

Abstract
The activities of cyclin D-dependent kinases serve to integrate extracellular signaling during G1 phase with the cell-cycle engine that regulates DNA replication and mitosis. Induction of D-type cyclins and their assembly into holoenzyme complexes depend on mitogen stimulation. Conversely, the fact that D-type cyclins are labile proteins guarantees that the subunit pool shrinks rapidly when cells are deprived of mitogens. Phosphorylation of cyclin D1 on a single threonine residue near the carboxyl terminus (Thr-286) positively regulates proteasomal degradation of D1. Now, we demonstrate that glycogen synthase kinase-3β (GSK-3β) phosphorylates cyclin D1 specifically on Thr-286, thereby triggering rapid cyclin D1 turnover. Because the activity of GSK-3β can be inhibited by signaling through a pathway that sequentially involves Ras, phosphatidylinositol-3-OH kinase (PI3K), and protein kinase B (Akt), the turnover of cyclin D1, like its assembly, is also Ras dependent and, hence, mitogen regulated. In contrast, Ras mutants defective in PI3K signaling, or constitutively active mitogen-activated protein kinase-kinase (MEK1) mutants that act downstream of Ras to activate extracellular signal-regulated protein kinases (ERKs), cannot stabilize cyclin D1. In direct contrast to cyclin D1, which accumulates in the nucleus during G1 phase and exits into the cytoplasm during S phase, GSK-3β is predominantly cytoplasmic during G1 phase, but a significant fraction enters the nucleus during S phase. A highly stable D1 mutant in which an alanine is substituted for the threonine at position 286 and that is refractory to phosphorylation by GSK-3β remained in the nucleus throughout the cell cycle. Overexpression of an active, but not a kinase-defective, form of GSK-3β in mouse fibroblasts caused a redistribution of cyclin D1 from the cell nucleus to the cytoplasm. Therefore, phosphorylation and proteolytic turnover of cyclin D1 and its subcellular localization during the cell division cycle are linked through the action of GSK-3β.
Keywords: Glycogen synthase kinase-3, cyclin D1, Ras signaling, proteolysis, nuclear transport
A family of cyclin-dependent kinases (CDKs) cooperatively regulates mammalian cell cycle progression (for review, see Sherr 1993). During G1 phase, D-type cyclins (D1, D2, and D3) are synthesized and assemble with either CDK4 or CDK6 in response to growth factor stimulation, thereby generating active holoenzymes that help inactivate the growth-suppressive function of the retinoblastoma protein (Rb) through its phosphorylation (for review, see Weinberg 1995). Cyclin D holoenzyme complexes also titrate CDK inhibitors, such as p27Kip1 and p21Cip1, facilitating the activation of cyclin E-CDK2 and subsequent entry into the DNA synthetic phase of the cell cycle (for review, see Sherr and Roberts 1995).
Ras-mediated pathways are important for cyclin D1 induction and its assembly with CDKs. Overexpression of activated oncogenic Ras alleles, but not wild-type Ras, initiates DNA synthesis independently of growth factor stimulation (Feramisco et al. 1984). Conversely, microinjection of antibodies that inactivate Ras or introduction of certain dominant-negative Ras alleles can block S-phase entry induced by mitogens (Mulcahy et al. 1985; Mittnacht et al. 1997; Peeper et al. 1997). Both cyclin D1 expression and assembly require the sequential activities of Raf1, mitogen-activated protein kinase-kinases (MEK1 and MEK2), and the sustained activation of extracellular signal-regulated protein kinases (ERKs; Albanese et al. 1995; Lavoie et al. 1996; Winston et al. 1996; Aktas et al. 1997; Kerkhoff and Rapp 1997; Weber et al. 1997; Cheng et al. 1998).
In turn, cyclin D1 degradation is mediated by phosphorylation-triggered, ubiquitin-dependent proteolysis (Diehl et al. 1997). Polyubiquitination of protein substrates involves the sequential action of three distinct enzymes termed E1, E2 (UBC; ubiquitin-conjugating enzyme), and E3 (ubiquitin ligase; Ciechanover 1994; King et al. 1996). Specificity of substrate recognition is dependent on several factors including E2 and E3 selectivity (King et al. 1996; Skowyra et al. 1997; Renny-Feldman et al. 1997), recognition motifs within the target proteins themselves (Glotzer et al. 1991), and, in some cases, a requirement for phosphorylation of specific residues within the substrate (Deshaies et al. 1995; Clurman et al. 1996; Lanker et al. 1996; Won et al. 1996). Ubiquitin-dependent degradation of cyclin D1 requires phosphorylation of a specific threonine residue (Thr-286) located near the protein carboxyl terminus, and this phosphorylation is not mediated by cyclin D-dependent kinases themselves (Diehl et al. 1997). Because the kinase that phosphorylates this residue has not yet been identified, it remains unclear whether cyclin D1 proteolysis, like its synthesis and assembly, is subject to mitogen regulation.
The subcellular distribution of D-type cyclins is also likely to be regulated by cell cycle-dependent events. Cyclin D1 accumulates in the nuclei of cells during G1 phase, but once DNA replication begins, it disappears from the nucleus (Baldin et al. 1993), despite the fact that its level of synthesis does not decrease markedly during S phase (Matsushime et al. 1991). The mechanisms that regulate the periodic subcellular redistribution of cyclin D1 during the cell division cycle have also not been defined.
We now demonstrate that glycogen synthase kinase-3β (GSK-3β) catalyzes the phosphorylation of cyclin D1 on Thr-286, thereby regulating cyclin D1 turnover in response to mitogenic signals. In turn, GSK-3β-mediated phosphorylation of cyclin D1 redirects the protein from the nucleus to the cytoplasm. Our results support a model in which phosphorylation of cyclin D1 on Thr-286 by GSK-3β links processes governing cyclin D1 subcellular localization with its proteasomal degradation.
Results
GSK-3β phosphorylates cyclin D1 on Thr-286 in vitro
Rapid turnover of cyclin D1 requires phosphorylation of Thr-286 (Diehl et al. 1997), and its location adjacent to Pro-287 suggested that this modification might be mediated by a proline-directed kinase. Hence, we tested several known proline-directed kinases including GSK-3β, ERK2, stress-activated protein kinase (SAPK, p54β1), and cyclin E–CDK2 (Fig. 1) for their ability to phosphorylate cyclin D1 on Thr-286. Previous work from our laboratory indicated that cyclin D1–CDK4 complexes expressed in mammalian cells were better substrates than D1 subunits alone (also see below). To prepare substrate, we expressed Flag epitope-tagged cyclin D1 in insect Sf9 cells together with a catalytically inactive mutant (K35M) of CDK4 and recovered both free and CDK4-bound D1 subunits with M2 monoclonal antibodies to the Flag tag. Kinase reactions performed in vitro demonstrated that purified GSK-3β efficiently phosphorylated wild-type cyclin D1 (Fig. 1A, lane 1), but not a cyclin D1 mutant containing an Ala for Thr-286 substitution [D1-(T286A), lane 2]. In contrast, ERK2 (lanes 3,4), SAPK (lanes 5,6), and cyclin E–CDK2 (lanes 7,8) all failed to phosphorylate cyclin D1, although each of the enzyme preparations was highly active on other substrates (data not shown).
Figure 1.
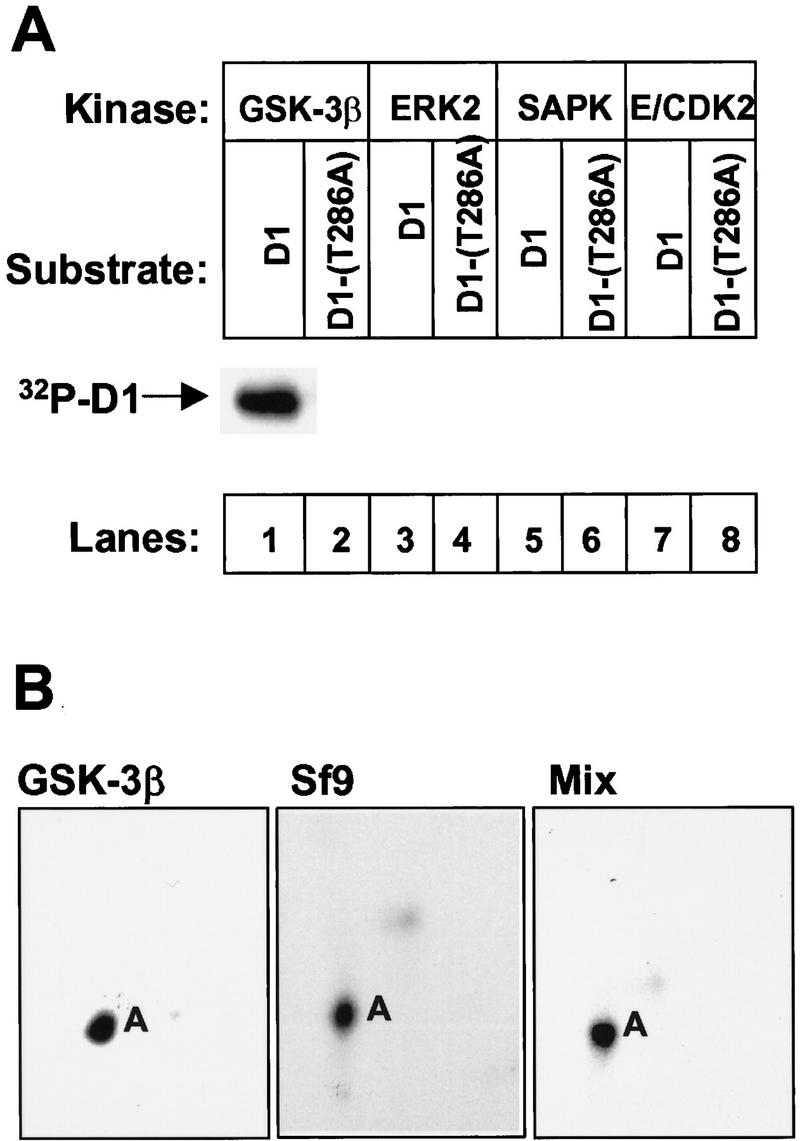
GSK-3β phosphorylates cyclin D1 on Thr-286. (A) Cyclin D1 (odd lanes) or cyclin D1-(T286A) (even lanes) immunoprecipitated from Sf9 cells infected with baculoviruses encoding D1 and CDK4 were mixed with recombinant GSK-3β (lanes 1,2), ERK2 (lanes 3,4), SAPK (lanes 5,6), or cyclin E–CDK2 (lanes 7,8) plus [γ-32P]ATP. After incubation at 30°C for 30 min, phosphorylated proteins were separated on a denaturing polyacrylamide gel, transferred to an Immobilon-P membrane, and visualized by autoradiography. The position of phosphorylated cyclin D1 is indicated. (B) Membrane slices containing cyclin D1 phosphorylated by GSK-3β in vitro (left), phosphorylated by endogenous Sf9 kinases (middle), or by a mixture of the two (right) were digested with trypsin and separated sequentially by electrophoresis and ascending chromatography. Phosphopeptides were visualized by autoradiographic exposure for 48 hr. The phosphopeptide containing Thr-286 was previously designated peptide A. Serine-containing peptides phosphorylated at lower stoichiometry can only be visualized after longer exposures (Kato et al. 1994; Diehl et al. 1997).
To confirm that the site of GSK-3β phosphorylation was Thr-286, radiolabeled cyclin D1 was electrophoretically separated on a denaturing gel, transferred to membrane, and digested with trypsin. Labeled peptides were sequentially separated in two dimensions by electrophoresis and ascending chromatography (Fig. 1B). The tryptic fingerprint derived from GSK-3β-phosphorylated cyclin D1 (left panel) revealed a single characteristic phosphopeptide. This peptide, previously designated A, appeared qualitatively similar to the major phosphopeptide derived from cyclin D1 phosphorylated in Sf9 cells (middle panel), which we know contains Thr-286, the only site of cyclin D1 threonine phosphorylation (Diehl et al. 1997). Mixing of these two phosphopeptides confirmed that they had indistinguishable mobilities in both dimensions (right panel). Therefore, GSK-3β can specifically phosphorylate cyclin D1 on Thr-286 in vitro.
To compare phosphorylation of cyclin D1 and D1–CDK4 complexes, Sf9 cells were infected with baculoviruses encoding cyclin D1 alone or cyclin D1 together with CDK4 in the presence or absence of GSK-3β, and infected cells were metabolically labeled with 32P-orthophosphate. Free cyclin D1 was isolated by direct precipitation with a monoclonal antibody directed against D1 itself (Fig. 2A, lanes 2,3), while cyclin D1 bound to CDK4 was isolated by precipitation with antiserum to the carboxyl terminus of CDK4 (Fig. 2A, lanes 4,5). Under conditions such as those used here in which CDK4 and cyclin D1 are expressed at similar levels, factors mediating their assembly in Sf9 cells are limiting and only about 10%–20% of the D1 molecules form binary complexes with the catalytic subunits (Kato et al. 1994). Therefore, lysates were normalized to yield roughly equivalent levels of D1 in each lane (bottom panel). In the absence of exogenous GSK-3β (Fig. 2A, lanes 2,4), both free cyclin D1 and CDK4-bound cyclin D1 were phosphorylated by endogenous insect kinases. Much of this background phosphorylation occurs on Thr-286 (Fig. 1B, middle) with the remainder occurring on multiple serine residues (requiring longer autoradiographic exposures to visualize). As shown previously, the introduction of CDK4 resulted in increased cyclin D1 phosphorylation (Fig. 2A, lane 4 vs. lane 2) on both Thr-286 and serine residues (Kato et al. 1994; Diehl et al. 1997). Coinfection with baculovirus encoding GSK-3β resulted in a further threefold increase in phosphorylation of CDK4-bound cyclin D1 (cf. lanes 5 and 4) without significantly affecting phosphorylation of unbound cyclin D1 (lane 3 vs. 2). Mapping of tryptic phosphopeptides again confirmed increased phosphorylation of D1 only on Thr-286. Therefore, the cyclin D1–CDK4 complex is a better substrate of GSK-3β than free cyclin D1.
Figure 2.

GSK-3β binds to D1 subunits and preferentially phosphorylates cyclin D1 in complexes with CDK4. (A) Sf9 cells infected with baculoviruses encoding cyclin D1 or cyclin D1 plus CDK4 with or without wild-type GSK-3β as indicated were metabolically labeled with [32P]orthophosphate. Lysates were subjected to precipitation with NRS (lane 1), antibody to cyclin D1 (lanes 2,3), or antiserum specific for the carboxyl terminus of CDK4 (lanes 4,5) as indicated. Phosphorylated proteins were resolved on denaturing polyacrylamide gels and transferred to a nitrocellulose membrane. Following autoradiography, the membrane was blotted with the antibody to cyclin D1 (bottom) and sites of antibody binding were visualized by enhanced chemiluminescence. (B) Sf9 cells infected with baculoviruses encoding cyclin D1 and CDK4 together with wild-type (wt) or kinase-defective (kd) GSK-3β were labeled with [32P]orthophosphate. Lysates were subjected to precipitation with NRS or anti-D1 as indicated, and phosphorylated proteins were resolved on a denaturing polyacrylamide gel followed by transfer to Immobilon-P membrane (autoradiographic exposure time 12 hr; top). Following autoradiography, the membrane was blotted with the antibody to cyclin D1 as in A. The relative ratio of 32P-labeled cyclin D1 versus total cyclin D1 in each lysate (densitometric scanning) is indicated between the two autoradiographs. (C) Sf9 lysates infected with baculoviruses encoding the proteins indicated were precipitated with the indicated antibodies and blotted with a monoclonal antibody to GSK-3β. Sites of antibody binding were visualized by enhanced chemiluminesence.
We also tested whether a kinase-defective GSK-3β mutant could inhibit phosphorylation of cyclin D1 in intact Sf9 cells. Sf9 cells were infected with baculoviruses encoding cyclin D1 and CDK4 together with different combinations of wild-type and kinase-defective GSK-3β. Infected cells were labeled with 32P-orthophosphate and cyclin D1 was isolated by immunoprecipitation (Fig. 2B). Cyclin D1 was again phosphorylated by endogenous insect kinases (Fig. 2B, lane 2), but expression of kinase-defective GSK-3β reduced background phosphorylation of cyclin D1 about fivefold (cf. lanes 2 and 3). As before, expression of wild-type GSK-3β with cyclin D1 induced an increase in cyclin D1 phosphorylation over background (cf. lanes 2 and 5). However, kinase-defective GSK-3β again interfered with D1 phosphorylation (lane 4 vs. 5), reducing it to a level below background (lane 4 vs. lane 2). Two-dimensional tryptic peptide mapping and phosphoamino acid analysis once again confirmed that GSK-3β phosphorylation occurred on Thr-286 (data not shown). Together, these data indicate that mutant kinase-defective GSK-3β can dampen phosphorylation of D1 by the wild-type form of the enzyme, as well as by endogenous D1 kinase(s) expressed in Sf9 cells.
One possible explanation of the latter results is that GSK-3β can interact stably with the cyclin D1–CDK4 complex directly and that binding of the kinase-defective mutant form of the enzyme occludes phosphorylation by the catalytically active form. Sf9 cells were infected with baculoviruses encoding cyclin D1, CDK4, and either wild-type or kinase-defective GSK-3β, and lysate proteins precipitated with nonimmune rabbit serum (NRS) or with antisera to GSK-3β, cyclin D1, or CDK4 were separated on denaturing gels and blotted with antibodies to GSK-3β (Fig 2C). Both baculovirus-encoded forms of GSK-3β were precipitated with the cognate antibody (lanes 5,7), whereas no endogenous form of the enzyme was recovered from uninfected insect cells (lane 3). A small but significant fraction of wild-type GSK-3β (estimated at 3%–5% in several such experiments) was coprecipitated with antibodies to cyclin D1 (lanes 4 vs. lane 5), and, surprisingly, almost half of the catalytically inactive form of the enzyme became stably associated with the cyclin (lanes 6,7). This suggested that phosphorylation of D1 by GSK-3β might destabilize the interaction, and consistent with this idea, removal of ATP from lysates containing wild-type GSK-3β by dialysis followed by addition of nonhydrolyzable ATP increased by threefold the level of wild-type GSK-3β coprecipitating with anti-D1 (data not shown). Catalytically inactive GSK-3β could not associate with CDK4 subunits alone (lanes 8,9) but did coprecipitate with unbound cyclin D1 subunits (lanes 10,11). Despite the ability of kinase-defective GSK-3β to bind free D1 subunits, D1–CDK4 complexes are the preferred substrate of wild-type GSK-3β (Fig. 2A). Therefore, kinase-defective GSK-3β can interact with free or CDK4-bound cyclin D1 and forms more stable complexes than its wild-type counterpart, consistent with the ability of the mutant form to interfere with D1 Thr-286 phosphorylation by catalytically active GSK-3β (Fig. 2B).
GSK-3β is a major cyclin D1 kinase in lysates of mouse NIH-3T3 fibroblasts
To verify that GSK-3β recovered from mammalian cells would also specifically phosphorylate cyclin D1 on Thr-286 in vitro, endogenous GSK-3β was immunoprecipitated from NIH-3T3 cell lysates, and recovered immune complexes were incubated in kinase reactions with GST–D1 fusion proteins plus [γ-32P]ATP. In an attempt to avoid complications of contamination of immune precipitates with adventitious serine kinases, we used fusion protein substrates containing only the carboxy-terminal 41 amino acids of cyclin D1. This D1 domain contains only two serine residues at codons 257 and 258 and two threonines at codons 286 and 288; as a control, we used a GST–D1 fusion protein containing the T286A mutation.
GST–D1 was efficiently phosphorylated by GSK-3β-containing immune complexes recovered from NIH-3T3 cells, but GST–D1–(T286A) was not (Fig. 3A, lanes 2,3). Immune complexes collected with NRS (lane 1) did not phosphorylate GST–D1. NIH-3T3 cells were lysed and subjected to two rounds of immunodepletion with either a control monoclonal antibody (9E10) or with the antibody to GSK-3β and depletion of GSK-3β was confirmed by immunoblotting (data not shown). When GST–D1 or GST–D1–(T286A) was added to the GSK-3β-depleted or mock-depleted lysates together with [γ-32P]ATP, both fusion proteins still underwent phosphorylation (data not shown). Because both fusion proteins contain other serine and threonine residues, their phosphorylation by kinases remaining in the lysates would likely obscure phosphorylation of Thr-286. Therefore, we prephosphorylated the fusion proteins with purified protein kinase A (PKA) in reactions performed with unlabeled ATP. Thr-286 does not fall into a PKA consensus site and should not be modified. Following removal of PKA by repurification of the fusion proteins, the ability of the immunodepleted extracts to phosphorylate GST–D1 or GST–D1-(T286A) was reassessed. Under these conditions, GST–D1 was efficiently phosphorylated by the mock-depleted lysate (Fig. 3B, lane 1), but depletion of GSK-3β reduced phosphorylation of GST–D1 by >90% (lane 3). GST–D1-(T286A) was not phosphorylated by either lysate (lanes 2,4). Therefore, GSK-3β is a major enzyme in NIH-3T3 cells that is able to catalyze the phosphorylation of cyclin D1 on Thr-286.
Figure 3.

Depletion of GSK-3β from mammalian cell lysates abrogates phosphorylation of cyclin D1 on Thr-286. (A) Detergent lysates prepared from NIH-3T3 cells were precipitated with either NRS (lane 1) or antibody to GSK-3β (lanes 2,3). Immune complexes were tested for their ability to phosphorylate GST-D1 (lanes 1,2) or GST–D1-(T286A) (lane 3) containing the carboxy-terminal 41 amino acids of cyclin D1. Phosphorylated GST-fusion proteins were resolved on polyacrylamide gels (exposure time 12 hr). (B) NIH-3T3 cell lysates were subjected to two rounds of depletion with either a control antibody, 9E10 (lanes 1,2), or with antibody to GSK-3β (lanes 3,4). Depleted lysates were mixed with [γ-32P]ATP in kinase reactions performed with GST–D1 (lanes 1,3) or GST–D1-(T286A) (lanes 2, 4) prephosphorylated by PKA. Radiolabeled GST–D1 proteins were collected on glutathione–Sepharose beads and resolved on a denaturing polyacrylamide gel (autoradiographic exposure time 12 hr).
Control of cyclin D1 turnover via the Ras–PI3K–Akt pathway
The Ras–Raf1–MEK–ERK kinase cascade regulates cyclin D1 expression and its assembly with CDK4 (Albanese et al. 1995; Lavoie et al. 1996; Winston et al. 1996; Aktas et al. 1997; Kerkhoff and Rapp 1997; Weber et al. 1997; Cheng et al. 1998). However, Ras signaling activates several other pathways, including one that inhibits the activity of GSK-3β. Specifically, Ras and phosphatidylinositol-3-OH kinase (PI3K) collaborate to activate the c-Akt proto-oncogene product (also designated protein kinase B; Rodriguez-Viciana et al. 1994, 1997; Boudewijn et al. 1995; Franke et al. 1995, 1997; Klinghoffer et al. 1996; Kauffmann-Zeh et al. 1997; Vanhaesebroeck et al. 1997). In turn, Akt down-regulates GSK-3β through site-specific phosphorylation (Cross et al. 1995; Dudek et al. 1997; Vanhaesebroeck et al. 1997), which should inhibit the rate of cyclin D1 turnover. Because Ras signaling depends on growth factor stimulation, the rate of cyclin D1 turnover in mouse fibroblasts should be influenced by serum stimulation. Therefore, we compared the half-life of cyclin D1 in cells rendered quiescent by serum starvation to that in cells proliferating in the presence of serum. Because synthesis of cyclin D1 also requires serum-derived growth factors, it was necessary to use NIH-3T3 cells engineered to overexpress cyclin D1. The half-life of ectopically expressed cyclin D1 in quiescent cells, as determined by pulse–chase analysis, was about 13 min, as compared with a half-life of 24 min in serum-stimulated cells (Table 1). Because of the rapidity of cyclin D1 turnover under both conditions, we had not noted this difference previously (Diehl et al. 1997), but in a series of independent experiments, the difference proved to be highly significant (see, Table 1, legend). In agreement with results of others (Welsh et al. 1996), the catalytic activity of GSK-3β isolated from serum-stimulated cells was reduced approximately threefold relative to the activity of GSK-3β isolated from serum-starved cells (data not shown), consistent with negative regulation of GSK-3β by mitogens (see above).
Table 1.
Cyclin D1 half-life in NIH-3T3 fibroblasts
| Treatment
|
D1 t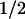 (min) (min)
|
|---|---|
| Proliferating in serum-containing medium | 24 ± 4 |
| Wortmannin (100 nm) plus complete medium | 10 ± 2 |
| Induced MEK1 (serum starved) | 11 ± 2 |
| RasV12 transfected (serum starved) | 31 ± 7 |
| RasV12.S35 transfected (serum starved) | 14 ± 2 |
| D1 overexpression (serum starved) | 13 ± 2 |
NIH-3T3 cells proliferating in the presence of serum or derivatives modified as indicated were labeled with l-[35S]methionine for 30 min and chased in the presence of excess unlabeled precursor. Radiolabeled cyclin D1 was precipitated from lysates collected after 0, 5, 10, 20, 40, 80, and 120 min using a D1-specific monoclonal antibody. Proteins were resolved on denaturing gels, detected by autoradiography, and the relative amount of radiolabeled cyclin D1 at each time point was determined by densitometric scanning of the film. The mean half-life of cyclin D1 (±s.d. from the mean) determined for each cell line was calculated from three to five independent experiments.
Next, we determined the consequences of overexpressing a constitutively active isoform of Akt (MyrAkt), which contains an amino-terminal myristoylation signal that enables Akt membrane association (Franke et al. 1995). NIH-3T3 cells were infected with a retrovirus encoding MyrAkt or with a control virus encoding the T cell coreceptor CD8, and 48 hr later, cells were pulse-labeled for 30 min with [35S]methionine and then chased in the presence of the unlabeled precursor for various times. In fibroblasts infected with the control CD8 vector, the observed half-life of metabolically labeled, immunoprecipitated cyclin D1 was about 20 minutes (Fig. 4A, lanes 2–6; Matsushime et al. 1992). In contrast, the half-life of cyclin D1 in cells expressing MyrAkt was extended two to threefold (lanes 7–11). MyrAkt expression was documented by immunoprecipitation with antiserum to a carboxy-terminal Akt epitope (Fig. 4B, lane 2). Because PI3K down-regulates GSK-3β through its activity on Akt, inhibition of PI3K should conversely accelerate cyclin D1 turnover. We therefore examined cyclin D1 stability in NIH-3T3 cells treated with wortmannin, a specific PI3K inhibitor (Ui et al. 1995). Treatment of intact cells with the inhibitor reproducibly accelerated cyclin D1 turnover more than twofold (10 min vs. 24 min, Table 1) and also reduced the steady-state levels of D1, as determined by immunoblotting (data not shown).
Figure 4.

Overexpression of MyrAkt stabilizes cyclin D1 in vivo. (A) NIH-3T3 cells infected with virus encoding the T-cell coreceptor (CD8; lanes 1–6) or MyrAkt (lanes 7–11) were labeled with [35S]methionine for 30 min and then chased in the presence of excess unlabeled precursor for the indicated times. Radiolabeled cyclin D1 was precipitated with antibody to cyclin D1 and resolved on denaturing polyacrylamide gels (autoradiographic exposure time, 16 hr). The position of labeled cyclin D1 is indicated. (B) NIH-3T3 cells infected with virus encoding CD8 (lane 1) or MyrAkt (lanes 2,3) were labeled with [35S]methionine. Detergent lysates were subjected to precipitation with either NRS (lane 3) or antiserum specific for a carboxy-terminal epitope of c-Akt (lanes 1,2). Immune complexes were resolved on a denaturing polyacrylamide gel, and MyrAkt was visualized by autoradiography (exposure time 16 hr).
Although Ras activity should stimulate both cyclin D1 synthesis and assembly through the Raf1–MEK–ERK pathway, a clear prediction is that it should also stabilize cyclin D1 through PI3K–Akt–GSK-3β signaling. Corollaries are that Ras mutants specifically defective in PI3K signaling, or constitutively active Raf1 or MEK1 (acting downstream of Ras in a parallel pathway), should not be able to stabilize cyclin D1, even though they should still support cyclin D1 synthesis and assembly. To determine the ability of Ras itself to modulate cyclin D1 turnover, we transfected NIH-3T3 cells with an oncogenic Ha-Ras allele (RasV12), or with a double mutant (RasV12.S35) that cannot activate PI3K (Rodriguez-Viciana et al. 1994, 1997; White et al. 1995; Kauffman-Zeh et al. 1997). Serum-deprived cell lines expressing these mutants synthesized cyclin D1 in direct contrast to parental NIH-3T3 cells, which require serum stimulation for cyclin D1 induction (Matsushime et al. 1992, 1994). Therefore, pulse–chase analyses were carried out in serum-starved transformants that manifest minimal endogenous Ras activity. The half-life of cyclin D1 in serum-starved RasV12 transformants was about 30 min, but was reduced to 14 min in cells expressing RasV12.S35 (Table 1), implying that PI3K signaling was required for the ability of Ras to stabilize cyclin D1.
Like Ras mutants defective in the PI3K pathway, constitutively active MEK1 was unable to extend the half-life of cyclin D1, despite its ability to promote cyclin D1 synthesis and assembly in serum-deprived cells (Cheng et al. 1998). For these experiments, we used NIH-3T3 cells harboring an active MEK1 mutant (previously designated MEK1*) expressed under the control of an inducible sheep metallothionein promoter. When these cells were treated for 6 hr with zinc in medium lacking serum, the half-life of induced cyclin D1 was only 11 min, versus 24 min for serum-treated control cells (Table 1). These results confirm that signaling through the Ras–Raf1–MEK1–ERK pathway is itself insufficient to extend the turnover of cyclin D1, although it facilitates cyclin D1 induction and assembly with CDK4 (Cheng et al. 1998).
GSK-3β redirects cyclin D1 to the cytoplasm
Cyclin D1 is a nuclear protein during the G1 phase of the cell cycle, but it exits the nucleus during S phase (Baldin et al. 1993). Because GSK-3β positively regulates NFAT nuclear export via site-specific phosphorylation (Beals et al. 1997), we considered the possibility that it might also affect the subcellular distribution of cyclin D1. NIH-3T3 cells were transfected with expression vectors encoding Flag-tagged cyclin D1 (Flag–D1) and CDK4 together with different forms of GSK-3β. Both the wild-type and kinase-defective forms of GSK-3β were engineered to express an amino-terminal Myc epitope to enable their detection by the 9E10 monoclonal antibody. Thirty-six hours post-transfection, the cells were fixed, and the cellular localization of Flag–D1 was visualized by immunofluorescent staining with the M2 antibody to the Flag tag. As shown previously (Diehl and Sherr 1997), most of the ectopically expressed Flag–D1 concentrated in the nucleus when it was coexpressed with CDK4 (Fig. 5A, left panels). In contrast, coexpression of wild-type GSK-3β with Flag–D1 and CDK4 resulted in the redistribution of Flag–D1 to the cytoplasm (middle panels). Cyclin D1 was detected primarily in the cytoplasm in >50% of the cells coexpressing wild-type GSK-3β, whereas it largely remained nuclear in its absence (Fig. 5B). Flag–D1 also localized to the nucleus when it was coexpressed with kinase-deficent GSK-3β (Fig. 5A, right panels, and B), consistent with the idea that cytoplasmic localization of cyclin D1 requires GSK-3β activity. In agreement, the D1-(T286A) mutant remained in the nucleus and was unaffected by GSK-3β (Fig. 5B, and see below). Although kinase-deficent GSK-3β has the potential to inhibit D1 phosphorylation in vitro by the wild-type enzyme (Fig. 2B), it did not act as a dominant-negative mutant when coexpressed with wild-type GSK-3β in mammalian cells. This may not be surprising, as inhibition likely requires stoichiometric binding whereas catalysis does not. Expression of either the wild-type or the kinase-defective forms of GSK-3β was confirmed by immunofluorescent staining with the 9E10 monoclonal antibody (data not shown). The dual requirement for GSK-3β activity and integrity of Thr-286 in cyclin D1 implies that its cytoplasmic localization is mediated by GSK-3β-dependent phosphorylation of D1 on this residue.
Figure 5.

GSK-3β overexpression redirects cyclin D1 to the cytoplasm. (A) NIH-3T3 cells transiently expressing CDK4 and either Flag-tagged cyclin D1 (left), Flag-tagged cyclin D1 plus wild-type GSK-3β (wtGSK–3β; middle), or Flag-tagged cyclin D1 plus kinase-defective GSK-3β (kdGSK-3β; right) were fixed and processed for immunofluorescence. Flag-tagged cyclin D1 was visualized by staining with the M2 monoclonal antibody to the tag (top), and cellular DNA was stained with Hoechst dye (bottom). (B) The subcellular localization of Flag-tagged cyclin D1 or Flag-tagged D1-(T286A) without ectopic GSK-3β or with either wtGSK-3β or kdGSK-3β was determined by immunofluorescent staining with the M2 monoclonal antibody as above. The percentage of cells expressing exclusively cytoplasmic cyclin D1 or D1-(T286A) is presented graphically and represents the average of at least four independent experiments. Vertical bars indicate standard deviations from the mean.
On the basis of these results, we considered the possibility that phosphorylation of cyclin D1 at Thr-286 might account for its relocalization from the nucleus during S phase. Therefore, we examined the subcellular distribution of wild-type cyclin D1 and the D1-(T286A) mutant throughout the cell cycle. NIH-3T3 fibroblasts engineered to stably overexpress either Flag-tagged cyclin D1 or cyclin D1-(T286A) were derived by coselection with puromycin and represented uncloned populations of transfectants expressing variable amounts of ectopic cyclin D1 per cell. The D1-(T286A) mutant does not act as a dominant-negative to inhibit cell cycle progression (Diehl and Sherr 1997), and like NIH-3T3 cells enforced to stably express wild-type D1 (Quelle et al. 1993), transfected cells appear to have a modestly contracted G1 interval and enter S phase about 2 hr earlier than their parental counterparts. Cell lines engineered to express D1 were rendered quiescent by contact inhibition and serum starvation for 30 hr and were then restimulated to synchronously enter the cell cycle by trypsinization and replating on glass coverslips in complete medium containing serum. At various times thereafter, fixed cells were examined by immunofluorescence with a monoclonal antibody to cyclin D1. Cyclin D1 accumulated in the nucleus of cells as they entered S phase (Fig. 6A; cf. 6 and 10 hr), but as S phase progressed (12 hr), increased cytoplasmic D1 staining was observed (Baldin et al. 1993). By G2 phase (18 hr), cyclin D1 was located almost entirely in the cytoplasm. In contrast, cyclin D1-(T286A) was retained in the nucleus throughout interphase (Fig. 6B). Thus, cell cycle-dependent redistribution of cyclin D1 depends on the integrity of Thr-286, presumably reflecting its phosphorylation status in response to GSK-3β.
Figure 6.
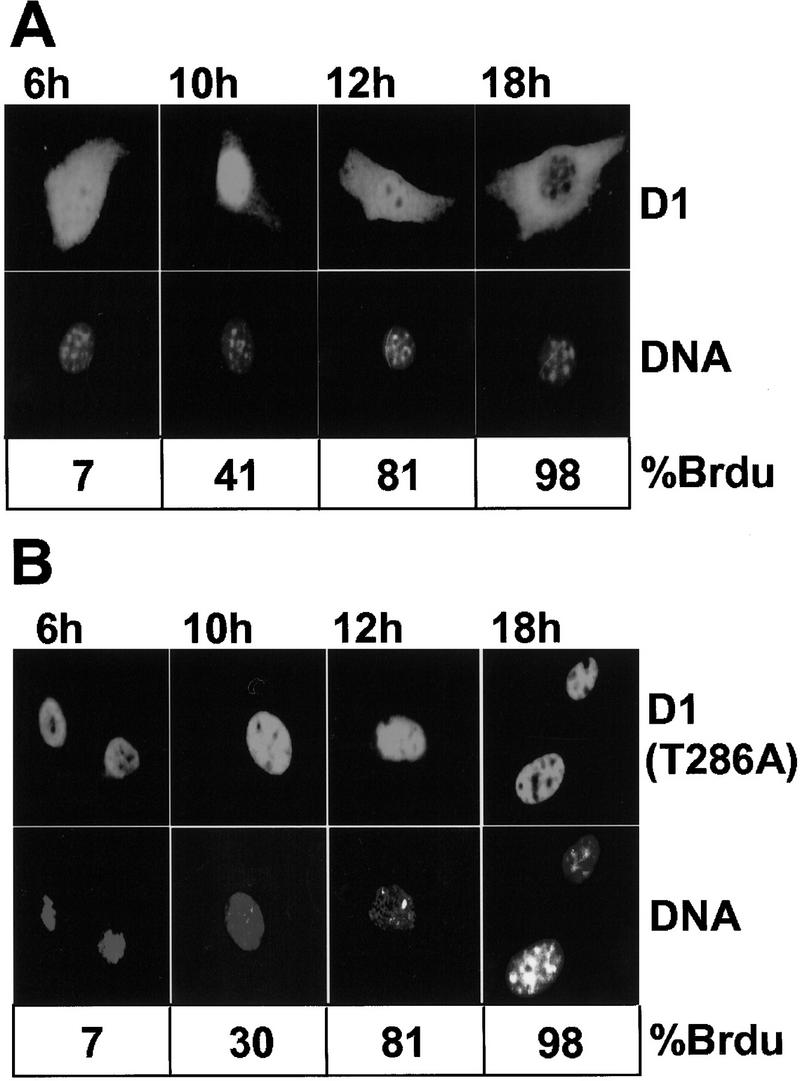
Cell cycle-dependent redistribution of cyclin D1 depends on the integrity of Thr-286. NIH-3T3 cells engineered to overexpress wild-type, Flag-tagged cyclin D1 (A) or Flag-tagged cyclin D1-(T286A) (B) were arrested in G0 by serum deprivation and contact inhibition and then stimulated to synchronously enter the cell cycle. Cells were fixed at the indicated times after serum addition. The subcellular localization of D1 proteins was determined by staining with the cyclin D1 monoclonal antibody (top), and total cellular DNA was visualized with Hoechst dye (middle). In parallel, cells released from G0 were stimulated to enter the cell cycle in the presence of BrdU to monitor S-phase entry. The percentage of cells incorporating BrdU at each time point is indicated below the panels. Because the levels of ectopic D1 expression were four- to eightfold above the endogenous background, a contribution of endogenous D1 to the staining pattern was negligible at the exposure chosen.
Cell cycle-dependent localization of GSK-3β
GSK-3β is thought to be a cytoplasmic kinase (Boyle et al. 1991a) whose activity is down-regulated by growth factor stimulation. However, it has been reported to mediate NFAT nuclear export through direct phosphorylation (Beals et al. 1997), implying that at least a fraction of GSK-3β must be nuclear. To address this issue, NIH-3T3 cells overexpressing cyclin D1 were rendered quiescent by contact inhibition and serum starvation for 26 hr and then allowed to synchronously re-enter the cell cycle by replating on glass coverslips at low density in medium containing serum. Immunofluorescent staining revealed that GSK-3β was exclusively cytoplasmic during G1 phase with predominant perinuclear staining (Fig. 7A, 6 and 8 hr). However, during S phase, GSK-3β staining was detected in the nuclei as well (16 and 18 hr). In parallel, G1-phase (6 hr) and S-phase cells (16 hr) were separated into nuclear and cytoplasmic fractions, which were blotted for GSK-3β (Fig. 7B). During G1, <5% of GSK-3β was detected in the nuclear fraction (cf. lanes 1 and 2), but during S phase, ∼25% of GSK-3β was detected in the nuclear fraction (cf. lanes 3 and 4). Thus, during S phase, a fraction of GSK-3β localizes to the nucleus, while cyclin D1 is redirected into the cytoplasm. Together, these data suggest that phosphorylation of cyclin D1 on Thr-286 by GSK-3β may occur in the nucleus to accelerate D1 export and cytoplasmic proteolysis.
Figure 7.

Subcellular localization of GSK-3β throughout the cell cycle. (A) NIH-3T3 cells engineered to overexpress Flag-tagged wild-type cyclin D1 were arrested by serum deprivation and contact inhibition and then dispersed and restimulated with serum to synchronously enter the cell cycle. Cells plated on glass coverslips in complete medium were fixed for immunofluorecence at the indicated times after serum addition, and were stained by use of a monoclonal antibody directed to GSK-3β (top). Cellular DNA was visualized with Hoechst dye (bottom). (B) At 6 hr (G1 phase) and 16 hr (S phase) after cell cycle entry, cells from parallel cultures were disrupted, and equal quantitites of protein from nuclear and cytoplasmic fractions were separated on denaturing gels, transferred to membrane, and blotted with antibody to GSK-3β. Sites of antibody binding were detected by enhanced chemiluminescence.
Discussion
GSK-3β is a cyclin D1 protein kinase
The turnover of cyclin D1 in proliferating cells is rapid (t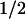 , ∼20 min), whereas that of its catalytic partner CDK4 is relatively slow (t
, ∼20 min), whereas that of its catalytic partner CDK4 is relatively slow (t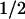 , ∼4 hr) (Matsushime et al. 1992). This creates a dynamic equilibrium in which regulatory cyclin D1 subunits exchange in and out of holoenzyme complexes with CDK4 as mitogen-stimulated cells move through G1 phase (Sherr 1993). Maintenance of active D1–CDK4 complexes requires persistent mitogenic signaling (Matsushime et al. 1991) through a Ras-dependent kinase cascade that utilizes Raf and MEK intermediates to target the ERKs (Albanese et al. 1995; Lavoie et al. 1996; Winston et al. 1996; Aktas et al. 1997; Kerkhoff and Rapp 1997; Weber et al. 1997; Cheng et al. 1998). In turn, mitogen withdrawal cancels Ras signaling and cyclin D1 synthesis, and the fast turnover of D1 ensures that D1–CDK4 complexes rapidly dissipate as cell proliferation ceases, usually within a single cycle.
, ∼4 hr) (Matsushime et al. 1992). This creates a dynamic equilibrium in which regulatory cyclin D1 subunits exchange in and out of holoenzyme complexes with CDK4 as mitogen-stimulated cells move through G1 phase (Sherr 1993). Maintenance of active D1–CDK4 complexes requires persistent mitogenic signaling (Matsushime et al. 1991) through a Ras-dependent kinase cascade that utilizes Raf and MEK intermediates to target the ERKs (Albanese et al. 1995; Lavoie et al. 1996; Winston et al. 1996; Aktas et al. 1997; Kerkhoff and Rapp 1997; Weber et al. 1997; Cheng et al. 1998). In turn, mitogen withdrawal cancels Ras signaling and cyclin D1 synthesis, and the fast turnover of D1 ensures that D1–CDK4 complexes rapidly dissipate as cell proliferation ceases, usually within a single cycle.
Cyclin D1 proteolysis is a regulated event that proceeds via the ubiquitin-dependent 26S proteasome. Degradation is facilitated by phosphorylation of a specific threonine residue located near the D1 carboxyl terminus, and elimination of this site through simple point mutation (e.g., T286A) markedly stabilizes the cyclin (t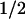 , 2–3 hr; Diehl et al. 1997). Although the identity of the D1 Thr-286 kinase has remained unknown, several lines of evidence now indicate that GSK-3β can carry out this function. First, purified GSK-3β or GSK-3β recovered from mouse fibroblasts was able to phosphorylate recombinant cyclin D1 only at this site in vitro. Second, immunodepletion of GSK-3β from mammalian cell extracts prevented phosphorylation of recombinant cyclin D1 on Thr-286. Therefore, not only can GSK-3β catalyze this phosphorylation event, but it appears to be a major kinase regulating this function, at least in lysates of NIH-3T3 cells. Third, a Ras-initiated kinase signaling cascade that negatively regulates GSK-3β activity in vivo was found to stabilize cyclin D1 in living cells. Manipulation of any of the components of this signaling pathway (Ras, PI3K, or Akt) affected cyclin D1 turnover in the predicted manner. Finally, phosphorylation of cyclin D1 at Thr-286 appears to facilitate its redistribution from the cell nucleus to the cytoplasm, and overexpression of catalytically active, but not kinase-defective, GSK-3β increased its cytoplasmic localization.
, 2–3 hr; Diehl et al. 1997). Although the identity of the D1 Thr-286 kinase has remained unknown, several lines of evidence now indicate that GSK-3β can carry out this function. First, purified GSK-3β or GSK-3β recovered from mouse fibroblasts was able to phosphorylate recombinant cyclin D1 only at this site in vitro. Second, immunodepletion of GSK-3β from mammalian cell extracts prevented phosphorylation of recombinant cyclin D1 on Thr-286. Therefore, not only can GSK-3β catalyze this phosphorylation event, but it appears to be a major kinase regulating this function, at least in lysates of NIH-3T3 cells. Third, a Ras-initiated kinase signaling cascade that negatively regulates GSK-3β activity in vivo was found to stabilize cyclin D1 in living cells. Manipulation of any of the components of this signaling pathway (Ras, PI3K, or Akt) affected cyclin D1 turnover in the predicted manner. Finally, phosphorylation of cyclin D1 at Thr-286 appears to facilitate its redistribution from the cell nucleus to the cytoplasm, and overexpression of catalytically active, but not kinase-defective, GSK-3β increased its cytoplasmic localization.
Although GSK-3β could phosphorylate Thr-286 in recombinant cyclin D1 or in a GST–D1 fusion protein containing only the carboxy-terminal 41 amino acids of the cyclin, it appeared to phosphorylate cyclin D1 in complexes with CDK4 more efficiently. This result is in general agreement with previous observations that phosphorylation of the D1 subunit increases as cyclin D1–CDK complexes achieve maximal levels and mammalian cells cross the G1/S boundary (Matsushime et al. 1991). Nonetheless, the rapid turnover of ectopically expressed, unassembled forms of cyclin D1 in fibroblasts rendered quiescent by serum starvation still requires their phosphorylation on Thr-286 (Diehl et al. 1997). Thus, the kinase that phosphorylates unbound cyclin D1 subunits must be active in quiescent cells. GSK-3β activity increases two- to threefold in cells deprived of growth factors (Cross et al. 1995; He et al. 1995; our data not shown). Under such conditions, we observed that ectopically expressed D1 subunits were degraded more rapidly than those expressed in proliferating cells or in serum-starved cells transformed by oncogenic Ras (see below). Thus, although unassembled cyclin D1 appears to be a poorer GSK-3β substrate relative to CDK4-bound subunits, the increased activity of GSK-3β in growth factor-deprived cells may compensate.
Phosphorylation of cyclin D1 on Thr-286 is dependent on the integrity of Pro-287 (data not shown) suggesting that a proline-directed kinase targets this site. Although cyclin D1 also contains a threonine at residue 288, Thr-286 is the only site of D1 threonine phosphorylation in mammalian cells (Diehl et al. 1997). GSK-3β can act as a proline-directed kinase, and the ability of the purified enzyme to phosphorylate a bacterially expressed, affinity-purified GST fusion protein containing only the carboxy-terminal 41 amino acids of D1 supports the principle. Several other proline-directed kinases, including ERK2, SAPK, cyclin E–CDK2, and cyclin D1–CDK4 itself were unable to phosphorylate cyclin D1 on Thr-286. GSK-3β can also act in a processive manner, requiring that its substrate first be phosphorylated by another kinase for maximal activity. The serines closest to Thr-286 lie some distance away at residues 257 and 258. Our data do not formally exclude processive phosphorylation, as a fraction of the cyclin D1 molecules expressed in proliferating mammalian cells (and in insect Sf9 cells) is phosphorylated on as-yet-unmapped serine residues.
An unexpected result was that recombinant GSK-3β and cyclin D1, whether free or bound to CDK4, were able to interact physically with one another in insect Sf9 cells. Only a small fraction of catalytically active GSK-3β associated with cyclin D1, but binding was significantly potentiated when GSK-3β was rendered catalytically inert. This suggested that phosphorylation of cyclin D1 by bound wild-type GSK-3β might destabilize such complexes, and in agreement, depletion of ATP from Sf9 extracts facilitated net complex formation two- to threefold (data not shown). Expression of the kinase-defective form of GSK-3β together with cyclin D1 in Sf9 cells blocked Thr-286 phosphorylation of D1 by endogenous kinase(s) and significantly inhibited the ability of ectopically expressed wild-type GSK-3β to phosphorylate the cyclin. Preferential binding of the inactive form of GSK-3β to cyclin D1 most likely accounts for its ability to suppress D1 phosphorylation by the wild-type kinase.
Cyclin D1 turnover is regulated via the mitogen-dependent Ras–PI3K–Akt pathway
Stimulation of cells with growth factors such as insulin or epidermal growth factor inhibits GSK-3β activity in a PI3K- and Akt-dependent manner (Saito et al. 1994; Boudewijn et al. 1995; Cross et al. 1995). If GSK-3β is a bona fide cyclin D1 kinase, then increased signaling through the PI3K–Akt pathway should stabilize cyclin D1 and vice versa. Enforced overexpression of a constitutively active form of Akt resulted in a threefold prolongation of cyclin D1 half-life (t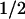 , ∼60 min) as well as a twofold decrease in D1 phosphorylation (data not shown). Conversely, inhibition of PI3K with wortmannin shortened the half-life of cyclin D1 from ∼20 to 10 min. Therefore, mitogenic signaling through the Ras–PI3K–Akt pathway can increase cyclin D1 stability by extinguishing GSK-3β activity, but if GSK-3β activity is not attenuated, cyclin D1 is actually turned over more rapidly than thought previously.
, ∼60 min) as well as a twofold decrease in D1 phosphorylation (data not shown). Conversely, inhibition of PI3K with wortmannin shortened the half-life of cyclin D1 from ∼20 to 10 min. Therefore, mitogenic signaling through the Ras–PI3K–Akt pathway can increase cyclin D1 stability by extinguishing GSK-3β activity, but if GSK-3β activity is not attenuated, cyclin D1 is actually turned over more rapidly than thought previously.
Transformation of NIH-3T3 cells with an oncogenic Harvey-Ras gene (RasV12) stabilized ectopically expressed cyclin D1 in cells deprived of serum (t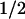 , ∼30 min vs. 13 min for untransformed cells). However, enforced expression of a double mutant (RasV12.S35) that is selectively defective in its ability to activate PI3K (Rodriguez-Viciana et al. 1994, 1997; White et al. 1995) had no such effect (t
, ∼30 min vs. 13 min for untransformed cells). However, enforced expression of a double mutant (RasV12.S35) that is selectively defective in its ability to activate PI3K (Rodriguez-Viciana et al. 1994, 1997; White et al. 1995) had no such effect (t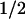 , 14 min). Ras independently regulates the Raf1–MEK–ERK kinase cascade. Although an inducible, constitutively active MEK1 mutant is able to trigger cyclin D1 synthesis and assembly with CDK4 in the complete absence of serum stimulation (Cheng et al. 1998), it was unable to extend the half-life of cyclin D1 (t
, 14 min). Ras independently regulates the Raf1–MEK–ERK kinase cascade. Although an inducible, constitutively active MEK1 mutant is able to trigger cyclin D1 synthesis and assembly with CDK4 in the complete absence of serum stimulation (Cheng et al. 1998), it was unable to extend the half-life of cyclin D1 (t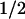 , 11 min). Therefore, cyclin D1 synthesis, assembly, and turnover are all regulated through Ras-dependent signaling, and while the Raf1–MEK1–ERK pathway guarantees cyclin D1 synthesis and assembly, the PI3K–Akt–GSK-3β pathway selectively affects cyclin D1 stability. Importantly, the identification of the latter pathway points to a previously unexpected mode of regulation of cyclin D1 by mitogenic signaling, further underscoring its role as a growth factor sensor.
, 11 min). Therefore, cyclin D1 synthesis, assembly, and turnover are all regulated through Ras-dependent signaling, and while the Raf1–MEK1–ERK pathway guarantees cyclin D1 synthesis and assembly, the PI3K–Akt–GSK-3β pathway selectively affects cyclin D1 stability. Importantly, the identification of the latter pathway points to a previously unexpected mode of regulation of cyclin D1 by mitogenic signaling, further underscoring its role as a growth factor sensor.
Unlike oncogenic Ras alleles such as RasV12, the double mutant, RasV12.S35, which does not activate PI3K, is attenuated in its ability to transform established fibroblast cell lines. However, its transforming potency can be rescued by other nontransforming Ras alleles, such as RasV12.C40, that do activate PI3K (White et al. 1995; Rodriguez-Viciana et al. 1997; Kauffmann-Zeh et al. 1997). Given that Ras transformation depends on cyclin D1 accumulation (Peeper et al. 1997; Mittnacht et al. 1997), stabilization of D1 may be necessary for the protein levels to rise to a threshold that enables G1 progression.
GSK-3β activity links cyclin D1 stability and subcellular localization
The critical functions of cyclin D1 during G1 phase, including phosphorylation of Rb and titration of p27Kip1, are nuclear events, and cyclin D1 is superfluous for completion of the cycle once cells enter S phase (Matsushime et al. 1991). Although cyclin D1 progressively accumulates in the nucleus during G1 phase, it redistributes into the cytoplasm as cells move through S phase, implying a periodicity of D1 function in proliferating cells that is independent of its rate of synthesis (Baldin et al. 1993). The stable D1-(T286A) mutant, unlike wild-type D1, remained in the nucleus throughout interphase, suggesting that redirection of cyclin D1 to the cytoplasm is mediated by Thr-286 phosphorylation. Conversely, overexpression of GSK-3β enforced cytoplasmic compartmentalization of D1 and was dependent on the kinase’s catalytic activity. Therefore, cyclin D1 proteolysis and relocalization are functionally linked through the status of Thr-286.
There are no amino acid sequences in cyclin D1 that show obvious homology to canonical nuclear import or export signals (NES), so other proteins likely play key roles in determining D1 compartmentalization (LaBaer et al. 1997; Diehl and Sherr 1997). One idea is that GSK-3β-mediated phosphorylation of nuclear cyclin D1 triggers its export to the cytoplasm, for example by facilitating an interaction between cyclin D1 and an exportin. Reminiscent of these findings, GSK-3β also phosphorylates the T cell transcription factor NFAT, causing its cytoplasmic redistribution (Beals et al. 1997). Alternatively, GSK-3β might phosphorylate cyclin D1 in the cytoplasm, preventing its association with proteins required for nuclear import and thereby targeting its cytoplasmic degradation. GSK-3β itself accumulates primarily in the cytoplasm of asynchronously proliferating cells (Boyle et al. 1991a). However, we observed that a subpopulation of GSK-3β becomes nuclear during S phase, the interval of the cycle in which cyclin D1 leaves the nucleus and enters the cytoplasm. Treatment of NIH-3T3 cells with leptomycin B (Nishi et al. 1994), an inhibitor of CRM1-dependent nuclear export (Fornerod et al. 1997), resulted in accumulation of the majority of GSK-3β in the nucleus (data not shown). Together, these results suggest that GSK-3β actively shuttles between the nucleus and cytoplasm and that its compartmentalization is cell cycle dependent. Therefore, nuclear accumulation of GSK-3β may be necessary for the relocalization of cyclin D1 observed during S phase.
Whether or not GSK-3β phosphorylates cyclin D1 in the nucleus, the cytoplasm, or both, the data imply that cyclin D1 degradation occurs preferentially in the cytoplasm. Proteasomes exist in both the cytoplasm and nucleus (Reits et al. 1997), but there are emerging precedents for compartment-specific degradation. For example, the p53 tumor suppressor is a nuclear transcription factor whose stability is regulated through ubiquitin-mediated proteolysis. p53 degradation depends on Mdm2 (Haupt et al. 1997; Kubbutat et al. 1997), which shuttles from the nucleus to the cytoplasm and appears to direct p53 to cytoplasmic proteasomes (Roth et al. 1998). Interfering with nuclear export of p53, either through mutation of the Mdm2 NES or through other manipulations that affect transport per se, stabilizes p53. By analogy, identification of proteins that interact specifically with the Thr-286 phosphorylated form of cyclin D1 will be necessary to enhance our understanding of its compartmentalization during the cell cycle.
A role for GSK-3β in cancer?
Overexpression of cyclin D1 is a common event in various forms of cancer. D1 can be overexpressed as a result of gene amplification or because it is targeted through chromosomal translocations (Hall and Peters 1996). However, in certain tumors, high levels of cyclin D1 expression have not been explained by such mechanisms, and events affecting cyclin D1 turnover might play some role. Although the p16INK4a–cyclin D1–Rb pathway is disabled in many forms of human cancer, colon carcinomas provide a conspicuous exception. Inactivation of the adenomatous polyposis coli (APC) tumor suppressor is the single most common event in colon cancer (Powell et al. 1992). APC is a target of Wnt signaling (for review, see Kinzler and Vogelstein 1996), and it regulates the proteolytic turnover of β-catenin in a manner that depends on phosphorylation of β-catenin by GSK-3β (Rubinfield et al. 1993; Yost et al. 1996). β-Catenin mutants that have lost GSK-3β phosphorylation sites remain constitutively active as coactivators of TCF/LEF-dependent transcription, and such mutations have now been found in the major fraction of colon cancers that lack mutated APC alleles (Korinek et al. 1997; Morin et al. 1997). The fact that GSK-3β can also regulate cyclin D1 turnover suggests that deregulation of Wnt signaling in colon cancer may target cyclin D1 in addition to the APC–β-catenin complex.
Materials and methods
Tissue culture conditions, cell lines, and transfections
NIH-3T3 cells were maintained in DMEM supplemented with 10% fetal calf serum (FCS), antibiotics, and glutamine (GIBCO BRL). Insect Sf9 cells were grown in Grace’s medium supplemented with 5% heat-inactivated FCS (Summers and Smith 1987). NIH-3T3 cells engineered to overexpress constitutively active MEK1 under control of the sheep metallothionein promoter were established previously, and their characteristics are described in detail elsewhere (Cheng et al. 1998). Derivatives of NIH-3T3 cells engineered to overexpress Flag-tagged cyclin D1, Flag-tagged cyclin D1-(T286A), RasV12, or RasV12.S35 were generated by cotransfection using the calcium phosphate coprecipitation protocol (Chen and Okayama 1987) with expression vectors encoding the appropriate cDNA plus the pJ6Ω-puro vector encoding the puromycin-resistance gene (Morgenstern and Land 1990). Transfected cell lines were selected and maintained in 7.5 μg/ml puromycin.
Myc-tagged GSK-3β, expression vectors, and GST-fusion proteins
For expression in insect Sf9 cells, the cDNAs encoding either wild-type GSK-3β or a kinase-defective GSK-3β mutant (provided by Jim Woodgett, Ontario Cancer Center, Canada) were inserted into pVL1393 (Pharmingen) as EcoRI fragments. Procedures for manipulation of baculoviruses were described previously (Summers and Smith 1987; Kato et al. 1994). For transient expression in mammalian cells, the cDNAs encoding either wild-type GSK-3β or a kinase-deficient GSK-3β mutant were inserted into the pJ3M expression vector (Sells and Chernoff 1995) as BclI–ClaI fragments, thereby creating an in-frame fusion with the Myc epitope tag. The retroviral vector encoding MyrAkt was provided by Philip Tsichlis (Fox Chase Cancer Center, Philadephia, PA). For construction of GST–D1 and the GST–D1-(T286A) fusion proteins, cyclin D1 cDNA was digested with XmnI and NotI and ligated into pGEX4T-1 (Pharmacia) digested with SmaI and NotI. This created an in-frame fusion between the carboxy-terminal 41 residues of cyclin D1 and GST. A 6× histidine-tagged ERK2 plasmid (NpT7-5-His–ERK2; Robbins et al. 1993) was provided by Melanie Cobb (Southwestern Medical School, Dallas, TX), and the plasmid harboring GST–SAPK (p54β1; Kyriakis et al. 1994) was provided by John Kyriakis (Harvard Medical School, Boston MA).
Purification of recombinant proteins from bacteria
Bacteria harboring GST–D1 or the GST–D1-(T286A) fusion proteins were induced to express recombinant proteins by addition of isopropyl β-d-thiogalactopyranoside (IPTG, 1 mm final concentration) to exponentially growing cultures. Bacteria were lysed in buffer containing 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 0.5% Nonidet P-40 (NP-40), and 1 mm phenylmethyl-sulfonyl fluoride (PMSF) by repeated cycles of freezing and thawing. GST-fusion proteins were absorbed to glutathione–Sepharose beads (Pharmacia) and eluted with kinase buffer [50 mm HEPES (pH 7.5), 10 mm MgCl2, 1 mm EGTA, 1 mm dithiothreitol (DTT), 1 mm PMSF, 0.4 mm NaF, and 0.4 mm NaV04] containing 4 mm reduced glutathione. Expression and purification of His–ERK2 (Robbins et al. 1993) and GST–SAPK (Kyriakis et al. 1994) was performed as described by these investigators.
Immunoprecipitation, immunoblotting, and immunofluorescence
Infected Sf9 cells used for coimmunoprecipitation analysis were lysed in 50 mm Tris HCl (pH 8.0), 150 mm NaCl, and 0.5% NP-40 containing protease and phosphatase inhibitors (1 mm PMSF, 20 U/ml aprotinin, 5 μg/ml leupeptin, and 0.4 mm NaV04). Lysates were cleared by sedimentation in a microcentrifuge for 5 min at 15,000 rpm and normalized for protein as indicated in the text prior to immune precipitation. GSK-3β-containing complexes were precipitated with either a commercially available mouse monoclonal antibody directed to GSK-3β (Transduction Laboratories, Lexington, KY), a mouse antibody raised directed against cyclin D1 (D1–72–13G, originally derived in our laboratory), or a mixture of two CDK4 antisera (Rz raised against a carboxy-terminal peptide and Ry, raised against full-length CDK4; Matsushime et al. 1992, 1994). Immune complexes were recovered with protein A–Sepharose (Pharmacia) or with protein A–Sepharose precoated with rabbit anti-mouse IgG.
Cells metabolically labeled with 200 μCi/ml l-[35S]methionine (1369 Ci/mmole; ICN) for pulse–chase analysis were lysed in 50 mm Tris HCl (pH 7.5), 150 mm NaCl, 1% NP-40, 1% sodium deoxycholate (DOC), 0.1% SDS, and protease and phosphatase inhibitors as indicated above. Radiolabeled cyclin D1 was precipitated with monoclonal antibody. For preparation of lysates containing active GSK-3β, cells were lysed in 50 mm Tris HCl (pH 7.5), 1 mm EDTA, 1 mm EGTA, 0.27 m sucrose, 1% Triton X-100, 1 mm DTT, and protease and phosphatase inhibitors. GSK-3β was precipitated with the cognate monoclonal antibody. Subcellular fractionation of NIH-3T3 was performed as described previously (Schreiber et al. 1989; Ostrowski et al. 1991). Proteins were resolved on denaturing polyacrylamide gels, electrophoretically transferred to nitrocellulose membranes (Millipore), and blotted with the indicated primary antibodies. Sites of antibody binding were visualized by use of protein A-conjugated horseradish peroxidase (EY Laboratories, San Mateo, CA) followed by chemiluminescence detection (ECL detection kit; Amersham). Immunofluorescent detection of cyclin D1 was carried out as described previously (Diehl and Sherr 1997) except in Figure 7, in which overexpressed cyclin D1 was detected with the mouse monoclonal antibody directed to cyclin D1 (1:10 dilution in TBS containing 5% FCS) rather than with the M2 monoclonal antibody.
Depletion of GSK-3β and detection of GSK-3β activity in mammalian cell lysates
For immunodepletion of GSK-3β, 5 × 106 NIH-3T3 cells made quiescent by serum starvation and contact inhibition for 36 hr were lysed in 50 mm HEPES (pH 7.5), 10 mm MgCl2, 1 mm EGTA, 1 mm DTT, 90 mm β-glycerophosphate, and protease and phosphatase inhibitors as indicated above. GSK-3β was removed from the lysates with a titered excess of mouse monoclonal antibody directed to the protein. For detection of GSK-3β activity in these lysates, GST–D1 or GST–D1–(T286A) prephosphorylated with PKA (Sigma) in the presence of unlabeled ATP was recovered on beads and incubated for 30 min at 30°C with lysate (corresponding to 2 × 106 cells) plus 20 μCi of [γ-32P]ATP (6000 Ci/mmole; NEN). Labeled products were denatured in SDS sample buffer and separated on denaturing polyacrylamide gels prior to autoradiography.
For detection of GSK-3β activity in immune complexes, 1 μg of bacterially expressed GST–D1 or GST–D1-(T286A) in 20 μl of kinase buffer was mixed with immune complexes containing GSK-3β. Reactions were initiated by addition of 10 μCi of [γ-32P]ATP (6000 Ci/mmole; NEN) and incubated at 30°C for 30 min. Labeled proteins were denatured in SDS sample buffer and separated on denaturing polyacrylamide gels prior to autoradiography.
In vitro phosphorylation of cyclin D1
Recombinant cyclin D1–CDK4 complexes were precipitated from programmed Sf9 lysates with monoclonal antibody to cyclin D1. Immune complexes were diluted into 20 μl of kinase buffer and mixed with recombinant GSK-3β (Calbiochem, La Jolla, CA), His–ERK2, or GST–SAPK plus 10 μCi of [γ-32P]ATP (6000 Ci/mmole; NEN). Reactions incubated at 30°C for 10 min were stopped by boiling in SDS sample buffer. Phosphorylated proteins were resolved on denaturing polyacrylamide gels, transferred to Immobilon-P membranes (Millipore), and visualized by autoradiographic exposure. The activity of other kinases tested for their ability to phosphorylate cyclin D1 was confirmed with other substrates. SAPK was scored for autophosphorylation, whereas ERK2 activity was monitored with myelin basic protein (Sigma) and baculovirus-produced cyclin E–CDK2 was tested with histone H1 (Boehringer Mannheim).
Metabolic labeling and two-dimensional phosphopeptide mapping of cyclin D1
NIH-3T3 cells were washed twice with PBS and refed with methionine-free medium containing 200 μCi/ml [35S]methionine (1369 Ci/mmole; ICN). Cells were labeled for 30 min except for detection of MyrAkt, in which cells were labeled for 2 hr. Cells were lysed and labeled cyclin D1 was precipitated with monoclonal antibody to cyclin D1 or with the M2 monoclonal antibody to the Flag epitope. MyrAkt was precipitated with an antiserum specific for a carboxy-terminal epitope of c-Akt, provided by Philip Tsichlis. Alternatively, Sf9 cells infected with the indicated baculoviruses were washed with phosphate-free Grace’s medium followed by a 1 hr pre-incubation in Grace’s phosphate-free medium containing 5% FCS. The cells were then labeled for 2 hr with 1 mCi/ml 32P-orthophosphate (ICN). Phosphorylated cyclin D1 was isolated by immunoprecipitation, resolved on denaturing polyacrylamide gels, transferred to Immobilon-P membranes (Millipore) and visualized by autoradiography. Membrane slices containing cyclin D1 were excised and subjected to trypsin digestion; phosphorylated cyclin D1 peptides were analyzed by electrophoresis in pH 1.9 buffer on a HTLE-7000 apparatus (CBS Scientific, Del Mar, CA) in the first dimension and ascending chromatography in the second dimension (Boyle et al. 1991b; Diehl et al. 1997).
Acknowledgments
We thank Jim Woodgett for GSK-3β cDNAs, Philip Tsichlis for MyrAkt cDNA and antibodies to the Akt protein, Melanie Cobb for ERK2, Michael White for Ras plasmids, John Kyriakis for SAPK cDNA, Natalie Ann for active MEK1 cDNA, and Jan van Deursen for a gift of leptomycin B. We also gratefully acknowledge the excellent technical assistance of Joseph Watson, Carol Bockhold, Esther Van de Kamp, Rose Mathew, and Zhen Lu, and helpful suggestions and criticisms from other members of our laboratory. This work was supported in part by National Institutes of Health grants CA-56819 (MFR), Cancer Center CORE grant CA-21765, and the American Lebanese Syrian Associated Charities (ALSAC) of St. Jude Children’s Research Hospital.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL sherr@stjude.org; FAX (901) 495-2381.
References
- Aktas H, Cai H, Cooper GM. Ras links growth factor signaling to the cell cycle machinery via regulation of cyclin D1 and the cdk inhibitor p27Kip1. Mol Cell Biol. 1997;17:3850–3857. doi: 10.1128/mcb.17.7.3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, Pestell RG. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995;270:23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- Baldin V, Lukas J, Marcote MJ, Pagano M, Draetta G. Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes & Dev. 1993;7:812–821. doi: 10.1101/gad.7.5.812. [DOI] [PubMed] [Google Scholar]
- Beals CR, Sheridan CM, Turck CW, Gardner P, Crabtree GR. Nuclear export of NF-ATc enhanced by glycogen synthase kinase-3. Science. 1997;275:1030–1033. doi: 10.1126/science.275.5308.1930. [DOI] [PubMed] [Google Scholar]
- Boudewijn M, Burgering T, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- Boyle WJ, Smeal T, Defize LH, Angel P, Woodgett JR, Karin M, Hunter T. Activation of protein kinase C decreases phosphorylation of c-Jun at sites that negatively regulate its DNA-binding activity. Cell. 1991a;64:573–584. doi: 10.1016/0092-8674(91)90241-p. [DOI] [PubMed] [Google Scholar]
- Boyle WJ, Van der Geer P, Hunter T. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Methods enzymol. 1991b;201:110–149. doi: 10.1016/0076-6879(91)01013-r. [DOI] [PubMed] [Google Scholar]
- Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M, Sexl V, Sherr CJ, Roussel MF. Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1) Proc Natl Acad Sci. 1998;95:1091–1096. doi: 10.1073/pnas.95.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- Clurman BE, Sheaff RJ, Thress K, Groudine M, Roberts JM. Turnover of cyclin E by the ubiquitin-proteosome pathway is regulated by CDK2 binding and cyclin phosphorylation. Genes & Dev. 1996;10:1979–1990. doi: 10.1101/gad.10.16.1979. [DOI] [PubMed] [Google Scholar]
- Cross DAE, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ, Chau V, Kirschner M. Ubiquitination of the G1 cyclin Cln2p by a Cdc34p-dependent pathway. EMBO J. 1995;14:303–312. doi: 10.1002/j.1460-2075.1995.tb07004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl JA, Sherr CJ. A dominant-negative cyclin D1 mutant prevents nuclear import of cyclin-dependent kinase-4 (CDK4) and its phosphorylation by CDK-activating kinase. Mol Cell Biol. 1997;17:7362–7374. doi: 10.1128/mcb.17.12.7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin–proteasome pathway. Genes & Dev. 1997;11:957–972. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DA, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Feramisco JR, Gross M, Kamata T, Rosenberg M, Sweet RW. Microinjection of the oncogenic form of human H-ras (T-24) protein results in rapid proliferation of quiescent cells. Cell. 1984;38:109–117. doi: 10.1016/0092-8674(84)90531-2. [DOI] [PubMed] [Google Scholar]
- Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051–1060. doi: 10.1016/s0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- Franke TF, Yang TO, Chan K, Datta A, Kazlauskas DK, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- Hall M, Peters G. Genetic alterations of cyclins, cyclin-dependent kinases, and cdk inhibitors in human cancer. Adv Cancer Res. 1996;68:67–108. doi: 10.1016/s0065-230x(08)60352-8. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- He X, Saint-Jeannet J-P, Woodgett JR, Varmus HE, Dawid IB. Glycogen synthase kinase-3 and dorsoventral patterning in Xenopus embryos. Nature. 1995;374:617–622. doi: 10.1038/374617a0. [DOI] [PubMed] [Google Scholar]
- Kato J-Y, Matsuoka M, Strom DK, Sherr CJ. Regulation of cyclin D-dependent kinase 4 (cdk4) by cdk4-activating kinase. Mol Cell Biol. 1994;14:2713–2721. doi: 10.1128/mcb.14.4.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- Kerkhoff E, Rapp UR. Induction of cell proliferation in quiescent NIH 3T3 cells by oncogenic c-Raf-1. Mol Cell Biol. 1997;17:2576–2586. doi: 10.1128/mcb.17.5.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RW, Deshaies RJ, Peters J-M, Kirschner MW. How proteolysis drives the cell cycle. Science. 1996;274:1652–1659. doi: 10.1126/science.274.5293.1652. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- Klinghoffer RA, Duckworth B, Valius M, Cantley L, Kazlauskas A. Platelet-derived growth factor-dependent activation of phosphatidylinositol 3-kinase is regulated by receptor binding of SH2-domain-containing proteins which influence ras activity. Mol Cell Biol. 1996;16:5905–5914. doi: 10.1128/mcb.16.10.5905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, Van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, Woodgett JR. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes & Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- Lanker S, Valdivieso MH, Wittenberg C. Rapid degradation of the G1 cyclin Cln2 induced by CDK-dependent phosphorylation. Science. 1996;271:1597–1601. doi: 10.1126/science.271.5255.1597. [DOI] [PubMed] [Google Scholar]
- Lavoie JN, L’Allemain G, Brunet A, Müller R, Pouysségur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem. 1996;271:20608–20616. doi: 10.1074/jbc.271.34.20608. [DOI] [PubMed] [Google Scholar]
- Matsushime H, Ewen ME, Strom DK, Kato J-Y, Hanks SK, Roussel MF, Sherr CJ. Identification and properties of an atypical catalytic subunit (p34PSKJ3/CDK4) for mammalian D-type G1 cyclins. Cell. 1992;71:323–334. doi: 10.1016/0092-8674(92)90360-o. [DOI] [PubMed] [Google Scholar]
- Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato J. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol. 1994;14:2066–2076. doi: 10.1128/mcb.14.3.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushime H, Roussel MF, Ashmun RA, Sherr CJ. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell. 1991;65:701–713. doi: 10.1016/0092-8674(91)90101-4. [DOI] [PubMed] [Google Scholar]
- Mittnacht S, Paterson H, Olson MF, Marshall CJ. Ras signaling is required for inactivation of the tumour suppressor pRb cell-cycle control protein. Curr Biol. 1997;7:219–221. doi: 10.1016/s0960-9822(97)70094-0. [DOI] [PubMed] [Google Scholar]
- Morgenstern JP, Land H. A series of mammalian expression vectors and characterization of their expression of a reporter gene in stably and transiently transfected cells. Nucleic Acids Res. 1990;18:1068–1077. doi: 10.1093/nar/18.4.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Mulcahy LS, Smith MR, Stacey DW. Requirement for ras proto-oncogene function during serum-stimulated growth of NIH-3T3 cells. Nature. 1985;313:241–243. doi: 10.1038/313241a0. [DOI] [PubMed] [Google Scholar]
- Nishi K, Yoshida M, Fujiwara D, Nishikawa M, Horinouchi S, Beppu T. Leptomycin B targets a regulatory cascade for crm1, a fission yeast nuclear protein, involved in control of higher order chromosome structure and gene expression. J Biol Chem. 1994;269:6320–6324. [PubMed] [Google Scholar]
- Ostrowski J, Sims JE, Sibley CH, Valentine MA, Dower ST, Meier KE, Bomsztyk K. A serine/threonine kinase activity is closely associated with a 65-kDa phosphoprotein specifically recognized by the κB enhancer element. J Biol Chem. 1991;266:12722–12733. [PubMed] [Google Scholar]
- Peeper DS, Upton TM, Ladha MH, Neuman E, Zalvide J, Bernards R, DeCaprio JA, Ewen ME. Ras signalling linked to the cell-cycle machinery by the retinoblastoma protein. Nature. 1997;386:177–181. doi: 10.1038/386177a0. [DOI] [PubMed] [Google Scholar]
- Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- Quelle DE, Ashmun RA, Shurtleff SE, Kato JY, Bar-Sagi D, Roussel MF, Sherr CJ. Overexpression of mouse D-type cyclins accelerates G1 phase in rodent fibroblasts. Genes & Dev. 1993;7:1559–1571. doi: 10.1101/gad.7.8.1559. [DOI] [PubMed] [Google Scholar]
- Reits EAJ, Benham AM, Plougastel B, Neefjes J, Trowsdale J. Dynamics of proteasome distribution in living cells. EMBO J. 1997;16:6087–6094. doi: 10.1093/emboj/16.20.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renny-Feldman RM, Correll CC, Kaplan KB, Deshaies RJ. A complex of Cdc4p, Skp1p, and Cdc53p/Cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell. 1997;91:221–230. doi: 10.1016/s0092-8674(00)80404-3. [DOI] [PubMed] [Google Scholar]
- Robbins DJ, Zhen E, Owaki H, Vanderbilt CA, Ebert D, Geppert TD, Cobb MH. Regulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitro. J Biol Chem. 1993;268:5097–5106. [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Dhand R, Gout I, Fry MJ, Waterfield M, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- Roth J, Dobbelstein M, Freedman D, Shenk T, Levine AJ. Nucleo-cytoplasmic shuttling of the hdm2 oncoprotein regulates the levels of the p53 protein via a pathway used by the human immunodeficiency virus rev protein. EMBO J. 1998;17:554–564. doi: 10.1093/emboj/17.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinfield B, Souza B, Albert I, Muller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P. Activation of the APC gene product with beta-catenin. Science. 1993;262:1731–1734. doi: 10.1126/science.8259518. [DOI] [PubMed] [Google Scholar]
- Saito Y, Vandenheede JR, Cohen P. The mechanism by which epidermal growth factor inhibits glycogen synthase kinase 3 in A431 cells. Biochem J. 1994;303:27–31. doi: 10.1042/bj3030027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octomer binding proteins with mini-extracts, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sells MA, Chernoff J. Epitope-tag vectors for eukaryotic protein production. Gene. 1995;152:187–189. doi: 10.1016/0378-1119(94)00685-l. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Mammalian G1 cyclins. Cell. 1993;73:1059–1065. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes & Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- Skowyra D, Craig KL, Tyers M, Elledge SJ, Harper JW. F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell. 1997;91:209–219. doi: 10.1016/s0092-8674(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Summers MD, Smith GE. A manual of methods for baculovirus vectors and insect culture procedures. 1987. [Google Scholar]
- Ui M, Okada T, Hazeki K, Hazeki O. Wortmannin as a unique probe for an intracellular signalling protein, phosphoinositide 3-kinase. Trends Biochem Sci. 1995;20:303–307. doi: 10.1016/s0968-0004(00)89056-8. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield M. Phosphoinositide 3-kinases: A conserved family of signal transducers. Trends Biochem Sci. 1997;22:267–272. doi: 10.1016/s0968-0004(97)01061-x. [DOI] [PubMed] [Google Scholar]
- Weber JD, Raben DM, Phillips PJ, Baldassare JJ. Sustained activation of extracellular-signal-regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G1 phase. Biochem J. 1997;326:61–68. doi: 10.1042/bj3260061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- Welsh GI, Wilson C, Proud CG. GSJ3: A shaggy frog story. Trends Cell Biol. 1996;6:274–279. doi: 10.1016/0962-8924(96)10023-4. [DOI] [PubMed] [Google Scholar]
- White MA, Nicolette C, Minden A, Polverino A, Van Aelst L, Karin M, Wigler MH. Multiple ras functions can contribute to mammalian cell transformation. Cell. 1995;80:533–541. doi: 10.1016/0092-8674(95)90507-3. [DOI] [PubMed] [Google Scholar]
- Winston JT, Coats SR, Wang Y-Z, Pledger WJ. Regulation of the cell cycle machinery by oncogenic ras. Oncogene. 1996;12:127–134. [PubMed] [Google Scholar]
- Won K-A, Reed SI. Activation of cyclin E/CDK2 is coupled to site-specific autophosphorylation and ubiquitin-dependent degradation of cyclin E. EMBO J. 1996;15:4182–4193. [PMC free article] [PubMed] [Google Scholar]
- Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of β-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes & Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]