

Abstract
Cell division in rod-shaped bacteria is initiated by formation of a ring of the tubulin-like protein FtsZ at mid-cell. Division site selection is controlled by a conserved division inhibitor MinCD, which prevents aberrant division at the cell poles. The Bacillus subtilis DivIVA protein controls the topological specificity of MinCD action. Here we show that DivIVA is targeted to division sites late in their assembly, after some MinCD-sensitive step requiring FtsZ and other division proteins has been passed. DivIVA then recruits MinD to the division sites preventing another division from taking place near the newly formed cell poles. Sequestration of MinD to the poles also releases the next mid-cell sites for division. Remarkably, this mechanism of DivIVA action is completely different from that of the equivalent protein MinE of Escherichia coli, even though both systems operate via the same division inhibitor MinCD.
Keywords: Bacillus subtilis, DivIVA, cell division, MinD, cytokinesis
A central component of division apparatus of bacteria is a highly conserved tubulin-like protein called FtsZ (Lutkenhaus and Addinall 1997). FtsZ can polymerize in vitro (Bramhill and Thompson 1994; Mukherjee and Lutkenhaus 1994; Erickson et al. 1996) and forms a ring-like structure at the division site in vivo (Bi and Lutkenhaus 1991; Wang and Lutkenhaus 1993; Levin and Losick 1996). Nucleation of this ‘Z ring’ is thought to be a primary point of control over the timing and positioning of the cell division septum. In most bacteria, several other proteins required for constriction and septal peptidoglycan synthesis associate with the Z ring, including divIB (Harry and Wake 1997); divIC (Katis et al. 1997); ftsA (Addinall and Lutkenhaus 1996; Ma et al. 1997); pbpB (Weiss et al. 1997); and zipA (Hale and De Boer 1997). In almost all cases targeting of these proteins to the division site requires ftsZ.
The Z ring is normally positioned precisely at the mid point of the cell, so that division gives rise to two equal daughters. Important insights into the mechanism responsible for identifying the mid-cell position have been obtained from studies of minicell producing (min) mutants. These mutants divide at approximately the normal frequency but many of the divisions are improperly positioned, occurring close to the cell pole to give spherical, usually anucleate minicells (Adler et al. 1967; Reeve et al. 1973). Thus, the min system seems to be required to inactivate used division sites (Teather et al. 1974). In Escherichia coli, the min locus contains three genes, minC, minD, and minE. Mutations of minC or minD give a minicell phenotype, whereas minE null mutations prevent division, giving rise to long aseptate filaments (De Boer et al. 1989). It is well established that MinC is an inhibitor of cell division, which in conjunction with MinD, prevents division at the cell poles (De Boer et al. 1990, 1992; Mulder et al. 1992). MinE controls the topological specificity of MinCD, allowing it or forcing it to inhibit division at the cell poles but not at the required mid-cell position (De Boer et al. 1989; Pichoff et al. 1995; Zhao et al. 1995).
The minCD division-inhibition system appears to be conserved across a broad range of bacteria. For example, homologs of minC and minD have been characterized in the Gram-positive bacterium Bacillus subtilis (Levin et al. 1992; Varley and Stewart 1992; Lee and Price 1993). Mutations in these genes give a typical minicell phenotype, consistent with their division inhibition function being conserved. However, the B. subtilis min locus lacks a minE homolog. Genetic evidence revealed that an unlinked gene, divIVA, fulfils the role of MinE in this organism (Cha and Stewart 1997; Edwards and Errington 1997). Thus, mutations in divIVA, or depletion of its protein product in a conditional mutant, result in the inhibition of division leading to formation of long aseptate cell filaments. This Sep− phenotype is, like that of minE, suppressed by mutations in minD, giving a minicell (Min−) phenotype indistinguishable from that of minD single mutants (Cha and Stewart 1997; Edwards and Errington 1997). The sequence of the predicted product of the divIVA gene shows no significant similarity to that of minE, raising the possibility that the two proteins have different functions or that they have arrived at the same function via convergent evolution.
The function of Escherichia coli MinE has been elucidated recently by the demonstration that this protein can localize in the form of a ring at mid-cell and that this localization is independent of FtsZ (Raskin and De Boer 1997). Thus, MinE must recognize a spatial cue independently at this site. In principle, this ‘topological target’ might be the same as the one that FtsZ putatively recognizes, or it could be some independent marker. The nature of the target is not yet clear, but interestingly, correct localization of MinE requires minD (Raskin and De Boer 1997).
Previous work with a DivIVA–GFP fusion protein revealed that this protein is also targeted to mid-cell division sites (Edwards and Errington 1997). Unlike MinE, however, DivIVA–GFP remains detectable at the cell poles well after division is completed. On the basis of these findings, and informed by previous proposals for the E. coli min system (De Boer et al. 1989; Pichoff et al. 1995; Zhao et al. 1995; Rothfield and Zhao 1996), we suggested two alternative classes of model to explain the action of DivIVA (Fig. 1; Edwards and Errington 1997). In the polar piloting model (Fig. 1A), DivIVA is recruited to the division site as division is in progress, or at least after the division apparatus has achieved some committing step that is sensitive to inhibition by MinCD. DivIVA then attracts the MinCD inhibitor to this mid-cell site. Sequestration of MinCD at the poles of the newborn cells prevents division from occurring again near these sites and also depletes the division inhibitor from mid-cell, allowing the next central division to occur. In the mid-cell inhibition model (Fig. 1B), DivIVA arrives at the mid-cell site early and sets up a zone of inhibition of MinCD (or otherwise prevents MinCD from acting here) so that the septum can form. Incorporation of DivIVA into the septum inactivates it or sequesters it away from MinCD, allowing the division inhibitor to prevent further divisions close to the poles. The latter model is probably most compatible with the recent localization results described for MinE (Raskin and De Boer 1997).
Figure 1.

Models for functioning of the Min system of B. subtilis. Possible models for functioning of the B. subtilis Min system (adapted from Edwards and Errington 1997). The solid triangles represent DivIVA molecules, and shaded circles the MinCD division inhibitor (we assume that MinC and MinD associate, but it is possible that they act separately). The symbols are solid if the proteins are active, and open if they are inactive. The shaded triangle represents a hypothetical early step in septation that is sensitive to MinCD inhibition. (A) The polar piloting model, in which DivIVA delivers active MinCD to the poles. (B) The mid-cell inhibition model, in which DivIVA blocks MinCD action at mid-cell.
We have now tested these models by examining the effects on DivIVA localization of mutations in various division genes. In striking contrast to the results of similar experiments with MinE (Raskin and De Boer 1997), we show that targeting of DivIVA requires most, if not all, of the components of the division apparatus, but not MinD. We have also examined the localization of B. subtilis MinD by use of a GFP fusion and by immunofluorescence. We show for the first time that MinD forms ring-like accumulations at mid-cell, and that the protein is retained at the newly formed poles after division. Also, this DivIVA-like localization requires divIVA. The results provide important insights into the mechanism of division site selection in B. subtilis and show that even though both E. coli and B. subtilis use a common molecule (MinCD) to inhibit division at the cell poles, the mechanisms used to control the topological specificity of this inhibitor are fundamentally different.
Results
DivIVA localization is a late step in the hierarchy of assembly of division proteins
Conditional mutants were used to test whether DivIVA bands can form in the absence of various division proteins. Because some of these experiments required the mutants to be incubated at high (nonpermissive) temperatures at which GFP fluorescence is reduced, we raised an antiserum against DivIVA so that localization of the protein could be determined by immunofluorescence microscopy (IFM). Figure 2A shows that the antiserum used in these experiments was highly specific for DivIVA. At a high dilution (1/5000) with extracts of the wild-type strain (168CA), it reacted with only a single protein of ∼19 kD, corresponding to the predicted mass for DivIVA (lane 1). In extracts of a strain with divIVA under the control of an IPTG-dependent promoter, the 19-kD band was detected after growth in the presence of (lane 2) but not the absence (lane 3) of inducer. The staining pattern obtained by IFM (Fig. 2B) was similar to that of DivIVA–GFP in unfixed cells (Edwards and Errington 1997). Thus, in these wild-type cells the fluorescence was localized to discrete bands near the mid points of cells and at spots or bands near the cell poles. Interestingly, some central bands had a doublet appearance. We assume that the lysozyme treatment used to permeabilize the cells enhances the separation of newly divided cells. These observations are consistent with DivIVA being targeted to nascent division sites and remaining there until well after the completion of division.
Figure 2.
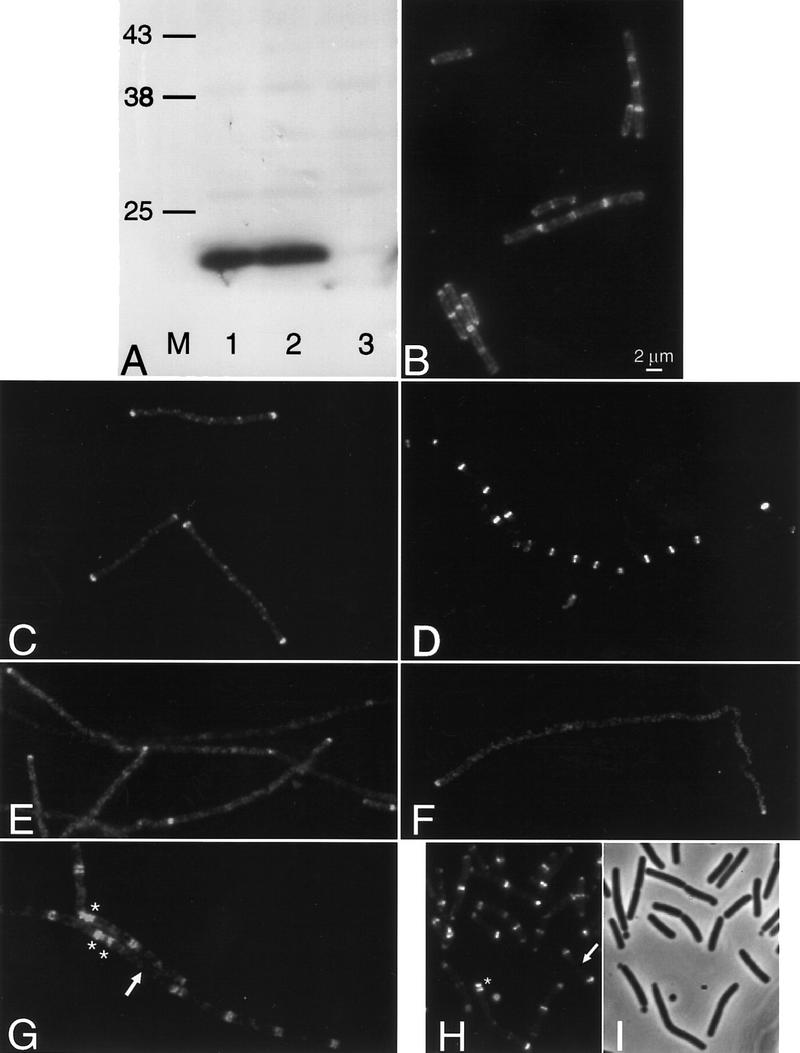
Localization of DivIVA in division mutants. (A) Western blot demonstrating the specificity of the crude anti-DivIVA antiserum. Extracts of wild-type B. subtilis (168CA; lane 1), and a divIVA repressible strain (1756) grown in the presence (lane 2) or absence (lane 3) of inducer. Lane M indicates the positions of prestained molecular mass markers (kD). (B–H) Fluorescence micrographs showing the localization of DivIVA by immunofluorescence (B,E–G) or a DivIVA–GFP fusion (C,D,H). (B) Wild-type cells (168CA); (C) FtsZ-repressed (strain 1759); (D) FtsZ-induced (strain 1759); (E) divIB mutant (1802); (F) divIC mutant (SU347); (G,H) minD mutant (strains 1901 and 1922, respectively). (I) Phase-contrast image of the cells in H. Arrows in G and H point to relatively long cells with no central band of DivIVA. Asterisks in G and H show examples of doublet or triplet bands of DivIVA.
Localization of DivIVA in the absence of FtsZ was tested with a strain carrying an IPTG-inducible allele of ftsZ and a divIVA–gfp fusion. Figure 2D shows the regular banding pattern for DivIVA–GFP in the presence of inducer. In the absence of inducer, repression of FtsZ synthesis leads to inhibition of division and hence formation of long aseptate filaments. In these cells, DivIVA–GFP showed a much more diffuse signal, with occasional spots scattered along the filament and major concentrations of fluorescence only apparent at the ends of the filaments, representing the sites at which the last cell divisions had occurred (Fig. 2C). Similar results were obtained by IFM using a strain with wild-type DivIVA (not shown). The dependence of DivIVA localization on divIB and divIC was tested using thermosensitive mutants. (Control experiments showed that DivIVA localization was not affected at the high nonpermissive temperature needed for the divIC mutant; not shown). Figure 2, E and F, shows that localization of DivIVA was lost in both of these mutants, apart from at pre-existing poles and at the rare division sites in the divIB mutant. Thus, DivIVA requires at least three components of the division machinery for targeting to the division site.
In contrast to these results, DivIVA readily formed bands at intermediate positions in cells of a minD mutant, whether examined by IFM (Fig. 2G) or with a DivIVA–GFP fusion (Fig. 2H,I). The pattern of bands in these cells was not normal but rather their placement appeared to follow the irregularly positioned division septa produced by the min mutant. Indeed, the DivIVA protein appeared to follow faithfully the division apparatus to either mid-cell sites or to aberrant polar sites, as indicated by the presence of double or triple rings at the poles of some cells in which minicells had been formed (stars in Fig. 2, G and H). Moreover, the formation of extra polar rings was accompanied by a tendency for rings to be missing in the body of longer cells (arrows in Fig. 2, G and H), just as for division septa in min mutants. We conclude that recruitment of DivIVA to sites of cell division requires at least partial assembly of the division apparatus but that it does not require minD.
Localization of MinD to division sites
To further distinguish between the two models shown in Figure 1, we examined the subcellular localization of MinD. B. subtilis MinD was purified and used to raise a polyclonal antiserum. The specificity of this antiserum was again examined by Western blotting (Fig. 3A). Two bands were detected in vegetative cells of B. subtilis (lane 1). The faster migrating band was of mobility expected for MinD and was absent from a minD null mutant (lane 2). Affinity purification resulted in a highly specific antibody preparation, which no longer detected the slower migrating band (lane 3).
Figure 3.
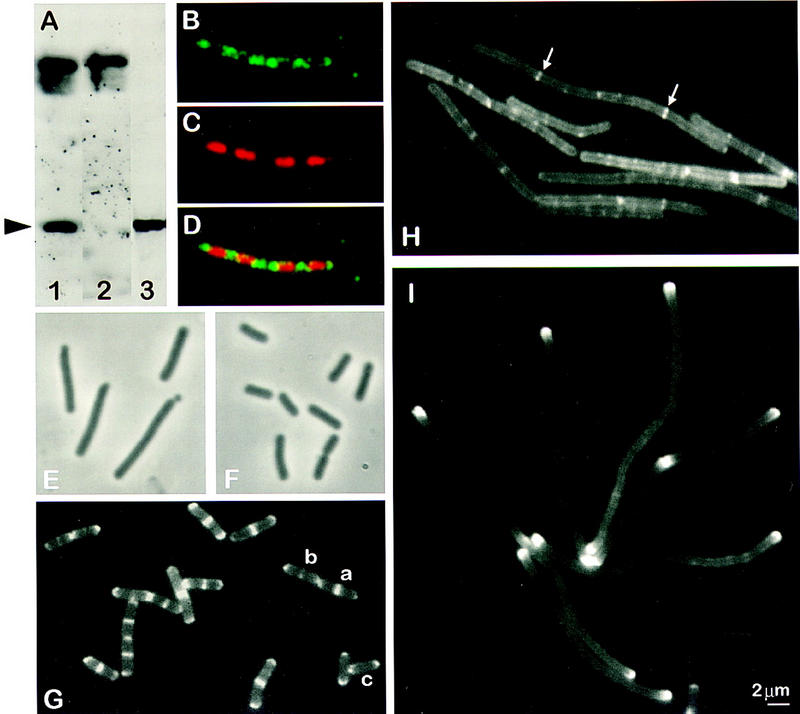
Subcellular localization of MinD and its dependence on divIVA and ftsZ. (A) Western blot, performed after SDS-PAGE (10%) and electrotransfer, demonstrating affinity purification of the anti-MinD antibodies. Crude anti-MinD antiserum was used to probe vegetative extracts of wild-type (SG38; lane 1) and minD (1901; lane 2) cells. After affinity purification, the anti-MinD antiserum recognized a single band in extracts of wild-type cells (SG38; lane 3). The arrowhead indicates the position of MinD protein. (B–D) Immunofluorescence micrographs showing detection of MinD at the cell poles and between nucleoids. The cells were stained for MinD protein (B) and DNA with DAPI (C), with the two images merged in D. (E,F) Phase-contrast images showing complementation of ΔminD by gfp–minD. Strain 1981 (ΔminD Pxyl–gfp–minD) grown in S medium (E) or S medium containing 0.5% xylose (F). (G) Fluorescence images showing the distribution of GFP–MinD in growing cells (S medium) of B. subtilis strain 1981. Examples of cells with different patterns of GFP–MinD are labeled a–c (see text). (H) Dependence of targeting of GFP–MinD to the poles on divIVA. Strain 1984 (ΔminD divIVA− Pxyl–gfp–minD) was grown in S medium and gfp–minD expression was induced by the addition of 0.5% xylose. (I) Polar targeting of GFP–MinD in FtsZ− filaments.Strain 1979 (ΔminD Pspac–ftsZ Pxyl–gfp–minD) was grown in S medium containing 0.5 mm IPTG and 0.5% xylose, and ftsZ was depleted by removing the IPTG.
The purified antibody was used to examine the localization of MinD by IFM (Fig. 3B–D). The protein showed a relatively uneven distribution in the cell, with scattered spots and patches of fluorescence. However, the strongest signals were almost always associated with the cell poles or at mid-cell regions between separated sister nucleoids. Because the fixation and permeabilization procedures required for IFM are damaging and do not completely preserve the structure of the cell, we also wished to examine MinD localization by use of GFP fusions. Attempts to fuse GFP to the carboxyl terminus of MinD were unsuccessful: The gene fusion proved to be nonfunctional as judged by its inability to complement a minD mutation (results not shown). However, fusion of gfp to the amino-terminal coding part of minD did give a functional fusion protein. B. subtilis strain 1981 was constructed to carry a null mutation in minD and, elsewhere in the chromosome, a gfp–minD fusion driven by a xylose-inducible promoter (Pxyl). Therefore, gfp–minD expression can be controlled by growing the cells in the presence or absence of xylose. In the absence of xylose the strain had a typical Min phenotype, with elongated cells and minicells (Fig. 3E). However, in the presence of 0.5% xylose the cells appeared normal (Fig. 3F), demonstrating that the gfp–minD fusion can complement the minD null mutation and thus that the GFP–MinD fusion protein retains MinD function.
Microscopic examination of the distribution of GFP–MinD in induced living cells revealed a similar overall distribution of MinD to that of IFM but a considerably improved image quality (Fig. 3G). The protein was indeed concentrated at the cell poles and at some mid-cell sites. At the cell poles, the prominent fluorescence was generally in the form of an arc following the line of the pole. The weaker signal in the rest of the cell tended to be strongest at the cell periphery, suggesting that most of the protein is close to the cell envelope. Some cells (generally the longer ones; see below) had prominent accumulations of GFP–MinD at mid-cell, either in the form of a band (e.g., cell marked a) or as one or two peripheral dots (e.g., cell marked b). Such patterns would be consistent with MinD forming a circumferential ring, as noted for other division proteins (e.g., Addinall and Lutkenhaus 1996; Harry and Wake 1997). In shorter cells, the GFP–MinD signal was particularly weak at mid-cell (e.g., cell marked c). Plots of signal intensity along lines drawn through the long axes of this and similar cells indicate that the protein concentration reduces gradually from the pole towards mid-cell (data not shown).
In summary, the pattern of localization of MinD was similar to that of DivIVA, in being mainly found at impending and old cell division sites. However, MinD seemed less precisely targeted than DivIVA, with a greater background signal diffusing away from the cell poles.
MinD requires divIVA for targeting to the cell poles
We reported previously that the inhibition of cell division that occurs in the absence of DivIVA is minD dependent (Edwards and Errington 1997). It was therefore interesting to test whether a divIVA mutation affects the targeting of MinD to division sites. To avoid the problem of the near-lethal filamentous phenotype of a null divIVA mutant (Cha and Stewart 1997; Edwards and Errington 1997) we again made use of the inducible gfp–minD construct, so that in the absence of xylose (effectively giving a minD divIVA double mutant), the mutant strain (1984) had a Min− phenotype (not shown). As expected, these uninduced cells did not fluoresce detectably (not shown), confirming that gfp–minD was repressed. Upon addition of xylose, synthesis of the functional GFP–MinD fusion protein resulted in cell filamentation, consistent with the idea that in the absence of DivIVA, the uncontrolled action of MinCD inhibits division at polar and mid-cell sites. Figure 3H shows that in these aseptate filaments the normal polar localization of GFP–MinD was abolished. Instead, the fluorescence was mainly diffusely distributed along the length of the filament. Interestingly, there were some bands of MinD–GFP at intermediate positions possibly corresponding to potential division sites (arrowed). However, these concentrations of MinD–GFP were much weaker than in the wild type and the lack of MinD–GFP concentrations at the completed cell poles was strikingly different from the wild type. Similar results were obtained in an experiment in which DivIVA protein was depleted by repression (using a Pspac–divIVA construct; strain 1756) and wild-type MinD protein was visualized by IFM (data not shown). It thus appears that targeting of MinD to the cell poles is dependent on DivIVA.
Given that targeting of DivIVA to the division site is in turn dependent on several components of the division machinery, MinD targeting should show a similar dependence. To test this, we examined localization of GFP–MinD in the absence of FtsZ (again by use of a repressible ftsZ allele). Figure 3I shows that the MinD protein in long ftsZ filaments was, as in the wild type, concentrated at the cell poles. However, in the absence of new divisions, there were no strong bands within the filaments, only a weak diffuse signal. This distribution was quite different from that of the DivIVA− filaments (Fig. 3H) and much more reminiscent of the localization of DivIVA itself in FtsZ-depleted cells (Fig. 2C). It thus appears that MinD localization at the cell poles requires DivIVA, though it may have a weak affinity for potential division sites in the absence of DivIVA.
DivIVA and MinD assemble approximately simultaneously at mid-cell before septal constriction
The above results suggested that MinD should assemble at mid-cell at about the same time or slightly later in the cell cycle than DivIVA. We therefore examined the formation of DivIVA and MinD bands as a function of cell length. Cells of strains 1918 (divIVA–gfp) and 1981 (gfp–minD), were grown to mid-exponential phase and samples from the cultures were fixed and examined microscopically. About 200 cells from each sample were scored for length and the presence or absence of a medial band of GFP (Fig. 4). Clearly, bands at mid-cell were only found in the longer cells. Not only were the proportions of cells with medial bands of DivIVA (Fig. 4A) and MinD (Fig. 4B) very similar, the average length at which bands appeared at mid-cell were also similar (calculated as 2.5 μm for both cultures, based on the methods of Sharpe et al. 1998).
Figure 4.
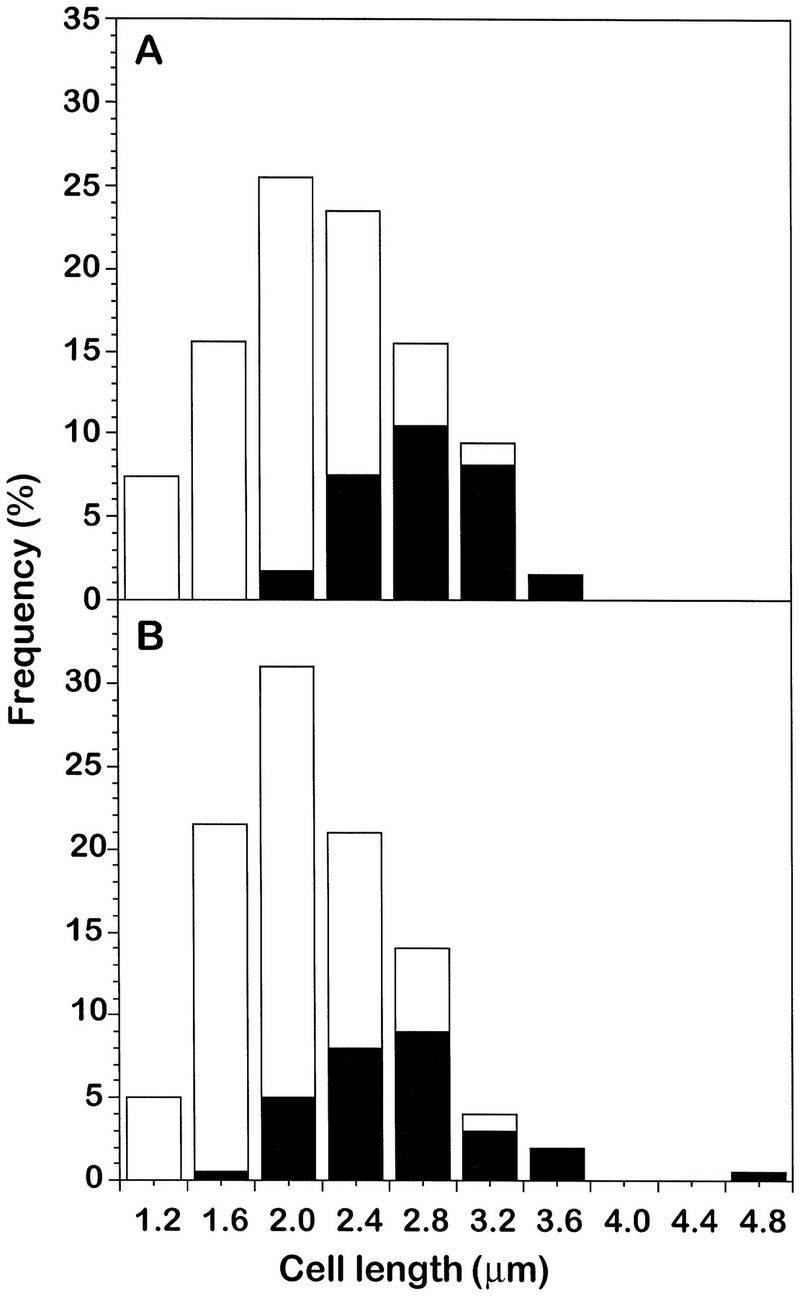
Assembly of DivIVA and MinD at mid-cell during cell-cycle progression. Strains containing GFP fusions to (A) divIVA (strain 1918) and (B) minD (strain 1981) were grown in S medium and viewed as shown in Figs. 2D and 3G. Cell-length frequency distributions for ∼200 cells were scored for the presence (solid bars) or absence (open bars) of a central ring of GFP.
Interestingly, these average cell lengths correspond to about the time of completion of DNA replication, ∼25 min before septal constriction begins (see Sharpe et al. 1998). Thus, the division inhibitor, or at least the MinD component of the inhibitor, assembles at mid-cell well before septation begins. In work to be described elsewhere, we have found that assembly of DivIVA, at least, seems to occur at about the time of termination of DNA replication under a range of other growth conditions (M.E. Sharpe, unpubl.).
Min–DivIVA control of division site selection acts atthe level of FtsZ ring assembly
To test whether the B. subtilis division inhibition system acts on FtsZ ring formation, we examined the localization of FtsZ protein in cells depleted for DivIVA. As discussed above, the cell filamentation that occurs in divIVA mutants seems to be caused by uncontrolled action of the MinCD inhibitor of cell division. As shown in Figure 5A, all but the shortest cells of strain 1756 grown in the presence of inducer (IPTG) had regularly positioned bands of FtsZ protein at mid-cell, corresponding to nascent and future division sites (see Levin and Losick 1996; Addinall et al. 1997). Although there was a diffuse signal throughout most of the cells, the finished cell poles generally did not show significant enrichment of FtsZ, indicating that, unlike DivIVA and MinD, the protein is not retained at the poles after division (Levin and Losick 1996). After growth in the absence of inducer, however, the pattern of FtsZ staining in the elongated cells was much more uniform with few prominent accumulations of protein (Fig. 5C). The occasional bright bands of FtsZ detected (e.g., arrowed in Fig. 5C) probably represent precursors to the rare divisions that occur in divIVA mutants, sometimes producing small oblique minicells (Reeve et al. 1973; Edwards and Errington 1997). In support of this interpretation, the bright bands usually coincided with larger spaces between the nucleoids (Fig. 5D). The fainter bands, again generally located between nucleoids, could correspond to the weak bands of MinD–GFP seen in similar cells (Fig. 3H). The near absence of FtsZ bands in these filaments shows that the MinCD inhibitor of B. subtilis acts, like that of E. coli (Bi and Lutkenhaus 1993), at the level of FtsZ ring formation.
Figure 5.

Localization of FtsZ in the presence and absence of DivIVA. FtsZ immunofluorescence (A,C) and DAPI fluorescence (B,D) micrographs of strain 1756 (Pspac–divIVA) grown in the presence (A,B) and absence (C,D) of inducer. Arrows in A and C point to FtsZ bands, which in C, are usually located where rare divisions have taken place.
Discussion
Targeting of DivIVA requires a late step in assembly of the division apparatus
Previous results showed that DivIVA–GFP is targeted to regularly spaced sites corresponding to mid-cell and polar positions. We have now characterized this targeting in more detail. Immunofluorescence microscopy showed a similar pattern of targeting to that of the GFP fusion, confirming that the GFP fusion provides a reliable indication of DivIVA localization. Characterization of DivIVA–GFP localization relative to cell length, showed that new DivIVA bands appear at mid-cell in a cell-cycle-dependent manner. After division, the protein remains present at the poles. The polar localizations must remain for a protracted period (or more DivIVA protein must continue to be recruited to the poles), because the poles of most cells had readily discernible accumulations of DivIVA (though always less than in the mid-cell bands). DivIVA bands appeared at a cell length corresponding approximately to the time of termination of DNA replication (Fig. 4; M.E. Sharpe, unpubl.). Interestingly, this is well before septation, confirming the notion that formation of an FtsZ ring and recruitment of late-division proteins to the ring occur well before the ring begins to contract (Addinall et al. 1996).
Although it now appears that DivIVA assembly occurs well before septation, its recruitment to the division site was dependent on FtsZ and on later assembling components of the division apparatus, DivIC and DivIB. So the topological target for DivIVA would appear to be a component of the division apparatus itself, possibly a relatively late-assembling component (or an early component that is modified or undergoes a change in conformation). In accordance with this idea, when the division-site specificity was relaxed, in a minD mutant, DivIVA continued to localize at the sites of division, whether they occurred at mid-cell or at the poles. Furthermore, this experiment showed that targeting of DivIVA to division sites is independent of MinD. The precise target for DivIVA in the division machinery is not yet clear, but it should be possible to resolve this by examining its requirements for other division proteins, such as ftsL and pbpB. Whatever the nature of the primary target directing the assembly of DivIVA at the division site, it is interesting to note that the protein remains at the pole after the septum has been completed and the primary target has presumably been removed. In this respect DivIVA differs from most other division proteins that have been studied. The results suggest that DivIVA may have at least two distinct targeting mechanisms.
MinD is targeted to the cell poles in a DivIVA-dependent manner
IFM and a GFP fusion were also used to determine the localization of MinD (Fig. 3). The images obtained with the GFP fusion gave a much clearer indication of the distribution of MinD in the cell. The less homogeneous signal detected by IFM probably reflects the poorer preservation of the samples after fixation and permeabilization. The pattern of localization observed was similar to that of DivIVA in some respects, most notably in the appearance of discrete bands at mid-cell (which formed at about the same point in the cell cycle as for DivIVA), and in the retention of the signal at the completed poles. MinD differed in exhibiting a greater signal at the cell poles and a much greater background signal, apparently in the form of a gradient of protein diminishing away from the main targeting sites at the poles. The fluorescence also seemed to follow the periphery of the cell, suggesting that most of the protein lies close to the cell envelope, perhaps associated with the inner face of the cytoplasmic membrane. Electron microscopic studies of E. coli MinD suggested an inner membrane localization (De Boer et al. 1991).
When division was blocked early (in FtsZ-depleted filaments), MinD was concentrated mainly at the cell poles. Thus, as for DivIVA, formation of bands at mid-cell requires assembly of at least part of the division apparatus. The distribution of MinD in these filaments was strikingly similar to that of DivIVA (cf. Figs. 2C and 3I). Furthermore, in filaments depleted for DivIVA, MinD clearly no longer targeted to the cell poles, indicating that polar targeting is dependent on DivIVA. Thus, MinD may be recruited to the septum as an even later step in the hierarchy than DivIVA. Given the functional relationship between these proteins, and that the only role of DivIVA in septation seems to be to control MinCD inhibition, it seems likely that MinD targeting involves a direct interaction with DivIVA. However, so far we have not been able to detect a direct interaction between these proteins. The weak MinD bands seen in Figure 3H show that in the absence of DivIVA, MinD may have weak affinity for some other component(s) of the division machinery.
A polar piloting mechanism for division-site-selection in B. subtilis
Figure 6 outlines in schematic form a possible mechanism for Min–DivIVA control of division site selection in B. subtilis. In Figure 6 we assume that the location of MinD coincides with the site of action of the MinC inhibitor. Yeast two-hybrid experiments with the E. coli genes suggest that MinC and MinD interact strongly (Huang et al. 1996), and we have obtained similar results with the B. subtilis genes (A.L. Marston and J. Errington, unpubl.). We also assume that nucleation of FtsZ rings is favored at the poles and mid-cell, perhaps by FtsZ or a factor needed for polymerization being excluded from the vicinity of the nucleoid. Wake and colleagues have shown that a minimum amount of DNA replication (∼70% of the chromosome) is needed before septation can occur at mid-cell in B. subtilis (McGinness and Wake 1979; Wu et al. 1995). After this amount of replication, the nucleoid undergoes a conformational change, becoming bilobed (Sharpe et al. 1998). This bilobation could produce a region deficient in DNA thereby facilitating the accumulation of sufficient FtsZ at mid-cell to allow polymerization to begin.
Figure 6.
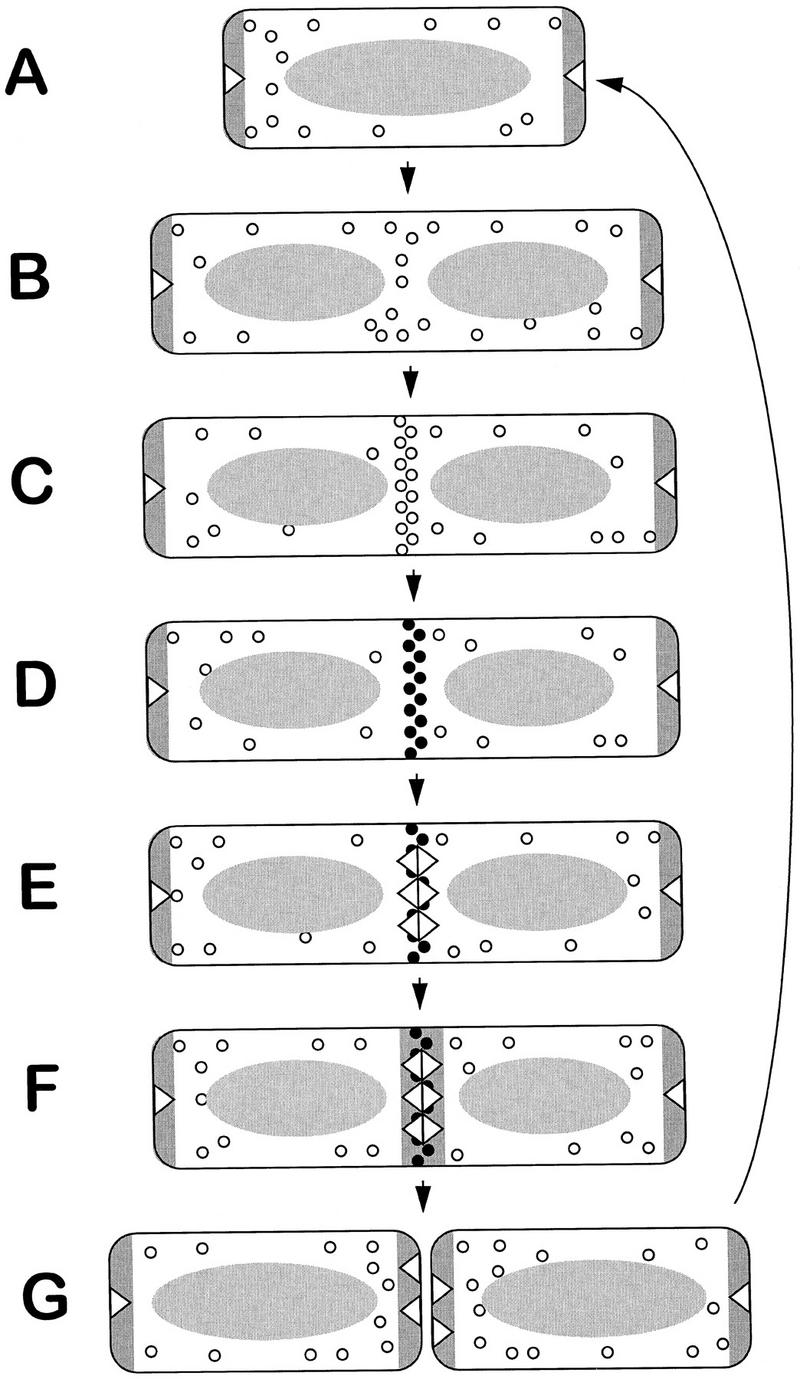
Model for division-site selection by the Min–DivIVA system. (A–G) Various steps in a cell cycle, beginning with a newborn cell (A). The oval shaded structure represents the nucleoid, which segregates into two separate nucleoids, following the completion of a round of DNA replication (B). Open triangles represent DivIVA protein, and the dark shaded stripes, MinD (probably associated with the MinC division inhibitor). The small open circles represent FtsZ monomers. These tend to be excluded from the vicinity of the nucleoid. Nucleation of FtsZ to produce the Z ring begins in the DNA-free zone between the nucleoids (C). The solid circles (D) indicate that the division apparatus has matured beyond the point at which its formation can be prevented by MinCD, possibly by recruitment of other division proteins that stabilize the Z ring. This maturation of the Z ring allows recruitment of DivIVA to mid-cell (E), which in turn allows targeting of MinD (F). Following cell division (G) both new cell poles have active MinD preventing further polar (minicell) divisions from taking place.
The model begins with a newborn cell (Fig. 6A) that contains DivIVA and MinD at both poles. Although these polar DNA-free zones would be favorable locations for nucleation of Z rings, they do not form there because of the presence of the MinD inhibitor. At about the time of completion of DNA replication, a new potential site for Z ring assembly is revealed at mid-cell (Fig. 6B). The depletion of MinD from this site, probably caused by its sequestration to the poles, allows assembly of the Z ring (Fig. 6C) and recruitment of other division proteins. The division machinery can thus mature beyond some point, as yet undefined, at which it becomes resistant to MinCD inhibition (Fig. 6D). DivIVA then assembles at the site of impending division (Fig. 6E), allowing MinD, in turn, to assemble there (Fig. 6F). All of this happens before contraction of the ring begins. After division, most of the division proteins leave the newly formed poles but DivIVA and MinD remain. The presence of MinD at the new poles prevents further (minicell) divisions from occurring there (Fig. 6G), thus explaining the inactivation of used division sites (Teather et al. 1974). It may be especially important to have MinCD at the newly formed cell poles because immediately after division there could be a high concentration of division proteins close to the poles, following their release from the constricting septum. The failure to retain MinD at the poles in divIVA mutant filaments could explain why these mutants make some minicells.
According to this model, based on the polar piloting model of Figure 1, DivIVA operates by assembling at the committed division site, acting to mark this as a used division site. By attracting MinD to this location (directly or indirectly), it both prevents these potential division sites from being used again and facilitates release of new mid-cell division sites.
B. subtilis and E. coli use different mechanisms to control the topological specificity of a common division inhibitor MinCD
Comparison of the B. subtilis and E. coli division site-selection system reveals some remarkable similarities and differences. The MinD protein is highly conserved between the two organisms, exhibiting 42% identical amino acids across the whole protein. In E. coli this protein has at least two functions (De Boer et al. 1992). It is required first as a coinhibitor for functioning of the MinC division inhibitor. Second, it is needed to make the division inhibitor sensitive to the topological specificity factor (MinE or DivIVA). The classical minicell phenotype produced by mutations in the B. subtilis minD gene (Levin et al. 1992; Varley and Stewart 1992; Lee and Price 1993) suggests that these functions are conserved. The MinC proteins of B. subtilis and E. coli are recognizable as likely homologs through the conserved position of their coding regions, immediately upstream of minD. The sequence conservation is much less; comprising only ∼29% of identical residues, restricted to the carboxy-terminal part of the protein. In E. coli, this domain of the protein is probably involved in interacting with MinD. Unfortunately, although a minC insertion produced a Min− phenotype (Varley and Stewart 1992), the possibility that this phenotype is caused by a polar effect on the downstream minD gene cannot be excluded. Nevertheless, it seems likely that MinC is also the division inhibitor in the B. subtilis system and we have shown above that the inhibition of division mediated by the B. subtilis Min system operates, as in E. coli (Bi and Lutkenhaus 1993), at the level of FtsZ ring formation (Fig. 5).
In contrast to MinC and MinD, the topological specificity factors, MinE of E. coli and DivIVA of B. subtilis, show no significant sequence similarity. Moreover, the minE gene lies immediately downstream of minD in E. coli, whereas divIVA lies in a distant locus in the B. subtilis chromosome. Raskin and De Boer (1997) recently showed that MinE, like DivIVA (Edwards and Errington 1997; Fig. 2) is targeted to mid-cell. However, the requirements for targeting of MinE to mid-cell are completely different from those of DivIVA. Thus, MinE targeting is independent of FtsZ and presumably all other later-assembling components of the division apparatus, whereas it requires minD. Moreover, MinE arrives at mid-cell relatively early and disappears before constriction is complete, whereas DivIVA arrives late and is retained at the cell pole after division. These observations strongly support the mid-cell inhibition model of Figure 1 for MinE, as described in detail by Raskin and De Boer (1997). Therefore, even though B. subtilis and E. coli use a common division inhibitor, MinCD, to control division-site selection, their respective topological-specificity factors operate by completely different molecular mechanisms.
Materials and methods
Bacterial strains and plasmids
The bacterial strains and plasmids used in this study are described in Table 1.
Table 1.
Bacterial strains and plasmids
| Strain/plasmid
|
Relevant characteristics
|
Source/construction
|
|---|---|---|
| B. subtilis | ||
| BB11 | chr::pJSIZΔpble (Pspac–ftsZ ble) | Beall and Lutkenhaus (1991) |
| SG38 | trpC2 amyE | Errington and Mandelstam (1986) |
| SU347 | trpC2 divIC355 | Katis et al. (1997) |
| 168CA | trpC2 | Kunst and et al. (1997) |
| 1306 | trpC2 Ω(amyE::gpr′–′lacZ cat) Ω(divIB::spc) | A. Feucht (unpubl.) |
| 1756 | trpC2 chr::pSG1043(Pspac–divIVA PdivIVA–lacZ ermC) | Edwards and Errington (1997) |
| 1757 | trpC2 chr::pSG1044 (divIVA–gfpS65T divIVA+cat) | Edwards and Errington (1997) |
| 1759 | trpC2 chr::pJSIZΔpble (Pspac–ftsZ ble) divIVA::pSG1044 (divIVA–gfpa divIVA+ cat) | 1757 transformed to phleomycin resistance with BB11 DNA |
| 1801 | trpC2 chr::pJSIZΔpble (Pspac–ftsZ ble) | 168CA transformed to phleomycin resistance with BB11 DNA |
| 1802 | trpC2 Ω(divIB::spc) | 168CA transformed to spectinomycin resistance with 1306 DNA |
| 1901 | trpC2 amyE Ω(minD::ermC)1901 | SG38 transformed to erythromycin resistance with pSG1704 DNA |
| 1918 | trpC2 chr::pSG1044 (divIVA–gfpadivIVA+ cat) | SG38 transformed to chloramphenicol resistance with 1757 DNA |
| 1920 | trpC2 amyE Ω(minD::ermC)1901 Ω(divIVA::tet)1042 | 1901 transformed to tetracycline resistance with pSG1042 DNA |
| 1922 | trpC2 Ω(minD::ermC)1901 chr::pSG1044 (divIVA–gfpadivIVA+ cat) | 1901 transformed to chloramphenicol resistance with pSG1044 DNA |
| 1979 | trpC2 Ω(minD::erm)1901 amyE::(spc Pxyl–gfpb–minD) chr::pJSIZΔpble (Pspac–ftsZ ble) | 1981 transformed to phleomycin resistance with BB11 DNA |
| 1981 | trpC2 Ω(minD::ermC)1901 amyE::(spc Pxyl–gfpb–minD) | 1901 transformed to spectinomycin resistance with pSG1730 DNA |
| 1984 | trpC2 Ω(minD::erm)1901 Ω(divIVA::tet)1042 amyE::(spc Pxyl–gfpb–minD) | 1920 transformed to spectinomycin resistance with pSG1730 DNA |
| E. coli | ||
| DH5α | F−endA1 hsdR17 supE44 thi-a1 λ− recA1 gyrA96 relA1 Δ(lacZYA–argF)U169 Φ80 dlacZΔM15 | GIBCO BRL |
| NM554 (pREP4) | F−araD139 Δ(ara–leu)7696 galE15 galK16 Δ(lac)X74 rpsL (Strr) hsdR2 (rk−mk−) mcrA mcrB1 recA13 (pREP4) kan laclq | Raleigh et al. (1988) |
| BL21(DE3) | F−ompT [lon] hsdSB(rB−mB−) λ(DE3) pol(T7) | Studier et al. (1990) |
| Plasmids | ||
| pET-3a | T7 expression vector | Studier et al. (1990) |
| pSG250 | bla ermC | Errington et al. (1992) |
| pSG1022 | pET3a containing minD+ | this work |
| pSG1042 | bla cat Ω(divIVA::tet)1042 | Edwards and Errington (1997) |
| pSG1044 | bla cat divIVA′–′gfpa | Edwards and Errington (1997) |
| pSG1151 | bla cat gfpb | P.J. Lewis (unpubl.) |
| pSG1613 | bla H6–divIVA | this work |
| pSG1704 | bla (minD::erm) | this work |
| pSG1717 | bla H6–minD | this work |
| pSG1729 | bla amyE3′ spc Pxyl–gfpbamyE5′ | P.J. Lewis and A.L. Marston (unpubl.) |
| pSG1730 | bla amyE3′ spcPxyl–gfpb–minD amyE5′ | this work |
| pQE-30 | vector for H6-tagged protein overproduction | Qiagen |
S65T variant of GFP (Heim et al. 1995).
F64L, S65T variant of GFP (Cormack et al. 1996).
Media and general methods
B. subtilis cultures were grown in either hydrolyzed casein (CH) medium (Sterlini and Mandelstam 1969), S medium (as described by Karamata and Gross 1970, but supplemented with 1% CH medium), or PAB (oxoid antibiotic medium no. 3). All media were supplemented with tryptophan (20 μg/ml). B. subtilis strains were transformed by the method of Anagnostopoulos and Spizizen (1961), as modified by Jenkinson (1983), or as described by Kunst and Rapoport (1995), except that 20 min after addition of DNA the transformed cultures were supplemented with 0.66% casamino acid solution. Transformants were selected on oxoid nutrient agar containing, as necessary, erythromycin (either 5 μg/ml or 1 μg/ml together with 25 μg/ml lincomycin), chloramphenicol (5 μg/ml), phleomycin (0.2 μg/ml), spectinomycin (100 μg/ml), or tetracycline (10 μg/ml). DNA manipulations and E. coli transformations were done by standard methods (Sambrook et al. 1989).
Construction of a null allele of minD
The ermC cassette from pSG250 was released by SstI digestion, gel purified, and inserted into the unique SstI site of pSG1022 to produce pSG1704. B. subtilis SG38 was transformed with pSG1704 with selection for erythromycin resistance. Disruption of minD was confirmed in the resulting strain (1901) by PCR analysis using primers 6780 and 6781 (see below).
Construction of a gfp–minD fusion
The B. subtilis minD coding sequence was amplified by PCR from the chromosome of strain SG38 using primers AM1 (5′-GGGAGGAACTCGAGTTGGGTGAGGC-3′) and AM2 (5′-GATTCTTCTCTTGAATTCTATCACATTAAG-3′), introducing XhoI and EcoRI sites, respectively. The XhoI–EcoRI minD fragment was inserted between these sites in pSG1729 to generate pSG1730, which places gfp–minD under the control of Pxyl.
gfp–minD induction experiments
To study the localization of GFP–MinD in a divIVA null mutant, strains 1981 and 1984 were grown to exponential phase (OD600 of 0.3) in the modified S medium at 30°C. gfp–minD expression was induced by addition of xylose to 0.5% and samples were taken at an OD600 of 0.6–0.8 for microscopy.
DivIVA depletion experiments
To check the anti-DivIVA antiserum for specificity by Western blotting, strain 1756 was grown at 30°C to late-log phase (OD600 of 1.0) in CH medium with or without 0.15 mm IPTG. Each culture was then diluted in the same medium to an OD600 of 0.05. Samples for Western blotting were collected when the cultures reached an OD600 of 0.4. The same procedure was followed for IFM with anti-FtsZ antibody except that S medium was used.
FtsZ depletion experiments
To examine the effect of FtsZ depletion on DivIVA localization, strain 1759 was grown at 30°C to an OD600 of 1.0 in PAB containing 0.1 mm IPTG. The cells were harvested, washed in PAB, and resuspended to an OD600 of 0.01. The culture was divided into two portions, to one of which was added 0.1 mm IPTG. The cultures were sampled for examination by fluorescence microscopy at an OD600 of 1.4.
To examine the effect of FtsZ depletion on MinD localization, strain 1979 was grown at 30°C in S medium containing 0.5 mm IPTG to an OD600 of 0.4. The cells were pelleted and IPTG was removed by washing them with fresh warm S medium. The culture was diluted back to an OD600 of 0.1 and grown to an OD600 of 0.4, then washed and diluted again. The cell filaments were examined after growth at 30°C overnight, reaching an OD600 of 0.9.
Temperature-shift experiments
Strains 1802 and SU347 were grown overnight at 30°C to late-log phase (OD600 ∼1.0) in CH medium. Each culture was then harvested and washed once in CH before being divided into two and diluted in CH to an OD600 of 0.05. For each strain one culture was left to grow at 30°C, whereas the other was placed at the restrictive temperature (37°C for 1802 and 45°C for SU347). All cultures were grown to a final OD600 of 0.7 before being sampled for IFM.
Overproduction and purification of His-tagged DivIVA
Primers 8618 (5′-GGAGGTGGATCCATGCCATTAACGCC-3′) and 8619 (5′-CAGAGAAGCTTTTCCTTTTCCTCAAATACAGCG-3′) were used to amplify divIVA and replace its stop codon with a HindIII restriction site, by PCR. Thus, divIVA was cloned into pQE-30 (Qiagen) to give pSG1613. E. coli strain NM554 (pREP4) (Qiagen) transformed with pSG1613 was induced to express the fusion protein, H6–DivIVA by addition of 1 mm IPTG. The cells were harvested and lysed in 6 m urea and extracts were fractionated by metal affinity chromatography (Qiagen), according to manufacturer’s instructions. Following washes with stringent buffer (8 m urea, 300 mm NaCl, 10 mm Tris, 100 mm NaHPO4) the H6–DivIVA was eluted with 300 mm imidazole. The pure H6–DivIVA was collected by precipitation with 50% acetone, and redissolved in deionized water.
Overproduction and purification of MinD protein
Plasmid pSG1022, for overproduction of B. subtilis MinD, was constructed by cloning a PCR amplified copy of the minD gene from strain 168 chromosomal DNA. The oligonucleotides used 6780 (5′-GGAATGTCATATGGGTGAGGCTATCG-3′) and 6781 (5′-GCTTTGATCAGATTCTTCTCTTTGATTC-3′) introduced NdeI and BclI sites for insertion into vector plasmid pET–3a, which was restricted with NdeI and BamHI.
To purify MinD for antibody production, pSG1022 was transformed into BL21 and the resultant strain grown in 50 ml of 2× TY containing 100 μg/ml ampicillin at 37°C. When the culture reached an OD600 of 0.5, IPTG (0.5 mm) was added. The induced cells were grown for a further 2 hr before harvesting. The culture pellet was resuspended in 2.5 ml of TE and 2.5 ml of 2× SDS loading buffer and heated to 95°C for 1 min. 600 μl of extract was separated by SDS-PAGE (10%) and the proteins were visualized by shaking with ice-cold KCl (0.25 m) for 5 min, followed by washing with cold water. Pure MinD was obtained by dialysis of a slice of gel containing the protein, followed by acetone precipitation.
For affinity purification of anti-MinD antibody, MinD with a 6× His affinity tag at the amino terminus (H6–MinD) was overproduced and purified. minD was amplified from the chromosome of strain SG38 using primers 8763 (5′-GAATTGGGTGGATCCATCGTAATAACTTCGGG-3′) and 8764 (5′-CTCTTTGATTCTATCACCTGCAGATCTTACTCCG-3′), and cloned between BamHI and PstI sites in pQE-30. The resultant plasmid, pSG1717, was transformed into E. coli strain NM554 (pREP4), and protein overproduction induced in a 50-ml culture in 2× TY by addition of 1 mm IPTG. After 4 hr, the culture pellet was harvested, dissolved in 2 ml of GuCl buffer (6 m guanidine chloride, 50 mm Na-phosphate at pH 7.4) and centrifuged (10 min). The cell extract was passed through a 200-μl Ni-chelated sepharose column, equilibrated in GuCl buffer. The column was washed with 2 ml of GuCl buffer before eluting H6–MinD with 20 mm EDTA. The eluate was dialyzed against ATP dialysis buffer (200 ml of PBS containing 2 mm DTT, 0.7 μg/ml pepstatin, 170 μg/ml PMSF, and 1 mm ATP).
Antibody production and Western blotting
Rabbit polyclonal antisera were raised by standard procedures (Harlow and Lane 1988) using H6–DivIVA or gel-purified MinD protein. Sheep anti-FtsZ antibodies were a gift of E.J. Harry (University of Sydney, Australia). The antibodies were affinity-purified as described by Reznekov et al. (1996). Anti-FtsZ antibodies were purified against native FtsZ (also from E.J. Harry), and eluted with 0.2 m glycine, 1 mm EDTA at pH 2.2, 50% glycerol. Anti-MinD antibodies were purified against H6–MinD using 4.5 m MgCl2, 0.1% BSA as eluant. The anti-DivIVA antibodies were used at a dilution of 1:5000 for Western blotting. Unpurified and affinity-purified anti-MinD antisera were used at dilutions of 1:500 and 1:250, respectively. Western blotting was carried out essentially as described by Wu and Errington (1994) using an enhanced chemiluminescence detection system (Amersham).
Immunofluorescence microscopy
Cells were fixed, permeabilized, and stained for immunofluorescence microscopy as described previously (Pogliano et al. 1995; Lewis et al. 1996; Reznekov et al. 1996), except that the glutaraldehyde concentration in the 2× fixative was reduced to 0.00005%. Crude anti-DivIVA, affinity-purified anti-MinD and affinity-purified anti-FtsZ antibodies were used at concentrations of 1:1500, 1:500, and 1:20, respectively. Images were grabbed, processed, and assembled as described previously (Lewis and Errington 1997). Exposure times were 1 sec for Cy3; 0.5 sec for DAPI; 3 sec for DivIVA–GFP, and 4 sec for GFP–MinD.
Cell-cycle analysis
The timing of DivIVA and MinD appearance at mid-cell was calculated as described previously (Glaser et al. 1997; Sharpe et al. 1998; Sharpe and Errington 1998).
Acknowledgments
This work was supported by grants from the Biotechnology and Biological Sciences Research Council, the BIOTECH Programme of the European Community, and the EPA Cephalosporin Research Fund. A.L.M. was the recipient of a Wellcome Trust Prize Studentship. We thank Ali Hullah for construction of plasmids and purification of His-tagged DivIVA, P.A.J. de Boer for helpful discussions, and David Sherratt and Ling Juan Wu for helpful comments on the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL erring@molbiol.ox.ac.uk; FAX 44-1865-275556.
References
- Addinall SG, Lutkenhaus J. FtsA is localized to the septum in an FtsZ-dependent manner. J Bacteriol. 1996;178:7167–7172. doi: 10.1128/jb.178.24.7167-7172.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addinall SG, Bi E, Lutkenhaus J. FtsZ ring formation in fts mutants. J Bacteriol. 1996;178:3877–3884. doi: 10.1128/jb.178.13.3877-3884.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addinall SG, Cao C, Lutkenhaus J. Temperature shift experiments with an ftsZ84(Ts) strain reveal rapid dynamics of FtsZ localization and indicate that the Z ring is required throughout septation and cannot reoccupy division sites once constriction has initiated. J Bacteriol. 1997;179:4277–4284. doi: 10.1128/jb.179.13.4277-4284.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler HI, Fisher WD, Cohen A, Hardigree AA. Miniature Escherichia coli cells deficient in DNA. Proc Natl Acad Sci. 1967;57:321–326. doi: 10.1073/pnas.57.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anagnostopoulos C, Spizizen J. Requirements for transformation in Bacillus subtilis. J Bacteriol. 1961;81:741–746. doi: 10.1128/jb.81.5.741-746.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall B, Lutkenhaus J. FtsZ in Bacillus subtilis is required for vegetative septation and for asymmetric septation during sporulation. Genes & Dev. 1991;5:447–455. doi: 10.1101/gad.5.3.447. [DOI] [PubMed] [Google Scholar]
- Bi E, Lutkenhaus J. FtsZ ring structure associated with division in Escherichia coli. Nature. 1991;354:161–164. doi: 10.1038/354161a0. [DOI] [PubMed] [Google Scholar]
- ————— Cell division inhibitors SulA and MinCD prevent formation of the FtsZ ring. J Bacteriol. 1993;175:1118–1125. doi: 10.1128/jb.175.4.1118-1125.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramhill D, Thompson CM. GTP-dependent polymerization of Escherichia coli FtsZ protein to form tubules. Proc Natl Acad Sci. 1994;91:5813–5817. doi: 10.1073/pnas.91.13.5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha J-H, Stewart GC. The divIVA minicell locus of Bacillus subtilis. J Bacteriol. 1997;179:1671–1683. doi: 10.1128/jb.179.5.1671-1683.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- De Boer PAJ, Crossley RE, Rothfield LI. A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in E. coli. Cell. 1989;56:641–649. doi: 10.1016/0092-8674(89)90586-2. [DOI] [PubMed] [Google Scholar]
- De Boer PAJ, Crossley RE, Rothfield LI. Central role for the Escherichia coli minC gene product in two different cell division-inhibition systems. Proc Natl Acad Sci. 1990;87:1129–1133. doi: 10.1073/pnas.87.3.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boer PAJ, Crossley RE, Hand AR, Rothfield LI. The MinD protein is a membrane ATPase required for the correct placement of the Escherichia coli division site. EMBO J. 1991;10:4371–4380. doi: 10.1002/j.1460-2075.1991.tb05015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boer PAJ, Crossley RE, Rothfield LI. Roles of MinC and MinD in the site-specific septation block mediated by the MinCDE system of Escherichia coli. J Bacteriol. 1992;174:63–70. doi: 10.1128/jb.174.1.63-70.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DH, Errington J. The Bacillus subtilis DivIVA protein targets to the division septum and controls the site specificity of cell division. Mol Microbiol. 1997;24:905–915. doi: 10.1046/j.1365-2958.1997.3811764.x. [DOI] [PubMed] [Google Scholar]
- Erickson HP, Taylor DW, Taylor KA, Bramhill D. Bacterial cell division protein FtsZ assembles into protofilament sheets and minirings, structural homologues of tubulin polymers. Proc Natl Acad Sci. 1996;93:519–523. doi: 10.1073/pnas.93.1.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington J, Mandelstam J. Use of a lacZ gene fusion to determine the dependence pattern of sporulation operon spoIIA in spo mutants of Bacillus subtilis. J Gen Microbiol. 1986;132:2967–2976. doi: 10.1099/00221287-132-11-2967. [DOI] [PubMed] [Google Scholar]
- Errington J, Appleby L, Daniel RA, Goodfellow H, Partridge SR, Yudkin MD. Structure and function of the spoIIIJ gene of Bacillus subtilis: A vegetatively expressed gene that is essential for σG activity at an intermediate stage of sporulation. J Gen Microbiol. 1992;138:2609–2618. doi: 10.1099/00221287-138-12-2609. [DOI] [PubMed] [Google Scholar]
- Glaser P, Sharpe ME, Raether B, Perego M, Ohlsen K, Errington J. Dynamic, mitotic-like behaviour of a bacterial protein required for accurate chromosome partitioning. Genes & Dev. 1997;11:1160–1168. doi: 10.1101/gad.11.9.1160. [DOI] [PubMed] [Google Scholar]
- Hale CA, De Boer PAJ. Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli. Cell. 1997;88:175–185. doi: 10.1016/s0092-8674(00)81838-3. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane DP. Antibodies: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- Harry EJ, Wake RG. The membrane-bound cell division protein DivIB is localized to the division site in Bacillus subtilis. Mol Microbiol. 1997;25:275–283. doi: 10.1046/j.1365-2958.1997.4581822.x. [DOI] [PubMed] [Google Scholar]
- Heim R, Cubitt AB, Tsien RY. Improved green fluorescence. Nature. 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- Huang J, Cao C, Lutkenhaus J. Interaction between FtsZ and inhibitors of cell division. J Bacteriol. 1996;178:5080–5085. doi: 10.1128/jb.178.17.5080-5085.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson HF. Altered arrangement of proteins in the spore coat of a germination mutant of Bacillus subtilis. J Gen Microbiol. 1983;129:1945–1958. doi: 10.1099/00221287-129-6-1945. [DOI] [PubMed] [Google Scholar]
- Karamata D, Gross JD. Isolation and genetic analysis of temperature-sensitive mutants of Bacillus subtilis defective in DNA synthesis. Mol & Gen Genet. 1970;108:277–287. doi: 10.1007/BF00283358. [DOI] [PubMed] [Google Scholar]
- Katis VL, Harry EJ, Wake RG. The Bacillus subtilis division protein DivIC is a highly abundant membrane-bound protein that localizes to the division site. Mol Microbiol. 1997;26:1047–1055. doi: 10.1046/j.1365-2958.1997.6422012.x. [DOI] [PubMed] [Google Scholar]
- Kunst F, Rapoport G. Salt stress is an environmental signal affecting degradative enzyme synthesis in Bacillus subtilis. J Bacteriol. 1995;177:2403–2407. doi: 10.1128/jb.177.9.2403-2407.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero MG, Bessieres P, Bolotin A, Borchert S, et al. The complete genome sequence of the Gram positive Bacillus subtilis bacterium. Nature. 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- Lee S, Price CW. The minCD locus of Bacillus subtilis lacks the minE determinant that provides topological specificity to cell division. Mol Microbiol. 1993;7:601–610. doi: 10.1111/j.1365-2958.1993.tb01151.x. [DOI] [PubMed] [Google Scholar]
- Levin PA, Margolis PS, Setlow P, Losick R, Sun D. Identification of Bacillus subtilis genes for septum placement and shape determination. J Bacteriol. 1992;174:6717–6728. doi: 10.1128/jb.174.21.6717-6728.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin PA, Losick R. Transcription factor Spo0A switches the localization of the cell division protein FtsZ from a medial to a bipolar pattern in Bacillus subtilis. Genes & Dev. 1996;10:478–488. doi: 10.1101/gad.10.4.478. [DOI] [PubMed] [Google Scholar]
- Lewis PJ, Errington J. Direct evidence for active segregation of oriC regions of the Bacillus subtilis chromosome and co-localization with the Spo0J partitioning protein. Mol Microbiol. 1997;25:945–954. doi: 10.1111/j.1365-2958.1997.mmi530.x. [DOI] [PubMed] [Google Scholar]
- Lewis PJ, Magnin T, Errington J. Compartmentalized distribution of the proteins controlling the prespore-specific transcription factor σF of Bacillus subtilis. Genes to Cells. 1996;1:881–894. doi: 10.1046/j.1365-2443.1996.750275.x. [DOI] [PubMed] [Google Scholar]
- Lutkenhaus J, Addinall SG. Bacterial cell division and the Z ring. Annu Rev Biochem. 1997;66:93–116. doi: 10.1146/annurev.biochem.66.1.93. [DOI] [PubMed] [Google Scholar]
- Ma X, Ehrhardt DW, Margolin W. Colocalization of cell division proteins FtsZ and FtsA to cytoskeletal structures in living Escherichia coli cells by using green fluorescent protein. Proc Natl Acad Sci. 1997;93:12998–13003. doi: 10.1073/pnas.93.23.12998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinness T, Wake RG. Division septation in the absence of chromosome termination in Bacillus subtilis. J Mol Biol. 1979;134:251–264. doi: 10.1016/0022-2836(79)90035-4. [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Lutkenhaus J. Guanine nucleotide-dependent assembly of FtsZ into filaments. J Bacteriol. 1994;176:2754–2758. doi: 10.1128/jb.176.9.2754-2758.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder E, Woldringh CL, Tétart F, Bouché J-P. New minC mutations suggest different interactions of the same region of division inhibitor MinC with proteins specific for minD and dicB coinhibition pathways. J Bacteriol. 1992;174:35–39. doi: 10.1128/jb.174.1.35-39.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichoff S, Vollrath B, Touriol C, Bouché J-P. Deletion analysis of gene minE which encodes the topological specificity factor of cell division in Escherichia coli. Mol Microbiol. 1995;18:321–329. doi: 10.1111/j.1365-2958.1995.mmi_18020321.x. [DOI] [PubMed] [Google Scholar]
- Pogliano K, Harry E, Losick R. Visualization of the subcellular location of sporulation proteins in Bacillus subtilis using immunofluorescence microscopy. Mol Microbiol. 1995;18:459–470. doi: 10.1111/j.1365-2958.1995.mmi_18030459.x. [DOI] [PubMed] [Google Scholar]
- Raleigh EA, Murray NE, Revel H, Blumenthal RM, Westaway D, Reith AD, Rigby PWJ, Elhai J, Hanahan D. McrA and McrB restriction phenotypes of some E. coli strains and implications for gene cloning. Nucleic Acids Res. 1988;16:1563–1575. doi: 10.1093/nar/16.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskin DM, De Boer PAJ. The MinE ring: An FtsZ-independent cell structure required for selection of the correct division site in E. coli. Cell. 1997;91:685–694. doi: 10.1016/s0092-8674(00)80455-9. [DOI] [PubMed] [Google Scholar]
- Reeve JN, Mendelson NH, Coyne SI, Hallock LL, Cole RM. Minicells of Bacillus subtilis. J Bacteriol. 1973;114:860–873. doi: 10.1128/jb.114.2.860-873.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznekov O, Alper S, Losick R. Subcellular localization of proteins governing the proteolytic activation of a developmental transcription factor in Bacillus subtilis. Genes to Cells. 1996;1:529–542. doi: 10.1046/j.1365-2443.1996.d01-262.x. [DOI] [PubMed] [Google Scholar]
- Rothfield LI, Zhao C-R. How do bacteria decide where to divide? Cell. 1996;84:183–186. doi: 10.1016/s0092-8674(00)80971-x. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sharpe ME, Errington J. A fixed distance for separation of newly replicated copies of oriC in Bacillus subtilis: Implications for co-ordination of chromosome segregation and cell division. Mol Microbiol. 1998;28:981–990. doi: 10.1046/j.1365-2958.1998.00857.x. [DOI] [PubMed] [Google Scholar]
- Sharpe ME, Hauser PM, Sharpe RG, Errington J. Bacillus subtilis cell cycle as studied by fluorescence microscopy: Constancy of the cell length at initiation of DNA replication and evidence for active nucleoid partitioning. J Bacteriol. 1998;180:547–555. doi: 10.1128/jb.180.3.547-555.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterlini JM, Mandelstam J. Commitment to sporulation in Bacillus subtilis and its relationship to the development of actinomycin resistance. Biochem J. 1969;113:29–37. doi: 10.1042/bj1130029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW, Rosenberg AH, Dunn JJ, Dubendorff JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- Teather RM, Collins JF, Donachie WD. Quantal behaviour of a diffusible factor which initiates septum formation at potential division sites in Escherichia coli. J Bacteriol. 1974;118:407–413. doi: 10.1128/jb.118.2.407-413.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varley AW, Stewart GC. The divIVB region of the Bacillus subtilis chromosome encodes homologs of Escherichia coli septum placement (MinCD) and cell shape (MreBCD) determinants. J Bacteriol. 1992;174:6729–6742. doi: 10.1128/jb.174.21.6729-6742.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lutkenhaus J. The FtsZ protein of Bacillus subtilis is localized at the division site and has GTPase activity that is dependent upon FtsZ concentration. Mol Microbiol. 1993;9:435–442. doi: 10.1111/j.1365-2958.1993.tb01705.x. [DOI] [PubMed] [Google Scholar]
- Weiss DS, Pogliano K, Carson M, Guzman L-M, Fraipont C, Nguyen-Disteche M, Losick R, Beckwith J. Localization of the Escherichia coli cell division protein FtsI (PBP3) to the division site and cell pole. Mol Microbiol. 1997;25:671–681. doi: 10.1046/j.1365-2958.1997.5041869.x. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Errington J. Bacillus subtilis SpoIIIE protein required for DNA segregation during asymmetric cell division. Science. 1994;264:572–575. doi: 10.1126/science.8160014. [DOI] [PubMed] [Google Scholar]
- Wu LJ, Franks AH, Wake RG. Replication through the terminus region of the Bacillus subtilis chromosome is not essential for the formation of a division septum that partitions the DNA. J Bacteriol. 1995;177:5711–5715. doi: 10.1128/jb.177.19.5711-5715.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C-R, De Boer PAJ, Rothfield LI. Proper placement of the Escherichia coli division site requires two functions that are associated with different domains of the MinE protein. Proc Natl Acad Sci. 1995;92:4313–4317. doi: 10.1073/pnas.92.10.4313. [DOI] [PMC free article] [PubMed] [Google Scholar]