

Abstract
Budding yeast RNA polymerase III (Pol III) contains a small, essential subunit, named C11, that is conserved in humans and shows a strong homology to TFIIS. A mutant Pol III, heterocomplemented with Schizosaccharomyces pombe C11, was affected in transcription termination in vivo. A purified form of the enzyme (Pol III Δ), deprived of C11 subunit, initiated properly but ignored pause sites and was defective in termination. Remarkably, Pol III Δ lacked the intrinsic RNA cleavage activity of complete Pol III. In vitro reconstitution experiments demonstrated that Pol III RNA cleavage activity is mediated by C11. Mutagenesis in C11 of two conserved residues, which are critical for the TFIIS-dependent cleavage activity of Pol II, is lethal. Immunoelectron microscopy data suggested that C11 is localized on the mobile thumb-like stalk of the polymerase. We propose that C11 allows the enzyme to switch between an RNA elongation and RNA cleavage mode and that the essential role of the Pol III RNA cleavage activity is to remove the kinetic barriers to the termination process. The integration of TFIIS function into a specific Pol III subunit may stem from the opposite requirements of Pol III and Pol II in terms of transcript length and termination efficiency.
Keywords: S. cerevisiae, RNA polymerase III, C11 subunit, RNA cleavage activity, termination
Transcription in eukaryotic cells is carried out by three functionally and structurally related but distinct forms of nuclear RNA polymerase (Pol I, II, and III). In terms of RNA synthesis, one of the major differences between Pol III and the two other enzymes is the length of the genes they have to transcribe. Whereas Pol III synthesizes RNAs of 100–150 nucleotides, Pol I and Pol II transcribe genes of several kilobases (kb). Transcription requirements in terms of processivity of elongation and termination efficiency are thus likely to be different for Pol III as compared to Pol I or Pol II. Recent studies have revealed the elongation and termination phases of transcription to be important targets for the regulation of eukaryotic gene expression (Spencer and Groudine 1990; Kerppola and Kane 1991; Kane 1994).
Transcribing RNA polymerase encounters diverse obstacles to elongation causing it to pause, to become arrested, or to terminate. Transcription arrest is attributable to specific DNA sites that trap a fraction of elongating RNA polymerase molecules, resulting in locked ternary complexes that can neither propagate nor dissociate (Levin and Chamberlin 1987; Reines et al. 1989; Sluder et al. 1989; Kerppola and Kane 1990; SivaRaman et al. 1990; Surratt et al. 1991). The cleavage of the nascent transcript in the ternary complex has emerged as a mechanism for countering elongation arrest. The 3′ → 5′ processive RNA hydrolytic activity is ubiquitous and most likely resides in the polymerase itself, but it is generally a factor-stimulated reaction. In Escherichia coli, two types of cleavage reactions were reported, a 3′ proximal cleavage that releases fragments of 2–3 nucleotides and depends on the cleavage factor GreA (Borukhov et al. 1992) and a 3′ distal cleavage occurring at a distance of 10–11 nucleotides from the transcript end mediated by the cleavage factor GreB (Borukhov et al. 1993). The anti-arrest mechanisms of these two factors appear to be different, although the two proteins display strong sequence homology. Whereas GreB restarts arrested complexes by way of RNA retraction, the action of GreA is more complex. GreA does not rescue arrested complexes but prevents the arrest if added before the blockage. It also induces an efficient cleavage on halted, but not arrested complexes (Borukhov et al. 1993). A parallel situation is observed for eukaryotic Pol II. The transcription elongation factor (TFIIS/SII) activates the nascent RNA cleavage activity of the enzyme. This factor enables Pol II to transcribe through a variety of transcription blocks including intrinsic arrest sites and nucleoprotein complexes (for review, see Reines 1994; Uptain et al. 1997). Quite similarly to the eubacterial enzyme, transcriptionally incompetent (arrested) or competent (halted) Pol II ternary complexes generate different cleavage products in a TFIIS-mediated cleavage reaction. Arrested complexes yield predominantly 7- to 14-nucleotide RNA fragments, whereas halted complexes generate primarily dinucleotide cleavage products (Izban and Luse 1993). Nascent RNA cleavage activity has been described for yeast Pol I as well. This activity is likely to be factor stimulated, but the protein responsible for the cleavage activation remains to be determined (Tschochner 1996; Labhart 1997). Highly purified yeast Pol III was found to possess an RNA chain retracting hydrolytic activity that cleaves the nascent RNA from its 3′ end (Whitehall et al. 1994). Cleavage occurs 5′ to an internal uridylate residue with released RNA fragments of 1–3 nucleotides depending on the sequence of the transcript (Bobkova and Hall 1997). This activity is likely factor independent (Whitehall et al. 1994) and thus appears to be more closely related to the intrinsic nucleolytic activity of vaccinia virus RNA polymerase (Hagler and Shuman 1993) than to the TFIIS-dependent cleavage activity of Pol II. The function generally attributed to the RNA polymerase cleavage activity is the restoration of a correct alignment of the transcript in the active site after retraction of the enzyme from a blocked situation, and possibly a role in transcriptional proofreading (Erie et al. 1993; Jeon and Agarwal 1996; Bobkova and Hall 1997). A puzzling observation, however, is the nonessential nature in E. coli or in yeast of the cleavage-stimulatory factors (Exinger and Lacroute 1992; Borukhov et al. 1993). Similarly, the Pol II subunit B12.6, which is involved in the cleavage activity (Awrey et al. 1997), is not an essential protein in yeast (Woychik et al. 1991). However, RpII15 protein, the Drosophila ortholog of subunit B12.6, is essential for viability (Harrison et al. 1992). Therefore, it would be interesting to know whether TFIIS is essential in higher organisms.
In this study, we characterize a new, small subunit of yeast Pol III, C11, homologous to Pol I A12.2 and Pol II B12.6 subunits and to elongation factor TFIIS. Subunit C11 is essential for cell viability and is required for the intrinsic 3′ exonucleolytic cleavage activity of Pol III ternary complexes. In vitro, a Pol III lacking C11 displays strong defects in elongation and termination properties. The data indicate that Pol III intrinsic cleavage activity is important for transcription termination. This observation may account for the essential character of the C11 subunit. A model is presented in which we propose that C11 allows the enzyme to switch between an RNA elongation and RNA cleavage mode, which facilitates termination, although transcript hydrolysis is unlikely to be a step in termination mechanism.
Results
C11 is an essential Pol III subunit
A small polypeptide with an apparent molecular mass of 11 kD is present in all purified Pol III preparations. We purified this polypeptide by SDS-PAGE and determined the sequence of one peptidic fragment obtained by trypsin digestion. An unambiguous amino acid sequence (KEVDDVLGG) was used for a gene data bank search. A 100% identical sequence was found in a unique hypothetical yeast protein of 110 residues (YDR045c) of 12,504 calculated molecular weight, which we named C11. The gene encoding this protein, named RPC11, is unique and maps on chromosome IV.
To demonstrate that C11 is a genuine Pol III subunit, we constructed a strain expressing the RPC11 open reading frame (ORF) under the control of the galactose-inducible GALS promoter. At various times after the shift to a glucose medium, cells were labeled with [3H]uracil for 30 min, and total RNA was extracted and analyzed by gel electrophoresis. Although the levels of rRNA species (28S, 18S, 5.8S, and 5S RNA) remained unchanged, the level of tRNAs decreased markedly after 12 hr of glucose repression of RPC11. Autoradiographic analysis of the labeled transcripts revealed a similar drop in tRNA synthesis followed by a decrease in 5.8S and 5S RNA synthesis after 24 hr of growth in glucose medium (data not shown). These results suggested that C11 is part of the Pol III transcription machinery, or is involved in its regulation.
A gene disruption experiment was performed to examine whether C11 was required for cell viability. A diploid yeast strain in which one of the two chromosomal copies of RPC11 was replaced by the HIS3 cassette was sporulated and several tetrads were dissected. Spore viability and His requirement of viable spores were examined at 30°C. All the tetrads examined produced two viable colonies that were His− (data not shown). Deletion of RPC11 did not result in defective germination as His+ spores germinated but failed to divide after two or three generations. Haploid cells containing the chromosomal disrupted allele and a plasmid harboring the wild-type RPC11 gene were viable. These results demonstrated that the protein encoded by RPC11 is essential for cell viability.
C11 is a TFIIS-like Pol III subunit conserved from yeast to humans
Sequence analysis indicated that C11 contains two potential zinc-binding motifs (an amino-terminal motif CX2CX17CX2C and a carboxy-terminal motif CX5CX24CX2C) separated by an intermediate domain without any particular motif. This tripartite structure is reminiscent of the structure of the A12.2 subunit of Pol I (Nogi et al. 1993) and of the B12.6 subunit of Pol II (Woychik et al. 1991). Sequence comparison of these three small subunits is shown in Figure 1A. Their sequence similarity is highest in the carboxy-terminal region (amino acid positions 67–110 of C11), which encompasses the second zinc-binding motif, but is also significant in the amino-terminal region (amino acid positions 1–39 of C11). The intermediate regions of the three proteins are unrelated. These observations, combined with their similarity in size (125, 122, and 110 residues, respectively), suggested that A12.2, B12.6, and C11 are structurally and functionally homologous proteins and strongly suggested that C11 is a genuine Pol III subunit.
Figure 1.
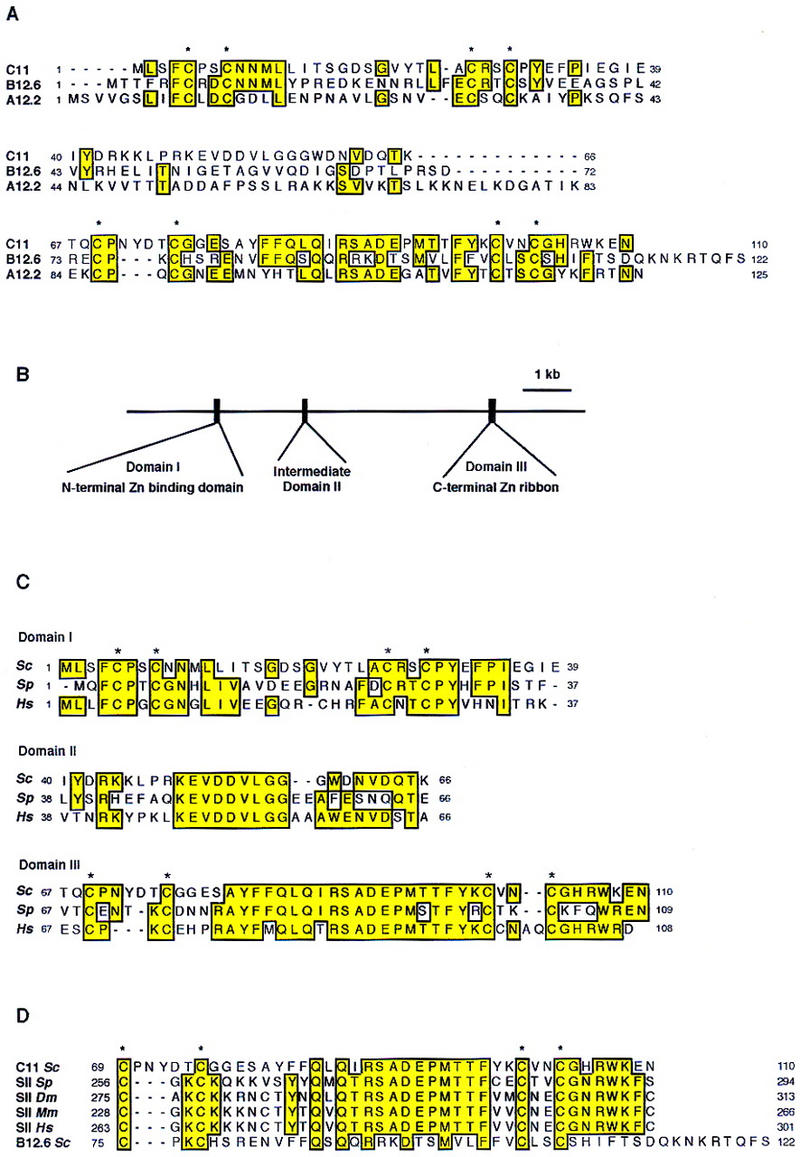
Sequence alignment of C11 with RNA polymerase subunits and factor TFIIS. (A) Alignment of the C11 sequence with Pol I A12.2 and Pol II B12.6 subunits from the yeast Saccharomyces cerevisiae. Amino acids identical in at least two sequences are boxed. Cysteines of the two potential zinc-binding domains are indicated by stars. (B) Schematic representation of the human gene encoding HsC11p. (C) Alignment of the sequences of C11 subunits from S. cerevisiae (C11), S. pombe (SpC11p), and Homo sapiens (HsC11p). Amino acids identical in at least two sequences are boxed. Cysteines of the two potential zinc-binding domains are indicated by stars. (D) Sequence alignment of the carboxy-terminal zinc-binding domains of Pol III C11 subunit, of Pol II B12.6 subunit, and of elongation factor TFIIS from S. cerevisiae (Sc), Drosophila melanogaster (Dm), Mus musculus (Mm), and H. sapiens (Hs). Amino acids identical in at least four sequences are boxed.
Two proteins with significant similarity to C11 were found in the National Center for Biotechnology Information (NCBI) data library. A Schizosaccharomyces pombe protein (accession no. Z99295) and a human protein (accession no. Z69719) that we named tentatively SpC11p and HsC11p, respectively. The three proteins are almost identical in size (110, 109, and 108 residues for the Saccharomyces cerevisiae, S. pombe, and human proteins, respectively) and their sequence alignment clearly demonstrate that they are orthologs (Fig. 1C). Whereas the two yeast genes do not contain intron, the human gene spreads over 7 kb on the short arm of chromosome 16 and contains two introns (Fig. 1B). Each of the three exons encodes a well-delineated domain of the HsC11p. The first and the third exons encode the amino-terminal and the carboxy-terminal potential zinc-binding domains of HsC11p. Sequence identity is particularly high in the carboxy-terminal zinc-binding motif. Interestingly, the 9-residue sequence KEVDDVLGG in the intermediate domain is strictly conserved between the three subunits. Because no homology was found between the intermediate domains of subunits A12.2, B12.6, and C11, we suggest that the sequence KEVDDVLGG is a Pol III ‘signature’ and that the intermediate domain plays a role in addressing these subunits to their cognate form of RNA polymerase.
Surprisingly, a striking sequence similarity was observed between the carboxy-terminal zinc-binding motif of C11 and the zinc ribbon of TFIIS, the eukaryotic elongation factor involved in Pol II transcription (48% identity and 67% similarity over amino acid positions 69–110 of C11; see Fig. 1D). The carboxy-terminal zinc-binding motif of B12.6 also shows some similarity to the TFIIS zinc ribbon (23% identity and 44% similarity over amino acid positions 75–122 of B12.6, see Fig. 1D). Using a randomizing test that evaluates the significance of pairwise similarities (Pearson 1990), we found that whereas C11 is significantly similar to TFIIS, B12.6 is not (data not shown). The term of elongation factor for TFIIS is a misnomer as this protein does not increase the elongation rate of Pol II, in contrast to TFIIF or TFIIX (Izban and Luse 1992). TFIIS is more specifically a cleavage stimulatory factor because it is required for cleavage of the nascent transcript in arrested Pol II transcription complexes (Reines 1994). Because cleavage of the nascent transcript is an intrinsic property of the Pol III enzyme (Whitehall et al. 1994) and as the C11 subunit harbors a TFIIS-like domain, it suggested to us that this subunit played a role in the transcript cleavage activity of Pol III. To test this hypothesis, we decided to analyze mutant enzymes defective in the C11 subunit.
Pol III Δ: a new form of enzyme lacking the C11 subunit
One of the ways to obtain informative mutants in RNA polymerase subunits is to take advantage of heterospecific complementation of a disrupted S. cerevisiae gene (Shpakovski et al. 1995). Therefore, we wondered whether the expression of SpC11p or of HsC11p could complement a disruption of RPC11 in S. cerevisiae and could possibly induce a useful mutant phenotype. Plasmid shuffle experiments (see Materials and Methods) indicated that SpC11p, but not HsC11p, was able to replace C11 functionally in S. cerevisiae. In addition, the heterocomplemented strain displayed a growth defect at 30°C (160 min generation time as compared to 110 min for the control strain) and was both cryosensitive at 16°C and strictly thermosensitive at 37°C (Fig. 2A). This result demonstrated that SpC11p is indeed the S. pombe ortholog of C11.
Figure 2.

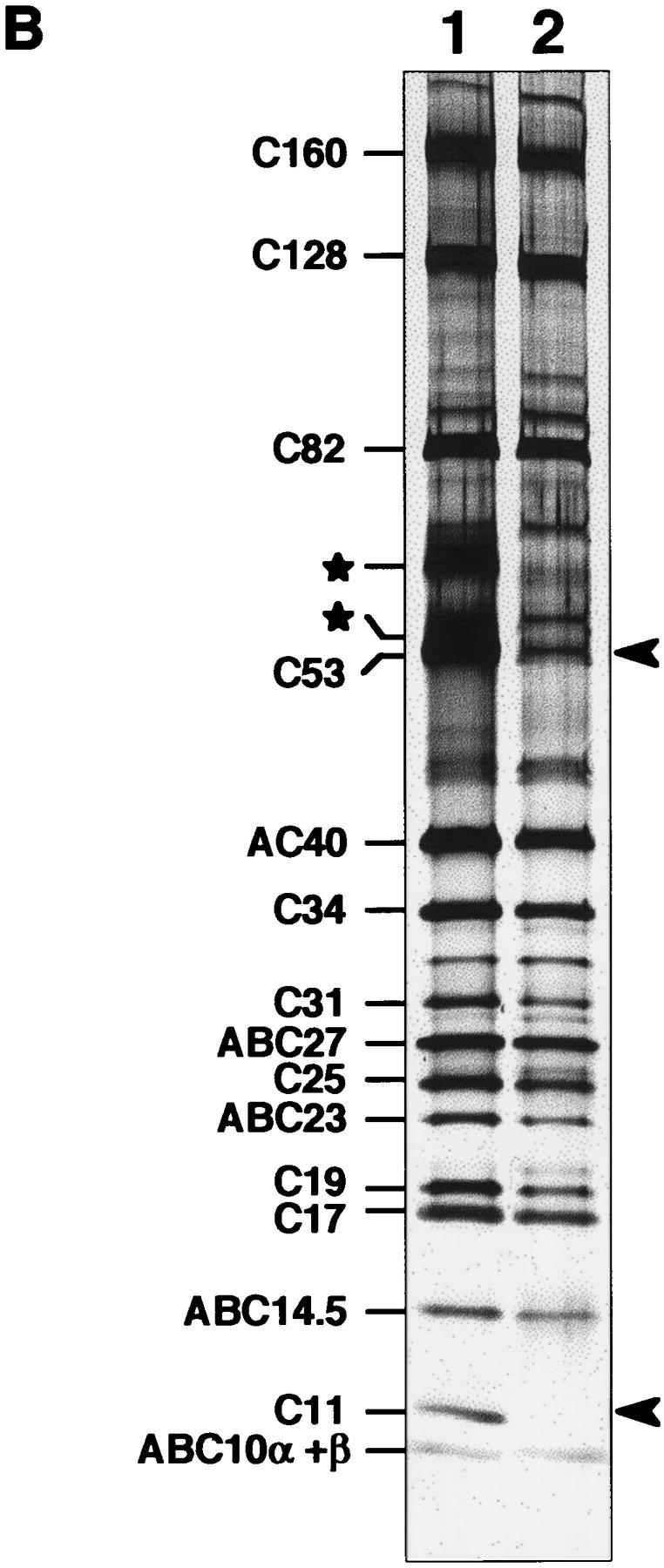
Heterocomplemention of C11 by its S. pombe ortholog SpC11p. (A) Interspecific complementation of rpc11::HIS3 by S. pombe gene encoding SpC11p. Haploid strains harboring the rpc11::HIS3 allele were complemented by pGEN–RPC11 (constitutively expressing the C11 subunit) or pGEN–SpC11 (constitutively expressing the S. pombe SpC11p subunit). These constructions were obtained by plasmid shuffling (see Materials and Methods). Growth was tested by spotting 5 μl of different dilutions of a liquid cell culture on YPD plates and incubating at 16°C, 30°C and 37°C for 7, 3, and 5 days, respectively. (B) Pol III Δ lacks subunit C11. Wild-type Pol III (lane 1) and Pol III purified from the heterocomplemented strain (Pol III Δ, lane 2) were analyzed by electrophoresis in a 4–15% SDS–polyacrylamide gel and silver stained. (*) Two major contaminants present in the wild-type enzyme preparation; (arrowheads) positions of C11 and C53 subunits in the wild-type enzyme.
Pol III was purified from the heterocomplemented strain according to the usual micropurification procedure (Huet et al. 1996). Half as much enzyme was obtained from mutant cells as compared to wild-type cells. The subunit composition of the wild-type and mutant enzymes differs only with regards to C11. It is likely that SpC11p was weakly associated with Pol III in vivo and was lost during purification. C53 subunit was also under-represented (Fig. 2B). Western blot analysis indicated that ∼40% of the C53 subunit was present in the mutant Pol III (data not shown), but variable stoichiometry has been previously reported for the C53 subunit in Pol III purified from wild-type cells (Sentenac 1985). The form of Pol III lacking C11 (hereafter called Pol III Δ) was active when assayed in a nonspecific transcription assay with poly [d(A–T)] as template, which demonstrated that the C11 subunit is not required for the basal process of RNA synthesis (data not shown). The availability of Pol III Δ prompted us to test the possible role of the C11 subunit in the intrinsic cleavage activity of Pol III.
Transcript cleavage requires the C11 subunit
The intrinsic RNA cleavage activity of Pol III Δ was studied initially by analyzing the time-dependent cleavage of the nascent transcript by stalled transcription complexes in the presence of MgCl2 (Whitehall et al. 1994). We took advantage of the SUP4-o tRNATyr (SUP4) gene sequence, which allows the synthesis of a 17-mer transcript in the absence of GTP (Fig. 3A). Stable preinitiation complexes were formed with TFIIIB and TFIIIC, then limiting amounts of wild-type or mutant Pol III were added together with ATP, CTP, and [α32P]UTP, to form halted ternary complexes containing the labeled 17-mer nascent RNA. The expected 17-mer RNA was synthesized by the wild-type enzyme, but surprisingly, a 18-mer transcript was made by Pol III Δ (Fig. 3, B and C, lanes 1,2). As several lines of evidence indicated that B12.6, the Pol II subunit homologous of C11, is required for accurate start site selection (Furter-Graves et al. 1994; Hull et al. 1995), we suspected that Pol III Δ initiated at position −1. To test whether initiation was correct ternary complexes were formed on the SUP4 gene in the presence of ATP, CTP, UTP, and 3′-O-methyl-GTP (3′-OMe-GTP). The GTP analog was expected to be incorporated into the RNA at position +18 and to block further elongation. Under these conditions, the same 18-mer transcript (18*-mer) was synthesized by both Pol III and Pol III Δ (Fig. 3B). This result demonstrated that both enzymes used the same transcription start site. Correlatively, we infer that Pol III Δ incorporated irreversibly one mispaired nucleotide at the 3′ end of the nascent RNA in the absence of GTP (we did not attempt to identify the extra nucleotide incorporated by Pol III Δ in the absence of GTP). We cannot formally exclude a more efficient incorporation of contaminating GTP by the Pol III Δ but this possibility appears unlikely as only a 18-mer transcript was synthesized quantitatively.
Figure 3.


Stalled Pol III Δ incorporates an additional nucleotide and does not cleave the nascent RNA transcript. (A) Sequence of the first 22 bases of the coding strand of SUP4 gene. The size of the 17-mer transcript obtained in the absence of GTP (3 XTPs) is indicated. (B) A stable initiation complex was formed on the SUP4 gene as described in Materials and Methods. Wild-type Pol III or Pol III Δ were then added together with ATP (600 μm), CTP (600 μm), and [α-32P] UTP (3 μm, 400 μCi/nmole) in the absence or in the presence of 3′-OMe-GTP (600 μM) as indicated. Reaction products were separated by electrophoresis on a 15% polyacrylamide, 7 m urea gel. The position of the 17-mer, 18-mer, and 18*-mer transcripts (see text) is indicated. (C) A stable initiation complex was formed as described above. Wild-type Pol III or Pol III Δ were then added together with ATP (600 μm), CTP (600 μm), and [α-32P] UTP (3 μm, 400 μCi/nmole) without recombinant C11 subunit (lanes 1,2, respectively). A 50 m excess of recombinant C11 subunit was incubated for 10 min with Pol III Δ before the transcription reaction (lane 3) or added for 2 min after the transcription reaction (lane 4). Reaction products were separated by electrophoresis on a 15% polyacrylamide, 7m urea gel. The positions of the 17-mer and 18-mer, transcripts (see text) are indicated. (D) Transcript cleavage by halted ternary complexes. Wild-type Pol III or Pol III Δ ternary complexes formed in the presence of 3 XTPs were isolated on Sepharose CL-2B columns as described in Materials and Methods. Pol III Δ ternary complexes were then incubated or not for 10 min with a 50 m excess of recombinant C11 subunit. Ternary complexes were then incubated in transcription buffer containing 8 mm MgCl2 in the absence of nucleotide for various periods of time. Sizes of transcripts are at left.
Importantly, when the recombinant C11 subunit (rC11) was preincubated for 10 min with Pol III Δ before the transcription reaction, a 17-mer transcript was observed instead of the 18-mer (Fig. 3C, lane 3). The same result was obtained when rC11 was added for 2 min to the reaction mixture after the synthesis of the 18-mer transcript (Fig. 3C, lane 4). Altogether, these data indicated that under our experimental conditions the RNA cleavage activity of Pol III was dependent on C11.
To confirm this conclusion, ternary complexes halted by omission of GTP in the transcription reaction were purified by gel permeation and incubated for various periods of time in the presence of MgCl2 and in the presence or in the absence of rC11 (Whitehall et al. 1994). As shown in Figure 3D, the wild-type enzyme cleaved the 17-mer in seconds and a 15-mer transcript accumulated in <1 min (Fig. 3D). No cleavage of the 18-mer transcript by Pol III Δ was detected, whereas an efficient cleavage was observed when rC11 was added to the purified ternary complexes (Fig. 3D). As described for the wild-type enzyme (Whitehall et al. 1994), cleavage mediated by rC11 was Mg2+ dependent (data not shown). Interestingly, no cleavage was observed if rC11 was incubated with the ternary complex before its purification by gel permeation (data not shown). This observation suggested a weak interaction between rC11 and Pol III Δ.
Taken together, these results demonstrated that the C11 subunit has a direct role in the intrinsic cleavage property of the Pol III enzyme.
Whereas purified wild-type ternary complexes containing either the 17-mer or the 18*-mer transcript were able to resume elongation in the presence of the four nucleoside triphosphates (Fig. 4A; data not shown), on the contrary, purified Pol III Δ ternary complexes containing the 18-mer RNA became transcriptionally incompetent (Fig. 4B). Pol III Δ was unable to resume elongation probably because a mispaired nucleotide was present at the 3′ extremity of the transcript. On the other hand, purified mutant ternary complexes resumed elongation of the 18-mer transcript when preincubated with rC11 (Fig. 4C). This result emphasized the critical role of the cleavage activity when elongating Pol III become trapped in a transcriptionally incompetent state.
Figure 4.

Stalled Pol III Δ ternary complexes are transcriptionally incompetent. Kinetic of elongation on the SUP4 gene by wild-type enzyme (A) or Pol III Δ (B,C). Wild-type Pol III-labeled 17-mer ternary complexes (A) or Pol III Δ-labeled 18-mer ternary complexes (B) were isolated and elongation was resumed for various periods of time as indicated, by adding unlabeled ATP, CTP, GTP (600 μm each) and UTP (30 μm). The size of the RNA transcripts and the four major transcriptional pause zones (P1–P4) are indicated at left. (C) Isolated Pol III Δ-labeled 18-mer ternary complexes were incubated for 10 min with a 50 m excess of recombinant C11 subunit, then incubated with unlabeled nucleotides as above for 5 or 60 sec, as indicated.
Pol III Δ is affected in pausing and termination
During transcription, Pol III carries out multiple cleavage and synthesis steps at intrinsic arrest sites and at terminators (Bobkova and Hall 1997). Therefore, it was of interest to analyze the behavior of Pol III Δ when full-length transcripts are being made. On a SUP4 gene as template, the wild-type enzyme synthesized one predominant transcript, whereas two discrete transcripts were generated by Pol III Δ (Fig. 5A, lanes 1, 2). One of them corresponded in size to the wild-type transcript and the second, approximately twice as abundant, was ∼10 nucleotides longer. Synthesis of these two transcripts was TFIIIB- and TFIIIC-dependent (data not shown). Because Pol III Δ had no defect in selection of the transcription initiation start site (see above), we attributed the altered transcript size to an effect on transcription termination. Nevertheless, a primer extension experiment was performed to confirm that the two transcripts synthesized by Pol III Δ differed at their 3′ end and were not attributable to a major initiation defect. Indeed, the same unique amplified DNA fragment was obtained with SUP4 tRNA synthesized by the wild-type or the mutant enzyme (data not shown). This result confirmed that Pol III Δ was affected in transcription termination.
Figure 5.

Pol III Δ is affected in pausing and transcription termination. (A) Termination and pausing on SUP4 gene. A schematic representation of the SUP4 gene terminator with the T1 and T2 blocks is shown. Stable initiation complexes were formed on the SUP4 gene as described in Materials and Methods. Wild-type enzyme (lanes 1,3) or Pol III Δ (lanes 2,4) was then added together with ATP, CTP, GTP (600 μm each) and [α-32P]UTP (30 μm, 20 μCi/nmole). Reaction products were separated by electrophoresis on a 6% polyacrylamide, 7 m urea gel. Lanes 3 and 4 represent a longer exposure time of lanes 1 and 2 to reveal paused transcripts. Large transcripts accumulating at the top of the gel are indicated as RNAs x. The positions of full-length transcripts terminated at T1 or T2 and RNAs corresponding to transitory paused ternary complexes (P2–P4) are indicated. (B) Pol III Δ reads through SNR6 gene terminator. Transcription was performed on a purified DNA fragment of 200 bp obtained after double digestion HindIII–SmaI of the pTaq6 plasmid that contains the truncated version of the yeast SNR6 gene (see Materials and Methods). The SmaI site is located 40 nucleotides downstream of the SNR6 gene terminator sequence. Wild-type Pol III or Pol III Δ were added together with ATP, CTP, GTP (600 μm each) and [α-32P] UTP (30 μm, 20 μCi/nmole) in the absence or in the presence of TFIIIB prealably assembled onto the DNA. Reaction products were separated by electrophoresis on a 6% polyacrylamide, 7 m urea gel. The positions of the full-length U6 RNA (U6) and of the run-off transcript (RO) are indicated.
This conclusion was strengthened by the similarity of the pattern of SUP4 gene transcripts synthesized by the Pol III Δ with those synthesized by Pol III mutants with decreased termination phenotypes (Shaaban et al. 1995). The SUP4 gene terminator is a T7GT6 sequence in the nontranscribed strand of DNA (Fig. 5A). Wild-type Pol III mostly terminates within the T7 stretch and SUP4 transcripts were found to be terminated with five, six, or seven U residues (Matsuzaki et al. 1994). Different mutants in the second largest subunit of Pol III with altered termination behavior have been described (Shaaban et al. 1995). Transcription termination on SUP4 gene by decreased termination mutants occurred predominantly within the T6 element of the terminator, giving rise to two sets of terminated RNAs differing by ∼7 nucleotides in length. Increased termination mutants required a longer time to traverse a template gene than wild-type Pol III; the converse holding true for most decreased termination mutants (Shaaban et al. 1996). The changes in elongation kinetics could be accounted for by a correspondingly longer or shorter dwell time at pause sites. A similar observation was made with Pol III Δ. When transcribing the SUP4 gene by Pol III Δ, no paused transcripts shorter than full-length RNA were detected under the conditions used (Fig. 5A, lane 4). In contrast, as described previously (Matsuzaki et al. 1994), wild-type Pol III paused at several defined sites (P1–P4) during transcription (Fig. 5A, lane 3). Altogether, these data demonstrated that Pol III Δ had clearly modified elongation properties such that many pause sites were not recognized.
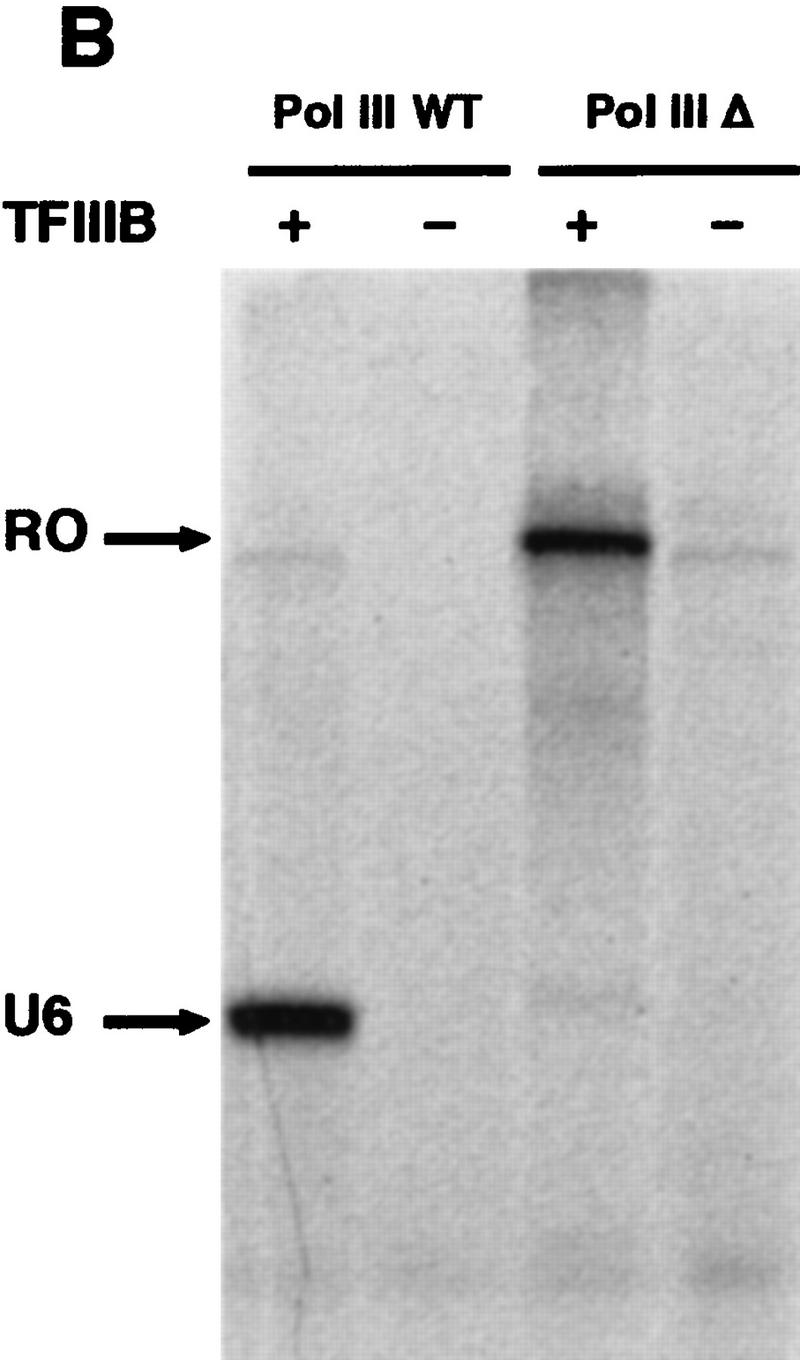
Remarkably in these experiments we observed transcripts that accumulated at the top of the gel (RNAs x) much more so with Pol III Δ than with the wild-type enzyme (Fig. 5A). Accumulation of such large transcripts was also observed when transcribing other DNA templates by Pol III Δ (data not shown). The presence of these long transcripts, which were transcription factor dependent (data not shown), suggested that a significant proportion of the elongating Pol III Δ molecules readthrough the terminator of class III genes.
To test the readthrough hypothesis, we used as template the SNR6 gene (encoding the U6 RNA) cleaved 40 nucleotides past the termination signal to evaluate the ratio of terminated versus runoff transcripts synthesized by wild-type Pol III or Pol III Δ. The truncated form of the yeast SNR6 gene lacking the extragenic B box (Brow and Guthrie 1988) can be transcribed in the presence of TFIIIB and Pol III only (Moenne et al. 1990). Transcription of this linear DNA template by the Pol III yielded a properly terminated U6 RNA molecules of normal length and a very faint amount of run-off transcript (Fig. 5B). In contrast, Pol III Δ totally ignored the terminator of the SNR6 gene and synthesized essentially the runoff transcript.
C11 is involved in transcription termination in vivo
To demonstrate that C11 plays a role in Pol III transcription termination in vivo, we took advantage of the genetic approach elaborated by Allison and Hall (1985). The yeast SUP4-o tRNATyr gene codes for a suppressor of ochre mutations. In a control strain overexpressing C11 and in the heterocomplemented strain overexpressing SpC11p, we tested the ability to suppress an ochre mutation in the ADE2 gene for different mutant SUP4 genes with progressive deletions in the 3′ flanking sequence (Fig. 6). As previously demonstrated, the suppression strength of each SUP4 mutant is highly correlated with the relative efficiency of transcription termination at the 3′ end of the gene (Allison and Hall 1985).
Figure 6.
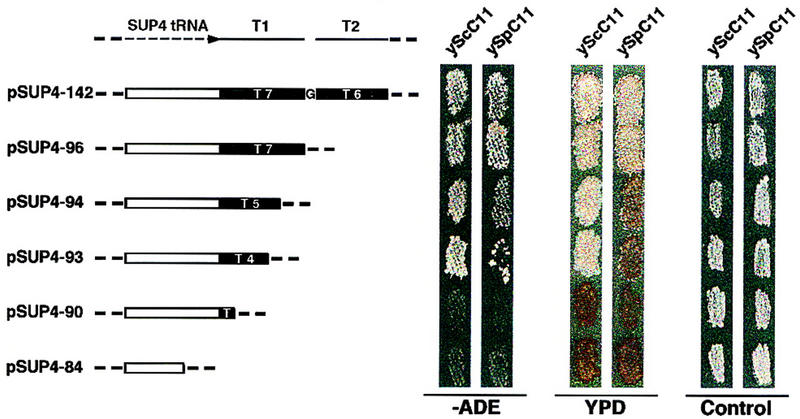
C11 is involved in class III genes termination in vivo. The heterocomplemented strain that carries the ocher allele of ade2-101, was transformed with one plasmid of the pSUP4 series (URA3, CEN6) that contained the ocher suppressing allele of SUP4-o tRNATyr and 3′ deletion mutants with altered terminators. A schematic representation of the pSUP4 serie is drawn on the left. Transformants were replicated in three different media. The growth of transformants was assayed on a media lacking Ade (−ADE). Cells in which termination of the SUP4 gene was defective were unable to grow. Color determination was performed on rich medium (YPD). Cells in which no or weak suppression of the ade2-101 allele occurred gave rise to red colonies. The controls show the growth of the cells on a medium containing Ade and Ura. Growth was scored after 4 days at 30°C. Eight independent transformants exhibited the same phenotype for growth and color assays.
Figure 6 shows the result of testing each 3′-deleted SUP4 gene for its ability to suppress ochre allele ade2-101 in yeast transformants. Total suppression of this mutation will result in Ade+ phenotype and in sectored white/red colonies on YPD medium, while decreasing levels of suppression will result in an Ade− phenotype and in an increasingly darker red colony color on YPD attributable to the accumulation of a colored product in the ade2-101 mutant. With both assays, suppression in the control strain was observed for SUP4 genes with deletions in the terminator up to 4T (pSUP4-93). On the contrary, as best measured by the color assay, suppression in the heterocomplemented strain required the T7 element of the terminator (pSUP4-142 and pSUP4-96, respectively). Suppression measured by growth of the transformants on a medium lacking adenine (Ade) appeared to be a less sensitive assay as a slight suppression was observed for SUP4 genes with deletions in the terminator up to 4T (pSUP4-93). However, a significant growth difference was observed between the control or the heterocomplemented cells carrying this construction.
These data indicated that there was a termination defect of the SUP4 gene in yeast cells heterocomplemented by SpC11p and strongly suggested that the C11 subunit is involved in termination efficiency in vivo as well as in vitro.
Is the cleavage of the nascent transcript required for termination?
We have shown above that Pol III Δ is not able to cleave the 3′ end of the nascent RNA in a ternary complex and that the enzyme hardly recognizes class III gene terminators. Although the molecular mechanisms of transcription termination remain to be elucidated, it has been reported recently that RNA cleavage occurs when Pol III transcription complexes reach the termination sites (Bobkova and Hall 1997). This observation prompted us to investigate whether cleavage of the nascent RNA transcript was one of the molecular mechanisms driving the termination step (i.e., transcription arrest and release of the RNA). Two lines of evidence indicated that this is not the case. (1) When using the same number of active Pol III and Pol III Δ (on the basis of the intensity of the 18*-mer transcript synthesized in the presence of ATP, CTP, UTP, and 3′-OMe-GTP), we quantified the amount of SUP4 tRNAs terminated within the T7 plus T6 sequences synthesized by the Pol III Δ molecules in the presence of the four XTPs. To determine whether these transcripts resulted from a single round- or multiround-specific transcription, we compared this amount to that of properly terminated RNAs synthesized by the wild-type enzyme in single round conditions (note that working in single-round conditions requires to add heparin on stalled complexes, which is not possible for stalled complexes containing Pol III Δ as they cannot resume elongation). We found a fourfold higher amount of specific terminated RNAs synthesized by the Pol III Δ compared to that synthesized by the wild-type enzyme (data not shown). This result indicates that Pol III Δ can perform multirounds of specific transcription. (2) Accordingly, using a size exclusion column that separates released transcripts from ternary complexes (excluded in the void volume), we found that the vast majority of full-length SUP4 tRNA transcribed by Pol III or Pol III Δ was eluted as free RNAs, well separated from the ternary complexes (data not shown). Altogether, these results demonstrated that cleavage of the RNA was not required for transcript release and reinitiation.
We then examined whether decreasing the polymerization rate of Pol III Δ could curtail its termination defect. Reduction of the elongation rate of RNA polymerases can be easily achieved by decreasing the nucleotide concentration (Wiest and Hawley 1990; Wiest et al. 1992; Gu and Reines 1995). Transcription of the SUP4 gene was thus performed with Pol III or Pol III Δ in the presence of UTP concentrations varying from 300 to 1 μm. Analysis of the transcripts by gel electrophoresis revealed that, at low UTP concentration (1 or 3 μm), the majority of the Pol III Δ terminated properly at the first block of the terminator (T1) (Fig. 7). We verified by gel permeation chromatography that the Pol III Δ transcripts synthesized in the presence of 3 μm UTP were released (data not shown). At the UTP concentration used for standard transcription reactions (30 μm), as observed previously, Pol III Δ mostly terminated at the second block of the terminator (T2) and long transcripts corresponding to a partial readthrough were synthesized. In the presence of 300 μm, the readthrough transcripts increased dramatically and only a small fraction of Pol III Δ terminated at T1 (Fig. 7). The influence of the UTP concentration on the transcription termination of the SUP4 gene by the wild-type enzyme was much different. Up to 30 μm UTP, termination occurred correctly at the T1 block (Fig. 7). At high UTP concentration (300 μm), T1 was still the major termination site of the enzyme but a small proportion of the transcripts terminated at T2 (Fig. 7). Some readthrough also occurred but to a much lesser extent than with Pol III Δ. This experiment showed that the termination transcription defect of Pol III Δ on the SUP4 gene could be fully corrected by decreasing the rate of polymerization of the RNA and thus strongly suggested that C11 is not required intrinsically for proper termination. One of the roles of the intrinsic Pol III cleavage property might be to increase the dwell time of the enzyme at the terminator to allow the termination mechanisms to take place.
Figure 7.

Low UTP concentrations corrects the termination defect of Pol III Δ. A stable initiation complex was formed on the SUP4 gene as described in Materials and Methods. Wild-type enzyme or Pol III Δ was then added together with ATP, CTP, and GTP (600 μm each) and [α-32P]UTP (400 μCi/nmole) at concentrations varying between 300 and 1 μm, as indicated. Reaction products were separated by electrophoresis on a 6% polyacrylamide, 7 m urea gel. Large transcripts accumulating at the top of the gel are indicated by RNAs x. The position of full length SUP4 tRNA transcripts terminated at T1 or T2 is indicated.
This model implies that the RNA cleavage activity of Pol III is essential for cell viability. Extensive mutational analysis of TFIIS indicated that the invariant Asp–Glu (DE) residues present in the conserved sequence RSADEP, within the disordered loop of the zinc ribbon, play critical roles in mediating Pol II RNA cleavage activity (Jeon et al. 1994; Cipres-Palacin and Kane 1995). Because the TFIIS-like carboxy-terminal zinc-ribbon of C11 contains the RSADEP sequence, we investigated the effect on cell viability of the double-point mutation D91A E92A. To this purpose, a yeast strain (YSC211) deleted at the RPC11 locus and complemented by a centromeric plasmid harboring RPC11 was transformed with a multicopy plasmid (pGEN) carrying the mutated RPC11 gene. No transformant was obtained among several independent transformation experiments (data not shown). This observation suggests that the overexpression of the mutated C11 was toxic to the cells. To obtain cells carrying the wild-type and the mutated RPC11 genes, YSC211 was transformed with a centromeric plasmid bearing the mutated RPC11 gene under the control of the weak, galactose-inducible GALS promoter and transformants were selected under repressive conditions. Transformants replicated to a galactose medium were viable, suggesting that the toxic effect of the mutated C11 was dose dependent. After a plasmid shuffle experiment on galactose medium, we observed that the cells were not able to lose the wild-type RPC11 allele, demonstrating that the DE mutation was lethal (data not shown). This result suggests that the intrinsic RNA cleavage activity of Pol III has a crucial role for the cell viability.
We then tested the effect of rC11 on the termination properties of Pol III Δ. The termination defect on the SUP4 gene of the mutant enzyme could not be corrected by the recombinant subunit under different transcription conditions (data not shown). Therefore, there was a differential influence of rC11 on the cleavage, pausing, and termination by Pol III Δ. Whereas cleavage activity was fully restored by rC11 (see Fig. 3C,D), RNAs corresponding to transitory pause sites were observed during a time course transcription–elongation by Pol III Δ in the presence of rC11, but were much less abundant than for wild-type Pol III (Fig. 4, cf. A with C). These observations suggested that rC11 was best able to restore the transcriptional properties of the Pol III Δ under static situations, (i.e., on halted or arrested complexes).
Localization of the C11-like subunit in the Pol I three-dimensional structure
C11 appears to be a key subunit for cleavage and proper elongation and termination processes. The positioning of this subunit in the Pol III structure would be of prime interest to correlate our functional data with structural features of the enzyme. Three-dimensional models of yeast Pol I (Schultz et al. 1993) and Pol II (Darst et al. 1991) are available, which both reveal a thumb-like projection surrounding a groove that is thought to bind double helical DNA (Kim et al. 1997). This key feature is also observed in the low resolution structure of the E. coli holoenzyme (Darst et al. 1989) and in the T7 DNA polymerase structure (Doublié et al. 1998). Because A12.2, B12.6, and C11 are homologous subunits (see Fig. 1A), it is reasonable to hypothesize that these three polypeptides are similarly located in the corresponding form of RNA polymerase. For this reason, subunit A12.2 was localized by immunoelectron microscopy on the three-dimensional model of Pol I (Schultz et al. 1993) using A12.2-specific polyclonal antibodies. The native enzyme adopts various orientations giving rise to characteristic views described previously (Klinger et al. 1996). For each enzyme view, RNA polymerase dimer images were averaged to define accurately the antibody interaction site (Fig. 8A–C). On three independent enzyme views, the antibodies were localized in the thumb region. In the case of the 90° view (Fig. 8A), the density difference map entered the outer contour of the enzyme by at least 2–3 nm indicating that the antibody was superimposed on the enzyme. In the case of the 30° dimer views (Fig. 8B,C), the smaller enzyme domain was labeled outside of the external contour of the enzyme indicating that the antibody was located outside of the groove on the domain from which the thumb-like structure protruded out. Altogether, these data indicate that the antibodies directed against A12.2 bind behind the finger-like stalk and outside of the groove (Fig. 8D).
Figure 8.
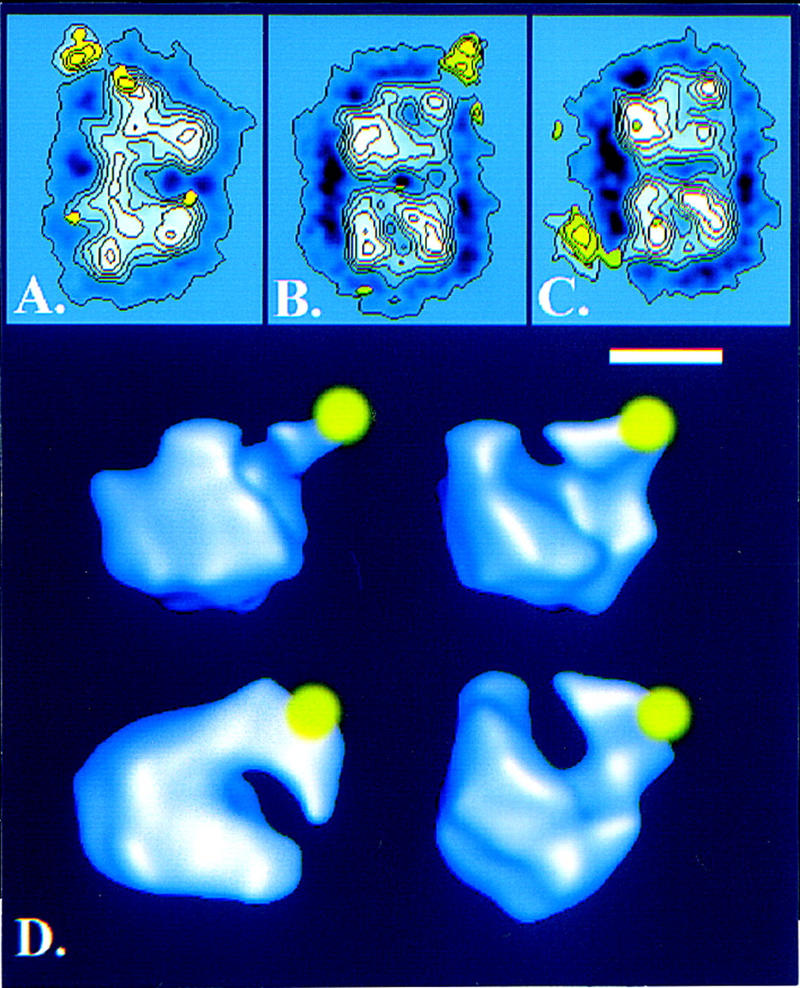
Immunolocalization of subunit A12.2 in the Pol I three-dimensional structure. (A–C) Average images of RNA Pol I dimers labeled with anti-A12.2 antibodies were obtained by analysis of 473 molecular images. The stain-excluding protein densities are outlined by contours of equal density. The difference image between the antibody-labeled and the unlabeled molecule is shown in yellow where the contours correspond to positive differences thresholded at a significance level of 5%. Each panel corresponds to a distinct dimer view identified by the angle of the internal two-fold axis with the normal to the plane. For the 90° dimer view (A), the images were transformed to show the binding in the upper molecule, thus the antibody interaction site is identified by comparing the upper to the lower monomer. For the 30° dimer view (B,C), two independent images were calculated where either the upper (B) or the lower monomer (C) was labeled. (D) Surface representation of the three-dimensional RNA Pol I model showing the position of A12.2 in yellow. Taking the monomeric envelope in the top left panel as a reference; the top right panel represents the envelope viewed after a counterclockwise tilt of 30° around a vertical axis. The reference is viewed from the top in the bottom left panel and from the bottom in the bottom right panel. (A–C) Bar, 10 nm; (D) bar, 5.5 nm.
Discussion
The work reported here reveals a link between intrinsic RNA cleavage activity and termination efficiency. C11, a newly discovered Pol III subunit that is involved in the cleavage activity, shares certain structural features with small conserved subunits of Pol I and Pol II and shares others with transcription factor TFIIS/SII. This chimeric structure suggests a double role for C11 in Pol III that is imparted to distinct polypeptides in the case of the Pol II enzyme. This contrasting situation may stem from the opposite requirements of Pol II and Pol III in terms of transcript length and termination efficiency.
A link between RNA cleavage and termination
The small Pol III subunit C11 is essential for yeast cell viability, which suggests an important role of this polypeptide in Pol III transcription. An incomplete form of Pol III lacking the C11 subunit (Pol III Δ) was found to have a number of enzymatic defects in vitro. Pol III Δ initiated correctly in specific transcription assays but totally lacked the RNA cleavage activity intrinsic to Pol III; it was deficient in termination and incorporated irreversibly one—and only one—additional (probably mispaired) nucleotide at the 3′ end of nascent RNA when halted on a DNA template in the absence of one nucleotide. All of these enzymatic defects are likely to be linked and are corrected, at least partly, by incubation of the enzyme with rC11.
The misincorporation reaction was not observed with the complete enzyme, probably because the mispaired nucleotide was removed efficiently or because its incorporation was slower than the multiple cleavage–synthesis cycles catalyzed by Pol III at arrest sites (Bobkova and Hall 1997). The fact that only one nucleotide was misincorporated argues against a global loss of substrate selectivity by the defective enzyme. Remarkably, after incorporation of the putative mispaired nucleotide, the halted complexes could not resume transcription elongation with the addition of the missing substrate GTP unless rC11 was added back. Without nuclease activity, Pol III Δ was unable to proofread the transcript and remained transcriptionally incompetent, perhaps because the 3′ end of the RNA was not properly inserted in the active site. The experiment with the 3′-OMe-GTP that blocks further chain elongation after being incorporated illustrates well the crucial role of the nuclease activity. The native enzyme could remove the incorporated elongation inhibitor and resumed transcription, whereas the incomplete enzyme remained irreversibly blocked (data not shown).
The loss of the RNA cleavage activity of Pol III Δ was accompanied by severe defects in chain termination. On the SNR6 gene, the incomplete enzyme totally ignored the termination signal, a T10 stretch, and synthesized long RNA chains that barely entered the polyacrylamide gel. The terminator of the SUP4 gene was also poorly recognized and more than half of the enzyme molecules that terminated did so at the second T-stretch (T6). This result indicated that the RNA cleavage activity was not absolutely required for correct termination, which includes release of the terminated transcript.
Importantly by using a genetic assay based on the suppressor strength of SUP4 genes with truncated terminators, we observed a termination defect in heterocomplemented cells expressing the S. pombe C11 subunit in place of the budding yeast C11. This result supports the conclusion derived from the in vitro experiments described above and demonstrates that the C11 subunit plays a critical role in transcription termination.
In keeping with its termination defect, Pol III Δ pauses less, leading to a nondetectable level of paused ternary complexes at transitory pause sites during elongation. We suggest that RNA cleavage activity might increase the dwell time of the enzyme at pause sites through multiple RNA cleavage–synthesis cycles at these sites. In agreement with this idea, the opposite behavior of a distinct RNA Pol III mutant confirms the existence of an indirect link between RNA cleavage and pausing. A mutant Pol III affected in the f domain (the conserved α-amanitin-binding domain in Pol II) of the largest subunit, which had ∼10-fold increased RNA cleavage rate, exhibited much extended pausing (Thuillier et al. 1996). Moreover, a double-point mutation in C11 of the invariant DE residues in the conserved sequence RSADEP, that are critical for the cleavage activity mediated by TFIIS (Jeon et al. 1994; Cipres-Palacin and Kane 1995), was found to be lethal. In addition, our data suggest that the mutated C11 has a dominant negative effect when overexpressed, consistent with the idea of a competition between the mutated and the wild-type subunits for their assembly into Pol III. This mutagenesis experiment suggested that the RNA cleavage activity mediated by C11 is essential for cell viability and that this activity plays a crucial role in one essential step of class III gene transcription. Our hypothesis is that RNA cleavage is important in removing the kinetic barriers to the termination step. The multiple cleavage synthesis cycles occurring at termination sites (Bobkova and Hall 1997) increase the dwell time of the enzyme at these sites and thereby favor the release of the nascent RNA and the dissociation of the complex. Accordingly, reducing the RNA synthesis rate by decreasing the UTP concentration fully corrected the termination defect of Pol III Δ. At 1–3 μm UTP, Pol III Δ displayed a normal termination behavior (arrest of the polymerase at the first T-stretch, transcript release, and reinitiation). This proposed role for RNA cleavage activity in Pol III termination is in agreement with the observations of Proudfoot and colleagues who showed that polymerase pausing over an extended region of template ensures termination of Pol II transcription close to the poly(A) site (Birse et al. 1997).
We noted that incubation of rC11 with Pol III Δ led to variable corrections of the transcriptional defects of the enzyme. Whereas the addition of a unique extra nucleotide in the presence of 3 XTPs on the SUP4 gene was totally prevented and the RNA cleavage activity of purified ternary complexes was fully restored by rC11, we observed only a weak effect of rC11 on the restoration of pausing during elongation and no influence of the recombinant subunit on the termination of Pol III Δ. However, the in vivo data presented in this paper suggest that subunit C11 is involved in the Pol III transcription termination process. One possible explanation for this apparent discrepancy is that rC11 does not stably reassociate with the elongating Pol III Δ. As indicated previously, rC11 was not found associated with Pol III Δ after a gel permeation chromatography. In this respect, to our knowledge, it is still unclear if the mode of action of TFIIS on Pol II or of GreA and GreB on eubacterial RNA polymerase is a transitory interaction possible only on static RNA polymerase molecules or if these elongation/cleavage factors may stay associated with elongating enzymes. Another possibility that we cannot formally exclude is that in conjunction with C11, another protein that is part of the Pol III transcriptional machinery, might be required for proper termination.
A dual role for C11
A simple two-state model for elongating RNA polymerase molecules accounts well for the atypical behavior of Pol III Δ (Price et al. 1989; Matsuzaki et al. 1994; Chen et al. 1996). On the basis of a quantitative analysis of RNA chain elongation on the SUP4 gene, Geiduschek and colleagues proposed that yeast Pol III is able to switch repeatedly between a fast and a slow stepping state, possibly at each elongation step (Matsuzaki et al. 1994). Assuming that the fast stepping state corresponds to an elongation only conformation and the slow stepping state to a cleavage only conformation of the ternary complex, as also suggested, a competition between these two catalytic states is expected to influence the duration of pausing and the termination efficiency.
With this model in mind, one could imagine that any mutation or inhibitor, or environmental condition interfering with one catalytic state may favor the other. Hence, the Pol III C160-270 mutant favors the cleavage mode with correlatively extended pausing and decreased mean elongation rate (Thuillier et al. 1996). The Pol III Δ enzyme described here, on the contrary, appears to be locked in the elongation mode at the expense of cleavage, pausing, and termination. Several mutations in the RET1 gene, encoding the β-like subunit of yeast Pol III, caused a reduced termination or increased termination phenotype and also affected pausing and the overall elongation kinetics (Shaaban et al. 1996). The model would predict that, at least for some of these mutants, the pausing and termination defects will be correlated with a corresponding change in the RNA cleavage activity of the enzyme.
The role proposed for C11 in the transition from the elongation mode to the cleavage mode is consistent with the mapping of the A12.2 subunit by immunoelectron microscopy on the thumb-like stalk of Pol I. Several observations (Darst et al. 1989, 1991; Sousa et al. 1994; Polyakov et al. 1995; Asturias et al. 1997) support the proposal that the thumb-like structure is flexible and can wrap around the bound template to form a topologically linked complex preventing the polymerase from dissociating from the template during processive synthesis. The location of A12.2 suggests that this subunit, which is the C11 homolog, could influence the thumb position and its mobility during the switch to the elongation mode. Consistently, thioredoxin, which acts as an elongation factor of T7 DNA polymerase, binds the enzyme at a domain structurally homologous to the Pol I domain where A12.2 was localized (Doublié et al. 1998).
A library displaying peptides derived from the gene encoding the Pol I subunit A135 was screened with monoclonal antibodies to map their epitope on the primary amino acid sequence (Klinger et al. 1996). A12.2 is located close to one particular epitope present in a domain of subunit A135 conserved in the C128 subunit, the second largest subunit of Pol III. Interestingly, a mutation in C128 affecting termination was mapped in this conserved domain (James and Hall 1990). The monoclonal antibody recognizing this epitope was found to inhibit only 50% of the Pol I enzyme (Huet et al. 1982). This puzzling observation could be interpreted in terms of a mixture of enzyme molecules in two conformational states.
We have shown that C11 is required for the intrinsic cleavage activity of Pol III and that, in its absence, the enzyme is blocked in the elongation mode. C11, therefore, appears to be involved both in RNA cleavage, probably performed by the largest subunits of the enzyme, and in the transition from the cleavage state to the elongation state. The implication of C11 in RNA cleavage is consistent with the striking conservation of the carboxy-terminal domains of C11 and TFIIS whose role as a cleavage cofactor is well documented (Reines 1994; Reines et al. 1996; Uptain et al. 1997). Because the C11 subunit shares structural features with both TFIIS and Pol II subunit B12.6, we suggest that C11 plays in Pol III the double function of the pair B12.6/TFIIS. Interestingly, Pol II purified from a yeast strain lacking B12.6 requires much higher TFIIS levels to effect cleavage and readthrough, indicating that B12.6 mediates TFIIS-dependent cleavage (Awrey et al. 1997). Along the lines of the two-state model discussed above, B12.6 is probably involved in the transition between the elongation and the cleavage modes. Pol II deprived of B12.6 transcribes through arrest sites with a higher efficiency and is globally more active than the complete Pol II (Awrey et al. 1997). As previously discussed (Awrey et al. 1997), the decreased stringency in start site selection in cells lacking B12.6 (Furter-Graves et al. 1994; Hull et al. 1995) may be a secondary effect of modifications in the conformational state of Pol II.
The double function of C11, shared by two different proteins (B12.6/TFIIS) in the Pol II machinery, might be inherited from archaebacteria. The unique archaeal RNA polymerase resembles in molecular complexity and structure the eukaryotic nuclear enzymes (Langer et al. 1995). TFIIS homologs have also been identified in several archaebacterial genomes (Langer and Zillig 1993; Kaine et al. 1994; Bult et al. 1996). However, as suggested by Kaine et al. (1994), we believe that the putative TFIIS homologs correspond instead to a RNA polymerase subunit orthologous to C11, because of the ORF size (∼110 residues), much smaller than TFIIS, and the presence of two potential zinc-binding domains instead of the single one found in TFIIS. This contention is further supported by the fact that archaebacteria do not possess homologs of other general eukaryotic transcription factors besides TBP- and TFIIB-related factors (Qureshi et al. 1997). Therefore, we suggest that the archaebacterial RNA polymerase has an intrinsic RNA cleavage activity, mediated by this subunit and would be, in this respect, similar to the eukaryotic Pol III machinery.
An intringuing question then is why Pol II, and probably also Pol I (Tschochner 1996; Labhart 1997), have evolved a two-component system to perform RNA cleavage. The answer to this question may lie in a major difference in transcription requirements of the Pol II and Pol III enzymes. Pol III transcribes very short genes (100–300 bp) and correct termination is essential for proper RNA processing and function (Allison and Hall 1985). In contrast, Pol II has to transcribe large genes up to the size of a full archaebacterial genome [the human dystrophin gene has 79 exons spanning at least 2.3 × 106 bp (Tennyson et al. 1995)]. An intrinsic RNA cleavage activity that favors termination would be an advantage to Pol III, but a strong impediment to Pol II. To transcribe long genes, Pol II needs instead a low sensitivity to pause sites and to potential terminators. The cleavage activity was retained by Pol II as an optional, cofactor-dependent activity, probably to reactivate accidentally arrested transcription complexes and possibly for transcriptional proofreading (Jeon and Agarwal 1996; Thomas et al. 1998).
Materials and methods
Cloning and disruption of RPC11
RPC11 was cloned from S. cerevisiae genomic DNA by PCR using two 45-mer oligonucleotides hybridizing downstream (−383) or upstream (+443) of the RPC11 ORF and containing a SacII and a BamHI restriction site, respectively . The amplified DNA fragment was inserted into the SacII–BamHI sites of pRS316 (URA3, CEN6) to give pRS316–RPC11.
The whole RPC11 ORF was disrupted by the direct-deletion method (Baudin et al. 1993). Two 57-mer oligonucleotides were used to amplify by PCR a DNA fragment containing the HIS3 gene and stop modules flanked by RPC11 promoter and terminator sequences. The 1102-bp PCR-amplified fragment was used to transform the diploid strain YPH501 (Sikorski and Hieter 1989). Correct integration of the HIS3 cassette in the resulting diploid disruptants (YSC200) was verified by PCR analysis. These diploid cells were then transformed with the pRS316–RPC11 plasmid. The haploid yeast strain YSC211, deleted at the RPC11 locus and complemented by the pRS316–RPC11 centromeric plasmid, was obtained after sporulation and tetrad dissection of the diploid strain.
RPC11 under the control of the GALS promoter
RPC11 was amplified by PCR on genomic DNA using a primer containing a BamHI restriction site (hybridizing at position −2) and a primer containing a SmaI restriction site (hybridizing at position +334). The amplified DNA fragment was cloned under the control of the galactose-inducible GALS promoter into p414GALS (TRP1, CEN6) (Mumberg et al. 1994) by insertion at BamHI and SmaI restriction sites to give p414GALS–RPC11. The haploid yeast strain YSC211, deleted at the RPC11 locus and complemented by the pRS316–RPC11 (URA3, CEN6) plasmid, was then transformed with the p414GALS–RPC11 plasmid. After selection of the transformants, the resident pRS316–RPC11 plasmid was eliminated by plasmid shuffling on a galactose medium containing 5-fluoro-orotic acid (5-FOA).
Heterocomplementation with SpC11 ORF
The ORF encoding SpC11p was amplified by PCR from S. pombe genomic DNA using 41-mer and 42-mer oligonucleotides hybridizing downstream (position −42) or upstream (position +391) of the SpC11 ORF and containing a BamHI and an EcoRI restriction site, respectively. The amplified DNA fragment was cloned under control of the phosphoglycerokinase (PGK) promotor between the EcoRI–BamHI sites of pGEN (TRP1, 2μ) to give pGEN–SpC11. Plasmid pGEN–SpC11 was introduced into the yeast strain YSC211 and the resident plasmid was eliminated by plasmid shuffling on 5-FOA medium. As a control, pRS316–RPC11 was also exchanged for pGEN–RPC11 by plasmid shuffling in the YSC211 strain.
Site-directed mutagenesis of RPC11
Residues D91E92 of C11 (see Fig. 1) were site-directed mutagenized to A91A92 by PCR on pGEN-RPC11. The amplified mutated DNA fragment, including both the unique PvuII restriction site of RPC11 and the unique XhoI restriction site of pGEN vector, was cloned into the unique PvuII–XhoI restriction sites of pGEN–RPC11 and the mutated RPC11 gene (RPC11*) was sequenced. To place the RPC11* gene under the control of the inducible-galactose GALS promoter, a BamHI–EcoRI fragment containing the RPC11* gene was obtained from pGEN–RPC11* and cloned into the unique BamHI–EcoRI restriction sites of p414GALS vector (see above), giving p414GALS–RPC11*.
Purification of RNA Pol III and class III transcription factors
Micropurification of RNA Pol III and purification of class III transcription factors were as described by Huet et al. (1996). Subunit composition of Pol III was analyzed by gel electrophoresis on a 4%–15% polyacrylamide gel (Bio-Rad) in denaturing conditions (Laemmli 1970) and proteins were silver stained (Blum et al. 1987).
Specific in vitro transcription assays, elongation, and cleavage assays
Plasmids used for specific in vitro transcription assays and reactions mixtures were as described by Dieci and Sentenac (1996) with minor modifications. For transcription of the SNR6 gene, pTaq6 containing a truncated form of the SNR6 gene lacking the extragenic B box (Guthrie and Patterson 1988) was digested with HindIII and SmaI restriction enzymes and the HindIII–SmaI fragment harboring the SNR6 gene was purified after gel electrophoresis. 3′-O-methyl-GTP (Pharmacia) was used at 500 μm and formation of the stalled complexes was performed with 50 ng of Pol III. Purification of the ternary complexes was carried out as described previously (Steinberg and Burgess 1992). Intrinsic transcript cleavage was carried out on purified ternary complexes in transcription buffer containing 8 mm MgCl2 as described by Thuillier et al. (1996). Elongation assays were performed on purified ternary complexes in transcription buffer by addition of ATP, CTP, GTP to 600 μm and UTP to 30 μm as described by Thuillier et al. (1996). For transcription assays after Pol III Δ reconstitution with rC11, recombinant protein was incubated at a molar ratio of 50:1 (rC11:Pol III Δ). Transcripts were analyzed by electrophoresis on denaturating polyacrylamide gel (6% for full length transcript and 15% for 17-mer, cleavage, and elongation assays). Gels were autoradiographed using X-OMAT AR film (Kodak) and quantitated using a PhosphorImager with ImageQuant software (Molecular Dynamics).
Expression and purification of rC11
The RPC11 coding region was subcloned by PCR to generate the coding region with a BamHI restriction site at the 5′ end of the sequence and a XhoI restriction site at the 3′ end. The amplified insert was then cloned between the BamHI–XhoI sites of the pGEX-4TK bacterial expression vector (Pharmacia) to give pGEX4TK–RPC11. XL1-blue cells containing the pGEX4TK–RPC11 plasmid were grown at 30°C to 0.8 OD600. Expression of the GST–RPC11 fusion protein was induced by addition of 0.1 mm of isopropyl-1-thio-β-d-galactopyranoside. Cells were harvested after 4 hr of induction by centrifugation and resuspended in PBS buffer containing 10 μm ZnSO4, 1% Triton, and a set of protease inhibitors (Complete without EDTA, Boehringer). After lysis by sonication at 4°C, the lysate was clarified by centrifugation in a Beckman JA20 rotor for 15 min at 15,000 rpm. The GST–RPC11 protein was purified by affinity chromatography on glutathione–Sepharose 4B according to the manufacturer’s protocol. Elution was done for 16 hr at room temperature by thrombin cleavage in PBS buffer containing 10 μm ZnSO4. The purified proteins were analyzed by PAGE on a 15% polyacrylamide gel in denaturating conditions, stained with Coomassie Blue and stored at −80°C.
Specimen preparation for electron microscopy and image processing
Polyclonal antibodies directed against subunit A12.2 were raised in rabbits upon injection of the isolated subunit and the IgG fraction was purified as described (Buhler et al. 1980). Formation of immune complexes and image analysis of the data were as described by Klinger et al. (1996).
Acknowledgments
We gratefully acknowledge Christine Conesa, Olivier Lefebvre, and Michel Werner for their advice in cloning and disruption strategies, Sylvie Camier for pertinent suggestions for the in vivo experiments, Ben Hall for providing us the pSUP4 plasmid serie, and Emmanuel Favry for his gift of transcription factors. We are very grateful to Caroline Kane, Ben Hall, Carl Mann, and Danny Reines for a critical reading of the manuscript and helpful suggestions. Stéphane Chédin was supported by a short term grant from the Association pour la Recherche contre le Cancer. This work was supported by a Human Frontier Science Program Organization grant.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Note added in proof
The GenBank accession no. for RPC11 is AF108188.
Footnotes
E-MAIL biochimi@jonas.saclay.cea.fr; FAX 33 1 69 08 47 12.
References
- Allison DS, Hall BD. Effects of alterations in the 3′ flanking sequence on in vivo and in vitro expression of the yeast Sup4-0 tRNATyr gene. EMBO J. 1985;4:2657–2664. doi: 10.1002/j.1460-2075.1985.tb03984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asturias FJ, Meredith GD, Poglitsch CL, Kornberg RD. Two conformations of RNA polymerase II revealed by electron crystallography. J Mol Biol. 1997;272:536–540. doi: 10.1006/jmbi.1997.1273. [DOI] [PubMed] [Google Scholar]
- Awrey D, Weilbaecher R, Hemming S, Orlicky S, Kane C, Edwards A. Transcription elongation through DNA arrest sites. J Biol Chem. 1997;272:14747–14754. doi: 10.1074/jbc.272.23.14747. [DOI] [PubMed] [Google Scholar]
- Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 1993;21:3329–3330. doi: 10.1093/nar/21.14.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birse CE, Lee BA, Hansen K, Proudfoot NJ. Transcriptional termination signals for RNA polymerase II in fission yeast. EMBO J. 1997;16:3633–3644. doi: 10.1093/emboj/16.12.3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum H, Beier H, Gross HJ. Improved silver staining of plant proteins, RNA and DNA in polyacrylamide gels. Electrophoresis. 1987;8:93–99. [Google Scholar]
- Bobkova E, Hall B. Substrate specificty of the RNase activity of yeast RNA polymerase III. J Biol Chem. 1997;272:22832–22839. doi: 10.1074/jbc.272.36.22832. [DOI] [PubMed] [Google Scholar]
- Borukhov S, Polyakov A, Nikiforov V, Goldfarb A. GreA protein: A transcription elongation factor from Escherichia coli. Proc Natl Acad Sci. 1992;89:8899–8902. doi: 10.1073/pnas.89.19.8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borukhov S, Sagitov V, Goldfarb A. Transcript cleavage factors from E. coli. Cell. 1993;72:459–466. doi: 10.1016/0092-8674(93)90121-6. [DOI] [PubMed] [Google Scholar]
- Brow DA, Guthrie C. Spliceosomal RNA U6 is remarkably conserved from yeast to mammals. Nature. 1988;334:213–218. doi: 10.1038/334213a0. [DOI] [PubMed] [Google Scholar]
- Buhler J-M, Huet J, Davies KE, Sentenac A, Fromageot P. Immunological studies of yeast nuclear RNA polymerases at the subunit level. J Biol Chem. 1980;255:9949–9954. [PubMed] [Google Scholar]
- Bult CJ, White O, Olsen GJ, Zhou L, Fleishmann RD, Sutton GG, Blake JA, FitzGerald LM, Clayton RA, Gocayne JD. Complete genome sequence of the methanogenic archaeon Methanococcus jannaschii. Science. 1996;273:1058–1073. doi: 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- Chen Y, Chafin D, Price DH, Greenleaf AL. Drosophila RNA polymerase II mutants that affect transcription elongation. J Biol Chem. 1996;271:5993–5999. [PubMed] [Google Scholar]
- Cipres-Palacin G, Kane CM. Alanine-scanning mutagenesis of human transcript elongation factor TFIIS. Biochemistry. 1995;34:15375–15380. doi: 10.1021/bi00046a046. [DOI] [PubMed] [Google Scholar]
- Darst SA, Kubalek EW, Kornberg RD. Three-dimensional structure of Escherichia coli RNA polymerase holoenzyme determined by electron crystallography. Nature. 1989;340:730–732. doi: 10.1038/340730a0. [DOI] [PubMed] [Google Scholar]
- Darst SA, Edwards AM, Kubalek EW, Kornberg RD. Three-dimensional structure of yeast RNA polymerase II at 16 Å resolution. Cell. 1991;66:121–128. doi: 10.1016/0092-8674(91)90144-n. [DOI] [PubMed] [Google Scholar]
- Dieci G, Sentenac A. Facilitated recycling pathway for RNA polymerase III. Cell. 1996;84:245–252. doi: 10.1016/s0092-8674(00)80979-4. [DOI] [PubMed] [Google Scholar]
- Doublié S, Tabor S, Long A, Richardson C, Ellenberger T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- Erie DA, Hajiseyedjavadi O, Young MC, Von Hippel PH. Multiple RNA polymerase conformations and GreA: Control of the fidelity of transcription. Science. 1993;262:867–873. doi: 10.1126/science.8235608. [DOI] [PubMed] [Google Scholar]
- Exinger F, Lacroute F. 6-Azauracil inhibition of GTP biosynthesis in Saccharomyces cerevisiae. Curr Genet. 1992;22:9–11. doi: 10.1007/BF00351735. [DOI] [PubMed] [Google Scholar]
- Furter-Graves EM, Hall BD, Furter R. Role of a small RNA pol II subunit in TATA to transcription start site spacing. Nucleic Acids Res. 1994;22:4932–4936. doi: 10.1093/nar/22.23.4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Reines D. Identification of a decay in transcription potential that results in elongation factor dependence of RNA polymerase II. J Biol Chem. 1995;270:11238–11244. doi: 10.1074/jbc.270.19.11238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Patterson B. Spliceosomal snRNAs. Annu Rev Genet. 1988;23:387–419. doi: 10.1146/annurev.ge.22.120188.002131. [DOI] [PubMed] [Google Scholar]
- Hagler J, Shuman S. Nascent RNA cleavage by purified ternary complexes of vaccinia RNA polymerase. J Bio Chem. 1993;268:2166–2173. [PubMed] [Google Scholar]
- Harrison DA, Mortin MA, Corces VG. The RNA polymerase II 15 kilodalton subunit is essential for viability in Drosophila. Mol Cell Biol. 1992;12:928–935. doi: 10.1128/mcb.12.3.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huet J, Manaud N, Dieci G, Peyroche G, Conesa C, Lefebvre O, Ruet A, Riva M, Sentenac A. RNA polymerase III and class III transcription factors from Saccharomyces cerevisiae. Methods Enzymol. 1996;273:249–267. doi: 10.1016/s0076-6879(96)73024-0. [DOI] [PubMed] [Google Scholar]
- Huet J, Phalente L, Buttin G, Sentenac A, Fromageot P. Probing yeast RNA polymerase A subunits with monospecific antibodies. EMBO J. 1982;1:1193–1198. doi: 10.1002/j.1460-2075.1982.tb00012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull MW, McKune K, Woychik NA. RNA polymerase II subunit RPB9 is required for accurate start site selection. Genes & Dev. 1995;9:481–490. doi: 10.1101/gad.9.4.481. [DOI] [PubMed] [Google Scholar]
- Izban M, Luse D. Factor-stimulated RNA polymerase II transcribes at physiological elongation rates on naked DNA but very poorly on chromatin templates. J Biol Chem. 1992;267:13647–13655. [PubMed] [Google Scholar]
- ————— The increment of SII-facilited transcript cleavage varies dramatically between elongation competent and incompetent RNA polymerase II ternary complexes. J Biol Chem. 1993;17:12874–12885. [PubMed] [Google Scholar]
- James P, Hall BD. ret1-1, a yeast mutant affecting transcription termination by RNA polymerase III. Genetics. 1990;125:293–303. doi: 10.1093/genetics/125.2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon C, Agarwal K. Fidelity of RNA polymerase II transcription controlled by elongation factor TFIIS. Proc Natl Acad Sci. 1996;93:13677–13682. doi: 10.1073/pnas.93.24.13677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon C, Yoon H, Agarwal K. The transcription factor TFIIS zinc ribbon dipeptide Asp-Glu is critical for stimulation of elongation and RNA cleavage by RNA polymerase II. Proc Natl Acad Sci. 1994;91:9106–9110. doi: 10.1073/pnas.91.19.9106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaine B, Mehr I, Woese C. The sequence, and its evolutionary implications, of a Thermococcus celer protein associated with transcription. Proc Natl Acad Sci. 1994;91:3854–3856. doi: 10.1073/pnas.91.9.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane C. Transcript elongation and gene regulation in eukaryotes. In: Conaway RC, Conaway JW, editors. Transcription: Mechanisms and regulations. New York, NY: Raven Press; 1994. pp. 279–296. [Google Scholar]
- Kerppola T, Kane C. RNA polymerase: Regulation of transcript elongation and termination. FASEB J. 1991;5:2833–2842. doi: 10.1096/fasebj.5.13.1916107. [DOI] [PubMed] [Google Scholar]
- Kerppola TK, Kane CM. Analysis of the signals for transcription termination by purified RNA polymerase II. Biochemistry. 1990;29:269–278. doi: 10.1021/bi00453a037. [DOI] [PubMed] [Google Scholar]
- Kim TK, Lagrange T, Wang YH, Griffith JD, Reinberg D, Ebright RH. Trajectory of DNA in the RNA polymerase II transcription preinitiation complex. Proc Natl Acad Sci. 1997;94:12268–12273. doi: 10.1073/pnas.94.23.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger J, Huet, Song D, Petersen G, Riva M, Sentenac A, Bautz EKF, Oudet P, Schultz P. Localization of yeast RNA polymerase I core subunits by immunoelectron microscopy. EMBO J. 1996;15:4643–4653. [PMC free article] [PubMed] [Google Scholar]
- Labhart P. Transcript cleavage in an RNA polymerase I elongation complex. J Biol Chem. 1997;272:9055–9061. doi: 10.1074/jbc.272.14.9055. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Langer D, Hain J, Thuriaux P, Zillig W. Transcription in archaea: Similarity to that in eucarya. Proc Natl Acad Sci. 1995;92:5768–5772. doi: 10.1073/pnas.92.13.5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer D, Zillig W. Putative tfIIs gene of Sulfolobus acidocaldarius encoding an archaeal transcription elongation factor is situated directly downstream of the gene for a small subunit of DNA-dependent RNA polymerase. Nucleic Acids Res. 1993;21:2251–2256. doi: 10.1093/nar/21.9.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin J, Chamberlin M. Mapping and characterization of transcriptional pause sites in the early genetic region of bacteriophage T7. J Mol Biol. 1987;196:61–84. doi: 10.1016/0022-2836(87)90511-0. [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Kassavetis GA, Geiduschek EP. Analysis of RNA chain elongation and termination by Saccharomyces cerevisiae RNA polymerase III. J Mol Biol. 1994;235:1173–1192. doi: 10.1006/jmbi.1994.1072. [DOI] [PubMed] [Google Scholar]
- Moenne A, Camier S, Anderson G, Margottin F, Beggs J, Sentenac A. The U6 gene of Saccharomyces cerevisiae is transcribed by RNA polymerase C (III) in vivo and in vitro. EMBO J. 1990;9:271–277. doi: 10.1002/j.1460-2075.1990.tb08105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M. Regulatable promoters of Saccharomyces cerevisiae: Comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res. 1994;22:5767–5768. doi: 10.1093/nar/22.25.5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogi Y, Yano R, Dodd J, Carles C, Nomura M. Gene RRN4 in Saccharomyces cerevisiae encodes the A12.2 subunit of RNA polymerase I and is essential only at high temperatures. Mol Cell Biol. 1993;13:114–122. doi: 10.1128/mcb.13.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR. Rapid and sensitive sequence comparison with FASTP and FASTA. Methods Enzymol. 1990;183:63–98. doi: 10.1016/0076-6879(90)83007-v. [DOI] [PubMed] [Google Scholar]
- Polyakov A, Severinova E, Darst SA. Three-dimensional structure of E. coli core RNA polymerase: Promotor binding and elongation conformations of the enzyme. Cell. 1995;83:365–373. doi: 10.1016/0092-8674(95)90114-0. [DOI] [PubMed] [Google Scholar]
- Price DH, Sluder AE, Greenleaf AL. Dynamic interaction between a Drosophila transcription factor and RNA polymerase II. Mol Cell Biol. 1989;9:1465–1475. doi: 10.1128/mcb.9.4.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi SA, Bell SD, Jackson SP. Factor requirements for transcription in the Archaeon Sulfolobus shibatae. EMBO J. 1997;16:2927–2936. doi: 10.1093/emboj/16.10.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reines D. Nascent RNA cleavage by transcription elongation complexes. In: Conaway RC, Conaway JW, editors. Transcription: Mechanisms and regulations. New York, NY: Raven Press; 1994. pp. 263–278. [Google Scholar]
- Reines D, Chamberlin MJ, Kane CM. Transcription elongation factor SII (TFIIS) enables RNA polymerase II to elongate through a block to transcription in a human gene in vitro. J Biol Chem. 1989;264:10799–10809. [PubMed] [Google Scholar]
- Reines D, Conaway JW, Conaway RC. The RNA polymerase II general elongation factors. Trends Biochem Sci. 1996;21:351–355. [PMC free article] [PubMed] [Google Scholar]
- Schultz P, Celia H, Riva M, Sentenac A, Oudet P. Three-dimensional model of yeast RNA polymerase I determined by electron microscopy of two-dimensional crystals. EMBO J. 1993;12:2601–2607. doi: 10.1002/j.1460-2075.1993.tb05920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sentenac A. Eukaryotic RNA polymerases. CRC Crit Rev Biochem. 1985;18:31–90. doi: 10.3109/10409238509082539. [DOI] [PubMed] [Google Scholar]
- Shaaban AA, Krupp BM, Hall BD. Termination-altering mutations in the second largest subunit of yeast RNA polymerase III. Mol Cell Biol. 1995;15:1467–1478. doi: 10.1128/mcb.15.3.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaaban S, Bobkova E, Chudzig D, Hall B. In vitro analysis of elongation and termination by mutant RNA polymerases with altered termination behavior. Mol Cell Biol. 1996;16:6468–6476. doi: 10.1128/mcb.16.11.6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpakovski GV, Acker J, Wintzerith M, Lacroix J-F, Thuriaux P, Vigneron M. Four subunits that are shared by the three classes of RNA polymerase are functionnaly interchangeable between Homo sapiens and Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:4702–4710. doi: 10.1128/mcb.15.9.4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SivaRaman L, Reines D, Kane CM. Purified elongation factor SII is sufficient to promote read-through by purified RNA polymerase II at specific termination sites in the human histone H3.3 gene. J Biol Chem. 1990;265:14554–14560. [PubMed] [Google Scholar]
- Sluder AE, Greenleaf AL, Price DH. Properties of a Drosophila RNA polymerase II elongation factor. J Biol Chem. 1989;264:8963–8969. [PubMed] [Google Scholar]
- Sousa R, Rose JP, Wang BC. The thumb’s knuckle. Flexibility in the thumb subdomain of T7 RNA polymerase. J Mol Biol. 1994;244:6–12. doi: 10.1006/jmbi.1994.1699. [DOI] [PubMed] [Google Scholar]
- Spencer C, Groudine M. Transcription elongation and eukaryotic gene regulation. Oncogene. 1990;5:777–785. [PubMed] [Google Scholar]
- Steinberg TH, Burgess RR. Tagetitoxin inhibition of RNA polymerase III transcription results from enhanced pausing at discrete sites and is template-dependent. J Biol Chem. 1992;267:20204–20211. [PubMed] [Google Scholar]
- Surratt CK, Milan SC, Chamberlin MJ. Spontaneous cleavage of RNA in ternary complexes of Escherichia coli RNA polymerase and its significance for the mechanism of transcription. Proc Natl Acad Sci. 1991;88:7983–7987. doi: 10.1073/pnas.88.18.7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennyson C, Klamut H, Worton R. The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nature Genet. 1995;9:184–190. doi: 10.1038/ng0295-184. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Platas AA, Hawley DK. Transcriptional fidelity and proofreading by RNA polymerase II. Cell. 1998;93:627–637. doi: 10.1016/s0092-8674(00)81191-5. [DOI] [PubMed] [Google Scholar]
- Thuillier V, Brun I, Sentenac A, Werner M. Mutations in the alpha-amanitin conserved domain of the largest subunit of yeast RNA polymerase III affect pausing, RNA cleavage and transcriptional transitions. EMBO J. 1996;15:618–629. [PMC free article] [PubMed] [Google Scholar]
- Tschochner H. A novel RNA polymerase I-dependent RNase activity that shortens nascent transcripts from the 3′ end. Proc Natl Acad Sci. 1996;93:12914–12919. doi: 10.1073/pnas.93.23.12914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uptain S, Kane C, Chamberlin M. Basic mechanisms of transcript elongation and its regulation. Annu Rev Biochem. 1997;66:117–172. doi: 10.1146/annurev.biochem.66.1.117. [DOI] [PubMed] [Google Scholar]
- Whitehall SK, Bardeleben C, Kassavetis GA. Hydrolytic cleavage of nascent RNA in RNA polymerase III ternary transcription complexes. J Biol Chem. 1994;269:2299–2306. [PubMed] [Google Scholar]
- Wiest DK, Hawley DK. In vitro analysis of a transcription termination site for RNA polymerase II. Mol Cell Biol. 1990;10:5782–5795. doi: 10.1128/mcb.10.11.5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiest D, Wang D, Hawley D. Mechanistic studies of transcription arrest at the adenovirus major late attenuation site: Comparison of purified RNA polymerase II and washed elongation complexes. J Biol Chem. 1992;267:7733–7744. [PubMed] [Google Scholar]
- Woychik NA, Lane WS, Young RA. Yeast RNA polymerase II subunit RPB9 is essential for growth at temperature extremes. J Biol Chem. 1991;266:19053–19055. [PubMed] [Google Scholar]