for quantitation is shown in Fig. 6f. Ten cells on the edge of the scratch with similar morphology from each cell line were chosen for quantitative analysis. Confocal images from those cells were obtained and the areas of each of the focal complexes (indicated by phosphorylated FAKY397 staining within a selected region on the cell front) were calculated. The total area (μm2) of focal complexes in T98G/shL1 cells was approximately 35% larger than control cells (Fig. 6d; *P = 0.001, bars are s.e.m.). This larger area of focal complexes can be explained by their slower turn-over rate in T98G/shL1 cells resulting from down-regulated FAK activity as indicated by a reduction in Y397 phosphorylation. The greater focal complex area was reversed in T98G/shL1 cells after incubation with the conditioned medium containing L1 ectodomain (Fig. 6e; *P < 0.01, bars are s.e.m.). Overall, our results demonstrate that the L1 ectodomain controls focal complex area in human glioma cells (as visualized by pFAK staining), and that focal complex area correlates with cell velocity.
Attenuation of L1 expression decreased T98G cell invasion into brain
T98G/shL1 and corresponding control cells were fluorescently labeled by GFP-encoding lentiviral vector infection. Matrigel was mixed with T98G cells before microinjection, which was necessary to be tumorigenic in mice [44]. The tumorigenicity of T98G in chicken embryonic brains was tested systematically and 30% Matrigel was found to be necessary for T98G to attach and invade into the OT (optic tectum) ventricle wall (data not shown). Here, 105 cells were injected into the midbrain ventricles of E5 chick embryos. At E9, the embryos were sacrificed and the brains were dissected. In order to visualize the injected cells, the brains were embedded and sectioned into 200 μm thick sections using a Vibratome. Nine embryonic brains injected with each cell line were examined. Nine out of 9 brains exhibited tumors inside the ventricle from T98G/control cell injection, and eight out of nine brains exhibited tumor formation from T98G/shL1 cell injection (Table 1). Although both cell lines formed tumors inside the ventricle, only T98G/control/GFP cells infiltrated and invaded into the brain wall of the OT (Fig. 7). On the other hand, T98G/shL1/GFP cells showed no detectable invasion into the brain, i.e. they were without cells migrating away from the tumor mass and infiltrating into the brain wall (Fig. 7). These results demonstrated that L1 not only stimulates human glioma cell motility in vitro, it also facilitates glioma cell invasion into the brain in vivo.
Table 1.
Summary results of T98G cell microinjection into chick embryos
| Cell type | # Embryos analyzed | # Attached tumors | # Brains with invasion |
|---|---|---|---|
| T98G/control | 9 | 9/9 | 9/9 |
| T98G/shL1 | 9 | 8/9 | 0/9 |
Fig. 7.
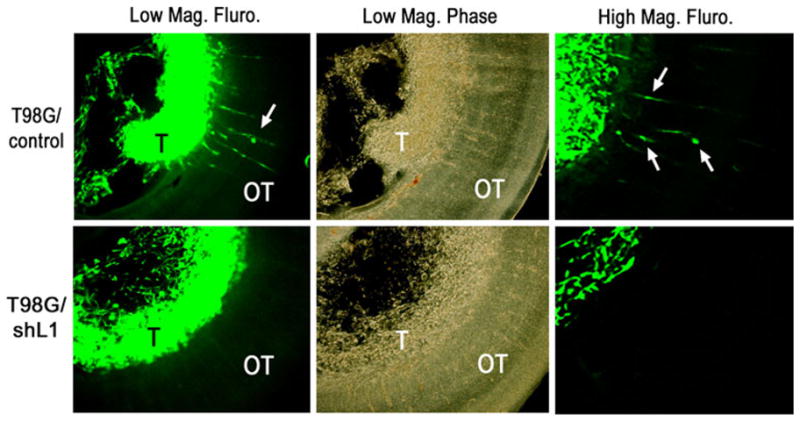
L1 attenuation decreased T98G cell invasion into brain. T98G/shL1 and T98G/control cells were labeled with a GFP retroviral vector, microinjected into E5 chick embryonic brain, and analyzed at E9 in Vibratome sections. Both T98G/control and T98G/shL1 cells formed tumors (T) inside the ventricle (low power mag., left upper and lower panels). However, only the T98G/control cells, but not the T98G/shL1 cells, showed infiltration inside the brain wall (OT). Phase contrast pictures are shown to demonstrate the location of the tumor (T) and the wall of embryonic OT (low power mag., middle upper and lower panels). The right upper and lower panels show images taken at higher magnification. Arrows denote invading cells
Discussion
In our study, the expression and proteolysis of L1 (L1CAM, CD171) was documented in the T98G human glioma cell line. The proteolyzed L1 ectodomain, likely by ADAM10 proteolysis, was released into the cell culture from those cells. L1 ectodomain shedding was clearly evident in migrating glioma cells, as opposed to non-migrating cells within in a cell mass. L1 also was found to be released into the cell culture medium from glioma cells as part of membrane-bound exosomal vesicles. T98G cells with attenuated L1 expression showed significantly decreased cell motility. Added soluble L1 ectodomain not only rescued cell motility in the L1-attenuated cells but also stimulated cell migration in a human glioma cell line that expressed L1-binding integrins but lacked endogenous L1 expression. L1-stimulated cell migration was coincident with FAK activation in glioma cells, thereby controlling cell migration through modulation of focal complex size and, thus, turnover rate. This supports the idea that an autocrine/paracrine stimulatory mechanism was utilized in human glioma cell migration. Attenuating L1 in human glioma cells also resulted in undetectable cell invasion in embryonic chick brain. Our findings provide evidence to support a model that L1 proteolysis and release stimulates glioma cell migration and invasion through binding to integrin receptors, therefore stimulating the FAK signaling pathway to regulate cell migration by increasing focal complex turnover rate.
Glioblastoma multiforme (GBM, grade IV) cells aggressively invade the brain, which can kill patients within months [6, 45, 46]. This extremely invasive nature of GBM is the main cause of therapeutic failure, whether it is after surgical resection or radiation therapy. Previous studies by other researchers suggested a function for L1 in rat glioma cell migration [47–51], but did not establish a mechanism. Our previous study [7] documented L1 expression and proteolysis in human and rat glioma cell lines, expression in high-grade human surgical samples, and proteolysis when surgical cells were placed in culture. L1 function was also demonstrated in a rat glioma cell line by using antisense sequences to attenuate L1 expression and L1 blocking antibodies against the L1 ectodomain. However the mechanism of this L1-facilitated glioma cell migration still needed to be established for human high-grade glioma cells, and a potential explanation needed to be sought for the difference in L1 proteolysis between surgical samples as fresh specimen versus being placed in culture.
This study demonstrated L1 expression and proteolysis in the T98G human glioma cell line and human surgical GBM cells by using RT-PCR, Western blotting, and immunofluorescent staining. The proteolyzed L1 ectodomain was found in the cell culture medium of the human glioma cell line. Further, L1 proteolysis was demonstrated in migrating glioma cells, as opposed to non-migrating cells, and this proteolysis was coincident with cell surface appearance of ADAM10 in the migrating cells. This finding is consistent with the studies done by Gavert et al. [42], where L1 and its protease ADAM10 have been shown to be expressed at the invasive front of colon cancer to stimulate cell migration. What we found demonstrated that L1 proteolysis, rather than expression of L1, is activated in migrating glioma cells, which is different from Gavert et al.’s finding.
Full-length L1 remains around the cell periphery in T98G cells behind the scratch edge where they are confluent, which indicates L1’s possible function there as a cell–cell adhesion molecule. Shtutman et al. [52] found that expression of full-length L1, but not soluble ectodomain, increased motility of epithelial-like MCF7 cells by disrupting adherens junctions. Not consistent with what occurs in MCF7 cells, our work clearly demonstrates a stimulatory role for soluble L1 ectodomain in glioma cells and is in agreement with other work done on highly malignant cancer cell types [19, 53, 54] including our work on breast cancer cells [36]. From our data here and previously [7], we propose a model whereby L1 proteolysis occurs preferentially in cells at the edge of a glioma cell mass to release soluble L1 ectodomain, which then is autocrine/paracrine stimulatory for peripheral cell motility. Such proteolysis at the edge could also explain why our surgical sample specimens taken from the inner bulk of the tumor did not exhibit L1 proteolysis [7], whereas those same cells placed in culture were induced to proteolyze L1. The mechanism responsible for upregulation of cell surface ADAM10 at the edge of the cell mass warrants further investigation.
L1 was found in exosomes released from the T98G human glioma cell line. Although ADAM10 also was located in exosomes, L1 proteolysis by ADAM10 was not detected, as the cleavage fragments (180 and 32 kDa) were not observed. This is unlike the findings from others [18, 20] and from our previously characterized U-87 MG glioma exosomes [7], where L1 was found and proteolyzed in released exosome vesicles. This apparent regulation of ADAM10 activity in T98G cell exosomes also needs further investigation. Nonetheless, the full-length L1 in the exosomal membrane would be accessible to bind integrin receptors on T98G cell surfaces, which makes exosomes potentially able to contribute to stimulation of glioma cell migration. It would be interesting to determine which form of released L1 (soluble ectodomain or exosomal) is more effective at stimulation.
The L1 shRNA lentiviral vector successfully attenuated L1 expression in the human glioma cell line and surgical cells. By using a highly quantitative Super Scratch time-lapse motility assay [39, 40], which allowed tracking of individual cell velocities throughout the time course of the experiment, it was clear that L1 attenuation caused a highly significant decrease of cell motility in T98G/shL1 cells. Using L1 ectodomain-conditioned medium and the time-lapse motility assay, we not only demonstrated the recovery of cell motility in L1-attenuated T98G/shL1 cells, but also showed the increase of cell motility in U-118 MG cells, which do not express endogenous L1 protein. The collective results indicate that L1 is important in stimulating glioma cell migration and is very likely due to the L1 ectodomain after ADAM10 proteolysis. This is similar to what others have found in ovarian, colon and melanoma cancer cells [17–21, 53–55]. The consistent finding of L1 function in cancer cell migration/invasion might provide a general target for malignant cancer treatment by inhibiting motility instead of cell proliferation.
We investigated the possible signaling pathway of L1 ectodomain-stimulated glioma cell migration and found that FAK activation was dramatically decreased in L1-attenuated T98G/shL1 cells. However, FAK activation was recovered after incubation in L1 ectodomain conditioned medium. This correlation suggests that L1 facilitates glioma cell migration through induced FAK activation. Induction might be through the L1-binding integrins that we demonstrated are expressed by T98G cells. FAK activation is known to occur by the binding of soluble molecules to integrins [25, 26] as well as after binding of ECM molecules. Discovering L1 ectodomain as one of the soluble molecules that can initiate FAK activation helps to elucidate the mechanism by which FAK activation regulates cell migration in cancer. The exact mechanism of how the L1 ectodomain activates FAK through integrin binding still requires further study.
To approach a mechanism of how L1-regulated FAK activation controls glioma cell migration, focal complex size was examined. Compared to control cells, L1-attenuated T98G cells showed larger average focal complex size (as indicated by pFAKY397 staining). The larger focal complex size suggests a slower focal complex turnover rate [25, 43] which is known to be controlled, at least in part, by FAK activation [25, 26]. The sizes of the focal complex were reversed after incubating the L1-attenuated T98G/shL1 cells with L1 ectodomain conditioned medium, which shows that the L1 ectodomain can, either directly or indirectly, modulate focal complex size along with modulating cell motility. Thus, we showed that the L1 ectodomain can coincidentally modulate FAK activity, focal complex size, and cell velocity. Although the role of FAK in cell migration has been well established [reviewed in 26] and L1-mediated cell migration also has been demonstrated in several types of cancers [19, 53–55], establishing the signaling pathway between these two molecules not only helps to obtain a better understanding of glioma migration but provides alterative protein targets for glioma treatment. As the L1-autocrine/paracrine signaling pathway suggests, targeting FAK might be a more efficient and fundamental approach than targeting L1 alone in prohibition of glioma cell migration.
To investigate whether these findings extended to the ability of L1 to stimulate human glioma cell invasion, we used our novel chick embryo xenograft model. We previously used this model to show rat and human glioma cell lines can establish tumor formation and infiltration inside the embryonic chick brain [39]. Here, we showed that using this model, L1-attenuated T98G cells exhibited dramatically reduced infiltration into the brain compared to the control cells. This finding confirmed the importance of L1 function in glioma cell invasion in vivo, at least in a xenograft model. The role of L1 in glioma cell proliferation and/or survival in a rat xenograft model has been shown previously, but Bao et al. [56] did not analyze effects on cell motility or invasiveness. The functions of L1 in promoting cell growth and survival were found to occur in human glioma stem cells and showed that L1 plays an important role in facilitating glioma progression through augmenting proliferation and survival. Our study did not distinguish if L1-stimulated glioma cell migration included the glioma stem cell population, as Bao et al. showed the proliferative and survival effects of L1 to be specific to stem cells. It is possible that the glioma stem cells also utilize L1 expression and proteolysis to migrate away from the tumor mass to cause eventual re-growth of the tumor. In light of our results and others, targeting L1 and the resultant FAK signaling in glioma cells will have the potentially synergistic effect of decreasing both glioma motility as well as tumor growth.
In conclusion, our collective data support a model of autocrine/paracrine stimulation of cell migration and invasion in human glioma cells by releasing the L1 ectodomain and/or L1 containing exosomal vesicles. This L1 ectodomain-stimulated glioma cell migration coincides with FAK activation and alteration of focal complexes. These data demonstrated that it is the released L1 (from proteolysis or exosomal L1) and not the full length cell membrane bound L1 that stimulates the cell migration in an autocrine/paracrine manner, possibly through binding to cell surface integrin receptors. The interaction between soluble L1 and receptors (presumably integrins) activates FAK inside focal complexes thereby controlling cell migration. By using our novel chick embryonic brain microinjection model, we further confirmed the function of L1-facilitated human glioma cell invasion in vivo. This mechanism can explain the aggressive migratory nature of high-grade human glioma cells in that it provides stimulation of migration even if cells are not stimulated to migrate by extracellular molecules they encounter in the brain. A possible scenario could be that diffuse glioma cell migration is driven by the L1-mediated autocrine/paracrine mechanism, whereas migration along blood vessels or axon tracts are driven by both the L1-mediated mechanism and stimulation by extracellular matrix molecules along these routes that they often follow.
Acknowledgments
This work was funded by grant Number 2 P20 RR016472 under the INBRE program of the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) and by research resources of the Genetically Defined Microbe and Expression Core of the UAB mucosal HIV and Immunology Center (R24DK64400). We thank Dr. Kirk Czymmek at the U.D. Bio-Imaging center for assistance with confocal microscopy, Dr. Ulhas Naik (U.D.) for MDA-MB-231 cells, Dr. Robert Sikes (U.D.) for 293T/17 cells, and Dr. Vance Lemmon (U. of Miami) for the hL1-pcDNA3 vector.
Abbreviations
- ADAM10
A disintegrin and metalloprotease 10
- FACS
Flow cytometry
- FAK
Focal adhesion kinase
- FN Repeats
Fibronectin-like repeats
- GBM
Glioblastoma multiforme
- Ig domains
Immunoglobulin-like domains
- RGD
Arg-Gly-Asp
- RIPA buffer
Radioimmunoprecipitation assay buffer
- RT-PCR
Reverse transcription-polymerase chain reaction
Contributor Information
Muhua Yang, Email: yangmugua@gmail.com, Department of Biological Sciences, University of Delaware, Wolf Hall, Newark, DE 19716, USA.
Yupei Li, Email: yupei34@gmail.com, Department of Biological Sciences, University of Delaware, Wolf Hall, Newark, DE 19716, USA.
Kalyani Chilukuri, Email: ckalyani@udel.edu, Department of Biological Sciences, University of Delaware, Wolf Hall, Newark, DE 19716, USA.
Owen A. Brady, Email: oab8@cornell.edu, Department of Biological Sciences, University of Delaware, Wolf Hall, Newark, DE 19716, USA
Magdy I. Boulos, Email: mboulos@christianacare.org, Helen F. Graham Cancer Center, Christiana Care Health System, Newark, DE 19713, USA
John C. Kappes, Email: kappesjc@uab.edu, Department of Medicine, University of Alabama, Birmingham, AL 35294, USA
Deni S. Galileo, Email: dgalileo@udel.edu, Department of Biological Sciences, University of Delaware, Wolf Hall, Newark, DE 19716, USA
References
- 1.Claes A, Idema AJ, Wesseling P. Diffuse glioma growth: a guerilla war. Acta Neuropathol. 2007;114(5):443–458. doi: 10.1007/s00401-007-0293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kleihues P, Soylemezoglu F, Schauble B, Scheithauer BW, Burger PC. Histopathology, classification, and grading of gliomas. Glia. 1995;15:211–221. doi: 10.1002/glia.440150303. [DOI] [PubMed] [Google Scholar]
- 3.Laerum OD. Local spread of malignant neuroepithelial tumors. Acta Neurochir. 1997;139:515–522. doi: 10.1007/BF02750993. [DOI] [PubMed] [Google Scholar]
- 4.Viola JJ, Martuza RL. Gene therapies for glioblastomas. Bailliere’s Clin Neurol. 1996;5:413–424. [PubMed] [Google Scholar]
- 5.Wen P, Fine HA, Black PM, Shrieve DC, Alexander E, Loeffler JS. High-grade astrocytomas. Neurol Clin. 1995;13:875–900. [PubMed] [Google Scholar]
- 6.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 7.Yang M, Adla S, Temburni MK, Patel VP, Lagow EL, Brady OA, Tian J, Boulos MI, Galileo DS. Stimulation of glioma cell motility by expression, proteolysis, and release of the L1 neural cell recognition molecule. Cancer Cell Int. 2009;9:27. doi: 10.1186/1475-2867-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faissner A, Teplow DB, Kubler D, Keilhauer G, Kinzel V, Schachner M. Biosynthesis and membrane topography of the neural cell adhesion molecule L1. EMBO J. 1985;4:3105–3113. doi: 10.1002/j.1460-2075.1985.tb04052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moos M, Tacke R, Scherner H, Teplow D, Gruth K, Schachner M. Neural adhesion molecule L1 as a member of the immunoglobulin superfamily with binding domains similar to fibronectin. Nature. 1988;334:701–703. doi: 10.1038/334701a0. [DOI] [PubMed] [Google Scholar]
- 10.Persohn E, Schachner M. Immunoelectron microscopic localization of the neural cell adhesion molecules L1 and N-CAM during postnatal development of the mouse cerebellum. J Cell Biol. 1987;105:569–576. doi: 10.1083/jcb.105.1.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stallcup WB, Arner L, Levine J. Antiserum against the PC12 cell defines cell surface antigens specific for neurons and Schwann cells. J Neurosci. 1983;3:53–69. doi: 10.1523/JNEUROSCI.03-01-00053.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brummendorf T, Kenwrick S, Rathjen FG. Neural cell recognition molecule L1; from cell biology to human hereditary brain malformation. Curr Opin Neurobiol. 1988;8:87–97. doi: 10.1016/s0959-4388(98)80012-3. [DOI] [PubMed] [Google Scholar]
- 13.Haspel J, Grumet M. The L1CAM extracellular region: a multidomain protein with modular and cooperative binding modes. Front Biosci. 2003;8:s1210–s1225. doi: 10.2741/1108. [DOI] [PubMed] [Google Scholar]
- 14.Hortsch M. The L1 family of neural cell adhesion molecules: old proteins performing new tricks. Neuron. 1996;17:587–593. doi: 10.1016/s0896-6273(00)80192-0. [DOI] [PubMed] [Google Scholar]
- 15.Schachner M. Neural recognition molecules and synaptic plasticity. Curr Opin Cell Biol. 1997;9:627–634. doi: 10.1186/1475-2867-9-27. [DOI] [PubMed] [Google Scholar]
- 16.Fransen E, Lemmon V, van Camp G, Vits L, Couchke P, Willems PJ. CRASH syndrome: clinical spectrum of corpus callo-sum hypoplasia, retardation, adducted thumbs, spastic paraparesis and hydrocephalus due to mutations in one single gene, L1. Eur J Hum Genet. 1995;3:273–284. doi: 10.1159/000472311. [DOI] [PubMed] [Google Scholar]
- 17.Mechtersheimer S, Gutwein P, Agmon LN, Stoeck A, Oleszewski M, Riedle S, Postina R, Fahrenholz F, Fogel M, Lemmon V, Altevogt P. Ectodomain shedding of L1 adhesion molecule promotes cell migration by autocrine binding to integrins. J Cell Biol. 2001;155:661–674. doi: 10.1083/jcb.200101099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutwein P, Mechtersheimer S, Riedle S, Stoeck A, Gast D, Joumaa S, Zentgraf H, Fogel M, Altevogt DP. ADAM10-mediated cleavage of L1 adhesion molecule at the cell surface and in released membrane vesicles. FASEB J. 2003;17:292–294. doi: 10.1096/fj.02-0430fje. [DOI] [PubMed] [Google Scholar]
- 19.Gutwein P, Stoeck A, Riedle S, Gast D, Runz S, Condon TP, Marmé A, Phong MC, Linderkamp O, Skorokhod A, Altevogt P. Cleavage of L1 in exosomes and apoptotic membrane vesicles released from ovarian carcinoma cells. Clin Cancer Res. 2005;11(7):2492–2501. doi: 10.1158/1078-0432.CCR-04-1688. [DOI] [PubMed] [Google Scholar]
- 20.Maretzky T, Schulte M, Ludwig A, Rose-John S, Blobel C, Hartmann D, Altevogt P, Saftig P, Reiss K. L1 is sequentially processed by two differently activated metalloproteases and presenilin/gamma-secretase and regulates neural cell adhesion, cell migration, and neurite outgrowth. Mol Cell Biol. 2005;25:9040–9053. doi: 10.1128/MCB.25.20.9040-9053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stoeck A, Keller S, Riedle S, Sanderson MP, Runz S, Le Naour F, Gutwein P, Ludwig A, Rubinstei E, Alevogt P. A role for exosomes in the constitutive and stimulus-induced ectodomain cleavage of L1 and CD44. Biochem J. 2006;393:609–618. doi: 10.1042/BJ20051013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Itoh K, Fushiki S, Kamiguchi H, Arnold B, Altevogt P, Lemmon V. Disrupted Schwann cell–axon interactions in peripheral nerves of mice with altered L1–integrin interactions. Mol Cell Neurosci. 2005;30(4):624–629. doi: 10.1016/j.mcn.2005.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liljelund P, Ghosh P, van den Pol AN. Expression of the neural axon adhesion molecule L1 in the developing and adult rat brain. J Biol Chem. 1994;269(52):32886–32895. [PubMed] [Google Scholar]
- 24.Yamasaki M, Thompson P, Lemmon V. CRASH syndrome: mutations in L1CAM correlate with severity of the disease. Neuropediatrics. 1997;28(3):175–178. doi: 10.1055/s-2007-973696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 2004;6(2):154–161. doi: 10.1038/ncb1094. [DOI] [PubMed] [Google Scholar]
- 26.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6(1):56–68. doi: 10.1038/nrm154. [DOI] [PubMed] [Google Scholar]
- 27.Ebeling O, Duczmal A, Aigner S, Geiger C, Schollhammer S, Kemshead JT, Moller P, Schwartz-Albiez R, Altevogt P. L1 adhesion molecule on human lymphocytes and monocytes: expression and involvement in binding to alpha v beta 3 integrin. Eur J Immunol. 1996;26:2508–2516. doi: 10.1002/eji.1830261035. [DOI] [PubMed] [Google Scholar]
- 28.Montgomery AM, Becker JC, Siu CH, Lemmon VP, Cheresh DA, Pancook JD, Zhao X, Reisfeld RA. Human neural cell adhesion molecule L1 and rat homologue NILE are ligands for integrins αvβ3. J Cell Biol. 1996;132:475–485. doi: 10.1083/jcb.132.3.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruppert M, Aigner S, Hubble M, Yagita H, Altevogt P. The L1 adhesion molecule is a cellular ligand for VLA-5. J Cell Biol. 1995;131:1881–1891. doi: 10.1083/jcb.131.6.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kadmon G, Altevogt P. The cell adhesion molecule L1: species- and cell-type-dependent multiple binding mechanisms. Differentiation. 1997;61(3):143–150. doi: 10.1046/j.1432-0436.1997. 6130143.x. [DOI] [PubMed] [Google Scholar]
- 31.Stein GH. T98G: an anchorage-independent human tumor cell line that exhibits stationary phase G1 arrest in vitro. J Cell Physiol. 1979;99(1):43–54. doi: 10.1002/jcp.1040990107. [DOI] [PubMed] [Google Scholar]
- 32.Pontén J, Macintyre EH. Long term culture of normal and neoplastic human glia. Acta Pathol Microbiol Scand. 1968;74(4):465–486. doi: 10.1111/j.1699-0463.1968.tb03502.x. [DOI] [PubMed] [Google Scholar]
- 33.Moscovici H, Moscovici MG, Jimenez H, Lai MM, Hayman MJ, Vogt PK. Continuous tissue culture cell lines derived from chemically induced tumors of Japanese quail. Cell. 1977;11(1):95–103. doi: 10.1016/0092-8674(77)90320-8. [DOI] [PubMed] [Google Scholar]
- 34.Tjio JH, Puck TT. Genetics of somatic mammalian cells. II. Chromosomal constitution of cells in tissue culture. J Exp Med. 1958;108:259–271. doi: 10.1084/jem.108.2.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7(8):2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Y, Galileo DS. Soluble L1CAM promotes breast cancer cell adhesion and migration in vitro, but not invasion. Cancer Cell Int. 2010;10(1):34. doi: 10.1186/1475-2867-10-34. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Tine BA, Kappes JC, Banerjee NS, Knops J, Lai L, Steenbergen RD, Meijer CL, Snijders PJ, Chatis P, Broder TR, Moen PT, Jr, Chow LT. Clonal selection for transcriptionally active viral oncogenes during progression to cancer. J Virol. 2004;78(20):11172–11186. doi: 10.1128/JVI.78.20.11172-11186.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen W, Wu X, Levasseur DN, Liu H, Lai L, Kappes JC, Townes TM. Lentiviral vector transduction of hematopoietic stem cells that mediate long-term reconstitution of lethally irradiated mice. Stem Cells. 2000;18(5):352–359. doi: 10.1634/stemcells.18-5-352. [DOI] [PubMed] [Google Scholar]
- 39.Cretu A, Fotos JS, Little BW, Galileo DS. Human and rat glioma growth, invasion, and vascularization in a novel chick embryo brain tumor model. Clin Exp Metastasis. 2005;22(3):225–236. doi: 10.1007/s10585-005-7889-x. [DOI] [PubMed] [Google Scholar]
- 40.Fotos JS, Patel VP, Karin NJ, Temburni MK, Koh JT, Galileo DS. Automated time-lapse microscopy and high-resolution tracking of cell migration. Cytotechnology. 2006;51(1):7–19. doi: 10.1007/s10616-006-9006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raposo G, Nijman HW, Stoorvogel W, Leijendekkerr R, Harding CV, Melief CJM, Geuze HJ. B Lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996;183:1161–1172. doi: 10.1084/jem.183.3.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gavert N, Conacci-Sorrell M, Gast D, Schneider A, Altevogt P, Brabletz T, Ben-Ze’ev A. L1, a novel target of beta-catenin signaling, transforms cells and is expressed at the invasive front of colon cancers. J Cell Biol. 2005;168:633–642. doi: 10.1083/jcb.200408051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302(5651):1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 44.Rubenstein M, Shaw M, Mirochnik Y, Slobodskov L, Glick R, Lichtor T, Chou P, Guinan P. In vivo establishment of T98G human glioblastoma. Methods Find Exp Clin Pharmacol. 1999;21(6):391. doi: 10.1358/mf.1999.21.6.541918. [DOI] [PubMed] [Google Scholar]
- 45.Smith JS, Jenkins RB. Genetic alterations in adult diffuse glioma: occurrence, significance, and prognostic implications. Front Biosci. 2000;5:D213–D231. doi: 10.2741/smith. [DOI] [PubMed] [Google Scholar]
- 46.Stiles CD, Rowitch DH. Glioma stem cells: a midterm exam. Neuron. 2008;58(6):832–846. doi: 10.1016/j.neuron,2008.05.031. [DOI] [PubMed] [Google Scholar]
- 47.Tsuzuki T, Izumoto S, Ohnishi T, Hiraga S, Arita N, Hayakawa T. Neural cell adhesion molecule L1 in gliomas: correlation with TGF-β and p53. J Clin Pathol. 1998;51:13–17. doi: 10.1136/jcp.51.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamanaka R, Tanaka R, Yoshida S. Effects of irradiation on the expression of the adhesion molecules (NCAM, ICAM-1) by glioma cell lines. Neurol Med Chir (Tokyo) 1993;33(11):749–752. doi: 10.2176/nmc.33.749. [DOI] [PubMed] [Google Scholar]
- 49.Izumoto S, Ohnishi T, Arita N, Hiraga S, Taki T, Hayakawa T. Gene expression of neural cell adhesion molecule L1 in malignant gliomas and biological significance of L1 in glioma invasion. Cancer Res. 1996;56(6):1440–1444. [PubMed] [Google Scholar]
- 50.Ohnishi T, Matsumura H, Izumoto S, Hiraga S, Hayakawa T. A novel model of glioma cell invasion using organotypic brain slice culture. Cancer Res. 1998;58(14):2935–2940. [PubMed] [Google Scholar]
- 51.Suzuki T, Izumoto S, Fujimoto Y, Maruno M, Ito Y, Yoshimine T. Clinicopathological study of cellular proliferation and invasion in gliomatosis cerebri: important role of neural cell adhesion molecule L1 in tumour invasion. J Clin Pathol. 2005;58(2):166–171. doi: 10.1136/jcp.2004.020909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shtutman M, Levina E, Ohouo P, Baig M, Roninson IB. Cell adhesion molecule L1 disrupts E-cadherin-containing adherens junctions and increases scattering and motility of MCF7 breast carcinoma cells. Cancer Res. 2006;66(23):11370–11380. doi: 10.1158/0008-5472.CAN-06-2106. [DOI] [PubMed] [Google Scholar]
- 53.Meier F, Busch S, Gast D, Göppert A, Altevogt P, Maczey E, Riedle S, Garbe C, Schittek B. The adhesion molecule L1 (CD171) promotes melanoma progression. Int J Cancer. 2006;119(3):549–555. doi: 10.1002/ijc.21880. [DOI] [PubMed] [Google Scholar]
- 54.Gavert N, Sheffer M, Raveh S, Spaderna S, Shtutman M, Brabletz T, Barany P, Notterman D, Domany E, Ben-Ze’ev A. Expression of L1-CAM and ADAM10 in human colon cancer cells induces metastasis. Cancer Res. 2007;67:16. doi: 10.1158/0008-5472.CAN-07-0991. [DOI] [PubMed] [Google Scholar]
- 55.Thies A, Schachner M, Moll I, Berger J, Schulze HJ, Brunner G, Schumacher U. Overexpression of the cell adhesion molecule L1 is associated with metastasis in cutaneous malignant melanoma. Eur J Cancer. 2002;38:1708–1716. doi: 10.1016/S0959-8049(02)00105-3. [DOI] [PubMed] [Google Scholar]
- 56.Bao S, Wu Q, Li Z, Sathornsumetee S, Wang H, McLendon RE, Hjelmeland AB, Rich JN. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res. 2008;68(15):6043–6048. doi: 10.1158/0008-5472.CAN-08-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]