

Abstract
Transcriptional activation by c-Myc through specific E box elements is thought to be essential for its biological role. However, c-MycS is unable to activate transcription through these elements and yet retains the ability to stimulate proliferation, induce anchorage-independent growth, and induce apoptosis. In addition, c-MycS retains the ability to repress transcription of several specific promoters. Furthermore, c-MycS can rescue the c-myc null phenotype in fibroblasts with homozygous deletion of c-myc. Taken together, our data argue against the paradigm that all of the biological functions of c-Myc are mediated by transcriptional activation of specific target genes through E box elements.
Keywords: c-Myc, transactivation, cell cycle, apoptosis, leaky scanning
Many studies have shown that the c-myc gene has a critical role in the control of cellular proliferation. Deregulated c-myc is associated with a variety of neoplasms in several species, including humans. Overexpression of c-Myc protein stimulates cell cycle progression, causes transformation, blocks differentiation, and induces apoptosis in low serum (Henrikson and Lüscher 1996). Despite numerous studies, the molecular mechanism responsible for these biological activities of c-Myc is unclear. c-Myc has been shown to both activate and repress transcription. Several genes involved in growth and apoptosis, including α-prothymosin, ornithine decarboxylase (ODC), p53, and cdc25A, have been identified as possible target genes transactivated by c-Myc through specific E box sequences (CACGTG) or E box Myc sites (EMS) (Grandori and Eisenman 1997). These studies have supported the prevailing hypothesis that transactivation through EMS is necessary and sufficient for the biological functions of the c-Myc protein. However, several studies have shown that c-Myc can repress specific promoters, such as the adenovirus-2 major late promoter (AdMLP) (Li et al. 1994), the growth arrest gene gadd45 (Marhin et al. 1997), C/EBPα (Mink et al. 1996), and the growth arrest gene gas1 (Lee et al. 1997).
The carboxyl terminus of c-Myc, containing the basic region (B) and helix–loop–helix/leucine zipper (HLH/LZ) domain, is necessary and sufficient for sequence-specific DNA binding and heterodimerization with Max (Fig. 1a). The amino terminus, containing two highly conserved regions termed Myc box (Mb) I and II, is necessary for transcriptional activation and repression (Fig. 1a). Both the transactivation domain (TAD) and the BHLH/LZ domain have been shown to be necessary for biological activity (Henriksson and Lüscher 1996).
Figure 1.

Regulation of transcription by c-Myc2 and c-MycS proteins. (a) Schematic diagram of c-Myc. (b) Transactivation of α-prothymosin and p53 promoters by c-Myc proteins. NIH-3T3 cells were cotransfected with 1 μg of the reporter plasmids, 0.5 μg of SV40–β-gal plasmid and 1 μg of either CMV–Myc2, CMV–MycS, or pcDNA3. The β-gal and CAT activities were determined after harvesting the cells 48 hr later as described in Materials and Methods. The graph represents the average of three separate experiments for fold CAT activity where reporter alone is one unit. (c) Transactivation of the EFII enhancer element by c-Myc proteins. NIH-3T3 cells were cotransfected with 0.5 μg of SV40–β-gal plasmid, 0.5 μg of the [EFII]6 CAT reporter plasmid, and either 2 μg of pcDNA vector, CMV–Myc1, or increasing amounts of CMV–MycS expression vector as indicated. Transfections were equalized with pcDNA3 to 10 μg of total DNA. The β-gal and CAT activities were determined after harvesting the cells 48 hr later as described above. The graph represents fold CAT activity in which reporter alone is 1 unit. (d) Repression by c-Myc2 and c-MycS proteins. NIH-3T3 cells were cotransfected with 3 μg of either MLP–Luc, GADD45–Luc or GAS1–Luc reporter plasmid, 0.5 μg of SV40–β-gal and 1 μg of either pcDNA3, CMV–Myc2, or CMV–MycS expression vector as indicated. The β-gal and luciferase activities were determined after harvesting 48 hr post-transfection as described. The graph represents average fold relative luciferase activity for four (MLP–Luc) or 3 (GADD45–Luc and GAS1–Luc) experiments where reporter alone represents 1 unit.
Our previous studies found that human, murine, and avian cells express different translational forms of c-Myc (Fig. 1a) that are regulated differentially during growth and differ in their abilities to transactivate (Hann et al. 1992, 1994; Spotts et al. 1997). The c-MycS proteins arise from a leaking scanning mechanism and initiate at two closely spaced downstream AUG codons, yielding c-Myc proteins lacking ∼100 amino-terminal amino acids, including the highly conserved MbI region. Synthesis of c-MycS increases to levels comparable to c-Myc2 during rapid cell growth, and constitutively high levels of c-MycS synthesis are found in some tumor cell lines (Spotts et al. 1997). Similar to full-length c-Myc proteins, c-MycS proteins are nuclear, unstable, and heterodimerize with Max. Unlike full-length c-Myc, c-MycS is unable to activate transcription through isolated EMS elements (Kretzner et al. 1992; Spotts et al. 1997). In this study we provide evidence that c-MycS retains many of the biological functions of full-length c-Myc, even though it is unable to transactivate.
Results and Discussion
To extend the previous findings that c-MycS does not transactivate isolated EMS elements (Kretzner et al. 1992; Spotts et al. 1997) we examined natural promoters containing EMS sites. CAT assays were performed using either the natural α-prothymosin or p53 promoters. Figure 1b shows that the α-prothymosin (PrT) CAT and p53 CAT reporters were activated by c-Myc2, but not by c-MycS. In addition, although the ODC CAT reporter was activated poorly by c-Myc2, c-MycS consistently failed to activate the ODCΔCAT reporter (data not shown). Western blot analysis confirmed that comparable levels of c-Myc2 and c-MycS were synthesized in these assays (data not shown). We have shown previously that c-Myc1, the non-AUG-initiated translational form of c-Myc, but not c-Myc2 transactivates through a noncanonical site contained in the EFII enhancer element of the RSV LTR (Hann et al. 1994). To determine whether c-MycS can transactivate through this element, we performed a CAT assay using CMV–Myc1 compared with increasing amounts of CMV–MycS. Figure 1c shows that although cMyc1 transactivated through this EFII element, c-MycS failed to transactivate. This suggests that the first 100 amino acids of c-Myc, including MbI (Fig. 1a), are necessary for transactivation through EMS or EFII enhancer sequences.
Because the MbI region has been shown to be dispensable for the ability of c-Myc to repress transcription through the Inr element of AdMLP (Li et al. 1994), we next determined if c-MycS could repress transactivation using the luciferase reporter vector MLP–Luc. We also examined the gadd45 and the gas1 promoters using the GADD45–Luc and GAS1–Luc reporters, both of which have been shown to be repressed by full-length c-Myc (Lee et al. 1997; Marhin et al. 1997). As shown in Figure 1d, similar to full-length c-Myc2, c-MycS also repressed transcription of the MLP–Luc, GADD45–Luc and GAS1–Luc reporter constructs. These results indicate that full-length c-Myc2 can both activate and repress transcription of specific target genes, whereas c-MycS can only repress transcription.
Although c-MycS behaves as a dominant-negative inhibitor of c-Myc2 transactivation through EMS elements, the synthesis of c-MycS during rapid cell growth and the constitutively high synthesis of c-MycS in some tumor cell lines (Spotts et al. 1997) suggests that c-MycS does not interfere with cell growth. To test the effects of c-MycS on cell growth, we first used a modified inducible Myc–estrogen receptor (Myc–ER) system, which allows expression and quantitation of an inactive c-Myc–ER fusion protein that can only be activated with hydroxytamoxifen (OHT) (Littlewood et al. 1995). We used c-MycΔ106–143–ER as a negative control. This protein, which contains an internal deletion spanning amino acids 106–143 and lacks the highly conserved MbII region, has been shown to be biologically inactive (Stone et al. 1987). The c-Myc–ER expression vectors are shown in Figure 2a. Stable Rat 1a cell lines were generated by infection, and expression of the c-Myc–ER proteins was determined by immunoprecipitation. Figure 2b demonstrates that the representative stable cell lines expressed comparable levels of the different c-Myc-ER proteins.
Figure 2.



c-MycS–ER enhances cellular proliferation. (a) Schematic of c-Myc–ER constructs. (b) Expression of c-Myc–ER proteins. Expression of the c-Myc–ER proteins in representative stable Rat 1a cell lines was determined by immunoprecipitation with anti-Mycfl as described previously (Spotts et al. 1997). (Lane 1) pMV7 vector alone; (lane 2) c-Myc2–ER; (lane 3) c-MycS–ER; (lane 4) c-MycΔ106–143–ER (ΔMyc–ER). (c) Growth of Rat-1a cells expressing c-Myc–ER proteins. Stable Rat 1a cell lines expressing the c-Myc–ER proteins described above were seeded at 5 × 104 cells in replicate 35-mm dishes in DMEM containing 10% CS. After 24 hr, the cells were transferred into fresh medium with or without 1 μm OHT. At the indicated time points, triplicate cultures were counted. Floating and adherent cell counts were combined to yield the total cell number. Independent stable cell lines are designated by different numbers.
To determine if c-MycS could stimulate cellular proliferation, the Rat 1a fibroblasts expressing c-Myc2–ER, c-MycS–ER, c-MycΔ106–143–ER, or pMV7 vector were used in cell growth assays. As shown in Figure 2c, when the cells were grown without OHT, no significant differences in growth rate were observed with any of the cell lines. When induced by OHT, the growth of cells expressing c-MycΔ106–143–ER or pMV7 vector was not affected. In contrast, both c-Myc2–ER and c-MycS–ER promoted proliferation of the Rat 1a cells when activated (Fig. 2c). These data demonstrate that c-MycS is sufficient to stimulate cellular proliferation comparable to full-length c-Myc.
One explanation for these results is that c-MycS altered the expression or activity of the endogenous c-Myc2. To address this question and determine if c-MycS can replace the endogenous full-length c-Myc, we used cells that had both c-myc gene copies disrupted by gene targeting. c-myc null cells (HO16.4c) grow two to three times slower than the parental TGR-1 cell line but regain normal growth upon reintroduction of c-Myc (Mateyak et al. 1997). We generated stable c-myc null cell lines expressing c-MycΔ106–143–ER or c-MycS–ER proteins. As shown in Figure 3a, the growth rate of cells expressing an unactivated c-MycS–ER or an OHT-activated c-MycΔ106–143–ER was not affected. In contrast, null cells expressing an OHT-activated c-MycS–ER grew at the same rate as the TGR-1 cells (Fig. 3a), suggesting that c-MycS rescued the growth rate of c-myc null cells. As an alternative approach and to verify that ER did not affect the growth rates, we generated myc null cells expressing constitutive Myc2 and MycS without ER in viral expression vectors. Figure 3b demonstrates that the stable cell lines expressing either c-Myc2 or c-MycS grew at rates comparable to the TGR-1 cells. These results indicate that MycS expression alone is sufficient to rescue the slow growth phenotype of c-myc null cell lines and can substitute for full-length c-Myc2.
Figure 3.

c-MycS rescues the slow growth phenotype of the c-myc null cells. (a) Growth of c-myc null cells expressing c-Myc–ER proteins. HO16.4c cells and HO16.4c cells expressing either c-MycS–ER or c-MycΔ106–143–ER (ΔMyc–ER), and the parental TGR-1 cell line were seeded at 2.5 × 104 in replicate 35-mm dishes. After 24 hr, the cells were transferred into medium with or without 1 μm OHT, and total cells were counted at the indicated time points. (b) Growth of c-myc null cells expressing constitutive c-Myc proteins. HO16.4c cells expressing c-Myc2, c-MycS, c-MycΔ150, or pBabepuro vector alone and TGR-1 cells were seeded at 5 × 104 in replicate 35-mm dishes, and total cells were counted at the indicated time points.
In addition to a stimulation of cell proliferation, a higher percentage of dead cells was observed with OHT-activated cells expressing c-Myc2 or c-MycS compared with the uninduced cells during the growth assays (Fig. 4a), suggesting that c-MycS induced apoptosis similar to full-length c-Myc. To further examine this possibility, the above cell lines were transferred to medium containing low serum with or without OHT. These conditions have been shown to induce a higher incidence of apoptosis using full-length c-Myc (Evan et al. 1992). As compared with the control cell lines, both OHT-activated c-Myc2–ER and c-MycS–ER enhanced cell death (Fig. 4b). DNA fragmentation assays were used to confirm that the cell death was due to apoptosis. The uninduced cell lines or the lines containing vector alone or c-MycΔ106–143–ER with OHT did not accumulate DNA fragments after starvation (Fig. 4c). In contrast, both OHT-activated c-MycS–ER and c-Myc2–ER induced apoptosis as evidenced by DNA laddering (Fig. 4c). In addition, a similar percentage of apoptosis was observed in Rat 1a cells constitutively expressing c-MycS or c-Myc2 without ER when the cells were placed in DMEM containing 0.25% calf serum (Fig. 4d).
Figure 4.


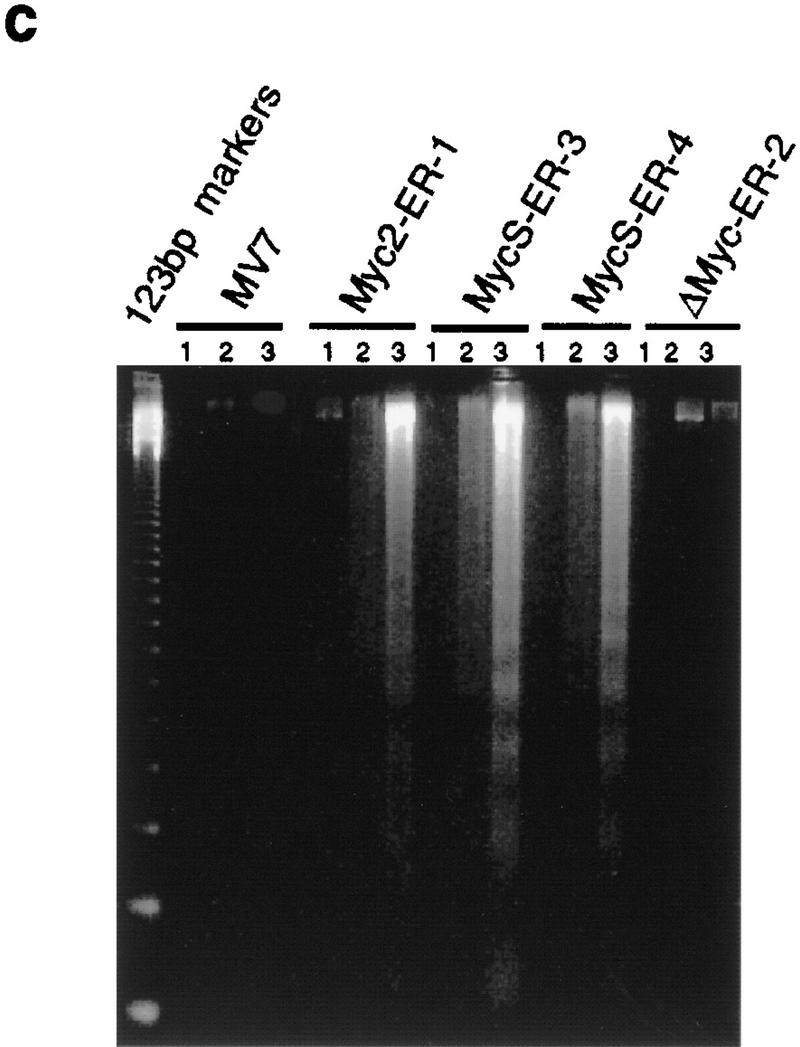
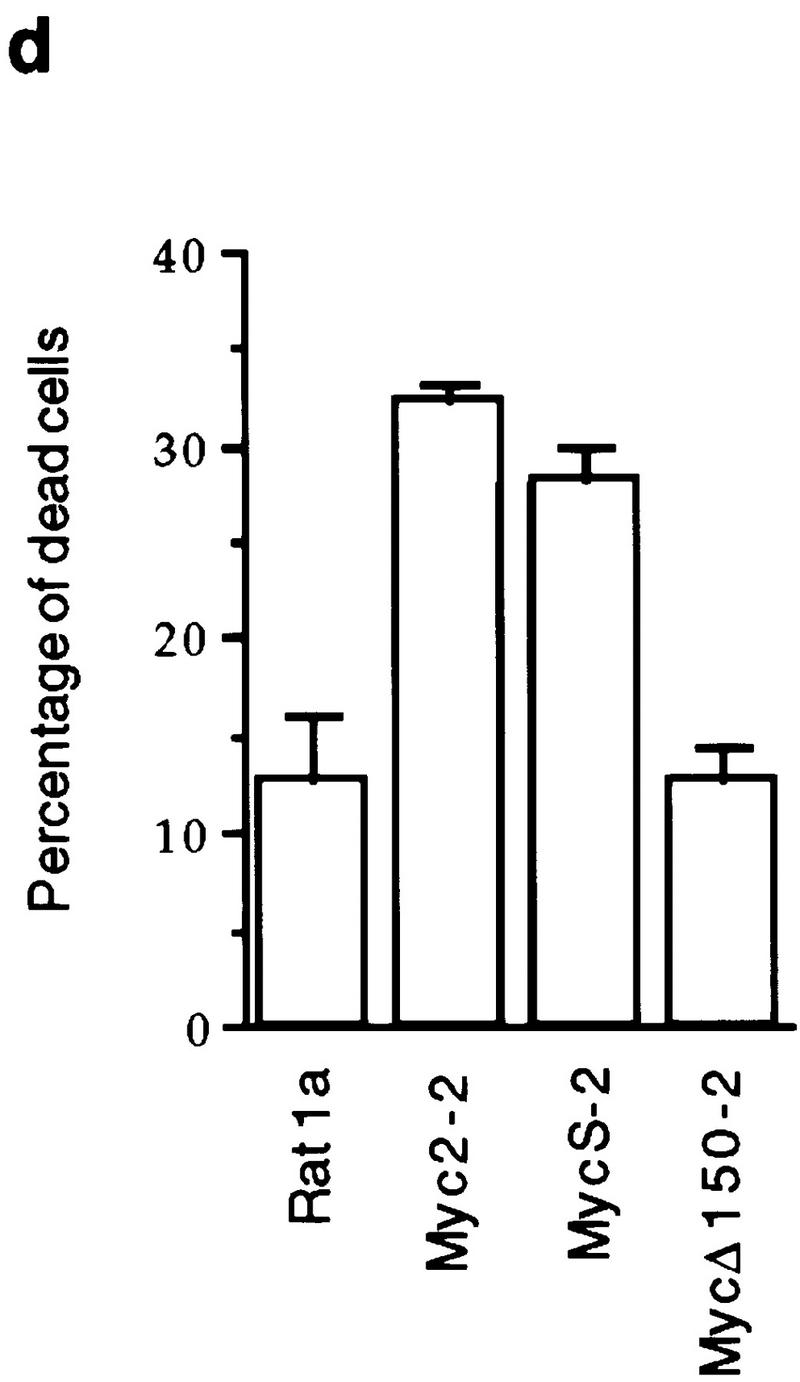
c-MycS induces apoptosis. (a) Apoptosis of Rat 1a cells induced by c-Myc–ER proteins. The stable Rat 1a cell lines expressing c-Myc2–ER, c-MycS–ER, c-MycΔ106–143 (ΔMyc–ER), or the pMV7 vector alone were seeded at 5 × 104 cells in replicate 35-mm plates. After 24 hr, the cells were transferred into DMEM containing 10% CS with or without 1 μm OHT. At the indicated time points, triplicate dishes were counted. The number of floating cells was divided by the total cell count to yield the percentage of dead cells. (b) Apoptosis induced by c-Myc–ER proteins in low serum. The cell lines described above were seeded at 5 × 104 cells in replicate 35-mm plates. After 24 hr, the cells were transferred into DMEM containing 0.25% CS with or without 1 μm OHT. At the indicated time points, the number of dead cells was determined as described above. (c) DNA fragmentation analysis of c-Myc–ER-expressing cells. The cell lines described above were plated at 1.5 × 106 cells/100-mm dish in DMEM containing 10% FCS. After 24 hr, the medium was changed to DMEM containing 0.25% FCS with or without 1 μm OHT. After 20 hr, the adherent cells were combined with the nonadherent cells and the DNA was extracted and analyzed by agarose gel electrophoresis as described. (Lanes 1) Cells in DMEM plus 10% FCS; (lanes 2) cells in DMEM plus 0.25% FCS without OHT; (lanes 3) cells in DMEM plus 0.25% FCS with OHT. (d) Apoptosis induced by constitutive c-Myc proteins in low serum. Rat 1a cells or Rat 1a stable cell lines expressing either c-Myc2, c-MycS or c-MycΔ150 were seeded at 5 × 104 cells in replicate 35-mm plates. After 24 hr, the cells were transferred into DMEM without serum, and the number of dead cells was determined as described above.
Our findings that c-MycS can stimulate proliferation and induce apoptosis are in conflict with earlier findings based on an internal deletion spanning amino acids 7–91 (c-MycΔ7–91). Several studies have shown that this mutated c-Myc protein cannot induce apoptosis or cooperate with Ras in the transformation of rat embryo fibroblast (REF) cells (Stone et al. 1987; Evan et al. 1992). One possible explanation for this discrepancy is that internal deletions in the amino-terminal domain of c-Myc confer an inactive conformation, perhaps due to improper folding. Thus, the possibility remains that neither MbI or MbII are necessary for some of the functions of c-Myc. In support of this idea, there is a report demonstrating that carboxy-terminal fragments of c-Myc without MbI or MbII are capable of inducing apoptosis in microinjection experiments (Kohlhuber et al. 1995). To address these possibilities without creating another internal deletion, we generated a c-Myc protein initiating at amino acid 150 (c-MycΔ150), which lacks both MbI and MbII. Figure 4d demonstrates that stable Rat 1a cell lines expressing c-MycΔ150 had a level of apoptosis comparable to the uninfected cells. This suggests that the MbII region is necessary for the function of c-MycS. In support of these results, overexpression of c-MycS also caused apoptosis of the human 293 kidney cells as efficiently as c-Myc2, whereas a c-MycS protein with a deletion of MbII failed to cause apoptosis (M. Cole, pers. comm.).
Finally, to determine if c-MycS could transform cells, we performed anchorage-independent growth assays. As shown in Figure 5a, the Rat 1a cell lines with vector alone or c-MycΔ106–143–ER did not grow in soft agar, with or without OHT. When cell lines expressing either c-Myc2–ER or c-MycS–ER were induced with OHT, multiple colonies of anchorage-independent cells were observed. We also examined Rat 1a cells constitutively expressing c-MycS, c-Myc2, or c-MycΔ150 without ER. As shown in Figure 5b, Rat 1a cells or stable Rat 1a cell lines expressing c-MycΔ150 or pBabe vector alone did not grow in soft agar, whereas comparable numbers of anchorage-independent colonies were observed with the cell lines expressing c-MycS or c-Myc2. Therefore, these data indicate that c-MycS causes transformation of Rat 1a cells as efficiently as full-length c-Myc and that MbII is necessary for this transformation.
Figure 5.

c-MycS causes anchorage-independent growth. (a) Anchorage-independent growth of Rat1a cells induced by c-Myc–ER proteins. The stable Rat 1a cell lines expressing c-Myc2–ER, c-MycS–ER, c-MycΔ106–143–ER (ΔMyc–ER) or pMV7 vector alone were plated at 1 × 104 cells/35-mm dish in soft agar containing DMEM plus 10% FCS with or without 2 μm OHT. Colonies (>100 μm) were counted by an Omnicon colony counter (Bausch & Lomb) on the tenth day after plating. (b) Anchorage-independent growth of Rat 1a cells caused by constitutive c-Myc proteins. Rat 1a cells or the stable Rat 1a cell lines expressing c-Myc2, c-MycS, c-MycΔ150, or the pBabepuro vector alone were plated at 1 × 104 cells/35-mm dish in soft agar containing DMEM plus 10% FCS. Colonies (>100 μm) were counted as described above.
Contrary to the prevailing model for the molecular function of c-Myc, which proposes that the transactivation of specific target genes through EMS elements is essential for all of its biological activities, our data suggest that transactivation through these elements is not necessary for at least some of the biological functions of c-Myc, including enhancement of proliferation, anchorage-independent growth, and apoptosis, in the model systems we have examined. These results are in agreement with those of Bush et al. (1998) demonstrating that the levels of the proposed transactivation target genes having EMS sites were not significantly different in the c-myc null fibroblasts compared with the parental fibroblasts. Studies showing that the biologically inactive c-MycΔ106–143 protein can still transactivate also suggest that c-Myc biological function can be uncoupled from transactivation (Kato et al. 1990). The ability of c-MycS to repress transcription suggests that repression of growth inhibitory genes, such as gadd45 and gas1, remains viable as an alternative model for c-Myc molecular function. This is supported further by the findings of Bush et al. (1998) that gadd45 levels are higher in the myc null fibroblasts compared to those in the parental cells.
Although we have shown that the transactivation-defective c-MycS protein can function in several biological assays and can substitute for the full-length c-Myc2 in myc null cells, c-MycS may not be able to function as full-length c-Myc2 in all assays. For example, c-MycS does not appear to cooperate with Ras in the transformation of REFs (McMahon et al. 1998). One explanation for these results is that the Myc/Ras cotransformation of REF cells requires transactivation of specific myc target genes through EMS sites that are not required for stimulation of proliferation, apoptosis, or anchorage-independent growth. Perhaps the ability of c-Myc2 to immortalize, which may be distinct from its ability to stimulate proliferation or induce apoptosis, is required to render REF cells susceptible to transformation by Ras, as Ras has been shown to induce senescence (Serrano et al. 1997). However, one caveat in the interpretation of these negative results is that in transient transfection assays c-MycS is expressed severalfold less in REF and other cells compared to c-Myc2 (data not shown). Demonstration of comparable levels of c-MycS in this assay or in any biological assay is important before definitive conclusions can be reached. Further experiments, such as determining the ability of c-MycS to cause tumorigenicity, block differentiation, or substitute for full-length c-Myc2 in mice, will define the requirements for MbI and transactivation through EMS sequences. The finding of new c-Myc target genes and perhaps new DNA-binding sites will also determine whether c-MycS has any transactivation capabilities. Comparison of c-Myc2 and c-MycS allows the separation of the transcriptional activation and repression abilities of c-Myc and will allow further insight into the molecular basis for the complex and diverse biological functions of c-Myc.
Materials and methods
Plasmids and vectors
To obtain the murine c-Myc–ER fusion constructs, a BamHI site was introduced at amino acid 439 of murine c-myc by site-directed mutagenesis using the oligonucleotide shown. The BamHI fragments containing the different c-myc coding sequences were then subcloned into the pBKS(+)ER vector, containing amino acids 281–599 of the murine ER harboring a point mutation at position 525 (Littlewood et al. 1995), which only allows induction with OHT. The chimeric constructs were inserted into the retroviral vector pMV7 or the retroviral pBabepuro vector. For constitutive expression, the c-Myc2 and c-MycS coding sequences were inserted into the retroviral pBabepuro vector or pWZLvector. To obtain the murine c-MycΔ150 expression vector, an ATG codon in optimal consensus sequence (ACCATGG) was introduced at amino acid 150 by site-directed mutagenesis. In addition, an XhoI site was placed 4 nucleotides upstream of the ATG for subcloning into the pWZL viral expression vector. The CMV–Myc expression vectors and CAT reporter vectors for [EFII]6 CAT ODCΔCAT, PrT–CAT, and human p53 promoter have been described previously (Bello-Fernandez et al. 1993; Reisman et al. 1993; Hann et al. 1994; Desbarats et al. 1996; Spotts et al. 1997). Luciferase reporter vectors pMLP–Luc, GADD45–Luc, and GAS1–Luc have been described previously (Li et al. 1994; Lee et al. 1997; Marhin et al. 1997).
Cell lines and infections
The HO16.4c c-myc null fibroblasts were generated as described in Mateyak et al. (1997). The pMV7 vector, pBabepuro vector, and the various c-Myc–ER and c-Myc constructs were transfected into the ϑ2 packaging cell line using the calcium-phosphate precipitation method and cell lines were selected in 400 μg/ml G418 for the pMV7 constructs or 2.5 μg/ml puromycin for the pBabepuro constructs. The different viral media were then collected and used to infect Rat 1a or HO16.4c c-myc null fibroblast cell lines with 6 μg/ml Polybrene, and stable cell lines were selected as described above. Expression of the c-Myc proteins was verified by immunoprecipitation using anti-Mycfl as described previously (Spotts et al. 1997).
Transient transfections and enzymatic assays
For CAT assays, NIH-3T3 cells were seeded at 5 × 105 cells/60-mm dish 24 hr prior to transfection. The quantity and type of DNA used are described in the figure legends. Cells were transfected using LipofectAmine (GIBCO BRL) in serum-free medium for 6 hr. After 48 hr lysates were prepared and analyzed for β-galactosidase and CAT activities as described in Hann et al. (1994). For luciferase assays, NIH-3T3 cells were plated at 7 × 105 cells/60-mm dish 24 hr prior to transfection. Samples were harvested 48 hr post-transfection by lysis in 400 μl of Reporter Assay buffer (Promega). Luciferase activity was measured in a luminometer with 100 μl room temperature Luciferase Assay Substrate (Promega) added by automatic injection to 20 μl of lysate. All readings were normalized by β-galactosidase activity.
DNA fragmentation assay
Adherent cells were trypsinized, combined with nonadherent cells and washed in cold PBS. The cells (3 × 106) were then lysed (50 mm Tris at pH 7.5, 20 mm EDTA, 1% NP-40) for 10 sec, and the supernatant was collected after centrifugation at 1600g for 5 min. After adjusting to 1% SDS and 2.5 μg/ml proteinase K, the supernatants were incubated for 2 hr at 37°C and the DNA was ethanol precipitated. Samples were loaded onto 1.6% TBE agarose gels and the DNA visualized by ethidium bromide staining.
Acknowledgments
We thank Linda Penn for the kind gift of GADD45–Luc, Ed Ziff for MLP–Luc, Theodore Lee for GAS1–Luc, John Cleveland for ODCΔCAT, Martin Eilers for PrT–CAT, David Reisman for human p53 CAT, and Trevor Littlewood and Gerard Evan for the modified ER construct. We also thank Mark Gregory, Gail Cornwall, Uri Vankin, and Michael Cole for reviewing the manuscript.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL steve.hann@mcmail.vanderbilt.edu; FAX (615) 343-5791.
References
- Bello-Fernandez C, Packham G, Cleveland JI. The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc Natl Acad Sci. 1993;90:7804–7808. doi: 10.1073/pnas.90.16.7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush, A., K. Dugan, M. Mateyak, A. Obaya, S. Adachi, J. Sedivy, and M. Cole. 1998. c-Myc controls cell cycle progression without transactivating known target genes. Genes & Dev. (this issue). [DOI] [PMC free article] [PubMed]
- Desbarats L, Gaubatz S, Eilers M. Discrimination between different E-box-binding proteins at an endogenous target gene of c-Myc. Genes & Dev. 1996;10:447–460. doi: 10.1101/gad.10.4.447. [DOI] [PubMed] [Google Scholar]
- Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Grandori C, Eisenman RN. Myc target genes. Trends Biochem Sci. 1997;22:177–181. doi: 10.1016/s0968-0004(97)01025-6. [DOI] [PubMed] [Google Scholar]
- Hann SR, Sloan-Brown K, Spotts GD. Translational activation of the non-AUG-initiated c-myc 1 protein at high cell densities due to methionine deprivation. Genes & Dev. 1992;6:1229–1240. doi: 10.1101/gad.6.7.1229. [DOI] [PubMed] [Google Scholar]
- Hann SR, Dixit M, Sears RC, Sealy L. The alternatively initiated c-Myc proteins differentially regulate transcription through a noncanonical DNA-binding site. Genes & Dev. 1994;8:2441–2452. doi: 10.1101/gad.8.20.2441. [DOI] [PubMed] [Google Scholar]
- Henriksson M, Lüscher B. Proteins of the Myc network: Essential regulators of cell growth and differentiation. In: Vande Woude G, Klein G, editors. Advances in cancer research. New York, NY: Academic Press; 1996. pp. 109–182. [DOI] [PubMed] [Google Scholar]
- Kato GJ, Barret J, Villa-Garcia M, Dang CV. An amino-terminal c-Myc domain required for neoplastic transformation activates transcription. Mol Cell Biol. 1990;10:5914–5920. doi: 10.1128/mcb.10.11.5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhuber F, Hermeking H, Graessmann A, Eick D. Induction of apoptosis by the c-Myc helix-loop-helix/leucine zipper domain in mouse 3T3-L1 fibroblasts. J Biol Chem. 1995;270:28797–28805. doi: 10.1074/jbc.270.48.28797. [DOI] [PubMed] [Google Scholar]
- Kretzner L, Blackwood EM, Eisenman RN. Myc and Max proteins possess distinct transactivational capabilities. Nature. 1992;359:426–429. doi: 10.1038/359426a0. [DOI] [PubMed] [Google Scholar]
- Lee TC, Linheng L, Lennart P, Ziff EB. Myc represses transcription of the growth arrest gene gas 1. Proc Natl Acad Sci. 1997;94:12886–12891. doi: 10.1073/pnas.94.24.12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LH, Nerlov C, Prendergast G, MacGregor D, Ziff EB. c-Myc represses transcription in vivo by a novel mechanism dependent on the initiator element and Myc box II. EMBO J. 1994;13:4070–4079. doi: 10.1002/j.1460-2075.1994.tb06724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marhin WW, Chen S, Facchini LM, Fornace AJ, Jr, Penn LZ. Myc represses the growth arrest gene gadd45. Oncogene. 1997;14:2825–2834. doi: 10.1038/sj.onc.1201138. [DOI] [PubMed] [Google Scholar]
- Mateyak MK, Obaya AJ, Adachi S, Sedivy JM. Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ. 1997;8:1039–1048. [PubMed] [Google Scholar]
- McMahon S, Van Buskirk HA, Dugan KA, Copeland TD, Cole M. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell. 1998;94:363–374. doi: 10.1016/s0092-8674(00)81479-8. [DOI] [PubMed] [Google Scholar]
- Mink S, Mutschler B, Weiskirchen R, Bister K, Klempnauer KH. A novel function for Myc: Inhibition of C/EBP-dependent gene activation. Proc Natl Acad Sci. 1996;93:6635–6640. doi: 10.1073/pnas.93.13.6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisman D, Elkind NB, Roy B, Beamon J, Rotter V. c-Myc transactivates the p53 promoter through a required downstream CACGTG motif. Cell Growth Differ. 1993;4:57–65. [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Spotts GD, Patel SV, Xiao Q, Hann SR. Identification of downstream-initiated c-Myc proteins which are dominant-negative inhibitors of transactivation by full-length c-Myc proteins. Mol Cell Biol. 1997;17:1459–1468. doi: 10.1128/mcb.17.3.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone J, de Lange T, Ramsay G, Jakobovits E, Bishop JM, Varmus H, Lee W. Definition of regions in human c-myc that are involved in transformation and nuclear localization. Mol Cell Biol. 1987;7:1697–1709. doi: 10.1128/mcb.7.5.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]