

Abstract
HIV-1 Tat activates transcription through binding to human cyclin T1, a regulatory subunit of the TAK/P-TEFb CTD kinase complex. Here we show that the cyclin domain of hCycT1 is necessary and sufficient to interact with Tat and promote cooperative binding to TAR RNA in vitro, as well as mediate Tat transactivation in vivo. A Tat:TAR recognition motif (TRM) was identified at the carboxy-terminal edge of the cyclin domain, and we show that hCycT1 can interact simultaneously with Tat and CDK9 on TAR RNA in vitro. Alanine-scanning mutagenesis of the hCycT1 TRM identified residues that are critical for the interaction with Tat and others that are required specifically for binding of the complex to TAR RNA. Interestingly, we find that the interaction between Tat and hCycT1 requires zinc as well as essential cysteine residues in both proteins. Cloning and characterization of the murine CycT1 protein revealed that it lacks a critical cysteine residue (C261) and forms a weak, zinc-independent complex with HIV-1 Tat that greatly reduces binding to TAR RNA. A point mutation in mCycT1 (Y261C) restores high-affinity, zinc-dependent binding to Tat and TAR in vitro, and rescues Tat transactivation in vivo. Although overexpression of hCycT1 in NIH3T3 cells strongly enhances transcription from an integrated proviral promoter, we find that this fails to overcome all blocks to productive HIV-1 infection in murine cells.
Keywords: HIV-1 transcription; HIV-1 Tat; TAR-, TAK/P-TEFb; cyclin T1; zinc-binding protein; RNA-binding protein
Cyclin domains and related structures that contain the classical cyclin fold are versatile motifs with multiple recognition surfaces that facilitate interactions between regulatory molecules (Martin-Castellanos and Moreno 1997; Noble et al. 1997). Therefore, the cyclin domain of cyclin A forms a trimeric complex with CDK2 and the CDK inhibitor p27Kip1 to control kinase activity through the cell cycle (Jeffrey et al. 1995; Russo et al. 1996), and the cyclin H subunit of the CDK-activating kinase (CAK) interacts with CDK7 and its various substrates in a coordinated process controlled by the MAT1 assembly factor (Tassan et al. 1995; Adamczewski et al. 1996; Rossignol et al. 1997; Yankulov and Bentley 1997). These different interactions serve to regulate kinase activity precisely and couple kinases to their substrates or inhibitors. Interestingly, diverse proteins such as the retinoblastoma (Rb) tumor suppressor and the RNAPII transcription initiation factor TFIIB use the cyclin-fold motif in an analogous manner to interact with their target proteins (Bagby et al. 1995; Nikolov et al. 1995; Lee et al. 1998). The cyclin-like structure of TFIIB recognizes the DNA-bound TATA-binding protein (TBP) through direct contacts with both TBP and the DNA backbone on both sides of the TATA box (Bagby et al. 1995; Nikolov et al. 1995), demonstrating that the cyclin fold can also participate in nucleic acid recognition.
We have recently isolated a new human C-type cyclin (called cyclin T) that binds to the transcriptional activation domain of the human immunodeficiency virus (HIV-1) Tat protein (Wei et al. 1998). Cyclin T (hereafter designated cyclin T1; Peng et al. 1998b) is a partner for CDK9 (originally called PITALRE; Grana et al. 1994), a CDC2-related kinase that functions at many genes as part of the positive transcription elongation factor complex, P-TEFb (Marshall and Price 1992, 1995; Marshall et al. 1996; Zhu et al. 1997). Cyclin T1 is the predominant cyclin associated with CDK9 in HeLa nuclear extracts, although CDK9 is also present in complexes with two minor cyclins, T2a and T2b (Peng et al. 1998b). The Tat-associated kinase (TAK) complex in nuclear extracts (Herrmann and Rice 1993, 1995; Yang et al. 1996) has been shown to contain cyclin T1, CDK9, and other as yet unidentified subunits of P-TEFb (Mancebo et al. 1997; Yang et al. 1997; Zhu et al. 1997; Wei et al. 1998). Studies using dominant-negative proteins and specific kinase inhibitors have shown that CDK9 kinase activity is critical for Tat transactivation (Mancebo et al. 1997; Zhu et al. 1997; Gold et al. 1998), and the multisubunit P-TEFb complex, but not hCycT1 and CDK9 alone, can restore Tat trans-activation to CDK9-depleted extracts in vitro (Mancebo et al. 1997; Zhu et al. 1997; Zhou et al. 1998). Consistent with a role for CDK9 as a kinase that phosphorylates the carboxy-terminal domain (CTD) of RNAPII (Zhu et al. 1997; Peng et al. 1998), the CTD has also been shown to be required for Tat activity (for review, see Jones 1997; Cullen 1998; Emerman and Malim 1998).
A key step in Tat-mediated trans-activation involves the specific binding to TAR, an RNA stem–loop structure that forms in the nascent viral transcript. Although Tat activity critically depends on bases in the loop and bulge of the RNA hairpin, purified Tat binds to the bulge without contacting the loop (for review, see Gait and Karn 1993). The interaction of Tat with hCycT1 strongly enhances binding to TAR RNA and confers a requirement for sequences in the loop of the hairpin (Wei et al. 1998). Within the hCycT1-Tat:TAR complex, the bulge is recognized by the arginine-rich RNA recognition motif (ARM) of Tat, whereas the loop is presumably contacted by the cyclin. It is possible that residues in the Tat trans-activation domain distinct from those required to bind hCycT1 might also recognize TAR RNA in the complex. Therefore, it appears that every residue in Tat that is important for transactivation in vivo is required to form a stable complex with hCycT1 on TAR RNA in vitro.
The region of Tat that interacts with hCycT1 includes a core sequence rich in hydrophobic amino acids as well as a motif containing six essential cysteine residues and a histidine. Previous studies have shown that the Cys-rich motif in Tat is capable of binding divalent cation metals such as zinc or cadmium, and that each monomer of Tat can coordinate two atoms of zinc (Huang and Wang 1996; Frankel et al. 1988a). The Cys-rich region does not fold into a conventional zinc finger structure, nor does it precisely match other well-characterized zinc-binding motifs. Interestingly, exogeneous zinc induces the formation of ‘metal-linked’ Tat dimers that are bridged through a shared coordination of zinc (Frankel et al. 1988a). Tat functions in transcription as a monomer, however (Dingwall et al. 1990; Rice and Chan 1991), and peptides of the Cys-rich subdomain disrupt dimer formation without affecting Tat trans-activation (Frankel et al. 1988b). Consequently, the potential role of zinc in Tat trans-activation has remained an important unresolved issue.
A critical role for a host cell cofactor in TAR-dependent Tat trans-activation was originally suggested from genetic studies of the defect in Tat transactivation in mouse cells. Therefore, it was shown that Tat only weakly activates the HIV-1 promoter in murine cell lines, and complementation studies established that this defect can be rescued by a factor encoded on human chromosome 12 (Hart et al. 1989, 1993; Alonso et al. 1994). The observation that cyclin T1 is encoded on Chromosome 12, promotes binding of Tat to TAR in vitro, and enhances Tat trans-activation in murine cells (Wei et al. 1998) strongly suggests that the murine cyclin T1 protein is unable to support efficient Tat activation of the HIV-1 promoter.
In this report, we have isolated the murine CycT1 gene and analyzed the interaction of Tat with both the human and murine cyclin T1 proteins. We identify a 13-amino-acid region at the carboxy-terminal boundary of the hCycT1 cyclin domain that contains residues important for binding to HIV-1 Tat and TAR RNA. Interestingly, the Tat–hCycT1 interaction requires zinc as well as an essential cysteine (C261) in cyclin T1. The murine CycT1 protein lacks C261 and forms a weak, zinc-independent complex with Tat that results in a dramatic (12-fold) reduction in binding to TAR RNA. Substitution of a cysteine for tyrosine at position 261 in murine CycT1 restores zinc-dependent binding to Tat as well as high-affinity binding of the complex to TAR RNA. Strikingly, this single change is also necessary and sufficient for mCycT1 to support high levels of Tat trans-activation in murine cells. Finally, we show that stable transduction of human cyclin T1 into murine cells significantly enhances HIV-1 transcription and increases virus production, but it is insufficient to overcome all of the species-specific barriers to HIV-1 replication in murine cells.
Results
HIV-1 Tat and CDK9 bind to independent sites within the cyclin domain of hCycT1
To better define the interaction between Tat and human cyclin T1, we sought to identify the minimal region of hCycT1 that can interact with Tat and enhance its binding to TAR RNA in vitro. A series of amino- and carboxy-terminal truncated GST–CycT1 proteins were expressed in bacteria, purified, and tested for their ability to bind cooperatively with HIV-1 Tat to TAR RNA. As summarized in Figure 1A, the minimal region of hCycT1 that binds to Tat and TAR RNA lies within the first 272 amino acids, which encompasses the entire cyclin domain. Further truncation to amino acid 254 completely eliminated the ability of the cyclin to form a complex with Tat and TAR. A mutant that lacks the first cyclin-fold repeat (amino acids 214–272) was also inactive in this assay, indicating that both cyclin repeats are required. To determine whether Tat is capable of binding to these truncation mutants, the GST–hCycT1 proteins were coupled to glutathione–S–Sepharose beads and incubated with purified (GST-cleaved; HA-tagged) HIV-1 and HIV-2 Tat proteins. The Tat that remained bound to the cyclin affinity resins after washing under stringent conditions was eluted by boiling of the beads in SDS sample buffer and visualized by immunoblot analysis of the SDS-PAGE. The wild-type HIV-1 and HIV-2 Tat proteins bound efficiently to hCycT1 proteins containing the extended cyclin domain (amino acids 1–303; amino acids 1–272) and did not interact with the cyclin mutant truncated to amino acid 254. Western blot analysis confirmed that the resins contained equivalent levels of the mutant GST–CycT1 proteins. Activation domain mutant Tat proteins (Tat-1 C22G; Tat-2 C59A) failed to bind to the GST–CycT1 beads, indicating that the interaction is specific. Therefore binding of Tat to hCycT1 requires the entire cyclin domain, and the carboxy-terminal boundary of the Tat:TAR recognition motif (TRM) lies between amino acids 254 and 272. We have not identified the amino-terminal boundary of the TRM, although most of the entire second cyclin fold can be required to mediate protein:protein interactions involving cyclins (e.g., see Lee et al. 1998).
Figure 1.



Identification of the TRM of hCycT1. (A) Summary of the ability of different truncated hCycT1 proteins to bind cooperatively with HIV-1 Tat to TAR RNA in gel-mobility shift experiments. The location of the TRM relative to the five helices (α1–5 and α1′–5′) of each fold of the cyclin domain are indicated. (B) Analysis of the ability of mutant hCycT1 proteins to interact directly with Tat. GST–hCycT1 (amino acids 1–303) proteins were coupled to beads and incubated with wild-type or mutant HIV-1 Tat (lanes 3–8) or HIV-2 Tat (lanes 11–16), as indicated above each lane. Aliquots corresponding to 10% of the input protein are shown for wild-type HIV-1 Tat (lane 1); HIV-1 Tat C22G (lane 2); wild-type HIV-2 Tat (lane 9); and HIV-2 Tat C59A (lane 10). Proteins were visualized by Western blot. (C) The cyclin domain of hCycT1 can interact simultaneously with HIV-1 Tat and CDK9. Binding of recombinant wild-type Tat, CDK9, and hCycT1 (amino acids 1–303) to HIV-1 TAR RNA was analyzed with gel-mobility shift experiments. Binding reactions contained 100 ng of (GST-cleaved) HIV-1 Tat (amino acids 1–86); 250 ng (GST-cleaved) hCycT1 (amino acids 1–303); and 200 ng of baculovirus-expressed CDK9, as indicated above each lane. Lanes 10, 13, 16, 19, 22, 25, and 28 contained a loop mutant TAR (+29/+34), and lanes 11, 14, 17, 20, 23, 26, and 29 contained a bulge mutant TAR RNA(U22A); all other lanes contained wild-type HIV-1 TAR RNA. Reactions in lanes 12–20 contained wild-type (WT) HIV-1 Tat, whereas lanes 21–29 contained an activation domain HIV-1 Tat mutant (C22G). (Right panel) CDK9 antibody alters the mobility of the Tat–hCycT1–CDK9 complex on HIV-1 TAR RNA. Reactions contained 100 ng (GST-cleaved) of HIV-1 Tat and 80 ng (GST-cleaved) of hCycT1 (amino acids 1–303) (lanes 30–33); 120 ng of CDK9 (lanes 31 and 33); and 100 ng of antisera specific to CDK9 (lanes 32 and 33). Arrows indicate the positions of the Tat:TAR, hCycT1–Tat:TAR, and CDK9–hCycT1–Tat:TAR complexes.
The recognition site for cyclin-dependent kinases lies within the first of the two cyclin repeats (alpha helices 1–5), whereas the portion of the TRM identified in this study lies at the carboxy-terminal edge of the second cyclin repeat (α helices 1′–5′) in a location that may overlap with α helix 5′. To determine whether CDK9 and Tat can bind simultaneously to the cyclin domain, the hCycT1 (amino acids 1–303) protein was incubated with purified Tat, CDK9, and TAR RNA, and the resulting complexes were analyzed with gel mobility shift experiments. As observed previously for the full-length hCycT1 protein, the truncated hCycT1 (amino acids 1–303) bound to TAR RNA in a highly cooperative manner with Tat, and did not recognize TAR RNA on its own (Fig. 1C). Purified CDK9, expressed and purified from recombinant baculovirus-infected Sf9 cells, also did not bind TAR RNA, either on its own or when mixed with hCycT1 (Fig. 1C, lanes 5 and 7), and did not influence the binding of Tat to TAR in the absence of the cyclin (Fig. 1C, lane 6), consistent with the fact that Tat does not interact directly with CDK9. On incubation of hCycT1(amino acid 1–303) with CDK9 and Tat, a slowly-migrating complex was observed that contains all three proteins (Fig. 1C, lanes 8 and 18). Antisera to CDK9 resulted in a further retardation in the mobility of this complex, without affecting the Tat–hCycT1:TAR complex (Fig. 1C, cf. lanes 30 and 31 with lanes 32 and 33). Whereas free Tat bound equivalently to wild-type and loop-mutant TAR RNA, the hCycT1–Tat and CDK9–hCycT1–Tat complexes bound only to the wild-type TAR RNAs (Fig. 1C, cf. lane 15 with 16 and lane 18 with 19). The bulge of TAR RNA was required for the formation of all complexes. Neither CycT1 nor CDK9 bound to a mutant Tat protein (Tat-1 C22G; Fig. 1C, lanes 21–29), confirming the specificity of these interactions. We conclude that Tat and CDK9 are likely to recognize distinct sites within the cyclin domain of hCycT1, and that CDK9 does not influence the RNA-binding specificity of the Tat–hCycT1 complex. In addition, these data show that cyclin T1 residues from amino acids 1–303 are sufficient to form a complex with Tat that can bind to TAR RNA in a loop- and bulge-specific manner.
We showed previously that overexpression of the human cyclin T1 protein enhances Tat activity in transfected murine cells (Wei et al. 1998), presumably through its ability to assemble into a functional P-TEFb complex with the murine CDK9. To assess whether the minimal region of hCycT1 that can bind Tat and TAR in vitro is also sufficient to support Tat transactivation in vivo, the activity of the truncated hCycT1 was examined by transient expression experiments in NIH3T3 cells. Remarkably, hCycT1 proteins containing the extended cyclin domain (amino acids 1–303 or 1–272) were just as effective as full-length hCycT1 in their ability to enhance Tat transactivation in vivo, and Tat activity in all cases required the loop of TAR RNA (Fig. 2A). Unlike the full-length hCycT1 protein, however, the truncated hCycT1 proteins (amino acids 1–303; 1–272) were unable to stimulate basal HIV-1 promoter activity in the absence of Tat (data not shown), consistent with previous reports that basal promoter activation by Drosophila CycT1 requires residues carboxy-terminal to the cyclin domain (Peng et al. 1998a). As a consequence, the truncated hCycT1 proteins supported much higher levels of Tat transactivation (200-fold) than that observed for the full-length cyclin T1 protein. Importantly, hCycT1 (amino acids 1–254), which lacks the TRM and is unable to bind Tat in vitro, also failed to stimulate Tat-dependent transcription from the HIV-1 promoter in vivo (Fig. 2A). Western blot analysis of the transfected cells demonstrated that the various hCycT1 proteins were expressed at comparable levels in vivo. Therefore, these data indicate that the Tat:TAR recognition motif defined in vitro is required to support TAR-dependent transactivation by Tat in vivo.
Figure 2.
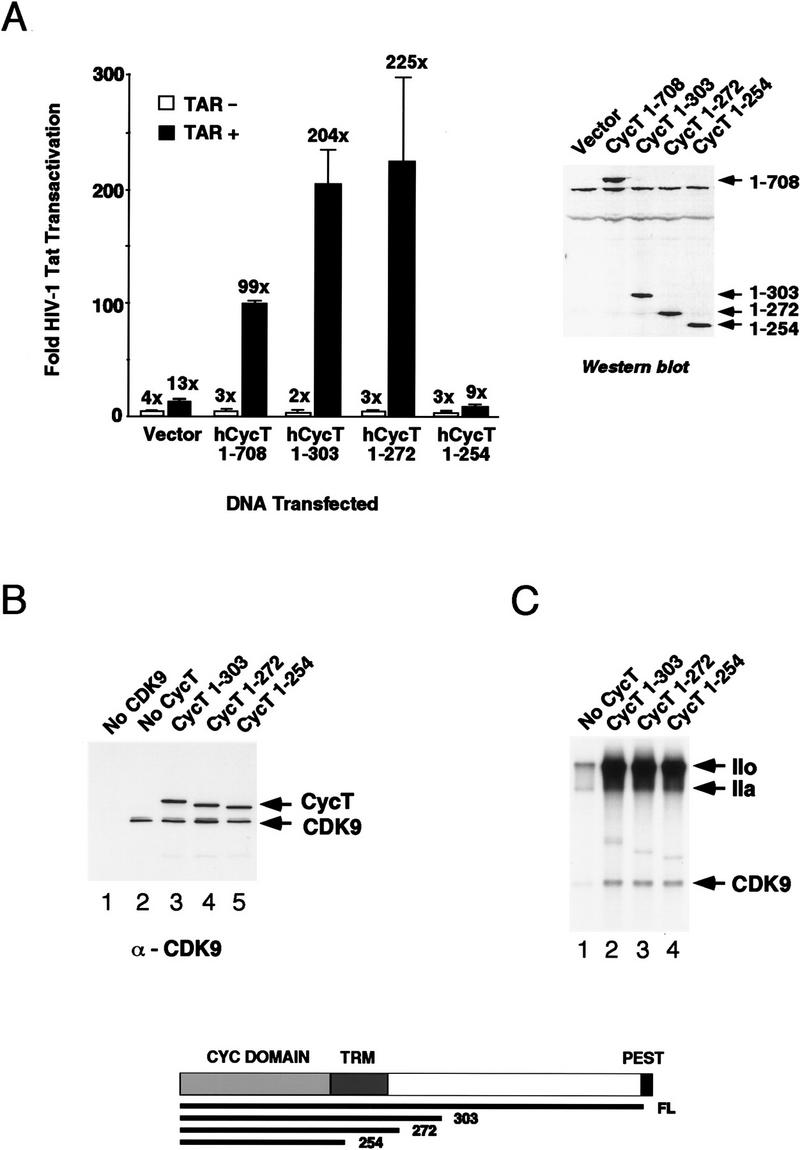
The TRM of hCycT1 is required for Tat transactivation in vivo but is not needed for hCycT1 to bind CDK9 or to enhance GST–CTD phosphorylation. (A) Analysis of the ability of hCycT1 truncation mutants to support HIV-1 Tat transactivation in murine (NIH-3T3) cells. NIH-3T3 cells were transfected with 100 ng of either pHIV-1/LUC (+TAR) or pHIV-1/LUC+30/+33 (−TAR), 50 ng of either pSV/Tat (+Tat) or pSV/TatZX (−Tat), 10 ng of pRL/CMV, 100 ng of either pCGN vector (vector) or the different pCMV/HA–hCycT1 truncation mutants, as indicated in the figure. The firefly luciferase activity produced from the HIV-1 promoter was normalized to Renilla luciferase activity from the CMV promoter to correct for transfection efficiency. The graph plots the fold HIV-1 Tat trans-activation as the ratio of corrected firefly luciferase activity observed in the presence and absence of Tat. Standard deviations were calculated from three independent transfections. The Western blot displays the relative level of expression of each cyclin T1 protein in transfected Chinese hamster ovary (CHO) cells. (B) Analysis of the ability of the hCycT1 truncation mutants to bind to purified CDK9 in vitro. Antisera specific to CDK9 was used to immunoprecipitate complexes formed between CDK9 and the truncated hCycT1 proteins. hCycT1 binding was visualized by Western blot using an anti-GST monoclonal antibody and CDK9 was visualized with an anti-FLAG monoclonal antibody. Reactions in lanes 3–5 each contained 200 ng of purified (FLAG-tagged) CDK9 protein and 500 ng of the different GST–hCycT1 proteins, as indicated above each lane. Control reactions contained 500 ng of GST–hCycT1 (amino acids 1–303) alone (lane 1), or 200 ng of CDK9 alone (lane 2). (C) Analysis of the ability of hCycT1 truncation mutants to enhance phosphorylation of GST–CTD by CDK9 in vitro. Reactions contained 250 ng of purified (FLAG-tagged) CDK9, 250 ng of the different hCycT1 truncation mutants (as indicated above each lane), and 200 ng of purified GST–CTD. Arrows indicate the position of autophosphorylated CDK9, and the hypophosphorylated (IIa) and hyperphosphorylated (IIo) forms of the CTD.
The part of the hCycT1 Tat:TAR recognition motif identified here lies at the extreme carboxyl terminus of the cyclin domain, and the corresponding region within cyclin H has been shown to be important for CTD phosphorylation by CDK7 in vitro (Andersen et al. 1997). Consequently, we investigated whether the TRM was also important for regulation of CDK9 activity by hCycT1 in vitro. Interestingly, hCycT1 (amino acids 1–254), which lacks this part of the TRM, was as active as the other hCycT1 proteins (amino acids 1–303; 1–272) in its ability to bind recombinant CDK9 in coimmunoprecipitation experiments in vitro (Fig. 2B), or enhance phosphorylation of GST–CTD by CDK9 (Fig. 2C). CTD phosphorylation was abrogated by mutation of the catalytic site of CDK9 (CDK9 mutant D167N), confirming that CTD phosphorylation is mediated by CDK9 in these experiments (data not shown). Therefore, hCycT1 sequences from amino acids 254 to 272 are critical for binding Tat, but are not essential for cyclin activity or for proper folding of the cyclin domain.
The hCycT1 TRM makes independent contacts with Tat and TAR RNA
To further characterize this part of the TRM, a series of clustered point mutations were generated spanning the region from amino acids 251–272, and tested for their ability to bind to Tat and TAR RNA in gel mobility-shift experiments. Mutant cyclins with substitutions of amino acids 251–254, or 256–259, failed to form a complex with Tat and TAR, whereas substitutions of amino acids 265–268 or 269–272 were tolerated (data not shown). These results refined the carboxy-terminal boundary of the TRM to the region between amino acids 250–265, which lies at the end of the last predicted alpha helix of the cyclin repeats. Critical residues within this region were then identifed by alanine-scanning mutagenesis (Fig. 3). Interestingly, single amino acid substitutions affecting four different cyclin residues (R251, L252, R254, and C261) completely abolished the formation of the hCycT1–Tat:TAR complex in vitro, and point mutations affecting four additional residues (N250, I255, W258, and R259) significantly reduced binding of the complex to TAR (Fig. 3A). Therefore this region of the TRM contains multiple residues that are critical for binding of the hCycT1–Tat complex to TAR.
Figure 3.

Alanine-scanning mutagenesis of the TRM of hCycT1. (A) Analysis of the ability of different alanine-substituted hCycT1 (amino acids 1–303) proteins to enhance the binding of HIV-1 Tat to TAR RNA in a gel-mobility shift experiment. Reactions contained either wild-type TAR RNA (odd-numbered lanes) or TAR loop mutant (+29/+34) RNAs (even-numbered lanes). Reactions included 175 ng (GST-cleaved) of Tat-1 (lanes 3–30) and 40 ng (GST-cleaved) of hCycT1 (amino acids 1–303). The position of each mutation is indicated above each lane. (WT) Wild-type hCycT1 (amino acids 1–303). (B) Analysis of the ability of the different hCycT1 mutants to interact directly with HIV-1 Tat in vitro. Reactions included 60 ng (GST-cleaved) of HIV-1 Tat and 250 ng of GST–hCycT1 (amino acids 1–303), either wild-type (WT) or mutant, as indicated above each lane. The hCycT1 protein was visualized with a monoclonal antibody to GST, and the (GST-cleaved, HA-tagged) Tat was visualized with an anti-HA monoclonal antibody. Lane 1 represents 10% of the input protein (200 ng) of the wild-type Tat protein. The TRM within hCycT1 is shown at the bottom. The residues designated with the ‡ symbol are required for TAR RNA recognition, whereas the residues indicated with the § symbol are needed to bind Tat.
To determine which residues in this part of the TRM are recognized by Tat, the mutant GST–hCycT1 proteins were analyzed using the protein:protein interaction assay described in Figure 1B. Importantly, C261A was the only mutation that completely abolished binding to Tat (Fig. 3B, cf. lanes 2 and 13). The R259A substitution also significantly reduced the interaction with Tat, and more modest effects were observed with mutants N250A, R251A, and E262A. Therefore, residues C261 and R259 have a key role in the association of cyclin T1 with Tat. In contrast, the two arginine residues at position 251 and 254, as well as L252, I255, and W258 are not critical contacts for Tat, but are essential for binding of the cyclin T1–Tat complex to TAR RNA. Therefore, the latter amino acids either contact TAR RNA directly within the complex, or are required indirectly to support a structure necessary for the hCycT1–Tat complex to interact with TAR. We conclude that TRM residues have separate roles in binding to Tat and TAR RNA. Importantly, a synthetic peptide containing the TRM failed to form a specific complex with Tat and TAR, as did other fragments of hCycT1 that lack small regions of the amino-terminal cyclin fold (data not shown), indicating that the entire cyclin domain is required to form the Tat:TAR complex. Therefore, Tat may need to make additional contacts with cyclin domain residues that lie outside of the TRM, or the overall structure and folding of the cyclin domain may be essential for Tat:TAR recognition by hCycT1.
The interaction between Tat and hCycT1 requires zinc
Given that binding of Tat to hCycT1 requires cysteines in each protein and Tat has been shown to be a metal-binding protein, we asked whether zinc is required for Tat to recognize hCycT1 and form a stable ternary complex on TAR RNA. As shown in Figure 4A, the binding of purified Tat to GST–hCycT1(amino acids 1–303) was destroyed by incubation of the proteins with EDTA. Tat was then incubated in the presence or absence of metal and mixed with metal-free GST–hCycT1 coupled to glutathione beads. Importantly, binding of Tat to GST–hCycT1(amino acids 1–303) was fully recovered by the addition of Zn(II) but not by Mg(II) or Co(II) (Fig. 4A; other data not shown). The inhibition of the hCycT1–Tat interaction by metal chelators and the dependence of the complex on zinc was also reflected in the binding of the Tat–hCycT1 complex to TAR RNA (Fig. 4B). In contrast, removal of zinc did not affect the binding of monomeric HIV-1 Tat to TAR RNA (Fig. 4B, cf. lanes 4 and 7), although zinc did enhance the binding of Tat dimers to TAR (Fig. 4B, right panel). The C261A mutation completely abolished binding of hCycT1 to Tat and TAR. A more conservative change replacing cysteine 261 with a histidine also failed to restore binding of hCycT1 to Tat (Fig. 4A, lanes 9–12) or TAR (Fig. 4B, lanes 26–29). We conclude that the interaction between hCycT1 and Tat requires zinc, and that histidine is not a tolerated substitution for C261 in hCycT1.
Figure 4.

The interaction between HIV-1 Tat and hCycT1 requires zinc. (A) Zinc is required for the binding of HIV-1 Tat to hCycT1 in vitro. The wild-type hCycT1 (WT amino acids 1–303; lanes 1–4), or mutant hCycT1 proteins that contain a substitution of cysteine 261 to either alanine (C261A; lanes 5–8), or to histidine (C261H; lanes 9–12) were coupled to beads and incubated with (GST-cleaved) HIV-1 Tat. The Tat and hCycT1 proteins were incubated together in buffer lacking EDTA (No EDTA; lanes 1, 5, and 9), or were treated with EDTA (EDTA; lanes 2, 6, and 10), or treated with EDTA and subsequently incubated with either zinc sulfate (Zn++; lanes 3, 7, and 11) or magnesium sulfate (Mg++; lanes 4, 8, and 12), as described in Materials and Methods. Proteins were visualized by Western blot. (B) Zinc-dependent binding of HIV-1 Tat and hCycT1 to TAR RNA. (Left panel) Zinc is required for the formation of hCycT1-Tat: TAR, but not Tat:TAR, complexes. Where indicated, reactions contained 150 ng of EDTA-treated HIV-1 Tat and 60 ng of EDTA-treated (GST-cleaved) hCycT1 (amino acids 1–303). Binding reactions contained either wild-type HIV-1 TAR RNA (lanes 1,4,7,10,13); loop-substituted HIV-1 TAR RNA (+29/+34; lanes 2,5,8,11,14); or bulge mutant RNA (U22A; lanes 3,6,9,12,15). Zinc sulfate was added to the EDTA-treated proteins (Zn++) in lanes 7–9 and lanes 13–15. (Right panel) Zinc-dependent binding of the hCycT1-Tat complex to wild-type HIV-1 TAR RNA. Reactions contained 150 ng of (GST-cleaved) HIV-1 Tat and 60 ng of either wild-type GST–hCycT1 amino acids 1–303 (WT; lanes 18–21); a cysteine to alanine mutant (C261A; lanes 22–25); or a cysteine to histidine mutant (C261H; lanes 26–29), as indicated above each lane. The HIV-1 Tat and GST–hCycT1 proteins were incubated in buffer lacking EDTA (no EDTA; lanes 18,22,26), or treated with EDTA (EDTA; lanes 19,23,27), or EDTA-treated and subsequently reconstituted with either zinc sulfate (Zn++; lanes 20,24,28) or magnesium sulfate (Mg++; lanes 21,25,29). The Tat preparations used in this experiment differed in the extent of dimer (Tat, top arrow) versus monomer (Tat, bottom arrow) formed, which had no effect on the results obtained.
The murine CycT1 protein lacks C261 and forms a weak, zinc-independent complex with Tat
To determine whether the low level of Tat trans-activation observed in murine cells reflects an inability of the murine CycT1 protein to support Tat trans-activation, we isolated a cDNA encoding the murine homolog of human cyclin T1. A database search identified a 1-kB expressed-sequence tag clone (I.M.A.G.E Consortium Clone ID 605445) that was highly homologous (86% identity) to amino acids 398–726 of the human cyclin T1 protein. A probe from this cDNA was isolated and used to screen a lambda ZAP C57B16 post-natal (day 21) mouse brain cDNA library for the full-length murine cyclin T1 gene. The intact murine cDNA clone was sequenced and found to encode a protein of 724 amino acids, which lacks residues 429 and 599 and is therefore two amino acids shorter than the human cyclin T1 protein. Overall, the murine homolog is 89.5% identical to the human cyclin T1, although the extent of homology is much higher (95.3% identity) within the cyclin domain (amino acids 1–300; Fig. 5A). Most interestingly, four amino acid differences were observed between the human and murine CycT1 proteins within the TRM, including residue cysteine 261, which is a tyrosine in the mouse (Fig. 5A).
Figure 5.



Analysis of the ability of the murine CycT1 protein to interact with HIV-1 Tat and TAR RNA in vitro. (A) Sequence comparison of the cyclin domains of the murine and human CycT1 proteins. Amino acid differences between the two proteins are shown in shaded boxes and the TRM at the carboxy-terminal boundary of the cyclin domain is indicated with brackets. (B) The murine CycT1 protein forms a weak, zinc-independent complex with HIV-1 Tat in vitro. Wild-type or mutant versions of the human and mouse GST–CycT1 proteins (amino acids 1–272) were coupled to beads and incubated with purified (GST-cleaved) HA-tagged HIV-1 Tat. Reactions either lacked EDTA (no EDTA; lanes 1,5,9), or contained additional zinc sulfate (Zn++; lanes 2,6,10), or were treated with EDTA in the absence (EDTA; lanes 3,7,11), or presence of exogeneous zinc sulfate (EDTA+Zn++; lanes 4,8,12). Tat was incubated with resins containing either wild-type hCycT1 (lanes 1–4), wild-type mCycT1 (lanes 5–8), or the mutants mCycT1 Y261C (lanes 9–12), or hCycT1 C261A (amino acids 1–303; lane 13). The (GST-cleaved; HA-tagged) HIV-1 Tat and various GST–CycT1 proteins were visualized by Western blot using monoclonal antisera specific to the hemagglutinin (HA) tag and GST, respectively. (C) Analysis of the ability of murine CycT1 to enhance the binding of HIV-1 Tat to TAR RNA. (Left panel) Wild-type or mutant mCycT1 (amino acids 1–272) proteins were incubated in the presence or absence of HIV-1 Tat with wild-type (odd-numbered lanes) or loop mutant (even-numbered lanes) HIV-1 TAR RNA probes. Where indicated, the reactions contained 50 ng of (GST-cleaved) HIV-1 Tat and 50 ng of (GST-cleaved) mCycT1 (amino acids 1–272). The mCycT1 YQ/CE is a double mutant containing both Y261C and Q262E substitutions. hWT refers to the GST–hCycT1 (amino acids 1–272) control. (Right panel) Zinc-dependent binding of the mCycT1–Tat complex to wild-type TAR-1 RNA. Reactions included 175 ng of HIV-1 (GST-cleaved) Tat and 50 ng of the various (GST-cleaved) CycT1 proteins, as indicated above each lane. The EDTA treatment and metal reconstitution conditions are described in B.
Because C261 is a critical contact for Tat, we first sought to determine whether Tat can bind to murine CycT1 in vitro. Despite the absence of C261, the murine cyclin T1 protein was recognized by Tat, although the interaction was weaker than that observed with hCycT1 (Fig. 5B, cf. lanes 1 and 5). This interaction was specific because an activation domain mutant Tat (C22G) did not bind to either the human or murine cyclin T1 proteins (data not shown). Replacement of Y261 with a cysteine (mCycT1 Y261C) enhanced binding to Tat (Fig. 5B, cf. lanes 5 and 9). Moreover, we observed a qualitative difference in the response of the human and murine cyclin T1 proteins to exogeneous zinc and metal chelators. Therefore, the addition of zinc stimulated binding of Tat to either the hCycT1 or mCycT1 Y261C, whereas the binding of Tat to mCycT1 was modestly inhibited by zinc (Fig. 5B, cf. lanes 2 and 10 with lane 6). Treatment of either hCycT1 or mCycT1 Y261C with EDTA destroyed binding to Tat, which was fully restored by incubation with zinc, whereas treatment of the wild-type murine cyclin T1 protein with EDTA only modestly reduced its ability to bind Tat, and binding was unaffected by subsequent addition of zinc (Fig. 5B, cf. lanes 7 and 8 for mCycT1 with lanes 3 and 4 for hCycT1). We conclude that Tat binds specifically to the mCycT1 protein, but forms a weaker, altered complex that no longer requires zinc. Most importantly, the introduction of a cysteine residue at position 261 restores zinc-dependent binding of the cyclin to Tat. We also noted that the wild-type murine CycT1 protein binds to Tat with greater affinity than the hCycT1 C261A mutant (Fig. 5B, cf. lanes 5 and 13), indicating that other residues in the mouse cyclin might have compensated for the loss of C261.
The murine CycT1–Tat complex bound weakly to TAR RNA in a loop-dependent manner in vitro. PhosphorImager scanning of the gel shown in Figure 5C revealed that the ternary complex formed on TAR with murine CycT1 is reduced 12-fold from that of the human CycT1 protein (Fig. 5C, cf. lanes 3 and 5). Interestingly, a single residue substitution of the mouse tyrosine 261 to alanine had no significant effect on the complex, indicating that, unlike the human cyclin T1 protein, amino acid 261 in mCycT1 is not an important contact for either Tat or TAR. Similarly, replacement of Q262 with a glutamate residue did not enhance binding of mCycT1 to Tat and TAR. In contrast, the Y261C point mutation, or the double mutant YQ/CE (261,262), which both restore cysteine to position 261, greatly enhanced the binding of the mCycT1-Tat complex to TAR RNA (Fig. 5C, cf. lanes 11 and 13 with lane 5). The specificity of the Tat–CycT1 Y261C complex for loop sequences in TAR was slightly reduced compared with the hCycT1 protein. Unlike the protein:protein interaction, the binding of the mCycT1–Tat complex to TAR RNA was found to require zinc (Fig. 5C, lanes 20–22), suggesting that Tat requires zinc to accommodate binding of the complex to TAR RNA. In contrast, the mutant hCycT1 C261A mutant failed to form a complex with Tat and TAR RNA (Fig. 5C, lane 26), which is consistent with the results obtained in the protein:protein interaction assay and indicates that residues in mCycT1 must partially compensate for the lack of C261.
A point mutation restoring cysteine 261 is sufficient to rescue HIV-1 Tat trans-activation by the murine CycT1 protein in vivo
We next examined whether the murine cyclin T1 protein could enhance TAR-dependent trans-activation by Tat in vivo. In contrast with the wild-type human CycT1 protein, which strongly enhances Tat-mediated HIV-1 transcription when transiently expressed in NIH3T3 cells, the murine CycT1 protein was unable to augment Tat trans-activation of the HIV-1 promoter in vivo, and even exerted a small inhibitory effect on Tat transactivation (Fig. 6). To test the importance of cysteine 261 in vivo, the Y261C mutation was introduced into both the full-length (706 amino acid) and truncated (272 amino acid) forms of murine cyclin T1 in mammalian expression vectors. Remarkably, both forms of mCycT1 Y261C dramatically increased Tat activity in NIH3T3 cells, and yielded trans-activation efficiencies comparable with those observed with human CycT1 (Fig. 6A). Overexpression of human cyclin H, which is unable to bind Tat and TAR in vitro (data not shown), neither enhanced nor interfered with HIV-1 Tat trans-activation in vivo (Fig. 6B). Western blot analysis of the transfected cells demonstrated that the various CycT1 and CycH proteins were expressed at similar levels. We conclude that replacement of mCycT1 tyrosine 261 with a cysteine is sufficient to rescue HIV-1 Tat-mediated transactivation in vivo.
Figure 6.
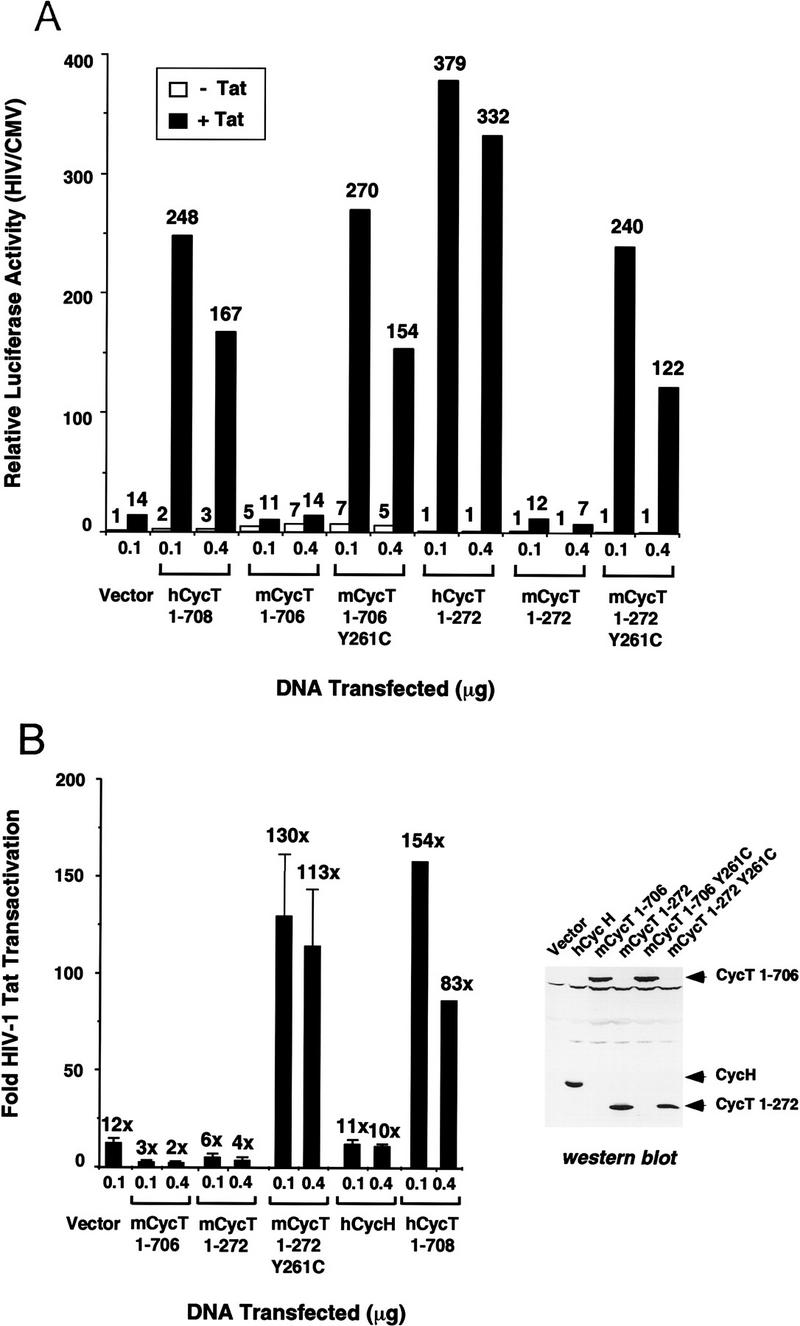
Analysis of the ability of wild-type and mutant (Y261C) murine cyclin T1 (mCycT1) proteins to support HIV-1 Tat trans-activation in vivo. (A) Overexpression of the murine cyclin T1 protein in NIH3T3 cells enhances basal, but not HIV-1 Tat-activated, transcription from the HIV-1 promoter. NIH3T3 cells were transfected with 100 ng of pHIV-1/LUC, and 50 ng of either pSV/Tat (+Tat) or pSV/TatZX (−Tat), 10 ng of pRL/CMV, and either 100 ng or 400 ng of pCGN (vector) or the different human or murine CycT1 expression vectors, as indicated at the bottom of the graph. The relative luciferase activity was calculated following normalization for Renilla luciferase activity expressed from the CMV promoter from the pRL/CMV internal control plasmid. Both the human (amino acids 1–708) and murine (amino acids 1–706) CycT1 constructs express the full-length proteins without the 18-amino-acid PEST sequence at the carboxyl terminus of each protein. (B) Comparison of the fold-increase in HIV-1 Tat transactivation in NIH3T3 cells on transient expression of human and murine CycT1. Standard deviations were calculated from three independent transfections. The Western blot displays the relative level of expression of each cyclin T1 protein in transfected CHO cells.
Interestingly, overexpression of full-length human and murine cyclin T1 proteins increased basal transcription from the HIV-1 promoter in vivo by a factor of two- to threefold for hCycT1 and five- to sevenfold for mCycT1 (Fig. 6A). Carboxy-terminal truncation of either the human or mouse cyclins (amino acids 1–272), however, destroyed their ability to activate basal HIV-1 promoter activity, consistent with a report that sequences carboxy-terminal to the cyclin domain in DmCycT1 are required to activate cellular genes in vivo (Peng et al. 1998a). For this reason, the fold increase in HIV-1 Tat trans-activation supported by the truncated hCycT1 protein is greater than that seen with the full-length cyclin T1 protein.
Stable overexpression of hCycT1 in murine cells enhances HIV-1 transcription and increases virus production, but does not rescue HIV-1 replication
Although hCycT1 enhances HIV-1 Tat-mediated trans-activation dramatically, it was unclear whether this would be sufficient for the efficient production of infectious HIV-1 from murine cells. To examine this question, NIH3T3 cells were transiently transfected with a molecular clone of an HIV-1 provirus in the presence and absence of hCycT1, and virus production was assayed 2 days post-transfection. In the presence of hCycT1, the murine cells produced ∼10-fold more HIV-1 (Fig. 7A). The increase in infectious virus produced also correlated with the amount of virus particles detected in these cultures by ELISA (data not shown). Given that Tat is present in all transfections involving the HIV-1 provirus, the increase in virus production seen with hCycT1 is similar to the increase in HIV-1 LTR-mediated transcription that is observed in murine cells that have been transfected with both hCycT1 and Tat, as compared with Tat alone.
Figure 7.
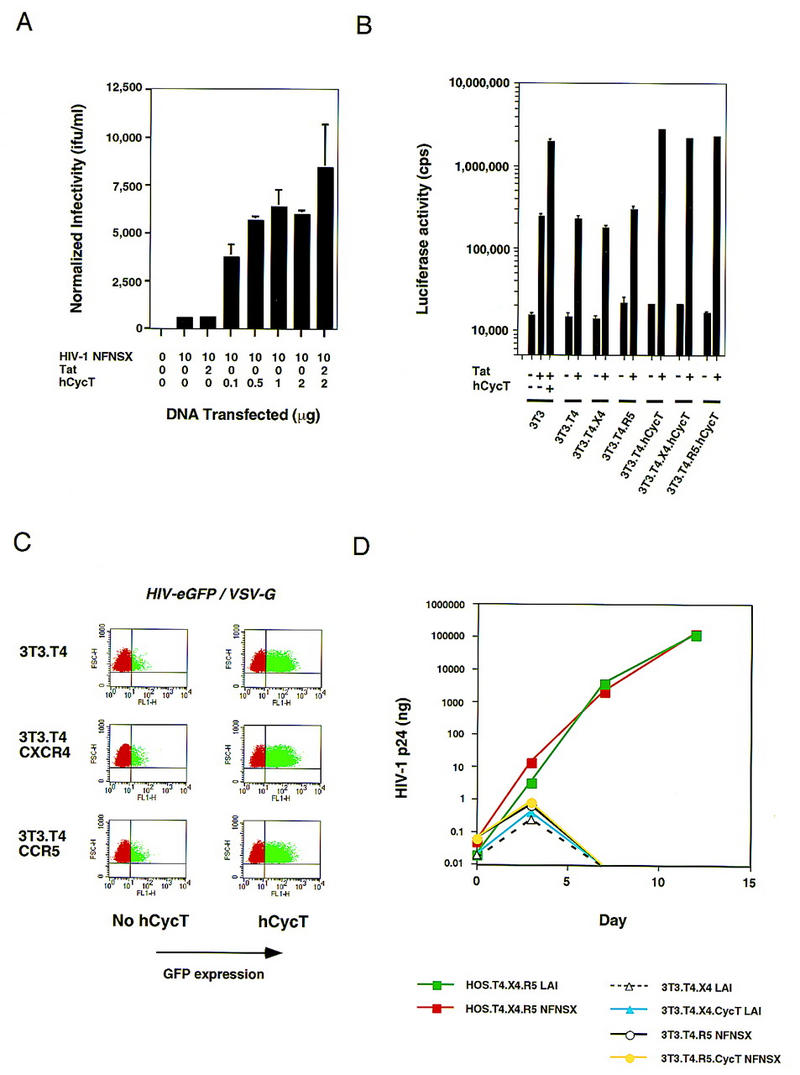
Effect of overexpression of hCycT1 on HIV-1 production in murine cells. (A) Expression of hCycT1 increases the production of infectious HIV-1 in transient expression experiments carried out in murine cells. NIH3T3 cells were transiently transfected with DNAs depicted along the abscissa. HIV-1 NFNSX is a full-length HIV-1 proviral construct; HIV-1 Tat is encoded by pCMV/Tat; and hCycT1 is encoded by pBABE–CycT. Cells were additionally transfected with pSV40/LUC to assess relative transfection efficiencies. Virus supernatants were titered on GHOST X4/R5 cells. Virus infectivity is depicted along the ordinate and reflects virus titers normalized to transfection efficiency. (B) Development of murine lines that stably express functional hCycT1. NIH3T3 lines expressing different HIV-1 receptor molecules, human CD4, CXCR4, and/or CCR5, were additionally stably transduced with hCycT1, as depicted beneath the panel. These stable cell lines were examined for their respective abilities to support transcription from an HIV-1 LTR construct (pHIV-1/LUC) that directs expression of the firefly luciferase. pHIV-1/LUC was transiently transfected in these lines in the presence and absence of pCMV/Tat. The parental NIH3T3 cell line was also transfected with pBABE–CycT as a positive control. (C) Increased gene expression of an HIV-1 provirus in hCycT1+ stable murine lines. NIH3T3 cells stably expressing hCycT1 (plots on right) vs. progenitors that do not (plots on left) were infected with a replication defective HIV-1 vector encoding eGFP. Dots in the upper right quadrants of the FACS profiles represent cells that detectably express eGFP. (D) The presence of hCycT1 and human receptors is not sufficient to enable spreading replication of HIV-1 in murine cell cultures. Different cell lines are depicted in the legend below the panel, (HOS) Human osteosarcoma line. HIV-1 replication after initial challenge on day 0 was measured by accumulation of HIV-1 CA (p24) antigen in the culture supernatants by ELISA. p24 values plotted represent averages of duplicate sets. (T4) Human CD4; (X4) human CXCR4; (R5) human CCR5; (NFNSX) R5-tropic HIV-1 isolate; (LAI) X4-tropic HIV-1 isolate.
To determine whether the enhanced ability of murine cells expressing human CycT1 to produce HIV-1 would enable efficient HIV-1 replication in murine cell culture, human CycT1 was first stably introduced into NIH3T3 cells that expressed human CD4 and either human CXCR4 or CCR5. Human CycT1 function was tested in these cells by transient transfection of a construct expressing firefly luciferase driven by the HIV-1 LTR. The presence of human CycT1 stably expressed in these cells enabled a greater than 100-fold increase in HIV-1 LTR-mediated transcription in the presence as compared with the absence of Tat (Fig. 7B). Expression of human CycT1 in these cells was also confirmed by immunoblot experiments (data not shown).
Our initial analyses had shown that transient cotransfection of DNA encoding an HIV-1 provirus and plasmids encoding human CycT1 would boost virus production in murine cells. We next examined whether HIV-1 infection in murine cells that stably express human CycT1 would enhance transcription from the integrated HIV-1 provirus in a chromatin environment. An HIV-1 vector lacking env coding sequence and possessing an enhanced Green Fluorescent Protein (eGFP) in place of the nef allele was pseudotyped with envelope glycoprotein of vesicular stomatitis virus (VSV) and used to challenge NIH3T3 cells stably expressing human CycT1. The expression of eGFP from this construct has been shown to depend on the HIV-1 LTR. Infection of NIH3T3 cells expressing human CycT1 enhanced expression of the proviral LTR-directed eGFP dramatically. In contrast, HIV-1 infection was barely detectable in NIH3T3 progenitor lines that do not express human CycT1 (Fig. 7C).
Given that the human CycT1 stable murine lines supported enhanced HIV-1 transcription, we next challenged these lines with small inoculums of replication competent X4- and R5-tropic HIV-1 (Fig. 7D). Virus production in the culture supernatant was monitored over the course of 12 days. Despite the presence of human CycT1 in NIH3T3 cells expressing appropriate HIV-1 coreceptors, very poor virus production was detected in these cultures, and the amount of virus released from human CycT1-expressing versus human CycT1-nonexpressing murine cells was indistinguishable. In contrast, human cells infected with either virus stock replicated the virus efficiently. Therefore, in addition to the well-characterized barriers to receptor-mediated entry and Tat-regulated transcription from the integrated provirus, strong species-specific restrictions exerted at post-transcriptional steps in the viral life cycle effectively block HIV-1 replication in murine cells.
Discussion
The data presented here provide strong support for the model that binding of HIV-1 Tat to the cyclin T1 subunit of the human TAK/P-TEFb transcription elongation complex is a critical first step in TAR RNA recognition and Tat-mediated trans-activation (Wei et al. 1998; for review, see Jones 1997; Cullen 1998; Emerman and Malim 1998). Through cloning and biochemical characterization of the human and mouse cyclin T1 proteins, we show that Tat forms a distinctive zinc-dependent complex with the cyclin domain of hCycT1. This interaction is mediated by residues in the core and cysteine-rich motifs of the Tat activation domain, and requires the entire cyclin structure as well as a key cysteine residue (C261) within the TRM. Our findings suggest that the low level of Tat transactivation observed in murine cells is attributable to the fact that the mouse CycT1 protein contains a tyrosine rather than a cysteine at position 261, which prevents mCycT1 from forming a specific zinc-dependent complex with Tat that is essential for binding to TAR RNA. Replacement of tyrosine 261 with a cysteine conferred zinc-dependent binding of mCycT1 to Tat and enabled high-affinity binding of the complex to TAR RNA. Strikingly, this single amino acid change also rescued Tat transactivation by mCycT1 in vivo. These findings have important implications for the structure of the Tat–hCycT1–TAR complex and for approaches to block complex formation in infected cells, as well as for the use of the human or modified murine cyclin T1 proteins in the development of transgenic mice capable of sustaining a productive HIV-1 infection.
Residues in human cyclin T1 protein are critical for binding both Tat and TAR RNA
Within the hCycT1–Tat:TAR complex, the arginine-rich motif of Tat interacts with the bulge of TAR but the residues responsible for RNA loop recognition are unknown. We show that binding of Tat to hCycT1 requires the entire cyclin domain as well as TRM sequences at the carboxy-terminal boundary of the cyclin fold, which is not needed for the cyclin to regulate CDK9 activity. Five TRM residues (R251, L252, R254, I255, and W258) were necessary for binding of the Tat–hCycT1 complex to TAR, but were not important for binding of hCycT1 to Tat. Therefore, these residues may directly contact the loop and upper stem of TAR RNA in the Tat–hCycT1 complex. Arginine residues are common determinants of RNA-binding specificity in proteins, and hydrophobic amino acids such as leucine, isoleucine, and tryptophan can provide important base-stacking and RNA-backbone contacts. Alternatively, these amino acids may be needed to impart a structure or flexibility to the cyclin that is critical for binding of the complex to TAR. In addition, it is also possible that Tat may contribute to the recognition of TAR loop sequences in the complex. We have shown that the Tat proteins from HIV-1 or HIV-2 bind weakly to HIV-2 TAR RNA in a loop-dependent manner in the absence of hCycT1, and that loop sequence-specificity is lost on truncation of the Tat activation domain (Garber et al. 1998; Wei et al. 1998). To date, however, no specific residues outside of the Tat ARM have been implicated in RNA-binding. Our data suggest that cyclin T1 is in direct contact with the loop of TAR in the complex, although RNA cross-linking and other structural studies will be needed to resolve this question fully.
It is remarkable that the cyclin domain of hCycT1 (amino acids 1–303) is sufficient to support Tat trans-activation in vivo (Fig. 2), given that this region is unable to stimulate basal HIV-1 promoter activity in the absence of Tat or regulate cellular gene transcription. The role of the carboxy-terminal half of hCycT1 is unknown, although the comparable region of the Drosophila CycT1 protein has been implicated in CTD substrate recognition (Peng et al. 1998a). Alternatively, the carboxy-terminal half of hCycT1 may have a role in recruiting the P-TEFb complex to RNAPII or to the initiation complex that assembles at the HIV-1 promoter in the absence of Tat. Whatever the function of this region, it appears to be dispensible or bypassed in the presence of Tat and TAR, and consequently the mechanism of Tat-mediated trans-activation must differ somewhat from the conventional mode of transcriptional activation by P-TEFb.
The interaction between Tat and human CycT1 may involve a zinc bridge
The observation that the Tat–hCycT1 interaction requires zinc as well as cysteine residues in both molecules raises the possibility of a connecting zinc bridge, in which cysteines in each protein coordinate a shared metal ion(s). The Tat activation domain contains six important cysteine residues, as well as two histidines, and coordinates two atoms of zinc per monomer (Frankel et al. 1998a). Incubation of Tat with zinc induces the formation of metal-linked dimers in vitro (Frankel et al. 1988a), although dimerization is unimportant for Tat function. Our data suggest that Tat acts instead to form a metal-linked heterodimer with the cyclin T1 subunit of the P-TEFb complex, as illustrated in Figure 8. A role for C261 in zinc-binding is strongly suggested from the observation that the murine cyclin T1 protein lacks C261 and binds to Tat in a zinc-independent manner, and that zinc-dependent binding to Tat is conferred by the Y261C point mutation in the mouse cyclin. In addition to the Cys-rich region, six residues in the Tat ‘core’ subdomain (FITKALGISYG) are essential for binding to hCycT1 (M.E. Garber, G. Caderas, K.A. Jones, unpubl.). This region is highly hydrophobic and unlikely to be involved in metal binding. A bipartite interaction surface would explain why peptides or protein fragments of Tat that contain only the Cys-rich motif are unable to disrupt the hCycT1–Tat complex or interfere with Tat-mediated transactivation.
Figure 8.
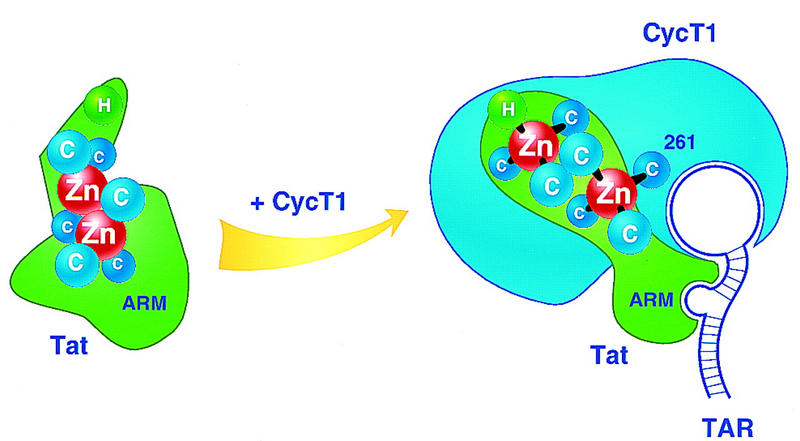
Hypothetical view of the metal binding site at the HIV-1 Tat–hCycT1 interaction surface. HIV-1 Tat contains six cysteine and one histidine residues that are essential for trans-activation and could have a role in metal-binding. Free Tat coordinates two atoms of zinc per monomer (Frankel et al. 1988a), potentially in an arrangement involving five cysteine residues (Huang and Wang 1996). Binding of hCycT1 to Tat is proposed to induce structural changes in Tat that allow high-affinity, loop-specific binding to TAR RNA. Part of the interaction surface between Tat and hCycT1 could involve the shared binding of a zinc atom that coordinates to residue C261 in the cyclin.
These findings suggest that Tat and hCycT1 might form a two-zinc module in which one of the zinc atoms is shared between the two proteins. Alternatively, zinc may have a separate role within each molecule to establish a structure necessary for their interaction. As has been seen for other proteins that bind metals through multiple closely-spaced cysteine residues, we find that histidine is not a tolerated substitution for cysteine 261 in the hCycT1–Tat interaction (Fig. 4), and comparable Cys to His substitutions are also not allowed in Tat (Sadaie et al. 1990). Interestingly, anti-viral agents such as azodicarbonamide and disulfiram, which function as zinc-ejection compounds (Rice et al. 1993, 1995; McDonnell et al. 1997; Huang et al. 1998) effectively block the binding of Tat to hCycT1 in vitro (M.E. Garber and K.A. Jones, unpubl.). Derivatives of compounds such as these, or others that disrupt the interaction between Tat and hCycT1, could be effective anti-viral agents that selectively block the binding of Tat to TAK/P-TEFb without disrupting CDK9 activity or the control of cellular gene expression.
Murine CycT1 forms an altered complex with Tat that impairs binding to TAR RNA
Although Tat transactivation through TAR is greatly reduced in murine cells, these cells can support activation by chimeric Tat proteins (e.g., Tat–Rev) that bypass the need to function through TAR RNA (Luo et al. 1993; Madore and Cullen 1993; Alonso et al. 1994). Consequently, these genetic studies predict that the murine CycT1 protein should bind to Tat but be unable to effectively enhance binding to TAR RNA. Our data, however, indicate that the key residue that is missing in the mouse cyclin (C261) is an amino acid that is normally used to bind Tat, rather than TAR RNA. This apparent discrepancy is reconciled by the fact that the absence of C261 in mCycT1 leads only to a slight decrease (three- to fourfold) in binding to Tat and a significantly greater (12-fold) decrease in binding of the mCycT1–Tat complex to TAR RNA. The weak binding of mCycT1 to Tat and TAR observed in vitro is consistent with the low but detectable (5- to 10-fold) level of Tat activity normally observed in NIH-3T3 cells, although we cannot exclude the possibility that Tat may interact with a distinct cyclin T1-related protein in murine cells.
Given the importance of residue C261 in the hCycT1–Tat interaction, it is striking that the murine CycT1 protein retains any ability to bind Tat, albeit with reduced affinity. In particular, the binding of the wild-type mCycT1 to Tat (and TAR) was significantly greater than that observed with the hCycT1 C261A mutant. This difference is not attributable to the presence of Y261 in the mouse, as converting the tyrosine to an alanine did not reduce binding of mCycT1 to Tat or TAR (Fig. 5C). Therefore some other residue(s) in mCycT1 must compensate for the lack of C261. Interestingly, the mCycT Y261C mutant was completely unable to bind Tat in the absence of zinc, even though its sequence is otherwise identical to the wild-type murine cyclin T1, which can bind Tat in the absence of zinc. Therefore, the presence of cysteine 261 may induce a conformational change in the cyclin, for example by forming an intramolecular disulfide bridge, which could sterically eliminate binding to Tat in the absence of zinc. In this model, the interaction of the human cyclin with Tat would disrupt the disulfide bridge and free C261 to coordinate the zinc bound to Tat.
Implications for the structure and function of the hCycT1(P-TEFb):Tat:TAR complex
We have shown recently that the full-length Tat protein binds poorly to TAR RNA in the absence of hCycT1, and that removal of the amino terminus greatly enhances RNA-binding activity in vitro (Garber et al. 1998). Consequently, native Tat may be poorly configured to bind RNA, and its interaction with hCycT1 may be accompanied by structural changes that allow efficient binding to TAR (Fig. 8). Taken together, the data indicate that high-affinity binding to TAR RNA is critically dependent on the exact three-dimensional positioning of the Tat and cyclin T1 subunits within the complex. Binding to zinc may impart a specific structure to the proteins or orient the two subunits in a manner that is critical for RNA-binding. Tat has been shown previously to enhance phosphorylation of the RNAPII CTD strongly during transcription (Parada and Roeder 1997; Garcia-Martinez et al. 1997), and consequently binding of the TAK/P-TEFb complex to TAR RNA may be important to activate CDK9 or direct the kinase to phosphorylate the CTD as a substrate.
The critical residues in hCycT1 that bind Tat and TAR, including C261, are absent in other C-type cyclins isolated to date, including the minor CDK9-associated cyclins (hCycT2a and T2b), and these cyclins are not found in nuclear TAK complexes (A.P. Rice and C.H. Herrmann, unpubl.). Therefore, the hCycT2 proteins, like mCycT1, may down-regulate Tat trans-activation by sequestering CDK9 in P-TEFb complexes that are not accessible to Tat. In support of this possibility, we find that overexpression of mCycT1 blocks Tat trans-activation in transient expression experiments (Fig. 6B). Therefore, variations in the relative levels of hCycT1 and other CDK9-associated cyclins might differentially affect Tat activity in different cells of the immune system.
Recent studies have shown that Tat transactivation can be mimicked by tethering hCycT1 or CDK9 to heterologous RNA targets (Fujinaga et al. 1998; Gold et al. 1998), indicating that binding of P-TEFb to the nascent transcript is sufficient to regulate RNAPII elongation at the HIV-1 promoter. It is interesting that Tat evolved to interact specifically with hCycT1, given that other cyclin partners for CTD kinases might have served such a function equally well. Importantly, TAK activity is induced strongly on activation of T cells and promonocytic cell lines, coincident with a dramatic increase in the level of hCycT1 (Yang et al. 1997; Herrmann et al. 1998). Levels of other cyclins, including CycT2 and CycH, were found to be unaffected by T cell activation, indicating that Tat selected a cyclin that is strongly induced under the cellular conditions that are most optimal for virus replication. As the other P-TEFb subunits and regulatory elongation factors (e.g., DSIF; Hartzog et al. 1998; Wada et al. 1998; Wu-Baer et al. 1998) are identified and characterized, it will be interesting to learn how they control Tat trans-activation and whether they are regulated in a manner that might influence HIV-1 infection.
Implications for a small animal model system to study HIV-1 replication
Although human CycT1 was capable of enhancing expression of an HIV-1 provirus in murine cells, its presence was not sufficient to enable a spreading infection of HIV-1 in cell culture, indicating that murine-specific restrictions to virus replication exist at multiple steps. Strong blocks to HIV-1 replication exist even in PBMC from the more closely related rhesus macaques, although SIVsm family viruses replicate efficiently in these cells. Studies with HIV-1/SIV chimeric viruses suggest that these primate-specific blocks are independent of HIV-1 Tat or Env (Shibata et al. 1991; Himathongkham and Luciw 1996). Post-transcriptional blocks to HIV-1 replication in murine cells have been observed previously (Canivet et al. 1990; Trono and Baltimore 1990). In particular, the nuclear to cytoplasmic transport of unspliced and singly spliced HIV-1 RNAs encoding the structural gene products appears to be inefficient in murine cells, and other post-transcriptional blocks such as those observed in primate cell lines may exist as well. Even when normalized for equivalent levels of HIV-1 transcription, murine cells expressing hCycT1 were found to produce nearly three orders of magnitude less infectious virus than do human cells, indicating that the post-transcriptional block is quite strong. The quality of the virus produced (relative infectivity per HIV-1 particle), however, was comparable between mouse and human cells (V.N. KewalRamani and D.R. Littman, unpubl.).
The identification of human cellular factors that participate in HIV-1 replication and that may surmount restrictions in murine cells is compelling not only because it provides a better understanding of the molecular aspects of HIV-1 replication but also because it increases the feasibility of developing a transgenic murine model for HIV-1 infection. Currently, there is no animal model with an intact host immune system that can be infected by HIV-1 such that a high persistent viremia can be established and can compromise host immune function. The closest existing animal model is infection of macaque monkeys with SIVsm family viruses, which has been valuable for studying pathogenesis as it correlates to virus spread and evolution, and for identifying pathogenic determinants encoded by the virus. To develop a transgenic murine model system to address similar issues as well as the role of host immune responses, it will be critical to identify the remaining human-specific factors, other than hCycT1 and the human receptors, which are needed to support productive HIV-1 infection in the mouse.
Materials and methods
DNA constructs
Bacterial expression vectors encoding the full-length or truncated human or mouse cyclin T1 fused to glutathione–S–transferase were subcloned into pGEX-2T as BamHI–EcoRI fragments. Mammalian transfection constructs expressing HA-tagged cyclin T1 proteins, under the control of the CMV promoter, were generated by insertion of human CycT1 (full-length or truncated) as an XbaI–BamHI fragment, or murine CycT1 (full-length or truncated) as an NheI–BamHI fragment, into the XbaI and BamHI sites of pCGN (Tanaka and Herr 1990). The full-length constructs lacked the carboxy-terminal PEST sequence, as indicated in Figure 6. For stable transduction of human CycT1 into NIH3T3 cells, full-length human CycT1 containing the carboxy-terminal PEST sequence was subcloned into the murine leukemia virus (MLV) expression vector, pBABEpuro (Morgenstern and Land 1990), to create pBABE–CycT.
Cloning of mouse cyclin T1 cDNA
The I.M.A.G.E Consortium Clone ID 605445, which contains a 1-kb cDNA fragment that is 86% identical to the carboxy-terminal region of human cyclin T1 (amino acids 398–726) was obtained from the American Type Culture Collection and used to screen a lambda ZAP C57B16 post-natal (day 21) mouse brain cDNA library. A single cDNA encoding the full-length murine cyclin T1 gene was isolated and sequenced.
Purification of CKD9, GST–CTD, and the CTD kinase assay
Full-length CDK9 and catalytic site mutant CDK9 (D167N) proteins were expressed in recombinant baculovirus vectors as amino-terminal FLAG-tagged fusion proteins. To construct the baculovirus CDK9-expression vectors, wild-type and D167N CDK9 cDNAs were inserted into pVL1392 (Pharmingen) and recombinant baculoviruses were generated in Sf9 cells with the BaculoGold Transfection Kit (Pharmingen). The plaque-purified recombinant baculovirus was used to infect Sf9 cells and the CDK9 protein was purified using the protocol described by Kim et al. (1996). CDK9 was dialyzed against buffer B (20 mm sodium phosphate, pH 7.2, 300 mm NaCl, 10% glycerol, 1 mm PMSF, 1 mm DTT) and quantified by SDS-PAGE. The 15 μl in vitro kinase reactions contained 100 ng of GST–CTD, 2 mm ATP, and 10 mCi [γ-32P]ATP (3000 Ci/mmole), 5 pmoles CDK9 and 5 pmoles GST–hCycT1 in kinase buffer (50 mm Tris-HCl, pH 7.4, 5 mm MnCl2, 5 mm DTT, as described by Hermann and Rice (1995). Reactions were incubated at 25°C for 30 min and analyzed on an 8% SDS–polyacrylamide gel. The GST–CTD was prepared as described by Peterson et al. (1992), and further purified following elution from glutathione–S–Sepharose beads by gel filtration chromatography on Superdex 75 resin (Pharmacia).
RNA-binding experiments
Gel mobility shift reactions (16 μl) were carried out in binding buffer (30 mm Tris-HCl, pH 8.0, 12% glycerol, 70 mm KCl, 0.03% NP-40, and 2 mm DTT) as described by Wei et al. (1998). Bacterially-expressed HIV-1 GST–Tat and GST–hCycT1 proteins were treated with thrombin prior to incubation with TAR, and the RNA-bound complexes were separated on a pre-run 6% Tris-Glycine gel. High specific-activity minimal HIV-1 TAR RNA probes (nucleotides +17 to +43) were uniformly labeled in vitro using a linearized DNA template and T7 RNA polymerase as described by Wei et al. (1998).
To remove zinc, purified preparations of GST–hCycT1 (2 mg/ml) and HIV-1 Tat (0.7 mg/ml) were incubated for 2 hr at 0°C in Tat elution buffer (50 mm Tris-HCl, pH 7.5, 100 mm NaCl, 10% glycerol) containing 5 mm EDTA/1 mm DTT and 10 mm EDTA/10 mm DTT, respectively. Where appropriate, Tat-1 (65 ng) was reconstituted with zinc or magnesium in 3 μl Tat elution buffer containing 1.3 mm EDTA, 1.3 mm zinc or magnesium sulfate (Fluka), and 8.7 mm DTT. Reconstitution was for 15 min at room temperature. Gel-shift reactions involving metal reconstitution were carried out in the above binding buffer with 0.3 mm EDTA (EDTA-treated lanes) or 0.3 mm EDTA containing 0.3 mm zinc sulfate. Neither the hCycT1 nor the Tat protein was treated with EDTA in the lanes marked no EDTA.
Protein–protein interaction experiments
GST-cleaved Tat (2 μg) was incubated with 3 μg GST–CycT1 pre-bound to glutathione–S–Sepharose beads (15 μl) in 500 μl of binding buffer (40 mm HEPES, pH 8.0, 0.5% NP-40, and 10 mm DTT) containing 120 mm NaCl. Incubation was for 2 hr at 4°C. Beads were washed extensively (3 × 15 min at 4°C) in 400 μl of binding buffer containing 500 mm NaCl before the addition of SDS sample buffer. One-half of the protein sample was analyzed by 12% SDS-PAGE and the Western blot was simultaneously probed with anti-HA (BMB) and anti-GST (Santa Cruz) monoclonal antibodies.
To examine the zinc requirement for the CycT–Tat interaction, each protein was treated with EDTA as stated above for the RNA-binding experiments. Where appropriate, Tat (2 μg) was reconstituted with zinc or magnesium in 100 μl of binding buffer containing 120 mm NaCl and 0.3 mm EDTA with or without 0.4 mm metal sulfate. Reconstitution was for 30 min at room temperature. The presence or absence of EDTA, zinc, or EDTA and zinc, as indicated above the lanes, refers only to the reconstitution of Tat. The GST–CycT1 used in all reactions was treated with 5 mm EDTA. Reactions (500 μl) were carried out in binding buffer with 120 mm NaCl, 75 μm EDTA, 80 μm metal sulfate, and 10 mm DTT, and the proteins were analyzed by Western blot as described above.
Immunoprecipitations
GST–CycT1 (500 ng) and CDK9 (250 ng) were diluted to 100 μl in EBC (40 mm Tris-HCl, pH 8.0, 120 mm NaCl, 0.5% NP-40, and 2 mm DTT) before the addition of CDK9 polyclonal antisera (60 ng; Santa Cruz). Complex formation was for 2 hr at 4°C followed by an additional 1-hr incubation with pre-blocked Protein A Sepharose beads (10 μl). Beads were washed three times in EBC containing 0.03% SDS and the proteins were analyzed by 12% SDS-PAGE. Western blot analysis was visualized with GST and FLAG (Kodak) monoclonal antisera.
Transient expression experiments
NIH-3T3 cells were grown in DMEM supplemented with 10% fetal calf serum and seeded at 2 × 105 cells/well in 6-well plates. Transfection experiments were carried out 18–24 hr later using a lipofectamine reagent (GIBCO BRL). The plasmids pHIV-1/LUC, pHIV-1/LUC+30/+33, pSV/Tat, and pSV/Tat-ZX have been described previously (Wei et al. 1998). Transfected cells were lysed and assayed for luciferase activity 48 hr post-transfection. HIV-1 luciferase activity was normalized to pRL/CMV, which encodes the Renilla luciferase from the CMV promoter, as an internal control.
Stable cell lines, virus preparations, and infections
NIH-3T3 cells that stably express human CD4 in conjunction with either human CXCR4 or CCR5 and are susceptible to HIV-1 infection have been described previously (Deng et al. 1997). Similar cells were transduced with MLV ecotropic pseudotypes of BABE–cycT, and puromycin-resistant populations were selected. NIH-3T3 cells expressing human CycT1 were maintained in DMEM supplemented with 10% FCS and 1 μg/ml puromycin. Human osteosarcoma (HOS) cells were sequentially transduced with retroviral vectors expressing human CD4, CCR5, and CXCR4 and populations that uniformly expressed these cell surface markers were sorted by FACS. HOS.T4.X4.R5 cells were maintained in DMEM supplemented with 10% FCS, 300 μg/ml G418, and 1 μg/ml puromycin.
HIV-1 was purified from the culture medium of transfected cells by initially removing cell debris by centrifugation at 1500g after which clarified supernatants were passed through 0.22 micron filters. Titrations of HIV-1 were performed on GHOST cells (Trkola et al. 1998). HIV-eGFP/VSV-G pseudotypes were prepared as described (Bartz and Vodicka 1997). Challenge of NIH-3T3 cells with HIV-eGFP/VSV-G was performed in the presence of 20 μg/ml polybrene. Virus and polybrene were removed 2 hr post-challenge, and cells were assayed for mean GFP fluorescence 2 days post-challenge by flow cytometry. To examine spreading infection, 2 × 105 NIH-3T3 or HOS cells seeded per well of 6-well plates were challenged with 5 ng of p24 of HIV-1 LAI or HIV-1 NFNSX in the presence of 20 μg/ml Polybrene for 2 hr, after which cells were washed three times with PBS and replenished with growth medium. All infections were performed in duplicate. Cell supernatants were harvested every several days. Infected cell cultures were split into fresh, uninfected cells on the same days (only 2nd and 3rd timepoints) immediately after cell supernatants were sampled. Clarified supernatants from HIV-1 infected and mock-infected cells were assayed for HIV-1 by p24 (CA) ELISA (Coulter Immunotech). Dilutions caused by infected cell passaging were taken into account in the calculation of p24 values.
Acknowledgments
We are grateful to Tom Alber and Joe Noel for the advice and ideas exchanged throughout the course of this work. We thank Michael Emerman, William Dynan, and Irvin Chen for various plasmid DNA constructs; Cary Lai for the mouse brain cDNA library; and Michelle Rhea for automated DNA sequencing. This work was funded by grants to K.A.J. from the National Institutes of Health (NIH) (AI33824), California Universitywide AIDS Research Program (R97-SAL-136), and the G. Harold and Leila Y. Mathers Foundation. M.E.G. is supported by a graduate student fellowship from The Chapman Foundation. P.W. was funded by a postdoctoral fellowship from the Universitywide AIDS Research Program. A.P.R. and C.H.H. are supported by the NIH (AI35381). V.N.K is a fellow of The Damon Runyon-Walter Winchell Foundation and D.R.L. is an investigator of the Howard Hughes Medical Institute supported in this project by the NIH (AI33856).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Note
The sequence of the cDNA encoding the full-length murine CycT1 protein reported in this paper will appear in the GenBank nucleotide sequence database under accession number AF087662.
Footnotes
E-MAIL jones@salk.edu; FAX (619) 535-8194.
References
- Adamczewski J, Rossignol M, Tassan J, Nigg E, Moncollin V, Egly J-M. MAT1, cdk7 and cyclin H form a kinase complex which is UV light-sensitive upon association with TFIIH. EMBO J. 1996;15:1877–1884. [PMC free article] [PubMed] [Google Scholar]
- Alonso A, Cujec T, Peterlin BM. Effects of human chromosome 12 on interactions between Tat and TAR of human immunodeficiency virus type 1. J Virol. 1994;68:6505–6513. doi: 10.1128/jvi.68.10.6505-6513.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen G, Busso D, Poterszman A, Hwang J, Wurtz J-M, Ripp R, Thierry J-C, Egly J-M, Moras D. The structure of cyclin H: Common mode of kinase activation and specific features. EMBO J. 1997;16:958–967. doi: 10.1093/emboj/16.5.958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagby S, Kim S, Maldonado E, Tong K, Reinberg D, Ikura M. Solution structure of the carboxy-terminal core domain of human TFIIB: Similarity to cyclin A and interaction with TATA-binding protein. Cell. 1995;82:857–867. doi: 10.1016/0092-8674(95)90483-2. [DOI] [PubMed] [Google Scholar]
- Bartz S, Vodicka M. Production of high-titer human immunodeficiency virus type 1 pseudotyped with vesicular stomatitis virus glycoprotein. Methods. 1997;12:337–342. doi: 10.1006/meth.1997.0487. [DOI] [PubMed] [Google Scholar]
- Canivet M, Hoffman AD, Hardy D, Sernatinger J, Levy JA. Replication of HIV-1 in a wide variety of animal cells following phenotypic mixing with murine retroviruses. Virology. 1990;178:543–551. doi: 10.1016/0042-6822(90)90352-r. [DOI] [PubMed] [Google Scholar]
- Cullen BR. HIV-1 auxiliary proteins: Making connections in a dying cell. Cell. 1998;93:685–692. doi: 10.1016/s0092-8674(00)81431-2. [DOI] [PubMed] [Google Scholar]
- Deng H, Unutmaz D, KewalRamani V, Littman D. Expression cloning of new receptors used by simian and human immunodeficiency viruses. Nature. 1997;388:296–300. doi: 10.1038/40894. [DOI] [PubMed] [Google Scholar]
- Dingwall C, Ernberg I, Gait MJ, Green SM, Heaphy S, Karn J, Lowe AD, Singh M, Skinner MA. HIV-1 Tat protein stimulates transcription by binding to a U-rich bulge in the stem of the TAR RNA structure. EMBO J. 1990;9:4145–4153. doi: 10.1002/j.1460-2075.1990.tb07637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerman M, Malim M. HIV-1 regulatory/accessory genes: Keys to unraveling viral and host cell biology. Science. 1998;280:1880–1884. doi: 10.1126/science.280.5371.1880. [DOI] [PubMed] [Google Scholar]
- Frankel A, Bredt D, Pabo C. Tat protein from human immunodeficiency virus forms a metal-linked dimer. Science. 1988a;240:70–73. doi: 10.1126/science.2832944. [DOI] [PubMed] [Google Scholar]
- Frankel A, Chen L, Cotter R, Pabo C. Dimerization of the tat protein from human immunodeficiency virus: A cysteine-rich peptide mimics the normal metal-linked dimer interface. Proc Natl Acad Sci. 1988b;85:6297–6300. doi: 10.1073/pnas.85.17.6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga K, Cujec TP, Peng J, Garriga J, Price DH, Grana X, Peterlin BM. The ability of positive transcription elongation factor b to transactivate human immunodeficiency virus transcription depends on a functional kinase domain, Cyclin T1, and Tat. J Virol. 1998;72:7154–7159. doi: 10.1128/jvi.72.9.7154-7159.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gait M, Karn J. RNA recognition by the human immunodeficiency virus Tat and Rev proteins. Trends Biochem Sci. 1993;18:255–259. doi: 10.1016/0968-0004(93)90176-n. [DOI] [PubMed] [Google Scholar]
- Garber, M.E., P. Wei, and K.A. Jones. 1998. HIV-1 Tat interacts with cyclin T1 to direct the P-TEFb CTD kinase complex to TAR RNA. Cold Spring Harbor Symp. Quant. Biol. 63: (in press). [DOI] [PubMed]
- Garcia-Martinez LF, Mavanka G, Neveu JM, Lane WS, Ivanov D, Gaynor RB. Purification of a Tat-associated kinase reveals a TFIIH complex that modulates HIV-1 transcription. EMBO J. 1997;16:2836–2850. doi: 10.1093/emboj/16.10.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MO, Yang X, Herrmann CH, Rice AP. PITALRE, the catalytic subunit of TAK, is required for human immunodeficiency virus Tat transactivation in vivo. J Virol. 1998;72:4448–4453. doi: 10.1128/jvi.72.5.4448-4453.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grana X, De Luca A, Sang N, Fu Y, Claudio P, Rosenblatt J, Morgan D, Giordano A. PITALRE, a nuclear CDC2-related protein kinase that phosphorylates the retino blastoma protein in vitro. Proc Natl Acad Sci. 1994;91:3834–3838. doi: 10.1073/pnas.91.9.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart C, Galphin J, Westhafer M, Schochetman G. TAR loop-dependent human immunodeficiency virus trans activation requires factors encoded on human chromosome 12. J Virol. 1993;67:5020–5024. doi: 10.1128/jvi.67.8.5020-5024.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart C, Ou C, Galphin J, Moore J, Bacheler L, Wasmuth J, Petteway S, Jr, Schochetman G. Human chromosome 12 is required for elevated HIV-1 expression in human-hamster hybrid cells. Science. 1989;246:488–491. doi: 10.1126/science.2683071. [DOI] [PubMed] [Google Scholar]
- Hartzog GA, Wada T, Handa H, Winston F. Evidence that Spt4, Spt5, and Spt6 control transcription elongation by RNA polymerase II in Saccharomyces cerevisiae. Genes & Dev. 1998;12:357–369. doi: 10.1101/gad.12.3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann CH, Rice AP. Specific interaction of the human immunodeficiency virus Tat proteins with a cellular protein kinase. Virology. 1993;197:601–608. doi: 10.1006/viro.1993.1634. [DOI] [PubMed] [Google Scholar]
- ————— Lentivirus Tat proteins specifically associate with a cellular protein kinase, TAK, that hyperphosphorylates the carboxyl-terminal domain of the large subunit of RNA polymerase II: Candidate for a Tat cofactor. J Virol. 1995;69:1612–1620. doi: 10.1128/jvi.69.3.1612-1620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann, C.H., R.G. Carroll, P. Wei, K.A. Jones, and A.P. Rice. 1998. Tat-associated kinase, TAK, activity is regulated by distinct mechanism in peripheral blood lymphocytes and promonocytic cell lines. J. Virol. (in press). [DOI] [PMC free article] [PubMed]
- Himathongkham S, Luciw PA. Restriction of HIV-1 (subtype B) replication at the entry step in rhesus macaque cells. Virology. 1996;219:485–488. doi: 10.1006/viro.1996.0276. [DOI] [PubMed] [Google Scholar]
- Huang HW, Wang KT. Structural characterization of the metal binding site in the cysteine-rich region of HIV-1 Tat protein. Biochem Biophys Res Comm. 1996;227:615–621. doi: 10.1006/bbrc.1996.1554. [DOI] [PubMed] [Google Scholar]
- Huang M, Maynard A, Turpin JA, Graham L, Janini GM, Covell DG, Rice WG. Anti-HIV agents that selectively target retroviral nucleocapsid protein zinc fingers without affecting cellular zinc finger proteins. J Med Chem. 1998;41:1371–1381. doi: 10.1021/jm9708543. [DOI] [PubMed] [Google Scholar]
- Jeffrey P, Russo A, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletich N. Mechanism of CDK activation revealed by the structure of a cyclin A-CDK2 complex. Nature. 1995;367:313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- Jones KA. Taking a new TAK on Tat transactivation. Genes & Dev. 1997;11:2593–2599. doi: 10.1101/gad.11.20.2593. [DOI] [PubMed] [Google Scholar]
- Kim KK, Chamberlin HM, Morgan DO, Kim S-H. Three-dimensional structure of human cyclin H, a positive regulator of the CDK-activating kinase. Nat Struct Biol. 1996;3:849–855. doi: 10.1038/nsb1096-849. [DOI] [PubMed] [Google Scholar]
- Lee J-O, Russo A, Pavletich N. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature. 1998;391:859–865. doi: 10.1038/36038. [DOI] [PubMed] [Google Scholar]
- Luo Y, Madore S, Parslow T, Cullen BR, Peterlin BM. Functional analysis of interactions between Tat and the trans-activation response element of human immunodeficiency virus type 1 in cells. J Virol. 1993;67:5617–5622. doi: 10.1128/jvi.67.9.5617-5622.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madore S, Cullen BR. Genetic analysis of the cofactor requirement for human immunodeficiency virus type 1 Tat function. J Virol. 1993;67:3703–3711. doi: 10.1128/jvi.67.7.3703-3711.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancebo H, Lee G, Flygare J, Tomassini J, Luu P, Zhu Y, Blau C, Hazuda D, Price D, Flores O. P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes & Dev. 1997;11:2633–2644. doi: 10.1101/gad.11.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall N, Peng J, Xie Z, Price DH. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J Biol Chem. 1996;271:27176–27183. doi: 10.1074/jbc.271.43.27176. [DOI] [PubMed] [Google Scholar]
- Marshall N, Price DH. Control of formation of two distinct classes of RNA polymerase II elongation complexes. Mol Cell Biol. 1992;12:2078–2090. doi: 10.1128/mcb.12.5.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem. 1995;270:12335–12338. doi: 10.1074/jbc.270.21.12335. [DOI] [PubMed] [Google Scholar]
- Martin-Castellanos C, Moreno S. Recent advances on cyclins, CDKs and CDK inhibitors. Trends Cell Biol. 1997;7:95–98. doi: 10.1016/S0962-8924(96)10055-6. [DOI] [PubMed] [Google Scholar]
- McDonnell N, De Guzman R, Rice W, Turpin J, Summers M. Zinc ejection as a new rationale for the use of cystamine and related disulfide-containing antiviral agents in the treatment of AIDS. J Med Chem. 1997;40:1969–1976. doi: 10.1021/jm970147+. [DOI] [PubMed] [Google Scholar]
- Morgenstern J, Land H. Advanced mammalian gene transfer: High titer retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolov D, Chen H, Halay E, Usheva A, Hisatake K, Lee DK, Roeder RG, Burley S. Crystal structure of a TFIIB-TBP-TATA element ternary complex. Nature. 1995;377:119–127. doi: 10.1038/377119a0. [DOI] [PubMed] [Google Scholar]
- Noble M, Endicott J, Brown N, Johnson L. The cyclin box fold: Protein recognition in cell-cycle and transcriptional control. Trends Biochem Sci. 1997;22:482–487. doi: 10.1016/s0968-0004(97)01144-4. [DOI] [PubMed] [Google Scholar]
- Parada C, Roeder RG. Enhanced processivity of RNA polymerase II triggered by Tat-induced phosphorylation of its carboxyl-terminal domain. Nature. 1996;384:375–378. doi: 10.1038/384375a0. [DOI] [PubMed] [Google Scholar]
- Peng J, Marshall N, Price DH. Identification of a cyclin subunit required for the function of Drosophila P-TEFb. J Biol Chem. 1998a;273:13855–13860. doi: 10.1074/jbc.273.22.13855. [DOI] [PubMed] [Google Scholar]
- Peng J, Zhu Y, Milton J, Price DH. Identification of multiple cyclin subunits of human P-TEFb. Genes & Dev. 1998b;12:755–762. doi: 10.1101/gad.12.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SR, Dvir A, Anderson CW, Dynan WS. DNA binding provides a signal for phosphorylation of the RNA polymerase II heptapeptide repeats. Genes & Dev. 1992;6:426–438. doi: 10.1101/gad.6.3.426. [DOI] [PubMed] [Google Scholar]
- Rice AP, Chan F. Tat protein of human immunodeficiency virus type 1 is a monomer when expressed in mammalian cells. Virology. 1991;185:451–454. doi: 10.1016/0042-6822(91)90797-f. [DOI] [PubMed] [Google Scholar]
- Rice W, Schaeffer C, Harten B, Villinger F, South T, Summers M, Henderson L, Bess J, Jr, Arthur L, McDougal J, et al. Inhibition of HIV-1 infectivity by zinc-ejecting aromatic C-nitroso compounds. Nature. 1993;361:473–475. doi: 10.1038/361473a0. [DOI] [PubMed] [Google Scholar]
- Rice W, Supko J, Malspeis L, Buckheit R, Jr, Clanton D, Bu M, Graham L, Schaeffer C, Turpin J, Domagala J, et al. Inhibitors of HIV nucleocapsid protein zinc fingers as candidates for the treatment of AIDS. Science. 1995;270:1194–1197. doi: 10.1126/science.270.5239.1194. [DOI] [PubMed] [Google Scholar]
- Rossignol M, Kolb-Cheynel I, Egly J-M. Substrate specificity of the cdk-activating kinase (CAK) is altered upon association with TFIIH. EMBO J. 1997;16:1628–1637. doi: 10.1093/emboj/16.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo A, Jeffrey P, Patten A, Massague J, Pavletich N. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- Sadaie M, Mukhopadhyaya R, Benaissa Z, Pavlakis G, Wong-Staal F. Conservative mutations in the putative metal-binding region of human immunodeficiency virus tat disrupt virus replication. AIDS Res Hum Retroviruses. 1990;6:1257–1263. doi: 10.1089/aid.1990.6.1257. [DOI] [PubMed] [Google Scholar]
- Shibata R, Kawamura M, Sakai H, Hayami M, Ishimoto A, Adachi A. Generation of a chimeric human and simian immunodeficiency virus infectious to monkey peripheral blood mononuclear cells. J Virol. 1991;65:3514–3520. doi: 10.1128/jvi.65.7.3514-3520.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Herr W. Differential transcriptional activation by Oct-1 and Oct-2: Interdependent activation domains induce Oct-2 phosphorylation. Cell. 1990;60:375–386. doi: 10.1016/0092-8674(90)90589-7. [DOI] [PubMed] [Google Scholar]
- Tassan J, Jaquenoud M, Fry A, Frutiger S, Hughes G, Nigg E. In vitro assembly of a functional human CDK7-cyclin H complex requires MAT1, a novel 36 kDa RING finger protein. EMBO J. 1995;14:5608–5617. doi: 10.1002/j.1460-2075.1995.tb00248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trkola A, Ketas T, KewalRamani V, Endorf F, Binley J, Katinger H, Robinson J, Littman D, Moore J. Neutralization sensitivity of human immunodeficiency virus type 1 primary isolates to antibodies and CD4-based reagents is independent of coreceptor usage. J Virol. 1998;72:1876–1885. doi: 10.1128/jvi.72.3.1876-1885.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trono D, Baltimore D. A human cell factor is essential for HIV-1 Rev action. EMBO J. 1990;9:4155–4160. doi: 10.1002/j.1460-2075.1990.tb07638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Toshiyuki T, Yamaguchi Y, Ferdous A, Imai T, Hirose S, Sugimoto S, Yano K, Hartzog GA, Winston F, Buratowski S, Handa H. DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes & Dev. 1998;12:343–356. doi: 10.1101/gad.12.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P, Garber ME, Fang S-M, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92:451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- Wu-Baer F, Lane WS, Gaynor RB. Role of the human homolog of the yeast transcription factor Spt5 in HIV-1 Tat-activation. J Mol Biol. 1998;277:179–197. doi: 10.1006/jmbi.1997.1601. [DOI] [PubMed] [Google Scholar]
- Yang X, Gold M, Tang D, Lewis D, Aguilar-Cordova E, Rice AP, Herrmann CH. TAK, an HIV Tat-associated kinase, is a member of the cyclin-dependent family of protein kinases and is induced by activation of peripheral blood lymphocytes and differentiation of promonocytic cell lines. Proc Natl Acad Sci. 1997;94:12331–12336. doi: 10.1073/pnas.94.23.12331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Herrmann CH, Rice AP. The human immunodeficiency virus Tat proteins specifically associate with TAK in vivo and require the carboxyl-terminal domain of RNA polymerase II for function. J Virol. 1996;70:4576–4584. doi: 10.1128/jvi.70.7.4576-4584.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankulov K, Bentley D. Regulation of CDK7 substrate specificity by MAT1 and TFIIH. EMBO J. 1997;16:1638–1646. doi: 10.1093/emboj/16.7.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Chen D, Pierstorff E, Luo K. Transcription elongation factor P-TEFb mediates Tat activation of HIV-1 transcription at multiple stages. EMBO J. 1998;17:3681–3691. doi: 10.1093/emboj/17.13.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Pe’ery T, Peng J, Ramanathan Y, Marshall N, Marshall T, Amendt B, Mathews M, Price DH. Transcription elongation factor P-TEFb is required for HIV-1 Tat transactivation in vitro. Genes & Dev. 1997;11:2622–2632. doi: 10.1101/gad.11.20.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]