

Abstract
TFIIH is a multisubunit complex, containing ATPase, helicases, and kinase activities. Functionally, TFIIH has been implicated in transcription by RNA polymerase II (RNAPII) and in nucleotide excision repair. A member of the cyclin-dependent kinase family, CDK7, is the kinase subunit of TFIIH. Genetically, CDK7 homologues have been implicated in transcription in Saccharomyces cerevisiae, and in mitotic regulation in Schizosaccharomyces pombe. Here we show that in mitosis the CDK7 subunit of TFIIH and the largest subunit of RNAPII become hyperphosphorylated. MPF-induced phosphorylation of CDK7 results in inhibition of the TFIIH-associated kinase and transcription activities. Negative and positive regulation of TFIIH requires phosphorylation within the T-loop of CDK7. Our data establishes TFIIH and its subunit CDK7 as a direct link between the regulation of transcription and the cell cycle.
Keywords: Transcription, TFIIH, CDK7, phosphorylation, cell cycle
Entry into mitosis is accompanied by repression of transcription by all three nuclear RNA polymerases (Johnson et al. 1965, 1987; Fink et al. 1977). In cultured cells, incorporation of RNA precursors ceases in prophase and resumes at the exit from mitosis. Mitotic repression has been associated with a number of regulatory mechanisms (Gottesfeld and Forbes 1997). Accumulating evidence indicates that mitotic repression involves the direct inactivation of key components of the transcription machinery (Gottesfeld et al. 1994). For example, the heptapeptide repeats present in the carboxy-terminal domain (CTD) of the largest subunit of RNA polymerase II (RNAPII) are hyperphosphorylated in meiotic and mitotic cells (Shermoen and O’Farrell 1991; Bellier et al. 1997; Parsons and Spencer 1997). Phosphorylation of the CTD is a critical regulatory step in the modulation of elongation competence (Lu et al. 1991; O’Brien and Lis 1991; Weeks et al. 1993; Dahmus 1996; Zhu et al. 1997). It was found that the CTD is hyperphosphorylated by MPF in vitro resulting in the dissociation of transcription complexes (Cisek and Corden 1989; Zawel et al. 1993). Additionaly, the TAF subunits of TFIID (Segil et al. 1996), and TFIIH are inactivated in mitosis (Long et al. 1998)
A key role in the regulation of the cell cycle is played by a family of cyclin-dependent serine/threonine protein kinases (CDKs) (Reed 1992; Coleman and Dunphy 1994; Elledge and Harper 1994; King et al. 1994). These enzymes are critical for initiation and completion of DNA replication and cell division in organisms from yeast to mammals (Reed 1992; King et al. 1994). The activity of CDKs is modulated through phosphorylation of their catalytic subunits and by their association with positive (cyclins) and negative regulatory proteins (Elledge and Harper 1994). Distinct cyclins perform different tasks in specific phases of the cell cycle. Entry into mitosis is mediated by M-phase-promoting factor (MPF; Labbe et al. 1989; Gautier et al. 1990). Activation of MPF results in the activation of different kinases and the inhibition of the phosphatases PP1 and PP2A (Kinoshita et al. 1990; Nigg 1993). As a result, multiple proteins are phosphorylated causing a reorganization of the nuclear envelope, the spindle apparatus, the chromosomes, and the regulation of transcription factors.
We have investigated the mechanisms of repression of transcription by RNAPII during mitosis. Transcription by RNAPII requires multiple factors. One family of factors functions to deliver RNAPII to the promoter (TFIIB, TFIID, TFIIF) (Conaway et al. 1990; Lu et al. 1991), whereas two other factors (TFIIE and TFIIH) mediate the escape of RNAPII from the promoter (Goodrich and Tjian 1994; Kumar et al. 1998). These factors are collectively known as the general transcription factors (GTFs) (Roeder 1991; Orphanides et al. 1996). We found that TFIIH and the CTD of RNAPII are hyperphosphorylated in mitosis. TFIIH is composed of nine polypeptides with four enzymatic activities (Drapkin and Reinberg 1994; Svejstrup et al. 1996). TFIIH contains a DNA-dependent ATPase activity, two ATP-dependent DNA helicase activities, and a kinase activity specific for the CTD of RNAPII (Drapkin and Reinberg 1994; Orphanides et al. 1996). TFIIH functions not only in transcription, but also in nucleotide excision repair (Shaeffer et al. 1993; Drapkin et al. 1994; Sancar 1996). TFIIH exists in two forms, a six subunit core complex that is active in nucleotide excision repair (Svejstrup et al. 1995) and holo-TFIIH that results from the association of core-TFIIH with the kinase complex composed of CDK7, cyclin H, and Mat1 (Drapkin et al. 1996; Reardon et al. 1996). This trimeric complex is known as the cdk-activation kinase (CAK) complex and was initially isolated as an activator of different CDKs (Fisher and Morgan 1994). Holo-TFIIH is necessary for transcription activity (Svejstrup et al. 1995; Drapkin et al. 1996; Reardon et al. 1996; Marinoni et al. 1997; LeRoy et al. 1998), yet the CDK7-kinase activity is dispensable for transcription of some genes in vitro (Akoulitchev et al. 1995; Makela et al. 1995). The regulation of CDK7 (CAK) activity has been the subject of extensive studies (Shuttleworth et al. 1990; Labbe et al. 1994; Tassan et al. 1994; Fisher et al. 1995; Martinez et al. 1997). The activity and substrate specificity of CDK7 is regulated by phosphorylation (Labbe et al. 1994) and/or association with other polypeptides like Mat1 and core TFIIH (Devault et al. 1995; Fisher et al. 1995; Adamczewski et al. 1996; Rossignol et al. 1997; Yankulov and Bentley 1997). Genetic studies demonstrate that the S. pombe CDK7 homolog, crk1/mcs6, is an essential gene with the same substrate specificity as human CDK7. Moreover, Δcrk1 cells undergo arrest in late mitosis (Buck et al. 1995). Crk1 was initially identified as the mitotic catastrophe suppressor, mcs6, and could be functionally complemented by Xenopus CDK7, but not by KIN28 (Cismowski et al. 1995; Valay et al. 1995) the S. cerevisiae homolog of CDK7 (Buck et al. 1995).
Here, we demonstrate that the CTD kinase and transcriptional activities of TFIIH are repressed as the cells enter mitosis. Mitotic repression of TFIIH is mediated through a regulatory phosphorylation of the CDK7 subunit of the CAK complex. This regulation takes place in the T-loop of CDK7 and it is presumed to induce a conformational change as was described for cdk2 (Russo et al. 1996).
Results
Mitotic extracts are transcriptionally impaired
To study mitotic repression of transcription and the possible role played by TFIIH, we used HeLa cells arrested with nocodazole. FACS analysis demonstrated that >95% of these cells arrested in G2/M (Fig. 1b). A mitotic hallmark is the induction of MPF, which is composed of cdc2 and its regulatory subunit cyclin B (Norbury and Nurse 1992). The extracts prepared from mitotic cells displayed ∼10-fold higher H1 kinase activity than extracts from asynchronous interphase cells (Fig. 1c, lanes 1–2). Affinity purification with immobilized p13 (suc1) protein (Fig. 1c, lanes 3–4; Labbe et al. 1991), followed by Western blot analysis (Fig. 1d), confirmed that almost the entire H1 kinase activity of the mitotic extract was attributed to an induction of cdc2 activity.
Figure 1.

FACS analysis of the asynchronous (interphase) (a) and nocodazole-treated (mitotic) (b) HeLa cells: Distribution of cells in the G1, S, and G2/M phases of the cell cycle (Y axis) on the basis of the cellular DNA content (X axis) is plotted on the graph. (c) Histone H1 kinase assay with extracts derived from the mitotic and interphase HeLa cells (lanes 1,2) or with Suc1(p13)-affinity purified cdc2 kinase from mitotic and interphase extracts (lanes 3,4). (d) Western blot analysis of the Suc1(p13)-affinity purified cdc2 kinase (lanes 2,3) and affinity control (lane 1). (e) Western blot analysis of the CTD of RNAPII by use of 8WG16 antibodies (Parsons and Spencer 1997) in the interphase (lane 1) and mitotic extracts (lanes 2,3). (Lane 3) Extracts were pretreated with 10 units of alkaline phosphatase. (f) Reconstituted basal (210 nucleotides) and activated (390 nucleotides) transcription with eTFIID immunopurified from interphase (lanes 1,2) and mitotic (lanes 3,4) LTR HeLa cells (Zhou et al. 1992). Reactions contained the activator Gal4–VP16 and the coactivators PC4 and TFIIA (lanes 2,4; Ma et al. 1996). The templates used were pG5MLP and pMLP that contain G-less cassettes of different sizes (390 and 210 nucleotides). Transcription was directed by the AdML promoter. The 390-nucleotide transcript was derived from pG5MLP, which contains five Gal4-binding sites upstream of the TATA box. pMLP is devoid of Gal4-binding sites.
Following an earlier report by us performed in vitro (Zawel et al. 1993), and reports of others performed in vivo (Shermoen and O’Farrell 1991; Parsons and Spencer 1997), demonstrating that the cdc2 kinase phosphorylates the CTD of RNAPII (Cisek and Corden 1989), we analyzed the state of phosphorylation of the CTD of RNAPII in interphase and mitotic extracts using monoclonal antibodies that recognize the hypophosphorylated form of RNAPII (IIA). Interphase extracts displayed a distinct band of ∼200 kD corresponding to the hypophosphorylated largest subunit of RNAPII, Rpb1 (Fig. 1e, lane 1). This band was not detected in mitotic extracts (Fig. 1e, lane 2). However, treatment of the mitotic extract with phosphatase restored the hypophosphorylated form (lane 3). It has been shown previously that the hyperphosphorylated form of RNAPII is functionally impaired in the formation of the preinitiation complex (Lu et al. 1992; Zawel et al. 1993; Dahmus 1996). Detection of hyperphosphorylation of CTD in mitotic somatic cells in vivo identifies it as one of the potential mechanisms for the mitotic block to transcription (Shermoen and O’Farrell 1991; Parsons and Spencer 1997).
To further characterize the mitotic extract, we analyzed the ability of TFIID to function in transcription activation. In agreement with earlier reports demonstrating that the TAFs are phosphorylated in mitosis resulting in the inhibition of TFIID to function in transcription activation (Segil et al. 1996), we observed that TFIID isolated from nocodazole treated cells by use of an immunopurification procedure (Zhou et al. 1992), was active in a reconstituted assay measuring basal transcription, however, TFIID was impaired in its ability to mediate activation by Gal4–VP16 (Fig. 1f).
The above analyses allow us to conclude that the extracts prepared from nocodazole-arrested cells have all the reported properties of mitotic extracts.
TFIIH activity is impaired in mitosis
We observed that the transcription activity of the mitotic extract was severely compromised compared with that of the interphase extract (Fig. 2a, cf. lanes 1 and 2). The transcription observed was mediated by RNAPII as it was sensitive to low concentrations (2 μg/ml) of α-amanitin (data not shown). Next, we analyzed whether the purified general transcription factors and/or RNAPII could reactivate the mitotically compromised extract. We observed that the addition of purified TFIIH and RNAPII together effectively restored transcriptional activity (Fig. 2a, lanes 7,8). We found that no other combination of GTFs/RNAPII complemented the mitotic extracts (Fig. 2a, lanes 3–6; data not shown). These finding strongly implicated TFIIH and RNAPII in mitotic inhibition of basal transcription.
Figure 2.

(a) Transcriptional activity of mitotic extracts (lane 2) as compared with the interphase extracts (lane 1) and complemented with different GTFs and RNAPII (lanes 3–8). Titration included all GTFs without RNAPII (lanes 3,4 ), GTFs and RNAPII without TFIIH (lanes 5,6), TFIIH and RNAPII (lanes 7,8). (Arrow) The specific transcript derived from the pMLP template. (b) Silver-staining of immunoaffinity purified TFIIH (LeRoy et al. 1998) (lane 2). (Lane 1) Molecular weight markers. (c) Reconstituted basal transcription (lane 1), dependent on TFIIH (lane 2) and TBP (lane 5). Reactions were reconstituted with immunoaffinity purified TFIIH or TFIID from interphase (lanes 3,6) or mitotic (lanes 4,7) extracts. (d) Western blot of the ERCC3 and p62 subunits of immunopurified TFIIH and of eTBP (Zhou et al. 1992), used in the reconstituted transcription in c.
Next, to directly investigate whether TFIIH was inactivated in the mitotic extracts, we isolated TFIIH from the mitotic extracts and assayed it in a reconstituted transcription system. TFIIH was isolated by an immuno-affinity purification method by use of monoclonal antibodies against ERCC3 (LeRoy et al. 1998). A silver staining of a polyacrylamide gel containing a representative TFIIH used in the experiments described below is shown in Figure 2b (see Fig. 2b; Materials and Methods). TFIIH isolated from interphase extracts was transcriptionally active (Fig. 2c, lane 3), whereas TFIIH isolated from the mitotic extract was severely compromised in its ability to reconstitute transcription (Fig. 2c, lane 4). This was not the result of the amounts of purified TFIIH added to the assay as demonstrated by Western blots (Fig 2d). Moreover, as an additional control, we isolated TBP from the same extracts using an immunopurification assay (Zhou et al. 1992), and found it to be active in its ability to reconstitute basal transcription (Fig. 2c,d). These results collectively demonstrate that TFIIH is impaired in its ability to function in transcription in mitosis. These results are in agreement with previous studies performed in vitro that demonstrated that the addition of MPF to interphase extracts resulted in the inactivation of TFIIH (Long et al. 1998).
The CAK complex is inactivated in mitotic extracts
Studies performed initially in yeast (Svejstrup et al. 1995), and recently extended to the mammalian factor (Drapkin et al. 1996; Reardon et al. 1996), demonstrate that TFIIH exists in at least two subcomplexes, a complex containing the core subunits of TFIIH (ERCC3, ERCC2, p62, p52, p44, and p34) and devoid of the kinase complex, referred to as core-TFIIH, and a complex associated with the kinase complex (CDK7, cyclin H, and MAT1), referred to as holo-TFIIH (Fig. 3a). Core TFIIH appears to be the form involved in nucleotide excision repair (Svejstrup et al. 1995), whereas holo-TFIIH functions in transcription. Previously, we (Drapkin et al. 1996) and others (Reardon et al. 1996; Marinoni et al. 1997) showed that the CAK complex could be dissociated from holo-TFIIH with high salt. The reassociation of CAK with core-TFIIH restores transcriptionally active TFIIH (holo-TFIIH) and requires the formation of the intermediary CAK/ERCC2 complex, which is present in HeLa cell extracts (Drapkin et al. 1996; Reardon et al. 1996; Marinoni et al. 1997). We exploited these features of TFIIH and analyzed whether the transcriptionally compromised mitotic TFIIH could be reactivated by the addition of excess ERCC2–CAK isolated from interphase extracts (Fig. 3c, lane 2). In agreement with the previous results, basal transcription reconstituted with mitotic TFIIH showed reduced transcriptional activity (Fig. 3b, lane 2). On the other hand, when the interphase CAK/ERCC2 complex was added in lieu of TFIIH, no transcription activity was observed (lane 1), although it was active as a CTD kinase (data not shown). Addition of excess purified interphase CAK/ERCC2 to reactions containing mitotic TFIIH led to restoration of basal transcription (Fig. 3b, lanes 3, 4). These results collectively establish that the CAK–ERCC2 complex is inactivated on entry into mitosis.
Figure 3.

(a) Schematic representation of the composition of core TFIIH, the ERCC2–CAK complex, and holo-TFIIH. (b) Basal transcription by the AdML promoter was reconstituted with affinity purified mitotic TFIIH (lanes 2–4), CAK/ERCC2 affinity purified from interphase extract (lane 1), or with increasing amounts of the CAK/ERCC2 complex in the presence of mitotic TFIIH (lanes 3,4). (Arrow) The specific transcript derived from the pMLP template. (c) Western blot analysis of the immunoaffinity preparation of mitotic TFIIH and interphase CAK/ERCC2 complexes.
The CDK7 subunit is hyperphosphorylated in mitosis
Phosphorylation is an important regulatory modification observed both in mitotic regulation and in regulation of cyclin-dependent kinases. Earlier reports established that CDK7 is phosphorylated in vivo at two major sites, Ser-164 and Thr-170 (Labbe et al. 1994; Fisher et al. 1995). Phosphorylation on Thr-170 is essential for CDK7 kinase activity in vivo (Labbe et al. 1994), whereas phosphorylation on Ser-164 is dispensible and even detrimental in stage VI oocytes (Labbe et al. 1994). The Ser-164 site matches the consensus sequence for cdks/MAP kinases (see Fig. 6a, below). Therefore, we analyzed whether the CDK7 subunit of TFIIH was specifically phosphorylated in mitosis. Interphase and nocodazole-arrested HeLa cells were labeled with [32P]orthophosphate in vivo. TFIIH was isolated by immunoprecipitation by use of the ERCC3 monoclonal antibodies described above (Fig. 2b). The isolated TFIIH complex was then treated with SDS to disrupt the interaction among the different subunits and the CDK7 subunit was immunoprecipitated with antibodies specific to CDK7 (Fig. 4a; Materials and Methods). The immunoprecipitates were separated by polyacrylamide gel electrophoresis. The autoradiograph displayed a single phospholabeled polypeptide with increased radioactivity in the mitotic-derived CDK7 sample (Fig. 4a). Western blot analysis revealed that the increased radioactivity observed in mitotic-derived CDK7 was not due to differences in the amount of CDK7 loaded on the gels (Fig. 4a, bottom). More importantly, a shift in the mobility of the mitotic-derived CDK7 was observed (Fig. 4a, bottom).
Figure 6.

(a) Amino acid sequence of a fragment of CDK7 spanning the sites of phosphorylation in vivo (Ser-164 and Thr-170). (Arrowheads) Positions of cleavage by trypsin. (b) Purified recombinant wild-type and mutant [S164D, T170D] CDK7 containing a carboxy-terminal-His-tag as detected by Western blot using antibodies against CDK7 (α-CDK7) and by Coomassie-blue staining (Ni2+). (c) Recombinant wild-type and mutant CDK7 were incubated with interphase or mitotic extracts in the presence of [γ-32]ATP. Labeled proteins were affinity purified, digested with trypsin, and resolved by one-dimension thin layer chromatography. Positions of Ser-164 and Thr-170 containing fragments are indicated.
Figure 4.
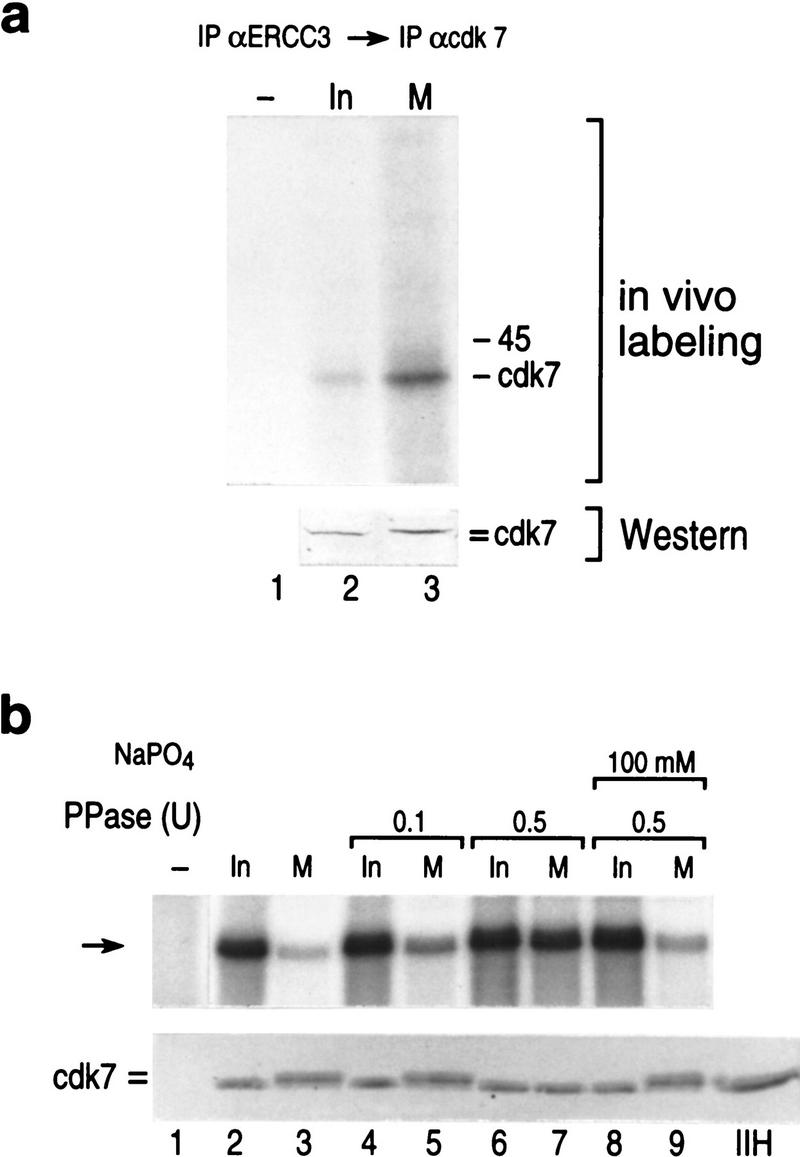
(a) In vivo-labeled CDK7 immunoprecipitated from affinity purified TFIIH and analyzed by Western blot (bottom) after autoradiography (top). (=) The resolved shift in the mobility of mitotic CDK7. (b). (Top) Basal transcription from the AdML promoter was reconstituted with affinity purified interphase TFIIH (lane 2), mitotic TFIIH (lane 3), mitotic or interphase TFIIH pretreated with alkaline phosphatase in the absence or presence of sodium phosphate as indicated. (Lane 1) The result using a mock affinity purification procedure. (Bottom) Western blot analysis of the samples analyzed in transcription at top using anti-CDK7 antibodies.
The above result prompted us to analyze whether transcriptional activity of mitotic TFIIH could be restored by phosphatase treatment. This approach was possible because in CDK7, the phosphate on Thr-170, which is essential for enzymatic activity (Labbe et al. 1994), is protected from nonspecific dephosphorylation in the CAK complex (Labbe et al. 1994). TFIIH immunopurified from the interphase and mitotic extracts was treated with phosphatase and then used in the reconstituted TFIIH-dependent transcription assay (Fig. 4b; see Materials and Methods). We observed that treatment of mitotic TFIIH with phosphatase restored transcriptional activity (Fig. 4b). Importantly, reactivation of TFIIH activity was dependent on the amount of phosphatase added and was blocked by sodium phosphate, a phosphatase inhibitor (Fig. 4b). Western blot analysis of the samples analyzed in transcription demonstrate a shift in the mobility of CDK7 associated with mitotic inactive TFIIH, suggestive that phosphorylation of CDK7 is associated with inactivation of TFIIH activity (Fig. 4b, bottom).
Phosphorylation of CDK7 impairs CTD-kinase and transcription activities
The CTD is one of the substrates of CDK7, and we found that TFIIH isolated from mitotic cell extracts displayed reduced CTD kinase activity (Fig. 5a, lanes 3,4). The reduced CTD-kinase activity was not due to the amount of factor analyzed as detected by Western blot analysis (Fig. 5b, lanes 4,5). We expanded this observation and asked whether inhibition of the TFIIH kinase activity could be reconstituted in vitro. Interphase TFIIH was attached to beads through monoclonal antibodies against ERCC3 and the beads were then incubated with mitotic- or interphase-derived extracts in the presence of ATP as described in Figure 5a. TFIIH was then recovered from the extracts and analyzed for its ability to phosphorylate the CTD. As shown in Figure 5a, mitotic extract specifically inactivated the TFIIH kinase activity, the interphase-derived extract was without effect (Fig. 5a). The inhibition observed was dependent on ATP (data not shown, see below).
Figure 5.

Mitotic TFIIH is deficient in CTD kinase activity. (a) CTD kinase activity of TFIIH directly purified from mitotic and interphase cells (in vivo, lanes 3,4). The kinase activity of conventionally purified TFIIH is shown in lane 1. Purified TFIIH was pretreated with mitotic or interphase extracts in the presence of ATP (in vitro, lanes 6–7, see diagram at right). (b) Western blot analysis of TFIIH subunits (p62 and CDK7) from samples analyzed in a. This blot shows that similar amounts of TFIIH were used in the assay performed in a. The electrophoretic mobility of CDK7 caused by phosphorylation in mitosis is not resolved in this analysis. The samples were separated on a Bio-Rad mini-gel.
Having established that CDK7 is hyperphosphorylated in mitosis and that this inhibitory effect can be reconstituted in vitro by incubating interphase TFIIH with mitotic extracts, we attempted to analyze the sites phosphorylated in CDK7 by the mitotic extract by incubating bacterially produced recombinant His-tagged CDK7 with interphase and mitotic extracts. Equal amounts (see Fig. 6b) of purified wild-type CDK7 or a mutant form of CDK7, in which the two major phosphorylation sites, Ser-164 and Thr-170, were substituted to alanine were incubated in the extracts in the presence of [γ-32P]ATP. Extensive digestion of the in vitro-labeled CDK7 polypeptides, followed by thin-layer chromatography identified two phosphopeptides (Fig. 6c; data not shown). The substitutions in CDK7 abrogated phosphorylation (Fig. 6c; data not shown). This result confirmed earlier reports that Ser-164 and Thr-170 are the two major sites of phosphorylation with particularly strong phosphorylation of Ser-164 in vivo (Labbe et al. 1994). Comparison of the levels of phosphorylation at these sites revealed ∼2-fold decrease for Thr-170, and 2.5- to 3-fold increase for Ser-164 in mitotic extracts.
To analyze more directly whether the phosphorylation of CDK7 is implicated in the regulation of TFIIH, we studied several mutants of CDK7 in vivo. Wild-type CDK7 and individual point mutations with Ser-164 (S) or Thr-170 (T) substituted to alanine were cloned into a mammalian expression vector with an in-frame triple c-Myc tag at the carboxyl terminus (Makela et al. 1997) and were transiently expressed in 293T cells. Following transfection, the cells were treated with nocodazole. c-Myc-TAG affinity-purified complexes were further selected for TFIIH by affinity purification using ERCC3 monoclonal antibodies (Fig. 7a). Interphase and mitotic TFIIH were isolated and assayed in transcription and kinase activities (Fig. 7b). In agreement with results presented above, mitotic-derived TFIIH was compromised in its ability to reconstitute transcription and in CTD phosphorylation (Fig. 7b, lanes 1,2). Analysis with mutant TFIIH reveals that both the transcriptional and kinase activities are dependent on phosphorylation of Thr-170 (lane 4). Importantly, however, the mutation in Ser-164 relieved mitotic repression of TFIIH (lane 3). This experiment demonstrates that phosphorylation on Ser-164 is detrimental for TFIIH activity. The studies collectively establish that in mitosis, Ser-164 is phosphorylated and impairs the transcription activity of TFIIH.
Figure 7.

(a) Schematic representation of the procedure used to isolate TFIIH from transfected cells containing mutated subunits of CDK7. TFIIH containing c-Myc-tagged CDK7 was isolated from interphase and mitotic cells. After cell lysis, the transfected CDK7 subunit was recovered by immunoprecipitation by use of antibodies recognizing the c-myc tag. The IPs were eluted and the transfected myc-tagged CDK7 subunits were selected for those subunits that were incorporated into TFIIH by performing a second immunoprecipitation with anti-ERCC3 monoclonal antibodies. The IPs were functionally analyzed in transcription and CTD phosphorylation. (b) TFIIH isolated from mitotic cells and containing a mutation in Ser-164 [Ser164A (S)] or a mutation in Thr-170 [Thr170A (T)] or a control vector were assayed for transcription and kinase activities. The amount of TFIIH recovered and used in the functional analysis was analyzed by Western blot by use of antibodies against the p62, cyclin H, and CDK7-[3xMyc] subunits (top). The samples were separated on a Bio-Rad minigel; therefore, the change in electrophoretic mobility due to phosphorylation of CDK7-[3xMyc] could not be resolved. Functionally, the mutant proteins were assayed in basal transcription (middle), and for CTD-kinase (bottom) activities. Minus (−) above the lane denotes the activity present in cells that were transfected with the Myc tag but devoid of the CDK7 coding sequences (empty vector).
Discussion
Cells arrested in the mitotic stage of the cell cycle display high levels of cdc2 kinase activity and a high level of protein phosphorylation. Under these conditions, basal transcription by RNAPII is repressed because of the modifications of two factors, RNAPII and TFIIH. Detection of the hyperphosphorylated form of RNAPII in mitotic extracts correlates with earlier observations in vitro (Cisek and Corden 1989) and studies in Xenopus oocytes (Bellier et al. 1997), Drosophila melanogaster (Shermoen and O’Farrell 1991) and HeLa cells (Parsons and Spencer 1997). It is currently unknown, however, whether inactivation of RNAPII activity in mitosis is due to the direct phosphorylation of the CTD by MPF, as is the case in vitro (Cisek and Corden 1989; Zawel et al. 1993), or whether it involves a downstream cascade of kinases. In metaphase II-arrested Xenopus oocytes, it was shown that inactivation of RNAPII results from the activation of the Xp42 MAP kinase (Bellier et al. 1997).
The second factor susceptible to mitotic block is TFIIH. Studies performed with the CDK7 homologs demonstrated its role in negative regulation during meiotic maturation in Xenopus (Shuttleworth et al. 1990), as well as a mitotic function in Schizosaccharomyces pombe (Buck et al. 1995) and in D. melanogaster (Larochelle et al. 1997). Earlier biochemical analysis of CDK7 regulation did not detect significant changes in its activity during the cell cycle in the context of the CAK complex (Tassan et al. 1994; Adamczewski et al. 1996). However, previous studies have demonstrated differences in substrate specificity between free CAK and CAK associated with core TFIIH (holoTFIIH) (Rossignol et al. 1997; Yankulov et al. 1997). Our studies uncovered that phosphorylation of CDK7 plays a critical regulatory role within the context of TFIIH. Similar conclusions were reached recently by Long et al. (1998), that demonstrated that the transcription and kinase activities of TFIIH are negatively regulated in in vitro reconstituted mitotic extracts. Their findings and our conclusions demonstrating that regulation of TFIIH dependent on the association of the CAK complex with core TFIIH are in agreement, yet in the studies of Long et al. (1998), the molecular mechanism of inhibition, through CDK7, was not analyzed.
Extensive studies of CDK7 regulation reveal pathways either common for other members of the CDK family or unique for CDK7. For example, as is the case with other CDKs, the kinase activity of CDK7 requires its association with a cyclin partner, cyclin H (Labbe et al. 1994; Martinez et al. 1997). Moreover, full activity in vivo requires phosphorylation of a specific residue within the T-loop (Thr-170; Labbe et al. 1994). On the other hand, in vitro, the association of the Mat1 subunit of CAK with the CDK7/cyclin H complex can confer activity to CDK7, not requiring prior phosphorylation (Fisher et al. 1995; Martinez et al. 1997). Ser-164 within the T-loop of CDK7 corresponds to the cdk/MAP kinase phosphorylation consensus site and is responsible for specific phosphorylation of CDK7 in mitosis and for negative regulation of TFIIH activity. The presence of a cdk/MAP kinase consensus sequence within the T-loop is unique to CDK7. As a phenomenon, inhibitory phosphorylation within the T-loop of a kinase has been described (Luo and Lodish 1997). Collectively, these results demonstrate that the transcriptional and kinase activities of TFIIH depend on the phosphorylation state of its CDK7 subunit in vivo.
Previous studies demonstrated that the CDK7-kinase activity is dispensable for transcription of one class of promoters (represented by the TATA-containing Ad-MLP) (Akoulitchev et al. 1995; Makela et al. 1995), but is required for transcription from other classes of promoters (represented by the TATA-less DHFR promoter) (Akoulitchev et al. 1995). Interestingly, in the case of the DHFR promoter, it was found that the requirement for the CTD (the substrate for the kinase) was during complex assembly and/or first bond formation, and preceded the requirement for the CDK7 kinase activity that was found to be at a later step. Regardless of the requirement for the CDK7-kinase activity, the CDK7 polypeptide, together with cyclin H and Mat1, are necessary for the transcriptional activity of TFIIH (Drapkin et al. 1996; Reardon et al. 1996; Marinoni et al. 1997). Although the inhibition of the CDK7-kinase activity, by mutations in the ATP-binding domain in the catalytic cleft, is not sufficient to compromise the transcriptional activity of TFIIH on the AdML promoter (Akoulitchev et al. 1995; Makela et al. 1995), we would like to suggest that negative regulation of the TFIIH transcription activity in mitosis is mediated via a conformational change of CDK7. The changes are mediated by phosphorylation of residues within the T-loop (Russo et al. 1997). It remains unclear as to which of the kinase(s) acts directly upstream from CDK7. Also, the relationship between the Thr-170 and Ser-164 phosphorylation pathways needs to be elucidated.
In conclusion, our studies demonstrate that mitotic repression of basal transcription results from the phosphorylation of RNAPII and TFIIH. Others have demonstrated previously that phosphorylation of the TAF subunits of TFIID in mitosis impairs activated transcription (Segil et al. 1996). These studies collectively demonstrate that the cell has developed mechanisms to silence transcription during mitosis, by affecting different steps of the transcryption cycle, that is, initiation via the TAF subunits of TFIID and CTD phosphorylation, promoter escape via the CDK7 subunit of TFIIH, and perhaps elongation, by extensive phosphorylation of the CTD by MPF or MPF-activated kinase.
Materials and methods
Cell culture and in vivo labeling
Long terminal repeat HeLa cells containing epitope-tagged TATA-binding protein (TBP; Zhou et al. 1992) were grown in suspension in 10% DMEM. Mitotic arrest was obtained by applying 400 ng/ml nocodazole to 30 liters of growing cell culture (2 × 1010 cells) 24 hr prior harvesting. [32P]Orthophosphate (1 mCi) was applied to actively growing cells 16 hr before harvesting.
Extract preparation
Whole-cell extract was prepared as described (Manley et al. 1980) from 2 × 1010 cells with addition of 20 mm EGTA, 80 mm β-glycerophosphate (pH 7.3), 10 mm sodium fluoride, 10 nm ocadaic acid, 100 nm calyculin A, and 1 mm ATP. Ocadaic acid and calyculin A were maintained in the extracts through purification.
Immunoprecipitation, immunoaffinity purification, and functional assays
Immunoaffinity purification of TFIIH with ERCC3 monoclonal antibodies was performed as described (LeRoy et al. 1998). In short, proteins from the second step of purification of TFIIH, the DEAE-cellulose bound fraction, were incubated with protein A-immobilized anti-ERCC3 monoclonal antibodies. The beads were washed extensively with BC400 (400 mm KCl, 10 mm Tris-HCl at pH 7.9, 0.1 mm EDTA, 20% glycerol) and TFIIH was eluted with a peptide. Immunoaffinity purification of CAK/ERCC2 was as follows: The same protein fraction used above was incubated with protein A-immobilized cyclin H monoclonal antibodies. The beads were washed extensively in BC900 (900 mm KCl; the rest as in BC400) to dissociate core TFIIH. The CAK complex was eluted with 1.0 m ammonium sulfate, 50% ethylene glycol, 10 mm Tris-HCl (pH 7.9). Double-immunoaffinity purification of in vivo labeled CDK7 was performed as follows: After immunoprecipitation of TFIIH with anti-ERCC3 antibodies the samples were heated at 100°C for 5 min in BC100, 1% SDS, diluted 10-fold in RIPA buffer (0.15 m NaCl, 50 mm Tris-HCl at pH 7.5, 0.5% NP-40, 0.1% SDS) and used in immunoprecipitation with anti-CDK7 antibodies immobilized on protein A beads. No other subunits of TFIIH coimmunoprecipitated with CDK7 in this procedure. Immunoprecipitates of TFIIH and epitope-tagged TBP (eTBP, eTFIID) used in transcription/kinase reactions were carried out as follows: 50 μl of protein A beads were saturated with the corresponding antibodies for 2 hr at 4°C, washed extensively with PBS, and incubated with 500 μl of the extracts for 2 hr at 4°C; washed four times with 20 mm Tris-HCl (pH 7.9), 150 mm NaCl, 2 mm EDTA, 10 mm β-glycerolphosphate, 5 mm sodium fluoride, 10 nm ocadaic acid, and 100 nm calyculin A; and washed two times in transcription or kinase buffer containing 10 nm ocadaic acid. Protein A beads (10 μl) were added to transcription/kinase reactions. Reconstituted transcription reactions with the AdMLP were performed as described (Akoulitchev et al. 1995). Reactions performed with the extracts were as described, but the interphase and mitotic extracts were pretreated with 10 units of hexokinase (Boehringer) to remove residual ATP. Reactions were performed under single-round transcription conditions by using a pulse-chase protocol: preinitiation complexes were formed for 1 hr, followed by the addition of ATP and UTP (500 mm), and [α-32P]CTP (1 μm) for 5 min; followed by the addition of 500 mm CTP for a 15 min chase.
Phosphatase treatment
Immunopurified TFIIH from 500 μl of interphase or mitotic extracts was incubated with 0.1–0.5 units of alkaline phosphatase for 15 min at 25°C in the absence or the presence of 100 mm sodium phosphate. Phosphatase treatment was performed on TFIIH attached to the beads. This allowed the removal of the phosphatase and/or phosphate prior to the addition of TFIIH to the assays.
Western blot
Western blots with antibodies against the CTD of RNAPII (8WG16), the HA tag of eTBP (12CA5), ERCC3, ERCC2, p62, cyclin H, or CDK7 were carried out with PVDF membranes (Bio-Rad) following the manufacturer’s recommendations.
Kinase assay
Kinase reactions were performed in a 40 μl volume containing 80 mm-glycerolphosphate (pH 7.5), 20 mm EGTA, 15 mm MgCl2, 1 mm DTT, 1 mm ATP, 10 μCi [γ-32P]ATP, and 1 mg/ml of CTD peptide or histone H1. Reactions were incubated for 15 min at 30°C. An aliquot (5 μl) was analyzed by electrophoresis on 15% polyacrylamide–SDS gels.
Acknowledgments
We thank Gary LeRoy for assistance in the affinity purification of TFIIH. We also thank the members of the Reinberg laboratory for helpful suggestions and to Drs. Mike Hampsey, Jim Manley, Ron Morris, and George Orphanides for comments on the manuscript. This work was supported by grants from the National Institute of Health (GM-37120 and GM-48518) and from the Howard Hughes Medical Institute to D.R.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL reinbed@umdnj.edu; FAX (732) 235-5294.
References
- Adamczewski JP, Rossignol M, Tassan J-P, Nigg EA, Moncollin V, Egly JM. MAT1, cdk7 and cyclin H form a kinase complex which is UV light-sensitive upon association with TFIIH. EMBO J. 1996;15:1877–1844. [PMC free article] [PubMed] [Google Scholar]
- Akoulitchev S, Mäkelä TP, Weinberg RA, Reinberg D. Requirement for TFIIH kinase activity in transcription by RNA polymerase II. Nature. 1995;377:557–560. doi: 10.1038/377557a0. [DOI] [PubMed] [Google Scholar]
- Bellier S, Dubois MF, Nishida E, Almouzni G, Bensaude O. Phosphorylation of the RNA polymerase II largest subunit during Xenopus laevis oocyte maturation. Mol Cell Biol. 1997;17:1434–1440. doi: 10.1128/mcb.17.3.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck V, Russel P, Millar JBA. Identification of a cdk-activating kinase in fission yeast. EMBO J. 1995;14:6173–6183. doi: 10.1002/j.1460-2075.1995.tb00308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisek LJ, Corden JL. Phosphorylation of RNA polymerase by the murine homologue of the cell-cycle control protein cdc2. Nature. 1989;339:679–684. doi: 10.1038/339679a0. [DOI] [PubMed] [Google Scholar]
- Cismowski MJ, Laff GM, Solomon MJ, Reed S. KIN28 Encodes a C-terminal domain kinase that controls mRNA transcription in Saccharomyces cerevisiae but lacks cyclin-dependent kinase-activating kinase (CAK) activity. Mol Cell Biol. 1995;15:2983–2992. doi: 10.1128/mcb.15.6.2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman TR, Dunphy WG. Cdc2 regulatory factors. Curr Opin Cell Biol. 1994;6:877–882. doi: 10.1016/0955-0674(94)90060-4. [DOI] [PubMed] [Google Scholar]
- Conaway JW, Reines D, Conaway RC. Transcription initiated by RNA polymerase II and purified transcription factors from liver. Cooperative action of transcription factors tau and epsilon in initial complex formation. J Biol Chem. 1990;265:7552–7558. [PMC free article] [PubMed] [Google Scholar]
- Dahmus ME. Reversible phosphorylation of the C-terminal domain of RNA polymerase II. J Biol Chem. 1996;271:19009–19012. doi: 10.1074/jbc.271.32.19009. [DOI] [PubMed] [Google Scholar]
- Devault A, Martinez A-M, Fesquet D, Labbe J-C, Morin N, Tassan J-P, Nigg EA, Cavadore J-C, Doree M. MAT1 a new RING finger protein subunit stabilizing cyclin H-cdk7 complexes in starfish and Xenopus CAK. EMBO J. 1995;14:5027–5036. doi: 10.1002/j.1460-2075.1995.tb00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drapkin R, Reinberg D. The multifunctional TFIIH complex and transcriptional control. Trends Biochem Sci. 1994;19:504–508. doi: 10.1016/0968-0004(94)90139-2. [DOI] [PubMed] [Google Scholar]
- Drapkin R, Reardon JT, Ansari A, Huang J-C, Zawel L, Ahn K, Sancar A, Reinberg D. Dual role of TFIIH in DNA excision repair and in transcription by RNA polymerase II. Nature. 1994;368:769–772. doi: 10.1038/368769a0. [DOI] [PubMed] [Google Scholar]
- Drapkin R, Le Roy G, Cho H, Akoulitchev S, Reinberg D. Human cyclin-dependent kinase-activating kinase exists in three distinct complexes. Proc Natl Acad Sci. 1996;93:6488–6493. doi: 10.1073/pnas.93.13.6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elledge SJ, Harper JW. Cdk inhibitors: On the threshold of checkpoints and development. Curr Opin Cell Biol. 1994;6:847–852. doi: 10.1016/0955-0674(94)90055-8. [DOI] [PubMed] [Google Scholar]
- Fink K, Turnock G. Synthesis of transfer RNA during the synchronous nuclear division cycle in Physarum polycephalum. Eur J Biochem. 1977;80:93–96. doi: 10.1111/j.1432-1033.1977.tb11860.x. [DOI] [PubMed] [Google Scholar]
- Fisher RP, Morgan DO. A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell. 1994;78:713–724. doi: 10.1016/0092-8674(94)90535-5. [DOI] [PubMed] [Google Scholar]
- Fisher RP, Jin P, Chamberlin HM, Morgan DO. Alternative mechanisms of CAK assembly require an assembly factor or an activating kinase. Cell. 1995;83:47–57. doi: 10.1016/0092-8674(95)90233-3. [DOI] [PubMed] [Google Scholar]
- Gautier J, Minshull J, Lohka M, Glotzer M, Hunt T, Maller JL. Cyclin is a component of maturation-promoting factor from Xenopus. Cell. 1990;60:487–494. doi: 10.1016/0092-8674(90)90599-a. [DOI] [PubMed] [Google Scholar]
- Goodrich JA, Tjian R. Transcription factors IIE and IIH and ATP hydrolysis direct promoter clearance by RNA polymerase II. Cell. 1994;77:145–156. doi: 10.1016/0092-8674(94)90242-9. [DOI] [PubMed] [Google Scholar]
- Gottesfeld JM, Forbes DJ. Mitotic repression of the transcriptional machinery. Trends Biochem Sci. 1997;22:197–202. doi: 10.1016/s0968-0004(97)01045-1. [DOI] [PubMed] [Google Scholar]
- Gottesfeld JM, Wolf VJ, Dang T, Forbes DJ, Hartl P. Mitotic repression of RNA polymerase III transcription in vitro mediated by phosphorylation of a TFIIIB component. Science. 1994;263:81–84. doi: 10.1126/science.8272869. [DOI] [PubMed] [Google Scholar]
- Johnson TC, Holland JJ. Ribonucleic acid and protein synthesis in mitotic HeLa cells. J Cell Biol. 1965;27:565–574. doi: 10.1083/jcb.27.3.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LH, White JH, Johnson AL, Lucchini G, Plevani P. The yeast DNA polymerase I transcript is regulated in both the mitotic cell cycle and in meiosis and is also induced after DNA damage. Nucleic Acids Res. 1987;15:5017–5030. doi: 10.1093/nar/15.13.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita N, Ohkura H, Yanagida M. Distinct, essential roles of type 1 and 2A protein phosphatases in the control of the fission yeast cell division cycle. Cell. 1990;63:405–415. doi: 10.1016/0092-8674(90)90173-c. [DOI] [PubMed] [Google Scholar]
- King RW, Jackson PK, Kirschner MW. Mitosis in transition. Cell. 1994;79:563–571. doi: 10.1016/0092-8674(94)90542-8. [DOI] [PubMed] [Google Scholar]
- Kumar KP, Akoulitchev S, Reinberg D. Promoter-proximal stalling results from the inability to recruit transcription factor IIH to the transcription complex and is a regulated event. Proc Natl Acad Sci. 1998;95:9767–9772. doi: 10.1073/pnas.95.17.9767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbe JC, Capony JP, Caput D, Cavadore JC, Derancourt J, Kaghad M, Lelias JM, Picard A, Doree M. MPF from starfish oocytes at first meiotic metaphase is a heterodimer containing one molecule of cdc2 and one molecule of cyclin B. EMBO J. 1989;8:3053–3058. doi: 10.1002/j.1460-2075.1989.tb08456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbe JC, Cavadore JC, Doree M. M phase-specific cdc2 kinase: preparation from starfish oocites and properties. Methods Enzymol. 1991;200:291–301. doi: 10.1016/0076-6879(91)00147-o. [DOI] [PubMed] [Google Scholar]
- Labbe J-C, Martinez A-M, Fesquet D, Capony J-P, Darbon J-M, Derancourt J, Devault A, Morin N, Cavadore J-C, Doree M. p40MO15 associates with a p36 subunit and requires both nuclear translocation and Thr 176 phosphorylation to generate cdk-activating kinase activity in Xenopus oocytes. EMBO J. 1994;13:5155–5164. doi: 10.1002/j.1460-2075.1994.tb06845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle S, Pandur J, Fisher RP, Salz HK, Suter B. Cdk7 is essential for mitosis and for in vivo Cdk-activating kinase activity. Genes & Dev. 1998;12:370–381. doi: 10.1101/gad.12.3.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoy G, Drapkin R, Weis L, Reinberg D. Immunoaffinity purification of the human multisubunit transcription factor IIH. J Biol Chem. 1998;273:7134–7140. doi: 10.1074/jbc.273.12.7134. [DOI] [PubMed] [Google Scholar]
- Long JJ, Leresche A, Kriwaski RW, Gottesfeld JM. Repression of TFIIH Transcriptional activity and TFIIH-associated cdk7 kinase activity at mitosis. Mol Cell Biol. 1998;18:1467–1476. doi: 10.1128/mcb.18.3.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Flores O, Weinmann R, Reinberg D. The nonphosphorylated form of RNA polymerase II preferentially associates with the preinitiation complex. Proc Natl Acad Sci. 1991;88:10004–10008. doi: 10.1073/pnas.88.22.10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Zawel L, Fisher L, Egly J-M, Reinberg D. Human general transcription factor IIH phosphorylates the C-terminal domain of RNA polymerase II. Nature. 1992;358:641–645. doi: 10.1038/358641a0. [DOI] [PubMed] [Google Scholar]
- Luo K, Lodish HF. Positive and negative regulation of type II TGF-beta receptor signal transduction by autophosphorylation on multiple serine residues. EMBO J. 1997;16:1970–1981. doi: 10.1093/emboj/16.8.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Olave I, Merino A, Reinberg D. Separation of the transcriptional coactivator and antirepression functions of transcription factor IIA. Proc Natl Acad Sci. 1996;93:6583–6588. doi: 10.1073/pnas.93.13.6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makela T, Parvin JD, Kim J, Huber LJ, Sharp PA, Weinberg RA. A kinase-deficient transcription factor TFIIH is functional in basal and activated transcription. Proc Natl Acad Sci. 1995;92:5174–5178. doi: 10.1073/pnas.92.11.5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley J, Fire A, Cano A, Sharp P, Gefter M. DNA-dependent transcription of adenovirus genes in a soluble whole-cell extract. Proc Natl Acad Sci. 1980;77:3855–3859. doi: 10.1073/pnas.77.7.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinoni J-C, Vermeulen W, Miniou P, Lutz Y, Weeda G, Seroz T, Gomez DM, Hoejimakers JHJ, Egly JM. Cloning and characterization of p52, the fifth subunit of the core of the transcription/DNA repair factor TFIIH. EMBO J. 1997;16:1093–1102. doi: 10.1093/emboj/16.5.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A-M, Afshar M, Martin F, Cavadore J-C, Labbe J-C, Doree M. Dual phosphorylation of the T-loop in cdk7: Its role in controlling cyclin H binding and CAK activity. EMBO J. 1997;16:343–354. doi: 10.1093/emboj/16.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA. Targets of cyclin-dependent protein kinases. Curr Opin Cell Biol. 1993;5:187–193. doi: 10.1016/0955-0674(93)90101-u. [DOI] [PubMed] [Google Scholar]
- Norbury C, Nurse P. Animal cell cycles and their control. Annu Rev Biochem. 1992;61:441–470. doi: 10.1146/annurev.bi.61.070192.002301. [DOI] [PubMed] [Google Scholar]
- O’Brien T, Lis JT. RNA polymerase II pauses at the 5′ end of the transcriptionally induced Drosophila hsp70 gene. Mol Cell Biol. 1991;11:5285–5290. doi: 10.1128/mcb.11.10.5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphanides G, Lagrange T, Reinberg D. The general transcription factors of RNA polymerase II. Genes & Dev. 1996;10:2657–2683. doi: 10.1101/gad.10.21.2657. [DOI] [PubMed] [Google Scholar]
- Parsons GG, Spencer CA. Mitotic repression of RNA polymerase II transcription is accompanied by release of transcription elongation complexes. Mol Cell Biol. 1997;17:5791–5802. doi: 10.1128/mcb.17.10.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon JT, Ge H, Gibbs E, Sancar A, Hurwitz J, Pan ZQ. Isolation and characterization of two human transcription factor IIH (TFIIH)-related complexes: ERCC2/CAK and TFIIH. Proc Natl Acad Sci. 1996;93:6482–6487. doi: 10.1073/pnas.93.13.6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SI. The role of p34 kinases in the G1 to S-phase transition. Annu Rev Cell Biol. 1992;8:529–561. doi: 10.1146/annurev.cb.08.110192.002525. [DOI] [PubMed] [Google Scholar]
- Roeder RG. The role of general transcription factors in transcription by RNA polymerase II. Trends Biochem Sci. 1991;16:402–408. [PubMed] [Google Scholar]
- Rossignol M, Kolb-Cheynel I, Egly JM. Substrate specificity of the cdk-activating kinase (CAK) is altered upon association with TFIIH. EMBO J. 1997;16:1628–1637. doi: 10.1093/emboj/16.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo A, Jeffrey PD, Pavletich NP. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat Struct Biol. 1996;3:696–700. doi: 10.1038/nsb0896-696. [DOI] [PubMed] [Google Scholar]
- Sancar A. DNA excision repair. Annu Rev Biochem. 1996;65:43–81. doi: 10.1146/annurev.bi.65.070196.000355. [DOI] [PubMed] [Google Scholar]
- Segil N, Guermah M, Hoffmann A, Roeder RG, Heintz N. Mitotic regulation of TFIID inhibition of activator-dependent transcription and changes in subcellular localization. Genes & Dev. 1996;10:2389–2400. doi: 10.1101/gad.10.19.2389. [DOI] [PubMed] [Google Scholar]
- Shaeffer L, Roy R, Humbert S, Moncollin V, Vermeulen W, Hoeijmakers JH, Chambon P, Egly JM. DNA repair helicase: A component of BTF2 (TFIIH) basic transcription factor. Science. 1993;260:58–63. doi: 10.1126/science.8465201. [DOI] [PubMed] [Google Scholar]
- Shermoen AW, O’Farrell PH. Progression of the cell cycle through mitosis leads to abortion of nascent transcripts. Cell. 1991;67:303–310. doi: 10.1016/0092-8674(91)90182-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuttleworth J, Godfrey R, Colman A. p40MO15, a cdc2-related protein kinase involved in negative regulation of meiotic maturation of Xenopus oocytes. EMBO J. 1990;9:3233–3240. doi: 10.1002/j.1460-2075.1990.tb07522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svejstrup JQ, Wang Z, Feaver WJ, Xu X, Bushnell DA, Donahue TF, Friedberg EC, Kornberg RD. Different forms of TFIIH for transcription and DNA Repair: Holo-TFIIH and a nucleotide excision repairosome. Cell. 1995;80:21–28. doi: 10.1016/0092-8674(95)90447-6. [DOI] [PubMed] [Google Scholar]
- Svejstrup JQ, Vichi P, Egly JM. The multiple roles of transcription/repair factor TFIIH. Trends Biochem Sci. 1996;21:346–350. [PubMed] [Google Scholar]
- Tassan J-P, Schultz S, Bartek J, Nigg EA. Cell cycle analysis of the activity, subcellular localization, and subunit composition of human CAK (CDK-activating kinase) J Cell Biol. 1994;127:467–478. doi: 10.1083/jcb.127.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valay J-G, Simon M, Dubois M-F, Bensaude O, Facca C, Faye G. The KIN28 gene is required both for RNA polymerase II mediated transcription and phosphorylation of the Rpb1p CTD. J Mol Biol. 1995;249:535–544. doi: 10.1006/jmbi.1995.0316. [DOI] [PubMed] [Google Scholar]
- Weeks JR, Hardin SE, Shen J, Lee LM, Greenleaf AL. Locus-specific variation in phosphorylation state of RNA polymerase II in vivo: Correlations with gene activity and transcript processing. Genes & Dev. 1993;7:2329–2344. doi: 10.1101/gad.7.12a.2329. [DOI] [PubMed] [Google Scholar]
- Yankulov KY, Bentley DL. Regulation of CDK7 substrate specificity by MAT1 and TFIIH. EMBO J. 1997;16:1638–1646. doi: 10.1093/emboj/16.7.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawel L, Lu H, Cisek LJ, Corden JL, Reinberg D. The cycling of RNA Polymerase II during transcription. Cold Spring Harbor Symp Quant Biol. 1993;58:187–198. doi: 10.1101/sqb.1993.058.01.023. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Lieberman PM, Boyer TG, Berk AJ. Holo-TFIID supports transcriptional stimulation by diverse activators and from a TATA-less promoter. Genes & Dev. 1992;6:1964–1974. doi: 10.1101/gad.6.10.1964. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Pe’ery T, Peng J, Ramanathan Y, Marshall N, Marshall T, Amendt B, Mathews MB, Price D. Transcription elongation factor P-TEFb is required for HIV-1 Tat transactivation in vitro. Genes & Dev. 1997;11:2622–2632. doi: 10.1101/gad.11.20.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]