

Abstract
The Ku heterodimer, consisting of the proteins Ku70 and Ku80, is the central component of the nonhomologous end joining (NHEJ) pathway of double strand break (DSB) repair. Ku is able to recognize and bind a DSB by virtue of its ring-like structure. Both pre-repair and topologically trapped post-repair Ku heterodimers are thought to be inhibitory to multiple cellular processes. Thus, a regulated mechanism for the removal of Ku from chromatin was predicted to exist. Recent evidence shows that Ku80 is removed from DNA through a ubiquitin-mediated process. Similar processes have been shown to be involved in the regulated dissociation of a host of other proteins from chromatin, and this appears to be a general and conserved mechanism for the regulation of chromatin-associated factors. A potential mechanism for this pathway is discussed.
Keywords: Ku70, Ku80, nonhomologous end joining, double strand break, VCP, proteasome
1. Introduction
DNA damage responses require the regulated association of multiple proteins with damaged DNA structures and the regions of chromatin surrounding those structures. Over the past several years, cytological and biochemical approaches have been used to make major progress in understanding how the recruitment of proteins to DNA damage is regulated. An equally important, yet much less well-understood, question concerns how proteins are removed from chromatin, either to make way for other damage binding factors or to return chromatin to an undamaged state following repair.
The conserved heterodimer formed between Ku70 and Ku80, known as Ku [1], is a particularly interesting candidate for the study of regulated protein removal from DNA damage. Ku is the central component of the nonhomologous end joining (NHEJ) pathway of DNA double strand break (DSB) repair [2], and its mode of binding to and high affinity for DSBs, abundance, and inhibitory roles in multiple repair pathways suggest that its removal from DNA is critically important for repair regulation and post-repair recovery. It has been shown recently that Ku80 can be removed from DNA using a K48-linked ubiquitin-dependent pathway [3], which a growing body of evidence suggests may be a widespread and conserved mechanism for the removal of proteins from DNA. Similar mechanisms may be involved in the removal of other proteins from the sites of DSBs during DNA repair responses.
2. Bringing it all together at a double strand break
DSBs occur as a result of programmed cellular events, as in V(D)J and class switch recombination in developing B- and T-cells, or due to genotoxic insults resulting from free radicals, ionizing radiation, or a failure to control nuclear enzymatic activities. Eukaryotic cells can call upon two major DSB repair pathways: homologous recombination (HR) [4] and NHEJ [2]. HR requires a homologous stretch of DNA, usually a replicated sister chromatid, and for this reason typically occurs during the late S or G2 phases of the cell cycle following replication. This pathway results in generally error-free repair of DSBs. NHEJ, in contrast, does not require a homologous piece of DNA and therefore can occur without a prior round of replication. The trade-off, however, is that NHEJ is an error-prone pathway that has the propensity to result in insertions or deletions at the site of the repaired DNA. The NHEJ pathway has been reviewed extensively elsewhere [2], and therefore this review will only touch on its basics.
NHEJ begins with the recognition of the DSB by the Ku complex. The toroidal ring-shaped structure of Ku (see below and Fig. 1) allows it to recognize and tightly bind to DSBs within seconds of their formation [5]. Upon binding to DSBs, Ku recruits additional factors in the NHEJ pathway: the PI3-related kinase DNA-PKcs, XLF, XRCC4, DNA ligase IV (LigIV), the nuclease Artemis, and the PolX family DNA polymerases pol μ and pol λ [2]. Ku itself has been reported to have deubiquitylating [6] and 5′-deoxyribose-5-phosphate lyase activities [7]. However, because its main function appears to be the recruitment of several factors with enzymatic activities to the DSB, it has been referred to as a “toolbelt” protein [2], much like the processivity factor PCNA, which recruits multiple proteins to the site of the replication fork. Following their recruitment, the various Ku-associated enzymes trim and/or extend the ends of the broken DNA in preparation for re-sealing by the NHEJ-dependent ligase LigIV.
Fig. 1.
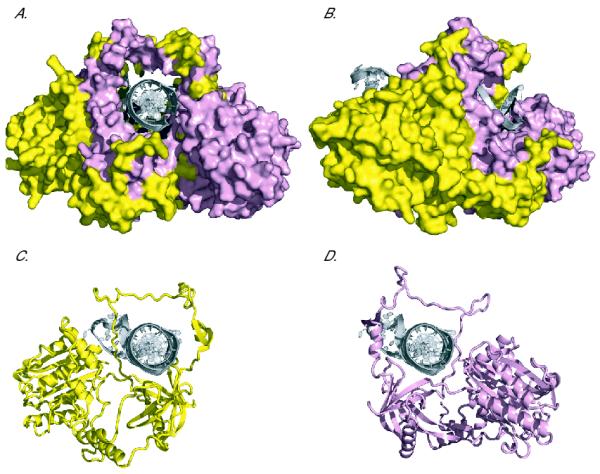
Ku70 and Ku80 are interconnected over a large surface, and both subunits encircle the DNA. (A) Front view and (B) side view surface representations of Ku bound to a DNA substrate. DNA is depicted as a cartoon in silver, Ku80 in yellow, and Ku70 in pink. (C) Front view cartoon representation of Ku80 bound to DNA. In this image, Ku70 has been removed to illustrate how Ku80 encircles the DNA on its own. (D) As in (C), but here Ku70 is represented alone. Structures [9] have been depicted using MacPyMOL.
For many years, it was assumed that the NHEJ pathway was unique to eukaryotes, largely because the best-studied bacterium, Escherichia coli, has no known mechanism to recircularize linear DNA. This assumption was disproved by bioinformatic analyses that revealed that many bacteria do, in fact, encode a version of the Ku protein [8]. Prokaryotic Ku homologs typically exist as homodimers, and Ku genes are frequently found adjacent on the chromosome to genes encoding an ATP-dependent ligase, LigD, with which it functions. While Ku and LigD are present in some form in many bacterial species, they are almost completely absent in archaea [8]. Therefore, whether the prokaryotic NHEJ pathway arose as the ancestor to eukaryotic NHEJ or is instead the result of horizontal gene transfer remains a mystery.
3. A ring without a clasp
The X-ray crystal structure of Ku demonstrated that Ku is able to recognize DSBs and initiate NHEJ by virtue of its toroidal, or donut-shaped, structure [9] (Fig. 1). A central channel exactly large enough to accommodate a B-form helix forms the Ku DNA binding surface, leading to an elegant model for the recognition of DSBs by Ku. A broken DNA end is able to thread through the hole at the center of Ku as through the eye of a needle, while unbroken DNA cannot possibly bind to Ku in this manner, ensuring exquisitely specific DSB recognition. Although the structure of prokaryotic Ku has not been determined, molecular threading studies have predicted that the basic ring structure is conserved [10]. Comparisons between DNA-bound and unbound crystal [9] and cryoelectron microscopy structures [11] of Ku have revealed that the C-termini of both Ku70 and Ku80 move upon DNA binding. Interestingly, the Ku70 and Ku80 N-terminal regions and the central ring-shaped DNA binding domains do not appear to change conformation significantly when DNA is present [9].
Ring-shaped proteins and complexes are widespread in biology, and several toroidal nucleic acid-binding factors are known [12]. Unlike Ku, however, most other toroidal proteins do not typically need to bind to DSBs to fulfill their functions and correspondingly employ distinct mechanisms of DNA binding. Instead of passively binding a DNA end, these ring-shaped complexes frequently open and re-close around DNA by virtue of conformational changes induced by interactions between DNA and exterior domains of the protein or the help of accessory factors [12]. Such ring opening requires the separation of adjacent but discreet domains or subunits.
There is no clear mechanism by which conformational changes in Ku subunits would result in ring opening. In contrast to most other ring-shaped complexes, the two subunits of Ku both encircle the DNA and are inter-connected over an extensive binding surface (Fig. 1C and D). Thus, while the toroidal structure of Ku provides an attractive mechanism for DSB binding and recognition, it presents a problem post-repair, as completion of NHEJ would topologically trap Ku on DNA (Fig. 2A and B). Such entrapment has been demonstrated in vitro, as the interaction between Ku and DNA is able to withstand 2 M salt without dissociation following recircularization, while such a high salt concentration induces Ku release from broken DNA [13]. Thus, the only possible conceivable mechanism for Ku removal after repair appeared to be via proteolysis or denaturation.
Fig. 2.
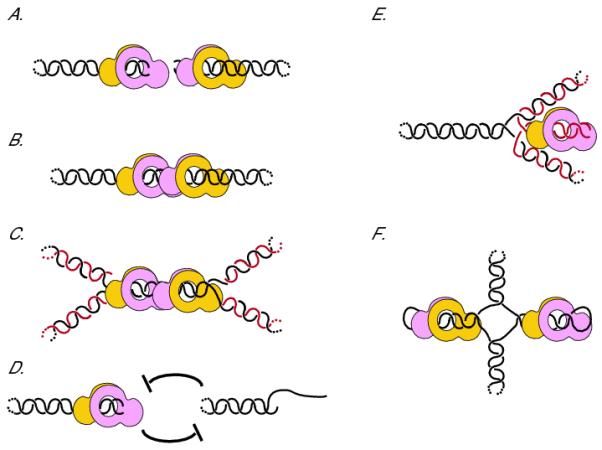
Ku has the ability to bind to multiple DNA structures, altering DNA metabolic processes. (A) A DSB: one heterodimer of Ku70 (pink) and Ku80 (yellow) is bound to each end of a DSB. Dotted lines denote a continuation of the DNA strand. (B) A repaired DSB: both Ku heterodimers are topologically attached to the DNA following NHEJ. (C) Post-repair replication: topologically trapped Ku heterodimers might inhibit replication fork progression when forks converge, preventing replication termination. Nascent daughter strands are depicted in red while parental strands are in black. (D) End resection: Ku bound to an unrepaired DSB may sterically block end resection enzymes from accessing the break. This could inhibit the early steps of end resection and, subsequently, HR. Similarly, the creation of a 3′-single stranded overhang inhibits Ku binding to the DSB because Ku binds poorly to single stranded DNA. (E) Chickenfoot: a regressed fork forms a four-way junction. The resultant daughter-daughter duplex resembles a DSB, and may be stabilized by binding to Ku. (F) Cruciform structures: inverted repeats in the genome have a tendency to form hairpin structures, to which Ku could also bind.
4. An inhibitor of many processes
It is difficult to imagine that Ku allowed to remain on DNA following repair would not impair multiple cellular processes, most notably transcription and DNA replication. There is a precedent for this type of inhibition in the case of Topoisomerase II topologically trapped on DNA after treatment with the drug ICRF-193, which inhibits the enzyme’s cleavage activity. ICRF-193 treatment results in a much more severe phenotype than a simple depletion of Topoisomerase II, presumably as a result of the physical blocks to transcription or replication progression imposed by the trapped enzyme [14].
Like Topoisomerase II, Ku’s central channel is only large enough to encircle a single duplex of DNA [9], and therefore Ku trapped on DNA would also presumably inhibit polymerase progression. It is possible that a moving polymerase is able to push Ku heterodimers along DNA, but even if earlier steps of replication or transcriptional elongation were not inhibited, Ku would minimally be predicted to prevent polymerase progression when two enzymes converge, as during replication termination (Fig. 2C).
Ku’s ability to inhibit DNA metabolism, however, is not restricted to post-repair processes. Instead, heterodimers bound to unrepaired DSBs may be inhibitory to multiple DNA repair pathways. Due to its high affinity for DNA ends (Kd ~ 2 nM) [15] and high concentration (~300 nM in HeLa cells) [16,17] Ku is positioned to be one of the first, if not the first, factors to bind to a DSB. Furthermore, there is ample evidence that Ku has a relatively slow kinetic off-rate from DNA ends under physiological conditions. In vitro studies using purified Ku protein in the presence of reducing agents demonstrate that a majority of Ku remains bound to double stranded oligo DNA for at least several minutes [18]. Similarly, in the absence of active removal mechanisms (see below) the majority of Ku80 remains associated with DNA over the course of at least an hour in Xenopus laevis egg extracts [3]. The high affinity, high concentration, and slow off-rate predict that Ku may sterically block other DNA binding proteins from accessing the break. In fact, in vitro studies have shown that Ku bound to a DSB inhibits enzyme activities at the break, including T4 DNA ligase-mediated ligation [15].
Several lines of evidence indicate that Ku bound to DSB ends impedes a variety of processes in vivo. First, data from Saccharomyces cerevisiae suggest that the degradation of 5′ ends in preparation for HR to free 3′ single stranded ends for strand invasion, a process known as end resection, is increased in the absence of Ku [19,20]. Ku inhibition of end resection is overcome by the initial clipping of the 5′ end that occurs prior to extensive 5′ degradation [21]. This suggests that at least one important role for this initial clipping step of resection, performed by the Sae2 enzyme and the Mre11-Rad50-Xrs2 complex in yeast, is to convert the DSB into a single-stranded overhang, a structure to which Ku cannot easily bind (Fig. 2D).
The role of Ku in inhibiting end resection and HR appears to be conserved in higher eukaryotes, as the absence of Ku from mammalian cells increases the frequency of HR at site-specific DSBs [22]. Induction of HR in the absence of the NHEJ factor XRCC4 is significantly less pronounced, suggesting that the bulky presence of Ku at DSBs competes with other factors required for end resection. Ku and LigIV are also inhibitory to a dangerous repair pathway known as alternative end joining (alt-EJ), in which DSBs are joined in a NHEJ-independent manner often involving microhomologies between the two repaired ends [23]. Because this pathway is not as well regulated as NHEJ, it frequently leads to chromosomal translocations [24]. With the caveat that more research is required to untangle the physical roles of Ku from enzymatic roles of other NHEJ components, such as the kinase activity of DNA-PKcs [25], it seems clear that Ku regulates repair pathway choice at least in part by restricting access to the break. A critical function of Ku in the cell may therefore be to quickly bind DSB ends and protect them from alternative repair pathways and enzymes.
Recently, an additional inhibitory role for Ku has been discovered in the repair of interstrand DNA crosslinks by the Fanconi anemia pathway. Experiments using Caenorhabditis elegans, mammalian cells [26], and chicken DT40 cells [27] have demonstrated that defects in crosslink repair resulting from depletion of Fanconi anemia pathway components can be rescued by Ku depletion. There are, however, some discrepancies in these results. In C. elegans and mammalian cells, depletion or inhibition of all NHEJ proteins tested, including DNA-PKcs and LigIV (LIG-4 in C. elegans), had the same effect as Ku depletion in suppressing Fanconi anemia pathway defects [26]. In the DT40 cells, however, only loss of Ku had this effect [27]. Therefore, it is unclear if the physical binding of Ku to a repair intermediate, a downstream toxic NHEJ pathway, or some combination of the two causes the inhibition of repair. One reasonable hypothesis is that the Fanconi anemia pathway acts in part by recruiting nucleases to the damage to generate a DNA substrate to which Ku can no longer bind, as in the case of initiators of end resection [27].
The high affinity of Ku for DNA ends could also cause problems that have not yet been explored. Ku requires only a double stranded end for binding and is thus capable of interacting with and stabilizing many DNA structures that are not actually damaged. For example, stalled replication forks are thought occasionally to regress, allowing displaced daughter strands to anneal and form a four-way junction known as a “chickenfoot” [28,29]. A chickenfoot contains a daughter-daughter “middle toe” that resembles a DSB and could be recognized by Ku (Fig. 2E). In addition, Ku could stabilize the cruciform hairpins that occur at sites of inverted repeats (Fig. 2F), which have a tendency to lead to replication fork stalling [30]. Removal of Ku from DNA may therefore be necessary for the efficient resolution of these structures as well as for the relief of Ku-induced inhibition of DNA processing.
5. Ku80 removal from DNA
The potential for Ku to interfere with replication and repair suggests that the cell may have mechanisms in place to remove Ku from DNA, a hypothesis supported by in vivo observations. First, Ku has been shown to dissociate rapidly from chromatin in vivo. Fluorescence recovery after photobleaching (FRAP) experiments in mammalian cells using GFP-tagged Ku80 show that Ku80 molecules assemble on laser-induced DNA damage within seconds, but are nearly completely replaced at the site of DNA damage within a few minutes of photobleaching [5]. The rapid exchange of Ku from DSBs in vivo contrasts with the slow in vitro off-rates discussed above and suggests that some active mechanism may be involved in Ku removal. However, it remains formally possible that this rapid removal is caused by the DNA passively unthreading from the protein.
Interestingly, chromatin immunoprecipitation (ChIP) experiments in yeast have shown that Ku is lost from sites around an endonuclease-induced DSB in a manner that is roughly coincident with the repair of the DSB [20]. These ChIP experiments do not allow the measurement of Ku exchange at the site of the unrepaired break, which presumably takes place with similar kinetics to those in mammalian cells. However, this approach does allow analysis of the fate of Ku molecules trapped on DNA at the moment of repair. The fact that topologically bound Ku is released from DNA so quickly after repair indicates that a mechanism for its active removal must exist.
How, then, might Ku be removed from DNA? Based on its structure and mode of binding, only a mechanism highly disruptive to protein integrity would plausibly suffice. Experiments conducted to analyze protein recruitment to and removal from model DNA damage in Xenopus laevis egg extract provide a satisfying potential answer to this mystery [3] (Fig. 3). Specifically, Ku80 bound to immobilized DNA modeling a DSB is rapidly modified with K48-linked polyubiquitylation, which marks proteins for proteasomal degradation. This polyubiquitylation is required for the dissociation of Ku80 from DNA. Surprisingly, however, although the proteasome inhibitor MG132 inhibits DSB- and polyubiquitin-dependent Ku80 degradation, it does not affect the removal of Ku80 from DSBs. This suggests that, while the proteolytic activity of the proteasome is required for the degradation of Ku80 once it is removed from DNA, it is not required for its removal per se.
Fig. 3.
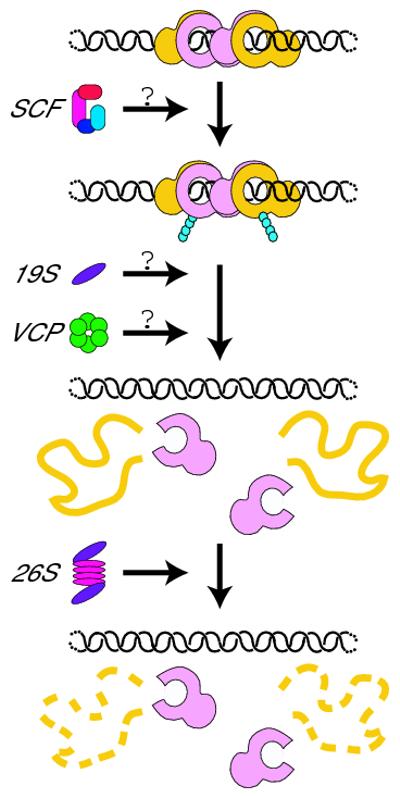
A model for Ku dissociation from DNA. Ku bound to DNA recruits an E3 ubiquitin ligase, possibly the SCF complex, which modifies Ku80 through K48-linked polyubiquitylation (blue circles). This ubiquitylation recruits another factor such as VCP, the 19S proteasome complex, or both, which uses its ATPase activity to unfold Ku80 and dissociate it from Ku70 and the DNA. After Ku80 dissociation, Ku70 loses its own ability to bind to DNA. Unfolded Ku80 is subsequently degraded by the 26S proteasome.
Interestingly, ubiquitylation and degradation occur with the same kinetics and specificity for full-length Ku80 as for truncation mutants missing both the N- and C-termini but retaining the minimal central DNA-binding domain [3]. However, such truncations are nonfunctional in NHEJ assays, suggesting that DNA binding, rather than NHEJ completion, triggers Ku80 ubiquitylation. This opens up the possibility that the ubiquitylation of Ku80 may be important for the removal of both topologically bound post-repair protein as well as protein on unrepaired DSBs or DSB-like structures.
It is unclear how Ku80 bound to DNA is specified for ubiquitylation while soluble protein remains unmodified. As described above, the only regions of Ku known to undergo substantial conformational changes upon binding to DNA are the C-termini of Ku70 and Ku80, but neither is required for Ku80 ubiquitylation ([3] and LP and Hironori Funabiki unpublished data). Therefore, it seems unlikely that a major DNA-dependent conformational change signals ubiquitylation. Two possibilities remain for the specific recognition of DNA-bound Ku80. First, Ku80 may be post-translationally modified upon DNA binding, and this modification may be recognized by the E3 ubiquitin ligase. Alternatively, the E3 ubiquitin ligase may act in a combinatorial manner, recognizing both Ku and either DNA itself or another DNA-bound factor and binding strongly only when both are present.
While the identity of the E3 ubiquitin ligase is still unknown, components of the Skp1-Cul1-F box (SCF) complex have been identified by mass spectrometry as preferentially associating with DSBs in Xenopus egg extract [3]. Intriguingly, this association is largely dependent on the presence of Ku, suggesting that Ku may be a substrate for the SCF. It is tempting to speculate that Ku may also serve as a platform for general recruitment of the SCF to DNA damage, aiding the ubiquitylation of additional factors during or following DSB repair.
6. Coming undone and letting go
There is an extensive body of literature linking the ubiquitin proteasome system (UPS) to the regulation of chromatin-bound proteins. Such mechanisms were first studied in depth in the context of several transcriptional regulators, where roles for the UPS have been implicated using diverse mechanisms that have been reviewed elsewhere [31,32]. In some cases, such as the regulation of the yeast mating type transcriptional repressor α2, ubiquitylation leads to protein removal from chromatin [33].
Recently, there has been a rash of examples of additional chromatin-bound proteins whose dissociation is regulated by the UPS [34]. Ubiquitin initiates cell cycle-dependent removal of the replication licensing factor Cdt1 [35,36] and the mitotic kinase Aurora B [37] from chromatin. In addition, ubiquitylation regulates the localization of the yeast H3 variant Cse4 to centromeres [38,39] and the JmjC family antisilencing protein Epe1 to heterochromatin boundaries [40] by inducing protein dissociation from other regions of the chromosome. At sites of DNA damage, ubiquitin-mediated processes remove the large subunit of stalled RNA polymerase II, Rpb1, during the transcription coupled repair process [41].
The observation that the removal of Ku80 from chromatin requires polyubiquitylation but not proteolytic activity of the proteasome is unexpected but not inexplicable. There are two known factors that can unfold ubiquitylated proteins, the valosin-containing protein (VCP, and also known as p97 or Cdc48 in yeast) hexameric AAA-ATPase complex and the 19S regulatory complex of the proteasome, and one or both of these may be responsible for releasing Ku80 from DNA. The 19S complex forms the 26S proteasome together with the 20S protease-containing complex [42]. A major role of the 19S complex is to recognize, unfold, and transfer proteins to the 20S complex for degradation, but it can also be found apart from the 20S complex and has the ability to function independently. Similarly, some ubiquitylated proteins require VCP to aid in their destruction. VCP, like the 19S complex, exhibits an ATPase-dependent chaperone activity, interacts with ubiquitylated proteins, and is thought to help with the destruction of ubiquitylated proteins by unfolding them for subsequent proteasomal degradation. This role of VCP has been best-studied in the context of degradation of misfolded proteins in the endoplasmic reticulum (ER) [43]. In addition to its function on the surface of the ER, VCP, like the proteasome, also localizes to the nucleus [44].
The exact roles for VCP and the proteasome in the removal of proteins from chromatin remain under active investigation. VCP ATPase activity is required for the removal of Aurora B from chromatin [37], but it is unclear whether the 19S or 20S complexes are also required. Similarly, Cdc48 mediates the chromatin dissociation and degradation of Rpb1 in yeast [41]. Proteasome components were found to be bound to ubiquitylated Rpb1 at the same time as Cdc48, suggesting that the two complexes might work together or concurrently. The Ku80 data [3] point to a potential general pathway in which proteasomal degradation is not required for the removal of polyubiquitylated proteins from chromatin, and instead VCP, the 19S complex, or both are employed to unfold proteins leading to dissociation (Fig. 3). This is a particularly attractive mechanism, because VCP has the ability to extract proteins from complexes or aggregates [45], an activity that may be required to disrupt the tight interaction between Ku70 and Ku80. As each monomer is unstable and unable to bind DSBs on its own [1], Ku70 left behind probably does not maintain its association with DNA. A non-proteolytic removal mechanism opens up the possibility that proteins can be re-folded post-dissociation, allowing for degradation to be regulated according to varying cellular conditions. It is also possible that, while 26S-dependent proteolysis is not absolutely required for Ku80 (or other protein) dissociation from chromatin, degradation is temporally linked to protein unfolding.
In light of the recent examples described above, it seems likely that the localization of many DSB-bound proteins in addition to Ku are regulated by UPS-dependent dissociation. Indeed, both 19S and 20S proteasome components have been localized to DSBs by ChIP [46], and Sem1, the yeast ortholog of the BRCA2-associated protein DSS1, is a component of the 19S proteasome [46,47]. Furthermore, the proteasome plays roles in both NHEJ and HR [46] and is involved in the activity of the Fanconi anemia pathway [48] and in DSB repair pathway choice [49,50]. The identification of additional DSB response proteins dissociated in this manner, and the mechanisms of their regulation, will be an important area of future study.
7. An ancient mechanism?
The mode of Ku binding to DNA and its role in NHEJ is presumably conserved from bacteria to mammals [10], and the problems posed by topologically bound Ku must therefore also be conserved. However, there is no ubiquitin homolog in bacteria. Could there nevertheless be a related mechanism for prokaryotic Ku removal? Despite the lack of ubiquitin, bacterial species do encode well-conserved ATP-dependent proteases, which are functional and mechanistic 26S proteasome analogs. For example, E. coli, the best-studied bacterial species, has five such ATP-dependent proteases: ClpAP, ClpXP, FtsH, HslUV, and Lon [51]. The ClpX subunit of ClpXP unfolds proteins in an ATP-dependent manner, much like VCP and the 19S proteasome, while ClpP degrades them. Like VCP and the proteasome, ClpXP has the ability to extract proteins from tightly bound complexes [52,53].
Recent evidence from E. coli indicates that ClpXP may be involved in removing proteins from the bacterial chromosome much like the UPS in eukaryotic cells. The prokaryotic nucleotide excision repair factor UvrA is induced following UV exposure and subsequently degraded by ClpXP [54]. Intriguingly, non-damaged DNA is required for UvrA degradation in vitro, suggesting that ClpXP may remove overexpressed protein nonspecifically interacting with undamaged DNA while allowing interactions with damage to remain [54]. Such a mechanism is reminiscent of the ubiquitin-dependent processes by which Cse4 and Epe1 are localized to particular sites on yeast chromosomes, during which the UPS performs an editing function by removing proteins from erroneous sites. While the particular example of UvrA regulation was discovered in E. coli, which does not encode Ku, the fact that ClpXP function is well-conserved among bacteria raises the tantalizing possibility that prokaryotic Ku, as well as other prokaryotic proteins, may be removed from DNA via a similar process.
8. Conclusions
It is becoming increasingly clear that Ku has a multitude of functions in the cell, from promotion of NHEJ to protection of DNA ends from various cellular enzymes that, if left unchecked, could lead to aberrant DNA processing and defects such as chromosomal translocations. In addition, topologically trapped Ku on the DNA may present complications during post-repair transcription and replication. The abundance of Ku and its high affinity for DNA ends, combined with its structure and mode of DNA binding, make it a particularly interesting candidate for the study of regulated protein dissociation from chromatin. The ubiquitin-dependent system recently discovered for the removal and degradation of Ku80 is an elegant process by which DNA is relieved of Ku either before or after repair completion. Future efforts will be required to determine the specifics and generality of the mechanism of Ku80 dissociation from chromatin and the full import of this pathway on the many functions of Ku.
Acknowledgments
I would like to thank Neil Osheroff and Allan Weissman for helpful discussions, J. Robert Hogg and Hironori Funabiki for reviewing the manuscript, and Brian Houck-Loomis for help with figure preparation. LP’s research has been supported with a postdoctoral fellowship from the Leukemia and Lymphoma Society of America and NIH training grant T32 CA09673.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Downs JA, Jackson SP. A means to a DNA end: the many roles of Ku. Nat Rev Mol Cell Biol. 2004;5:367–78. doi: 10.1038/nrm1367. [DOI] [PubMed] [Google Scholar]
- [2].Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Postow L, Ghenoiu C, Woo EM, Krutchinsky AN, Chait BT, Funabiki H. Ku80 removal from DNA through double strand break-induced ubiquitylation. J Cell Biol. 2008;182:467–79. doi: 10.1083/jcb.200802146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–57. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- [5].Mari PO, et al. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc Natl Acad Sci U S A. 2006;103:18597–602. doi: 10.1073/pnas.0609061103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Amsel AD, Rathaus M, Kronman N, Cohen HY. Regulation of the proapoptotic factor Bax by Ku70-dependent deubiquitylation. Proc Natl Acad Sci U S A. 2008;105:5117–22. doi: 10.1073/pnas.0706700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Roberts SA, Strande N, Burkhalter MD, Strom C, Havener JM, Hasty P, Ramsden DA. Ku is a 5′-dRP/AP lyase that excises nucleotide damage near broken ends. Nature. 2010;464:1214–7. doi: 10.1038/nature08926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pitcher RS, Brissett NC, Doherty AJ. Nonhomologous end-joining in bacteria: a microbial perspective. Annu Rev Microbiol. 2007;61:259–82. doi: 10.1146/annurev.micro.61.080706.093354. [DOI] [PubMed] [Google Scholar]
- [9].Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412:607–14. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- [10].Bowater R, Doherty AJ. Making ends meet: repairing breaks in bacterial DNA by non-homologous end-joining. PLoS Genet. 2006;2:e8. doi: 10.1371/journal.pgen.0020008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rivera-Calzada A, Spagnolo L, Pearl LH, Llorca O. Structural model of full-length human Ku70-Ku80 heterodimer and its recognition of DNA and DNA-PKcs. EMBO Rep. 2007;8:56–62. doi: 10.1038/sj.embor.7400847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hingorani MM, O’Donnell M. A tale of toroids in DNA metabolism. Nat Rev Mol Cell Biol. 2000;1:22–30. doi: 10.1038/35036044. [DOI] [PubMed] [Google Scholar]
- [13].Paillard S, Strauss F. Analysis of the mechanism of interaction of simian Ku protein with DNA. Nucleic Acids Res. 1991;19:5619–24. doi: 10.1093/nar/19.20.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jensen LH, et al. A novel mechanism of cell killing by anti-topoisomerase II bisdioxopiperazines. J Biol Chem. 2000;275:2137–46. doi: 10.1074/jbc.275.3.2137. [DOI] [PubMed] [Google Scholar]
- [15].Blier PR, Griffith AJ, Craft J, Hardin JA. Binding of Ku protein to DNA. Measurement of affinity for ends and demonstration of binding to nicks. J Biol Chem. 1993;268:7594–601. [PubMed] [Google Scholar]
- [16].Mimori T, Hardin JA. Mechanism of interaction between Ku protein and DNA. J Biol Chem. 1986;261:10375–9. [PubMed] [Google Scholar]
- [17].Zhao L, Sukstanskii AL, Kroenke CD, Song J, Piwnica-Worms D, Ackerman JJ, Neil JJ. Intracellular water specific MR of microbead-adherent cells: HeLa cell intracellular water diffusion. Magn Reson Med. 2008;59:79–84. doi: 10.1002/mrm.21440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Andrews BJ, Lehman JA, Turchi JJ. Kinetic analysis of the Ku-DNA binding activity reveals a redox-dependent alteration in protein structure that stimulates dissociation of the Ku-DNA complex. J Biol Chem. 2006;281:13596–603. doi: 10.1074/jbc.M512787200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- [20].Wu D, Topper LM, Wilson TE. Recruitment and dissociation of nonhomologous end joining proteins at a DNA double-strand break in Saccharomyces cerevisiae. Genetics. 2008;178:1237–49. doi: 10.1534/genetics.107.083535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mimitou EP, Symington LS. Ku prevents Exo1 and Sgs1- dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 2010;29:3358–69. doi: 10.1038/emboj.2010.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pierce AJ, Hu P, Han M, Ellis N, Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001;15:3237–42. doi: 10.1101/gad.946401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol. 2010;17:410–6. doi: 10.1038/nsmb.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].McVey M, Lee SE. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–38. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Neal JA, Dang V, Douglas P, Wold MS, Lees-Miller SP, Meek K. Inhibition of Homologous Recombination by DNA-Dependent Protein Kinase Requires Kinase Activity, Is Titratable, and Is Modulated by Autophosphorylation. Mol Cell Biol. 2011;31:1719–33. doi: 10.1128/MCB.01298-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Adamo A, et al. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell. 2010;39:25–35. doi: 10.1016/j.molcel.2010.06.026. [DOI] [PubMed] [Google Scholar]
- [27].Pace P, Mosedale G, Hodskinson MR, Rosado IV, Sivasubramaniam M, Patel KJ. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science. 2010;329:219–23. doi: 10.1126/science.1192277. [DOI] [PubMed] [Google Scholar]
- [28].Higgins NP, Kato K, Strauss B. A model for replication repair in mammalian cells. J Mol Biol. 1976;101:417–25. doi: 10.1016/0022-2836(76)90156-x. [DOI] [PubMed] [Google Scholar]
- [29].Postow L, Ullsperger C, Keller RW, Bustamante C, Vologodskii AV, Cozzarelli NR. Positive torsional strain causes the formation of a four-way junction at replication forks. J Biol Chem. 2001;276:2790–6. doi: 10.1074/jbc.M006736200. [DOI] [PubMed] [Google Scholar]
- [30].Zhao J, Bacolla A, Wang G, Vasquez KM. Non-B DNA structure-induced genetic instability and evolution. Cell Mol Life Sci. 2009;67:43–62. doi: 10.1007/s00018-009-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Auld KL, Silver PA. Transcriptional regulation by the proteasome as a mechanism for cellular protein homeostasis. Cell Cycle. 2006;5:1503–5. doi: 10.4161/cc.5.14.2979. [DOI] [PubMed] [Google Scholar]
- [32].Lipford JR, Deshaies RJ. Diverse roles for ubiquitin-dependent proteolysis in transcriptional activation. Nat Cell Biol. 2003;5:845–50. doi: 10.1038/ncb1003-845. [DOI] [PubMed] [Google Scholar]
- [33].Wilcox AJ, Laney JD. A ubiquitin-selective AAA-ATPase mediates transcriptional switching by remodelling a repressor-promoter DNA complex. Nat Cell Biol. 2009;11:1481–6. doi: 10.1038/ncb1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].O’Connell BC, Harper JW. Ubiquitin proteasome system (UPS): what can chromatin do for you? Curr Opin Cell Biol. 2007;19:206–14. doi: 10.1016/j.ceb.2007.02.014. [DOI] [PubMed] [Google Scholar]
- [35].Arias EE, Walter JC. PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat Cell Biol. 2006;8:84–90. doi: 10.1038/ncb1346. [DOI] [PubMed] [Google Scholar]
- [36].Arias EE, Walter JC. Replication-dependent destruction of Cdt1 limits DNA replication to a single round per cell cycle in Xenopus egg extracts. Genes Dev. 2005;19:114–26. doi: 10.1101/gad.1255805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ramadan K, Bruderer R, Spiga FM, Popp O, Baur T, Gotta M, Meyer HH. Cdc48/p97 promotes reformation of the nucleus by extracting the kinase Aurora B from chromatin. Nature. 2007;450:1258–62. doi: 10.1038/nature06388. [DOI] [PubMed] [Google Scholar]
- [38].Hewawasam G, Shivaraju M, Mattingly M, Venkatesh S, Martin-Brown S, Florens L, Workman JL, Gerton JL. Psh1 is an E3 ubiquitin ligase that targets the centromeric histone variant Cse4. Mol Cell. 2010;40:444–54. doi: 10.1016/j.molcel.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ranjitkar P, Press MO, Yi X, Baker R, MacCoss MJ, Biggins S. An E3 ubiquitin ligase prevents ectopic localization of the centromeric histone H3 variant via the centromere targeting domain. Mol Cell. 2010;40:455–64. doi: 10.1016/j.molcel.2010.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Braun S, Garcia JF, Rowley M, Rougemaille M, Shankar S, Madhani HD. The Cul4-Ddb1(Cdt)(2) ubiquitin ligase inhibits invasion of a boundary-associated antisilencing factor into heterochromatin. Cell. 2011;144:41–54. doi: 10.1016/j.cell.2010.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Verma R, Oania R, Fang R, Smith GT, Deshaies RJ. Cdc48/p97 mediates UV-dependent turnover of RNA Pol II. Mol Cell. 2011;41:82–92. doi: 10.1016/j.molcel.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pickart CM, Cohen RE. Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol. 2004;5:177–87. doi: 10.1038/nrm1336. [DOI] [PubMed] [Google Scholar]
- [43].Raasi S, Wolf DH. Ubiquitin receptors and ERAD: a network of pathways to the proteasome. Semin Cell Dev Biol. 2007;18:780–91. doi: 10.1016/j.semcdb.2007.09.008. [DOI] [PubMed] [Google Scholar]
- [44].Madeo F, Schlauer J, Zischka H, Mecke D, Frohlich KU. Tyrosine phosphorylation regulates cell cycle-dependent nuclear localization of Cdc48p. Mol Biol Cell. 1998;9:131–41. doi: 10.1091/mbc.9.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kobayashi T, Manno A, Kakizuka A. Involvement of valosin-containing protein (VCP)/p97 in the formation and clearance of abnormal protein aggregates. Genes Cells. 2007;12:889–901. doi: 10.1111/j.1365-2443.2007.01099.x. [DOI] [PubMed] [Google Scholar]
- [46].Krogan NJ, et al. Proteasome involvement in the repair of DNA double-strand breaks. Mol Cell. 2004;16:1027–34. doi: 10.1016/j.molcel.2004.11.033. [DOI] [PubMed] [Google Scholar]
- [47].Funakoshi M, Li X, Velichutina I, Hochstrasser M, Kobayashi H. Sem1, the yeast ortholog of a human BRCA2-binding protein, is a component of the proteasome regulatory particle that enhances proteasome stability. J Cell Sci. 2004;117:6447–54. doi: 10.1242/jcs.01575. [DOI] [PubMed] [Google Scholar]
- [48].Jacquemont C, Taniguchi T. Proteasome function is required for DNA damage response and fanconi anemia pathway activation. Cancer Res. 2007;67:7395–405. doi: 10.1158/0008-5472.CAN-07-1015. [DOI] [PubMed] [Google Scholar]
- [49].Gudmundsdottir K, Lord CJ, Ashworth A. The proteasome is involved in determining differential utilization of double-strand break repair pathways. Oncogene. 2007;26:7601–6. doi: 10.1038/sj.onc.1210579. [DOI] [PubMed] [Google Scholar]
- [50].Murakawa Y, et al. Inhibitors of the proteasome suppress homologous DNA recombination in mammalian cells. Cancer Res. 2007;67:8536–43. doi: 10.1158/0008-5472.CAN-07-1166. [DOI] [PubMed] [Google Scholar]
- [51].Gottesman S. Proteases and their targets in Escherichia coli. Annu Rev Genet. 1996;30:465–506. doi: 10.1146/annurev.genet.30.1.465. [DOI] [PubMed] [Google Scholar]
- [52].Moore SD, Baker TA, Sauer RT. Forced extraction of targeted components from complex macromolecular assemblies. Proc Natl Acad Sci U S A. 2008;105:11685–90. doi: 10.1073/pnas.0805633105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zolkiewski M. A camel passes through the eye of a needle: protein unfolding activity of Clp ATPases. Mol Microbiol. 2006;61:1094–100. doi: 10.1111/j.1365-2958.2006.05309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pruteanu M, Baker TA. Controlled degradation by ClpXP protease tunes the levels of the excision repair protein UvrA to the extent of DNA damage. Mol Microbiol. 2009;71:912–24. doi: 10.1111/j.1365-2958.2008.06574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]