

Abstract
Nearly all human gliomas exhibit alterations in one of three genetic loci governing G1 arrest: INK4a–ARF, CDK4, or RB. To discern the roles of CDK4 amplification and INK4a–ARF loss in gliomagenesis, we compared the behavior of astrocytes lacking a functional INK4a–ARF locus with astrocytes overexpressing CDK4. Either a deficiency of p16INK4a and p19ARF or an increase in Cdk4 allows cultured astrocytes to grow without senescence. Astrocytes overexpressing CDK4 grow more slowly than INK4a–ARF-deficient astrocytes and convert to a tetraploid state at high efficiency; in contrast, INK4a–ARF-deficient cells remain pseudodiploid, consistent with properties observed in human gliomas with corresponding lesions in these genes.
Keywords: G1 arrest, CDK4 amplification, INK4a–ARF loss, gliomagenesis, mouse astrocytes
Over half of high-grade human gliomas lack a functional INK4a–ARF locus (Jen et al. 1994; Schmidt et al. 1994) and hence can produce neither p16INK4a nor p19ARF, the two proteins encoded by this locus (Quelle et al. 1995). Most of the remaining gliomas either lack the RB gene or demonstrate a 10- to 100-fold amplification of the CDK4 locus (He et al. 1994, 1995; Ichimura et al. 1996). We are developing animal models for gliomagenesis in hopes of understanding these patterns and discerning the contributions made to tumor formation by each abnormality (Holland et al. 1998). We have taken advantage of two genetic alterations in mice: disruption of the INK4a–ARF locus by targeted mutation (Serrano et al. 1996) and astrocyte-specific expression of a transgene encoding TVA, the receptor for subgroup A avian leukosis viruses (ALV) (Holland and Varmus 1998). Production of TVA molecules by these cells makes them susceptible to infection by RCAS vectors carrying coding domains for CDK4 and other genes (Holland et al. 1998).
We have used this gene transfer system to investigate the effects of INK4a–ARF loss and CDK4 overexpression in astrocyte cell culture and to determine whether similarities exist between the cultured cells and human gliomas with similar abnormalities.
Results and Discussion
Loss of INK4a–ARF and overexpression of CDK4 both immortalize astrocyte cultures
To compare the growth properties of INK4a–ARF-deficient and CDK4-overexpressing astrocytes, we subjected appropriately selected cultures to repeated passage at standard density and counted the number of cells at each passage. Astrocytes overexpressing CDK4 were prepared by infecting primary brain cultures from Gtv-a mice [carrying a tv-a transgene under the control of the astrocyte-specific glial fibrillary acidic protein (GFAP) promoter] with an RCAS vector carrying the human CDK4 cDNA (RCAS–cdk4). Cultures of INK4a–ARF-deficient astrocytes were prepared by infecting primary brain cell cultures from Gtv-a transgenic; INK4a–ARF−/− mice with an RCAS vector bearing the puro-R gene (RCAS–puro) (Holland et al. 1998) and selecting for resistance to puromycin. Parallel cultures were prepared by infecting brain cells from Gtv-a transgenic mice with RCAS vectors carrying the alkaline phosphatase (AP) or the basic fibroblast growth factor (bFGF) coding sequences.
As illustrated in Figure 1A, control astrocytes underwent about three or four cell doublings, gradually entered senescence, and failed to survive beyond ∼50 days. Astrocytes infected with RCAS–bFGF, which induce proliferation and migration of glial cells in vivo (Holland and Varmus 1998; also see Fig. 4, below), grew to slightly greater numbers than control astrocytes, as expected in view of the known mitogenic effects of bFGF on cultured astrocytes (Hou et al. 1995) but did not survive significantly longer. In marked contrast, INK4a–ARF−/− astrocytes exhibited no loss of growth potential with passage and could be propagated indefinitely, consistent with the behavior of mouse embryo fibroblasts (MEFs) with lesions in this locus (Alcorta et al. 1996; Nobel et al. 1996) or in ARF alone (Kamijo et al. 1997). Furthermore, astrocytes infected with RCAS–cdk4 also escaped senescence, implying that both INK4a–ARF deficiency and excess CDK4 allow immortalization of astrocytes. However, the growth rates for these two populations were markedly different; INK4a–ARF−/− cultures grew at a rapid and constant rate, whereas the CDK4-immortalized cultures initially grew slowly and then increased in rate over time.
Figure 1.
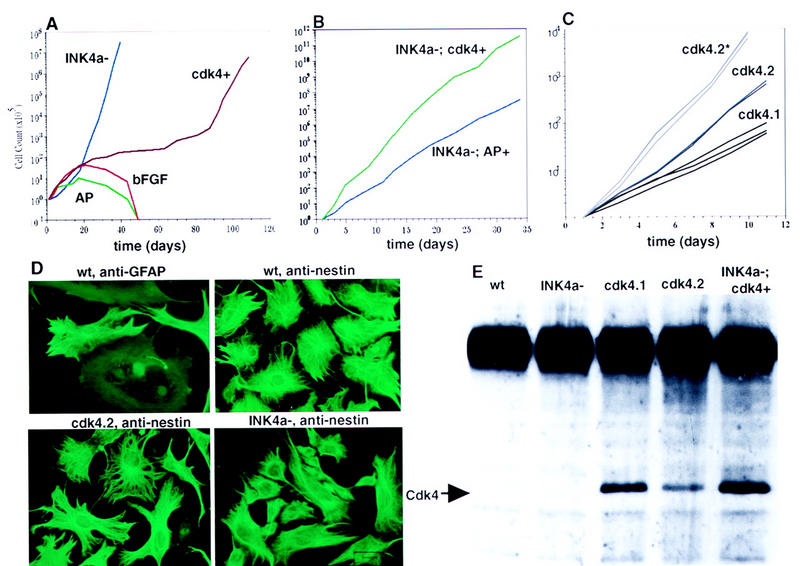
Immortalization of astrocytes by INK4a–ARF loss and cdk4 overexpression. (A) Growth curves comparing INK4a–ARF−/− astrocytes (INK4a−) with wild-type tv-a+ astrocytes infected with RCAS–CDK4 (cdk4+), RCAS–bFGF (bFGF+), and RCAS–AP (AP). (B) Comparison of growth rates between INK4a–ARF−/− astrocytes infected with RCAS–AP (INK4a−; AP+) and RCAS–CDK4 (INK4a−; cdk4+). (C) Growth curves comparing two independent Gtv-a astrocyte populations infected with RCAS–CDK4 and maintained in culture for 3 months (cdk4.1 and cdk4.2) relative to the cdk4.2 population after continuous maintenance in culture for a total of 9 months (cdk4.2*). (D) Immunofluoresence staining for GFAP and nestin in INK4a–ARF+/+ astrocytes. Anti-nestin immunofluoresence in cdk4 immortalized and INK4a–ARF−/− astrocytes. Magnification, 100×. (E) IP–Western blot analysis of cultured cells for expression of Cdk4. The arrow indicates the position of the 34-kD CDK4 gene product. (wt) Wild-type astrocytes; (cdk4.1, cdk4.2, INK4a−; cdk4+) as above.
Figure 4.
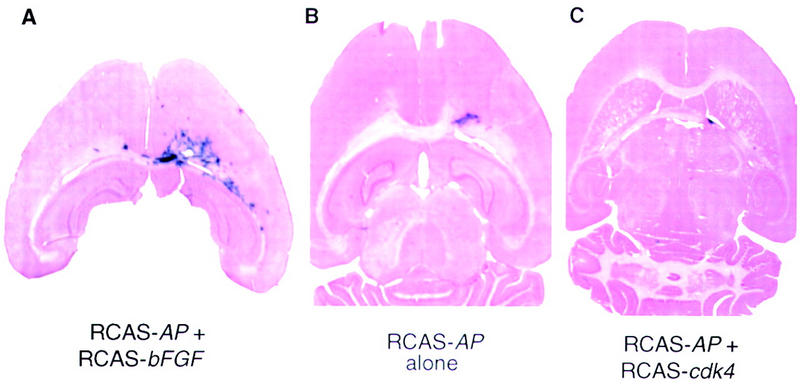
Glial-specific CDK4 gene transfer does not result in proliferation in vivo. An equal mixture of cells producing RCAS–AP and RCAS–bFGF (A), RCAS–AP alone (B), or RCAS–AP and RCAS–cdk4 (C) was injected into the right frontal lobe of Gtv-a mice. The mice were sacrificed and brains (40-μ sections) analyzed for AP activity at 10 weeks of age and counterstained.
We demonstrated that the INK4a–ARF−/− and CDK4-immortalized cultures were glia by staining cells with antibodies to GFAP (Bignami and Dahl 1976) and to nestin, an intermediate filament protein expressed in CNS progenitors (Tohyama et al. 1992). Gtv-a transgenic primary brain cultures infected with RCAS–puro initially demonstrated a significant percentage of GFAP+ cells (Fig. 1D), and virtually all cells expressed nestin as has been observed previously in rat astrocyte cultures (Gallo and Armstrong 1995). To better quantify the population of cells initially infected with RCAS vectors, Gtv-a transgenic primary brain cultures were infected with RCAS–GFP, carrying the gene for green fluorescence protein and analyzed for green fluorescence, GFAP, and nestin. All of the GFP+ cells were nestin-positive (data not shown). Importantly, 90% of the GFP+ cells also expressed GFAP, as detected by immunocytochemistry and the remaining 10% of GFP+ cells not expressing GFAP still displayed an astrocytic morphology. With continued passage, INK4a–ARF−/− and CDK4-immortalized cultures displayed a polygonal, flat morphology characteristic of cultured astrocytes and lost GFAP expression, as observed with astrocytes immortalized by other techniques (Bernard et al. 1994; Frisa et al. 1994). All cells in both populations expressed large amounts of nestin, consistent with a neuroectodermal origin (Frederiksen and McKay 1988). Very large nestin-positive cells were found in both the wild-type and cdk4-immortalized populations; in contrast, the INK4a–ARF−/− cells were primarily small and morphologically immature.
The concentration of Cdk4 in RCAS–cdk4-immortalized astrocytes maintained for 3 months in culture was ∼5- to 10-fold higher than in wild-type astrocytes (Fig. 1D). These astrocytes grew less rapidly than INK4a–ARF−/− cells, raising the possibility that excess Cdk4 is a less potent stimulator of growth than a combined deficiency of p19ARF and p16INK4a. Although we have been unable to examine astrocytes producing higher levels of Cdk4, we have asked whether the growth stimulation provided by excessive Cdk4 would occur in the absence of p19ARF and p16INK4a. Cultures from brains of INK4a–ARF−/−; Gtv-a transgenic mice were infected with RCAS–cdk4 and also produced ∼10-fold higher than normal levels of Cdk4 (Fig. 1E). These cells grew significantly faster than INK4a–ARF−/−; Gtv-a control infected cells (Fig. 1B). Thus, the growth stimulus provided by CDK4 overexpression is limited by both the CDK4 concentration and the presence of the INK4a–ARF products.
CDK4 overexpression induces hyperploidy that is INK4a–ARF dependent
Tumor cells, including glioma cells, often contain an abnormal number of chromosomes (Bigner and Mark 1984), and hyperploidy occurs when mechanisms for control of cell cycle progression have been disrupted (Tahanda et al. 1995). Therefore, we asked whether euploidy is maintained in cultured astrocytes with a deficiency of INK4a–ARF or an excess of Cdk4, and whether any abnormalities in ploidy are correlated with specific genetic mutations, as reported in human gliomas (van Meyel et al. 1994). Flow cytometry was used to assess DNA content in astrocytes with different passage histories after maintenance at confluence or after addition of nocodazole.
By these measures, six of six astrocyte cultures independently infected with RCAS–cdk4 were mostly converted to tetraploid status within 15–20 population doublings (PD), at a rate approaching 10% of cells per generation (Fig. 2A). After 25 PDs, virtually all CDK4-expressing cells had twice the normal amount of DNA at confluence, and nearly half of the nocodazole-treated cells were arrested in G2/M with an 8N DNA content (Fig. 2B). In sharp contrast, INK4a–ARF-deficient cells were very similar to wild-type astrocytes, even after >50 PDs. In nocodazole, a higher proportion of INK4a–ARF-deficient cells than wild-type cells were arrested in G2/M, but <10% of the cells had an 8N DNA content (Fig. 2B, right). These findings were confirmed by direct inspection of metaphase chromosomes (Fig. 2D); chromosome numbers in metaphase spreads from INK4a–ARF-deficient cells were ∼40 and thus diploid or pseudodiploid, whereas CDK4-overexpressing cells had approximately twice the normal number of chromosomes.
Figure 2.

RCAS–cdk4-infected astrocytes shift to tetraploidy. Flow cytometry analysis of the indicated cultures after propidium iodide staining. (A) CDK4-Immortalized cells analyzed after 4 and 15 population doublings (PDs). (B) Cultures analyzed at confluence (G1 arrest) after the indicated number of PDs and after treatment for 16 hr with 0.12 μg/ml nocodazole (right). (C) Flow cytometry of cell cultures at confluence illustrating the pseudodiploid RCAS–cdk4-infected INK4a–ARF−/−; Gtv-a astrocytes maintained for 9 months in culture compared with the hyperploid cdk4.1, cdk4.2, and cdk4.2* populations described above. (D) Metaphase spreads of diploid INK4a–ARF−/− and tetraploid CDK4-immortalized cells as indicated.
The number of recognized genetic alterations capable of immortalizing cells in culture is relatively small; and of those, only loss of p53 function and hyperproduction of Myc protein have been reported previously to induce hyperploidy. The mechanism by which elevated levels of Cdk4 induce hyperploidy is unknown but in some way must result from endoduplication of chromosomes and aberrant cell cycle arrest in G2.
We next examined the DNA content of INK4a–ARF−/−; Gtv-a astrocytes infected with RCAS–cdk4 and producing at least 10-fold more Cdk4 than normal cells (Fig. 1E). Surprisingly, these cells maintained pseudodiploidy, implying a requirement for one or both of the products of the INK4a–ARF locus for induction of inappropriate rounds of DNA replication by Cdk4. p19ARF functions by binding to Mdm2 and thereby inactivating p53 (Kamijo et al. 1997; Pomerantz et al. 1998; Zhang et al. 1998) and is involved in both G1 and G2 arrest (Quelle et al. 1997). Therefore, it may be a more likely candidate to promote Cdk4-induced hyperploidy than p16INK4a, which is known only to inhibit Cdk4 and block passage from G1 to S phase. Overexpression of CDK4 in astrocytes derived from mice with targeted mutations specific for p16INK4a or p19ARF would help identify the INK4a–ARF product required for Cdk4-induced hyperploidy.
Curiously, although mutations in p53 lead to aneuploidy in culture, a mutation that specifically eliminates the production of p19ARF results in pseudodiploid immortalized cells (Kamijo et al. 1997). It is not known whether the shift in ploidy seen in p53−/− cells is dependent on the presence of wild-type p19ARF or p16INK4a as appears to be the case for the ploidy shift due to cdk4 overexpression.
Cdk4-induced immortalization and hyperploidy can occur independent of p53 mutations
Mutations in p53 are associated with loss of growth control and chromosomal instability (Levine 1993). Thus, secondary mutations of p53 in astrocytes infected with RCAS–cdk4 could be responsible for the properties described above. To address this possibility we analyzed two independent populations of RCAS–cdk4-infected Gtv-a astrocytes that had been maintained in culture for 3 months. Both populations (cdk4.1 and cdk4.2) showed elevated levels of Cdk4 by Western analysis (Fig. 1D). Flow cytometry of these two populations (performed on day 6 of the experiment shown in Fig. 3) demonstrated that cdk4.1 cells are mostly tetraploid and cdk4.2 cells mostly octaploid.
Figure 3.
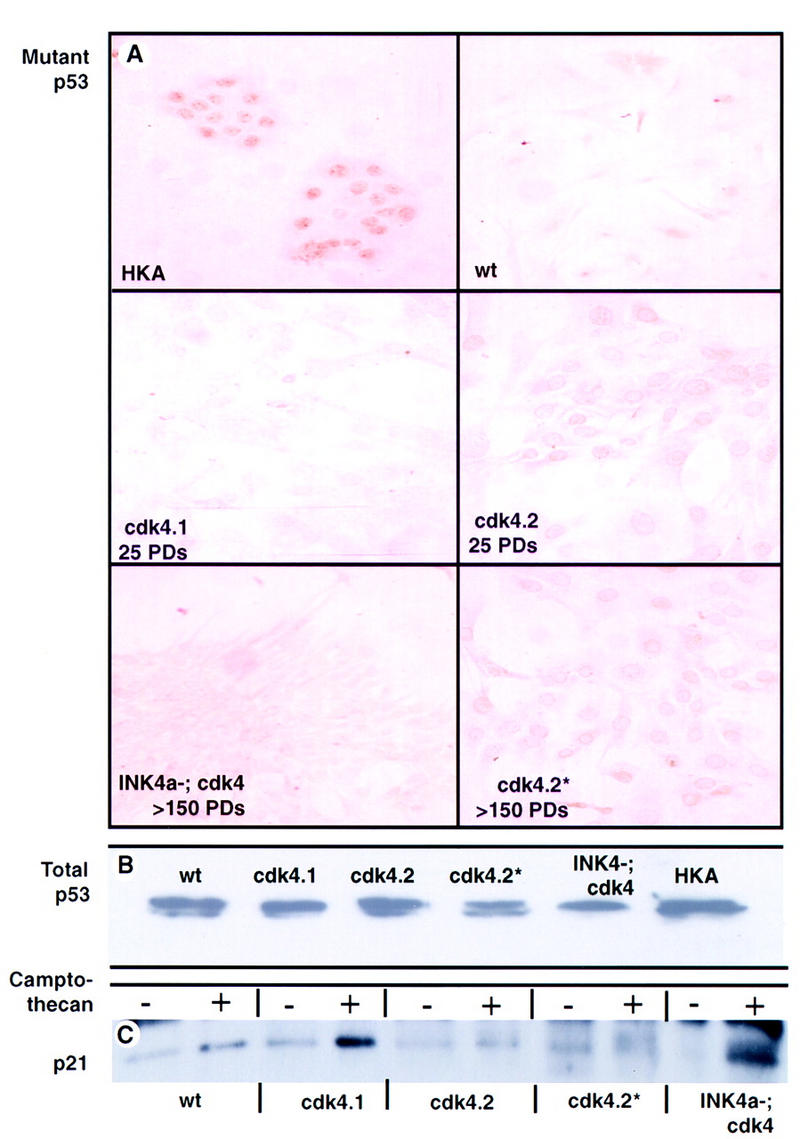
Mutations in p53 can, but do not necessarily, arise in CDK4-immortalized populations. (A) Immunoperoxidase staining of cell cultures using a monoclonal antibody to the mutant conformation of p53. Wild type, cdk4.1, cdk4.2, cdk4.2*, and INK4a−; cdk4+ are as in Fig. 1, (HKA) human keratinocyte cell line known to harbor a p53 mutation. (B) Western analysis for p53 in the indicated cultures. (C) Western analysis for p21 in cultured cells with and without camptothecan-induced DNA damage.
We judged p53 status using three criteria: immunohistochemical staining with antibodies specific for the mutant conformation of p53; the level of total p53 by Western blot analysis; and the induction of p21 after DNA damage. By all three criteria the cdk4.1 population showed no evidence of mutant p53 (Fig. 3). These data imply that in the cdk4.1 culture, immortalization and hyperploidy occurred in the presence of wild-type p53 protein and normal p53 function.
The cdk4.2 culture, in contrast, stained positively with the antibody for the mutant p53 and failed to induce p21 after camptothecan treatment. The cdk4.2 population was maintained in culture for an additional 6 months (cdk4.2*) and analyzed for ploidy, growth rate, and p53 status. The cdk4.2* cells demonstrated increased growth rate relative to that seen at 3 months (Fig. 1C), maintained evidence of mutant p53 by immunocytochemical criteria, and lacked p21 induction after camptothecan treatment. Surprisingly, flow cytometry showed that the population shifted from being mainly octaploid at 3 months to mainly tetraploid at 9 months. Presumably, the increased growth rate observed between the cdk4.2 population after 6 additional months in culture reflects the further occurrence of mutations, epigenetic events, or both.
The existence of a CDK4-immortalized and hyperploid culture with wild-type p53 function (cdk4.1) implies that CDK4-overexpressing astrocytes do not require mutations in p53 to achieve extended proliferation and genomic instability. The extended life span of the cdk4.1 population could be formally explained by a secondary mutation in INK4a–ARF or ARF alone. However, this is unlikely, as INK4a–ARF; CDK4-overexpressing astrocytes are pseudodiploid and rapidly proliferating, whereas the cdk4.1 population is polyploid and grows more slowly. Furthermore, Southern analysis of the cdk4.1 population with the p19ARF cDNA did not demonstrate deletions or alterations in the genomic structure of the INK4a–ARF locus (data not shown). These data imply that additional mutations in p53 or INK4a–ARF are not required for immortalization of CDK4-overexpressing astrocytes. In contrast, immortalization of MEFs by myc overexpression selects for those that lose either p53 or p19ARF function (Zindy et al. 1998) and MEF cultures selected for spontaneous immortalization develop mutations in p53 or ARF (Kamijo et al. 1997). Although mutations in p53 have long been known to result in both immortalization of cells in culture and hyperploidy, the pathways leading to these effects are not completely understood. One of the effects of p53 is to increase the concentration of p21, which, in turn, inhibits Cdk4. Therefore, loss of p53 function might be expected to result in higher Cdk4 activity. It would be valuable to know whether the immortalization and ploidy shifts seen in p53-deficient cells are a result of, or dependent on, inappropriately elevated Cdk4 activity.
Overexpression of cdk4 by astrocytes in vivo does not induce proliferation
To gauge the physiological significance of Cdk4 overproduction in astrocytes in vivo, we infected newborn Gtv-a transgenic mice with RCAS–cdk4. With this method, cells lining the injection track are coinfected with RCAS–AP to monitor cells (Holland and Varmus 1998). AP+ cells are no more numerous or widely dispersed after coinfection with a mixture of RCAS–AP and RCAS–cdk4 than after infection with RCAS–AP alone (Fig. 4B,C). In contrast, as described previously (Holland and Varmus 1998), AP+ cells are highly abundant and spread over a large expanse of the brain after coinfection with a mixture of RCAS–AP and RCAS–bFGF (Fig. 4A). Thus, by itself, excessive levels of Cdk4 do not appear to perturb the proliferative or migratory behavior of astrocytes in vivo.
The absence of gliomas in INK4a–ARF-deficient mice (Serrano et al. 1996) suggests that a lack of both p16INK4a and p19ARF is insufficient to produce grossly abnormal growth of glial cells in vivo; this is similar to our results with gene transfer of cdk4 to astrocytes in Gtv-a transgenic mice (Fig. 4). However, either INK4a–ARF loss or CDK4 overexpression can immortalize astrocytes in vitro; thus, additional limitations on proliferation may be mediated by unknown mechanisms in vivo. Of note, cultures of both INK4a–ARF−/− and CDK4-immortalized cells arrest in G1 upon reaching confluence, implying the existence of an intact G1-arrest pathway in these cells. Identification of factors and pathways causing growth arrest of either INK4a–ARF−/− or CDK4-immortalized astrocytes in culture may help elucidate the mechanism for arrest of these cells in vivo.
Recently, we have reported the utility of transgenes expressing tv-a in the glial lineage in intact animals (Holland and Varmus 1998; Holland et al. 1998). Here we demonstrate that primary cell lines from such tv-a transgenic animals can be used to perform high efficiency gene transfer to defined cells within a mixture of cell types. Selection for specific, infectable cell types is possible if the population is initially infected with RCAS–puro and subsequently grown in puromycin. The ability to manipulate populations of primary astrocytes genetically in culture has allowed us to study the effects of individual alterations in the G1 arrest pathways, something not possible in established cell lines in which the endogenous G1 arrest pathway has already been altered.
In Holland et al. (1998) we describe the contributions that excess Cdk4 or loss of the INK4a–ARF gene products make to gliomagenesis. We find that a constitutively active, mutant EGFR is insufficient to induce gliomas in mice, but mutant EGFR can induce glioma formation either in INK4a–ARF−/− mice or, less often, in combination with excess Cdk4. Furthermore, mutant EGFR does not induce gliomas in p53-deficient mice unless CDK4 is overexpressed. Our results demonstrate that INK4a–ARF loss and CDK4 overexpression not only immortalize astrocytes in culture, as shown here, but are also important components of gliomagenesis in mice. The fact that p53 mutations arise in some CDK4-immortalized astrocyte cultures implies that p53 loss can provide a growth advantage to these cells. The results in Holland et al. (1998) suggest that CDK4 overexpression and p53 loss also cooperate in gliomagenesis. These apparent interactions between components of the cell cycle arrest pathways in mice and in cultured mouse astrocytes resemble the genetic abnormalities found in human gliomas. Most notably, results in the two species illustrate the importance of disrupting the p16INK4a–Cdk4–Rb pathway, the nonequivalence of mutations in the pathways that govern G1–S transition, and the apparent synergy between CDK4 overexpression and p53 loss. Taken as a whole, these observations indicate that behavior of genetically defined primary astrocyte cultures reflects many aspects of gliomagenesis both in mice and man.
Materials and methods
Transgenes and viral vectors
Construction of the Gtv-a transgene and RCAS–AP and RCAS–bFGF have been described (Holland and Varmus 1998). The Gtv-a mouse line was originally generated from an FVB/N crossed with a C57B6 × BALB/c F1. The Gtv-a founder was then bred to an FVB/N to generate F1 progeny that have sunsequently been interbred to maintain the transgenic line. RCAS–puro was obtained from Steve Hughes (National Cancer Institute). RCAS–cdk4 was constructed by ClaI digestion of RCAS–puro to remove the Escherichia coli puromycin resistance gene and replacement with a BstBI–ClaI fragment from pcdk4.1 (gift from Robert Sikorski), which contains the complete human CDK4 cDNA (Matsushime et al. 1992). RCAS–GFP was a gift of Connie Cepko (Harvard University, Cambridge, MA).
Cell culture
Primary brain cell cultures from newborn transgenic mice were obtained by mechanical dissociation of the whole brain, followed by digestion with 0.25% trypsin for 15 min at 37°C. Large debris was allowed to settle, and single cells were plated and grown in DMEM with 10% FCS (GIBCO BRL). DF-1 cells (gift from D. Foster; Schaefer-Klein et al. 1998) were grown in DMEM with 5% FCS, 5% calf serum, 1% chicken serum, and 10% tryptose phosphate broth (GIBCO BRL).
Infection with RCAS vectors
The supernatant from DF-1 cells infected with and producing RCAS vectors was filtered through a 0.45-μ filter and plated directly onto primary brain cells cultures from Gtv-a mice. INK4a–ARF−/−; Gtv-a cultures were generated from the F2 progeny of Gtv-a mated with mice having targeted deletions of INK4a–ARF (gift of Ron DePinho, Harvard Medical School, Boston, MA). These primary brain cultures were infected with filtered medium from RCAS–puro-producing cells and then selected in 4 μg/ml puromycin. To infect cells in Gtv-a transgenic mice, DF-1 cells infected with RCAS vectors were harvested by trypsin digestion and pelleted by centrifugation, the cell pellets were resuspended in ∼50 μl of medium, and placed on ice. Using a 10-μl gas-tight Hamilton syringe, a single intracranial injection of 1 μl (containing 104 cells) was made in the right frontal region, just anterior to the striatum, with the tip of the needle just touching the skull base.
Brain sectioning and staining
Animals were sacrificed at 10 weeks of age, the brains fixed in 4% formaldehyde, 0.4% glutaraldehyde, 1× PBS for 36 hr, and dehydrated in 20% sucrose, 2% glycerol, and 1× PBS. Frozen sections (40 μm) were obtained using a sledge microtome (Zeiss) and stained in solution for alkaline phosphatase activity using 5-bromo-4-chloro-indolyl-phosphate and 4-nitro-blue-tetrazolium-chloride (Boehringer), after treatment at 65°C (pH 9.5) for 30 min to remove endogenous alkaline phosphatase activity. The sections were then mounted on glass slides and counterstained with hematoxylin and eosin.
Flow cytometry
Cultures were either grown to confluence and maintained for 24 hr or treated with 0.12 μg/ml nocodazole (Sigma) for 16 hr. Cells (5 × 105) were harvested by trypsin digestion, centrifuged, disbursed in 500 μl of propidium iodide solution (Electa), incubated for 20 min at 37°C, and analyzed on a Beckman FaxScan using ModFit LT software (Verity).
Immunofluorescence and immunohistochemistry
Cell cultures used for immunostaining were grown on glass coverslips precoated with 0.1 mg/ml poly-d-ornithine (Sigma). For staining with anti-GFAP antibodies, cells were fixed in 4% paraformaldehyde (pH 7.4 in PBS) for 15 min, permeabilized in 95% ethanol/5% acetic acid for 10 min, and incubated with 1:300 diluted rabbit anti-human GFAP antibody (Chemicon) for 1 hr. After incubation with fluorescein- or rhodamine-conjugated goat anti-rabbit (GAR; Cappel-Organon Teknika) for 45 min, cells were washed extensively in PBS and mounted in Vectashield (Vector Laboratories). For staining with anti-nestin antibody, cells were fixed and permeabilized as described above and incubated overnight at 4°C with an anti-nestin polyclonal rabbit antibody (gift from Ron McKay; Tohyama et al. 1992) (1:1000; in 1% fetal bovine serum + 0.5% bovine serum albumin). After incubation with fluorescein- or rhodamine-conjugated GAR for 45 min at room temperature, cells were washed extensively in PBS and mounted in Vectashield. The immunofluorescence micrographs presented are representative of two to three experiments and were taken on a Zeiss Axiophot fluorescence microscope (40× Neofluar objectives).
For detecting mutant p53, cells were initially fixed with 100% methanol and incubated with Tris-buffered saline (pH 8.0), 0.1% Tween 20, (TBST) 5% dried milk, and 1% goat serum (TBST). A mouse monoclonal antibody recognizing mutant p53 in nondenaturing conditions (Ab-3, Oncogene Science) was incubated at a 1:200 concentration in TBST for 1 hr at room temperature. The cells were washed with PBS and the antibody detected with a biotin-conjugated anti-mouse antibody and avadin-horseradish peroxidase (ABC kit, Vector Labs).
Metaphase spread
Cells were treated with 0.02 mg/ml Colcemid (Sigma) for 6 hr and harvested by trypsin digestion and centrifugation. The cells were resuspended in 0.06 m KCl, fixed in methanol/acetic acid (3:1), dropped onto glass slides, stained with 1 μg/ml DAPI (Polyscience), and visualized by fluorescence microscopy.
Western blot analysis
Total cell proteins (0.6 mg) isolated in RIPA buffer were precipitated overnight at 4°C with 0.4 μg of anti-Cdk4 antibody (Santa Cruz) using protein A–Sepharose (Sigma). The products were separated by SDS–PAGE and transferred to nitrocellulose. The blot was incubated with the same anti-Cdk4 antibody, washed extensively in TBST and incubated with a HRP-conjugated GAR antibody (Boehringer) and detected with ECL.
p21 was inducted by treating cultures at 70% confluence with 300 nm camptothecan (Sigma) for 24 hr. Total cellular protein was isolated in RIPA buffer, separated by SDS–PAGE transferred to nitrocellulose, and probed with p21 monoclonal antibodies (Ab-5, Calbiochem). The antibodies were visualized using HRP-conjugated, anti-mouse antibody (Boehringer), and ECL. For the p53 Western blot, total protein from untreated cultured cells was isolated and analyzed as for p21, using an anti-p53 antibody that recognizes both mutant and wild-type p53 proteins after denaturation under SDS–PAGE (Ab-3, Oncogene Science).
Acknowledgments
We thank Ron DePinho for the INK4a–ARF−/− mouse line, Doug Foster for the DF-1 cells, Robert Sikorski for the CDK4 cDNA, Steve Hughes for RCAS–puro vector, Connie Cepko for RCAS–GFP vector, Ron McKay for the anti-nestin antibody, Stacie Anderson for excellent assistance with the flow cytometry analysis, Zoe Weaver for expertise and help with the metaphase spread analysis, Bart Williams for many helpful discussions, and Tony Wynshaw-Boris and Yi Li for critical reading of this manuscript. E.C.H. was a Howard Hughes Physician Postdoctoral Fellow.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL eholland@notes.mdacc.tmc.edu; FAX (713) 794-4950.
References
- Alcorta DA, Xiong Y, Phelps D, Beach D, Barrett JC. Involvement of the cycline-dependent kinase inhibitor p16(INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci. 1996;93:13742–13747. doi: 10.1073/pnas.93.24.13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard R, LeBert M, Borde I, Galiana E, Evrard C, Rouget P. Immortalization of different precursors of glial cells with a targeted and temperature-sensitive oncogene. Exp Cell Res. 1994;214:373–380. doi: 10.1006/excr.1994.1270. [DOI] [PubMed] [Google Scholar]
- Bignami A, Dahl D. The astroglial response to stabbing. Immunofluorescence studies with antibodies to astrocyte-specific protein (GFA) in mammalian and submammalian species. Neuropathol Appl Neurobiol. 1976;2:99–100. [Google Scholar]
- Bigner SH, Mark J. Chromosomes and chromosomal progression of human gliomas in vivo, in vitro, and in athymic nude mice. Prog Exp Tumor Res. 1984;27:67–82. doi: 10.1159/000408223. [DOI] [PubMed] [Google Scholar]
- Frederiksen K, McKay RDG. Proliferation and differentiation of rat neuroepithelial precursor cells in vivo. J Neurosci. 1988;8:1144–1151. doi: 10.1523/JNEUROSCI.08-04-01144.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisa PS, Goodman MN, Smith GM, Silver J, Jacobberger JW. Immortalization of immature and mature mouse astrocytes with SV40 T antigen. J Neurosci Res. 1994;39:47–56. doi: 10.1002/jnr.490390107. [DOI] [PubMed] [Google Scholar]
- Gallo V, Armstrong RC. Developmental and growth factor-induced regulation of nestin in oligodendrocyte lineage cells. J Neurosci. 1995;15:394–406. doi: 10.1523/JNEUROSCI.15-01-00394.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Allen JR, Collins VP, Allalunis-Turner MJ, Godbout R, Day RS, James CD. CDK4 amplification is an alternative mechanism to p16 gene homozygous deletion in glioma lines. Cancer Res. 1994;54:5804–5807. [PubMed] [Google Scholar]
- He J, Olson JJ, James CD. Lack of p16INK4 or retinoblastoma protein (pRb) or amplification-associated overexpression of cdk4 is observed in distinct subsets of malignant glial tumors and cell lines. Cancer Res. 1995;55:4833–4836. [PubMed] [Google Scholar]
- Holland EC, Varmus HE. Basic fibroblast growth factor induces cell migration and proliferation after glia-specific gene transfer in mice. Proc Natl Acad Sci. 1998;95:1218–1223. doi: 10.1073/pnas.95.3.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland, E.C., W.P. Hively, R.A. DePinho, and H.E. Varmus. 1998. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell cycle arrest pathways to induce glioma-like lesions in mice. Genes & Dev. (this issue). [DOI] [PMC free article] [PubMed]
- Hou YJ, Yu ACH, Garcia JMRZ, Aotaki-Keen A, Lee YL, Eng LF, Hjelmeland LJ, Menon VK. Astrogliosis in culture. IV. Effects of basic fibroblast growth factor. J Neurosci Res. 1995;40:359–370. doi: 10.1002/jnr.490400310. [DOI] [PubMed] [Google Scholar]
- Ichimura K, Schmidt EE, Goike HM, Collins VP. Human glioblastomas with no alterations of the CDKN2A (p16INK4a, MTS1) and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene. 1996;13:1065–1072. [PubMed] [Google Scholar]
- Jen J, Harper JW, Bigner SH, Bigner DD, Papadopoulos N, Markowitz S, Willson JK, Kinzler KW, Vogelstein B. Deletion of p16 and p15 genes in brain tumors. Cancer Res. 1994;54:6353–6358. [PubMed] [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Dowling JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- Levine AJ. The tumor suppressor genes. Annu Rev Biochem. 1993;62:623–651. doi: 10.1146/annurev.bi.62.070193.003203. [DOI] [PubMed] [Google Scholar]
- Matsushime H, Ewen ME, Strom DK, Kato J, Hanks SK, Roussel MF, Sherr CJ. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992;71:323–334. doi: 10.1016/0092-8674(92)90360-o. [DOI] [PubMed] [Google Scholar]
- Nobel JR, Rogan EM, Neumann AA, Maclean K, Bryan TM, Reddel RR. Association of extended in vitro proliferative potential with loss of p16INK4 expression. Oncogene. 1996;13:1259–1268. [PubMed] [Google Scholar]
- Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee H, Cordon-Cardo C, DePinho RA. The INK4a tumor suppressor gene product, p19ARF, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 1998;92:713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encoding two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Quelle DE, Cheng M, Ashmun RA, Sherr CJ. Cancer-associated mutations at the INK4a locus cancel cell cycle arrest by p16INK4a but not by the alternative reading frame protein p19ARF. Proc Natl Acad Sci. 1997;94:669–673. doi: 10.1073/pnas.94.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer-Klein J, Givol I, Barsov EV, Witcomb JM, VanBrocklin M, Foster DN, Federspiel MJ, Hughes SH. The EV-O-derived cell line DF-1 supports the efficient replication of avian leukosis-sarcoma viruses and vectors. Virology. 1998;248:305–311. doi: 10.1006/viro.1998.9291. [DOI] [PubMed] [Google Scholar]
- Schmidt EE, Ichimura K, Reifenberger G, Collins VP. CDKN2 (p16/MTS1) gene deletion or cdk4 amplification occurs in the majority of glioblastomas. Cancer Res. 1994;54:6321–6324. [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell motility. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Tahanda AM, Bruner JM, Donehower LA, Morrison RS. Astrocytes derived from p53-deficient mice provide a multistep in vitro model for development of malignant gliomas. Mol Cell Biol. 1995;15:4249–4259. doi: 10.1128/mcb.15.8.4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohyama T, Lee V, Rorke LB, Marvikn M, McKay RD, Trojanowski JQ. Nestin expression in embryonic human neuroepithelium and in human neuroepithelial tumors. Lab Invest. 1992;66:303–313. [PubMed] [Google Scholar]
- van Meyel DJ, Ransay DA, Casson AG, Keeney M, Chambers AF, Cairncross JG. p53 mutation, expression and DNA ploidy in evolving gliomas: evidence for two pathways of progression. J Natl Cancer Inst. 1994;86:1011–1017. doi: 10.1093/jnci/86.13.1011. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes & Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]