

Abstract
The molecular origin of nucleotide insertion catalysis and fidelity of DNA polymerases is explored by means of computational simulations. Special attention is paid to the examination of the validity of proposals that invoke prechemistry effects, checkpoints concepts and dynamical effects. The simulations reproduce the observed fidelity in Pol β, starting with the relevant observed x-ray structures of the complex with the right (R) and wrong (W) nucleotides. The generation of free energy surfaces for the R and W systems also allowed us to analyze different proposals about the origin of the fidelity and to reach several important conclusions. It is found that the potential of mean force (PMF) obtained by proper sampling does not support QM/MM based proposals of prechemistry barriers. Furthermore, examination of dynamical proposals by the renormalization approach indicates that the motions from open to close configurations do not contribute to catalysis or fidelity. Finally we discuss and analyze the induced fit concept and show that, despite its importance, it does not explain fidelity. That is, the fidelity is apparently due to change in the preorganization of the chemical site, due to its response to the binding site reorganization in the binding of the W instead of the R base. Furthermore, since the issue is the barrier associated with the enzyme-substrate (ES)/DNA complex at the chemical transition state and not the path to this complex formation (unless this path involves rate determining steps), it is also not useful to invoke checkpoints while discussing fidelity.
Keywords: enzyme catalysis, renormalization approach, replication fidelity, conformational change, free energy landscape, induced fit, check points
I. Introduction
The reproduction of all forms of life depends on the accurate replication of the genome, which is facilitated by DNA polymerases.1 These enzymes increase the rate of phosphodiester bond formation by many orders of magnitude compared to the corresponding reaction in water.2 The rate of incorporation of an incoming wrong nucleotide (W) is drastically slower than the corresponding rate of the right nucleotide (R) for DNA polymerases that exhibit high replication fidelity (for reviews see refs.3-6). Thus it is important to quantify the factors that control the fidelity.
Despite significant experimental progress in studies of the fidelity of various DNA polymerases (e.g., refs.3-10), we still do not have a clear quantitative picture of the energetics of this process. A significant progress has been made in recent theoretical studies, which explored the contributions to fidelity from the binding and chemical steps,11-18 as well as on the possible roles of conformational changes,16,19,20 and protein motions.21 However, the progress in these key areas is still at a relatively early stage.
Further insight about the fidelity issue has been provided in a recent study,21 which used computer simulations to model the effect of mutations on the structure of Pol β, and examined the corresponding electrostatic effects by a qualitative analysis. Additionally, theoretical studies of related problems have been reported recently.22-25 Molecular dynamics simulations have helped to delineate a possible sequence of conformational change events after dNTP binding in the catalytic cycle of Pol β.19,20,26 Simulation studies also enabled to test various catalytic proposals of nucleotide transfer reactions.12,27 Finally, an attempt to progress in a more quantitative direction in the comparison of calculated and observed mutational effects has been reported.28 This study also introduced a useful approach of constructing interaction matrices and using them in probing the transfer of information between the binding and catalytic sites.
One of the intriguing aspects of the action of DNA polymerases is the role of substrate-induced conformational changes in the catalytic process and the influence of these changes on DNA replication fidelity. This aspect is related to the general issue of the role of the enzyme conformational landscape in catalysis29-31 and to proposals that dynamical effects and coupled motions play a major role in enzyme catalysis.32,33 Although careful studies (for review, see refs.34,35) have questioned the validity of such proposals, there are aspects that were not explored in a conclusive way and one of these aspects is the relationship between DNA polymerase conformational changes and replication fidelity, that will be explored in this work. More specifically, based on structural and kinetic studies of Pol β, it is generally believed that the first step of the nucleotide insertion pathway includes the N–subdomain’s (equivalent to the fingers subdomain of right-handed DNA polymerases) closing triggered by correct dNTP binding, while binding of an incorrect dNTP will hamper this closing, consistent with an “induced fit” mechanism.36 It has been suggested that conformational changes that occur prior to the chemical step play a major role in establishing the fidelity of Pol β (e.g., ref.37) and DNA polymerases in general.4-6 Instead, we believe that it is considerably more likely that fidelity is determined by the binding energy plus the activation barrier of the chemical step, and that the barrier for conformational changes between the open and closed forms is not likely to determine the fidelity in Pol β.14,15 Experimental evidence also indicates that the open to closed subdomain transition is too rapid to be rate determining.38-41
A recent computational study16 has attempted to generate the catalytic landscape for Pol β and obtained insightful conclusions including capturing some of the structural changes, that were not known at the time of that work (see discussion). However, after the crucial elucidation of the structure of R (ref.42) and W (ref.43) we can move to a more concrete ground. In fact, the advances in structural studies led to several interesting theoretical studies.17,44,45 However, some of these studies17,27,44,46,47 involve some questionable conclusions. For example, a QM/MM study on the prechemistry barrier, before the nucleotide transfer reaction, in human DNA pol β with a G:A mismatch in the active site44 was estimated to be about 14 kcal/mol. Based on this, the authors suggested that the free energy required for formation of the prechemistry state is the major contributing factor to the decrease in the rate of incorrect nucleotide incorporation compared with correct nucleotide insertion and therefore to the fidelity enhancement. Also in a more recent QM/MM study on DNA polymerase IV (Dpo4),48 it was argued that the prechemistry reorganization of the catalytic site ( which brings the enzyme to an active conformation from its x-ray position prior to the chemical reaction) would cost approximately 4.0 to 9.0 kcal/mol. However, both these studies employed a problematic treatment, using basically energy minimization without proper relaxation and sampling.49 Such treatments can lead to artificial barriers as they will be illustrated in the present work.
In a complex enzymatic reaction, involving the association of the enzyme with more than one substrate, it is unlikely that the highest energy barrier will be approached in a single step from the ground state reactants. Instead there will be one or more intermediate steps along the path way. This fact and other considerations led different workers46,50 to suggest that the intermediate points can serve as kinetic checkpoints. Basically it was argued that although the presence of such checkpoints will not change the overall fidelity of the reaction , they will define the pathway by which that fidelity would be realized. In the polymerase reaction, the intermediate steps or checkpoints will allow rejection of inappropriately paired dNTPs before the polymerase attempts phosphodiester bond formation of such substrates.50 Unfrotunetly, these concepts are not based on clear molecular concepts and have not been supported by consistent simulations. In fact, even the common suggestion that fidelity is due to the induced fit effect, is not based on clear structure-energy analysis.16 ( see also discussion in section IV. 2)
Fortunately, with the new available structural information42,43 we are entering a stage where one can apply consistent theoretical analysis and establish the molecular origin of replication fidelity, while analyzing (and disproving if needed) problematic proposals. Such an analysis is the purpose of the present work.
In order to set the stage for our discussion and analysis we start with the general working hypothesis of Figure 1. This conceptual figure has emerged from our previous studies of DNA polymerases15,16 where we concluded (based on calculations of the barrier for the rate determining chemical step) that the path along the coordinate for the chemical process pass at a somewhat different value of the “orthogonal” coordinate of the protein structural changes (typically considered as the motion between the open and closed forms). We would like to emphasize that, this view is fundamentally different than related assumptions19 that the barriers for the conformational changes between the open and closed form contribute to fidelity or to the reaction rate. The barriers for the motion from the open to closed form in the reactant state of R (from rORS(R) to rCRS(R) in Figure 1) is not likely to influence fidelity, or for that matter, the reaction rate for both W and R, as long as they are lower than the chemical barriers.35 Similarly the interesting possibility that the pre-chemistry barriers are different for W and R (e.g., ref.19) is not likely to influence fidelity as long as those barriers are lower than the chemical barrier. In other words, we are focusing here on the path between the bound incoming dNTP to the product (rCRS(R) to rCP(R) and rC’RS(W) to rCP(W)) and on its dependence on the protein conformation, assuming (based on experimental findings) that the chemical step is rate-determining (this point will be further considered in the Discussion section IV).
Figure 1.
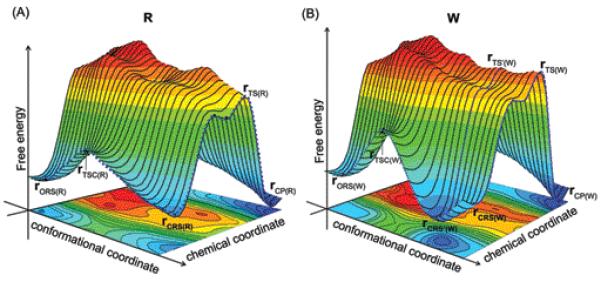
Schematic free energy surfaces that illustrate possible coupling between the conformational and chemical coordinates for the insertion of (A) correct and (B) incorrect dNTP by a DNA polymerase. rO, and rC, designate respectively the open and closed configurations. TSC designate the transition state associated with the conformational motion. RS, TS, and P designate respectively the reactant state (ES), the TS, and the product sate. (R) and (W) designate the right and wrong systems, respectively.
With the above perspective in mind we try to move forward from our previous study (using now the actual structures of R and W of Pol β) and address the following questions: (i) the possible relationship between the chemical barrier for R and W and the orthogonal protein conformational coordinate; (ii) The general relationship between the landscape of the protein conformations and the catalytic power of enzymes; (iii) the molecular meaning of the “induced fit” effect and its relationship, if any, to fidelity; (iv) the problems with the prechemistry concept; and (v) the idea of coupled motions and check points.
II. Computational Methods
II.1 Potential surfaces
II.1a General Considerations
To simulate the chemical steps in the reaction of DNA polymerase it is essential to have a reasonable potential energy surface that describes the bond-breaking and bond-making processes. The empirical valence bond51 approach provides a powerful tool. The EVB method has been described extensively elsewhere (e.g., refs.51-53) provides a powerful way of obtaining reliable activation free energies, while taking into account the full protein flexibility and configurational space. Key details about this method are given in the SI.
The EVB calculations require information about the reaction mechanism and potential surface for the solution reaction as well as the most likely mechanism for the reaction in the protein. In the case of the reaction catalyzed by Pol β, there is still debate (and some confusion) whether phosphodiester hydrolysis reaction follows an associative, dissociative or concerted mechanism (these mechanisms are depicted in Figure 2). Thus we have emphasized for a long time the need for a careful analysis of the relevant experimental and theoretical information about this type of reaction as well as related phosphate monoester hydrolysis reactions in solution.54-58 Our studies demonstrated repeatedly that the available experimental information (including linear free energy relationships (LFER) studies) cannot tell what is the mechanism, but that all careful and consistent computational studies (that in fact reproduce the available experimental results) lead to associative and/or concerted surfaces, unless we deal with extremely strong leaving group. Our most careful recent ab initio solution study of phosphate diester hydrolysis56 produced the surface shown in Figure 3A where the reaction proceeds through a concerted path. Using a similar surface to calibrate our EVB we concluded previously12 that an associative mechanism with some concerted character is the most likely reaction mechanism for correct nucleotide insertion. Thus for chemistry in the active site of Pol β, we assumed the reaction takes place in the three generic steps (see Figure 2); a proton transfer from the primer 3′OH is followed by in-line nucleophilic attack of 3′O− to the dNTP α-phosphate, and the reaction is completed by the departure of the pyrophosphate leaving group (as discussed here, these steps can involve a concerted process). For the proton transfer step there are also several possibilities: the proton of the primer 3′OH could either transfer to Asp256, 5′-oxygen or a non-bridging oxygen of the dNTP α-phosphate, or a surrounding water. Based on previous simulations, it seems most likely that the dominant mechanism involves a proton transfer to Asp25612,28 or water followed by nucleophilic attack on Pα by the deprotonated 3′OH and formation of a pentavalent intermediate in an associative/concerted mechanism. The concerted EVB surface calibrated on the ab initio solution reaction appears to reproduce the main features of this surface as shown in Figure 3B.
Figure 2.
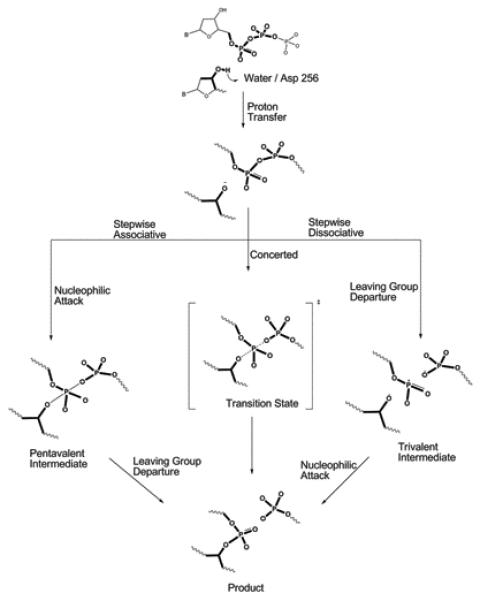
A schematic description of the chemical steps of the nucleotide incorporation reaction of Pol β. Deprotonation of the primer 3′-hydroxyl is followed by nucleophilic attack on the Pα of the incoming dNTP. Formation of O3′- Pα bond results in releasing a pyrophosphate group (leaving group). As shown in the Figure, after the initial proton transfer, the reaction may proceed either via a stepwise (associative or dissociative) or concerted mechanism. However, previous studies (ref.103) suggested that the hydrolysis of a phosphate diester is unlikely to proceed via a stepwise dissociative mechanism. In the stepwise associative model, a pentacovalent intermediate is formed between the bond-forming and breaking steps, resulting in two separate activation barriers. In the concerted model, reaction proceeds through a single transition state with partial bond forming and bond breaking character at the Pα.
Figure 3.
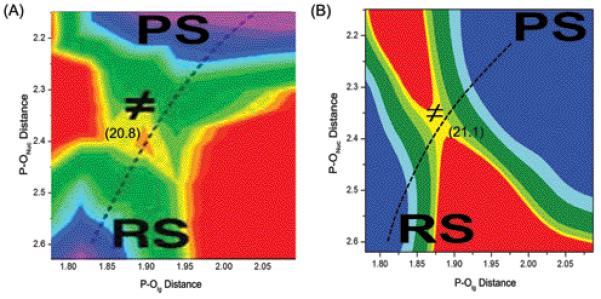
The ab initio free energy surface (A) for the solution reaction (adapted from ref.56) and the corresponding calibrated EVB surface (B). The reproduction of the activation barrier (20.8 vs 21.1 kcal/mol) and the corresponding transition state geometry (P-Onuc: 2.33 vs 2.32 Å and P-Olg: 1.83 vs 1.88 Å) establishes our ability to model the concerted mechanism by the EVB approach.
Although the above concerted surface provides the most reliable representation of the true surface, we found out that we can obtain very similar results (see below) with a stepwise surface that allow for significantly more efficient calculations and thus more effective sampling. More specifically, in our study, we constructed the EVB surfaces by exploring both the stepwise associative and the concerted path with a single TS. In the associative mechanism, we considered the initial proton transfer to both bulk water and Asp 256. The corresponding EVB resonance structures are depicted in the Figure 4A. Both calibrated EVB surfaces reproduce the barrier of the solution surface and can be used for studies in proteins by replacing the solvent with the protein environment (after demonstrating that the two EVB representations produce similar calculated barriers in the protein).
Figure 4.
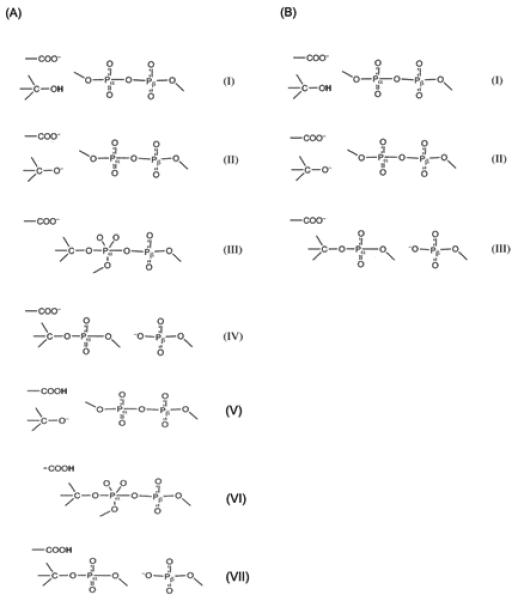
The valence bond structures representing the reactants, intermediates, and products, used for modeling the pol-β catalyzed nucleotide insertion reaction with (A) stepwise associative mechanism and (B) a concerted mechanism. Resonance states (I-IV) of A, corresponds to the initial proton transfer (PT) to bulk water mechanism and the resonance states (V-VII) of A corresponds to the PT to Asp256 mechanism. However, the concerted mechanism (B) is explored only for the PT to bulk water mechanism.
The modeling of the different mechanistic options is described in the SI, and here we only address key points.
II.1b Proton transfer to the bulk water
In order to have a complete reaction free energy profile, we need to calculate the proton transfer activation barrier, Δg‡PT. However, since this step is not likely to be the rate limiting in the nucleotide incorporation reaction of Pol β, it is sufficient (in most cases) just to calculate the proton transfer reaction energy, ΔGPT, which in turn requires calculating the pKa shift of the proton donor. This was done as described in the SI (II. 1a). In this respect we like to point out that as much as reactions in proteins are concerned we believe that the calibrated EVB provided by far the most reliable tool59 as well as analysis of the requirements for converging pKa calculations by ab-initio QM/MM approaches.60
II.1c The mechanisms considered
All the steps associated with the associative (i.e., initial PT to bulk water, or Asp256, nucleophilic attack, leaving group departure) and concerted pathways explored in this study are described in SI (II. 1 – II. 2).
II.1d What about PT to the phosphate
A mechanism of a PT to the phosphate has been considered in our previous works for phosphomonoesters (eg., refs.55,61). This type of mechanism can operate in polymerases12 and a version of this mechanism has been used recently in a QM/MM energy minimization study of Pol β27,47 considering PT from the O3′ to the O1γ of pyrophosphate through two water molecules. It was further argued that this mechanism might be the most favorable pathway even for other polymerases.47,48 Unfortunately, the study of refs.47,48 and the corresponding conclusions involve major problems. First, the energetics of the PT process has not been calibrated or validated by any pKa or related calculations in solution. Second, the evaluation of proton transfer barriers and paths by energy minimization has been show by us (e.g., refs.59,62) to be extremely problematic, including cases with relatively careful treatments (e.g. refs.63,64). Overall we have shown (and others now started to reach similar conclusions65) that electrostatics rather than the orientational effects would drive the proton transfer in biological systems and that a proper sampling is essential to obtain the relevant activation free energies. Similarly we also have shown that when the distance requirements are not extreme (namely when the energy of bringing the donor and acceptor to close distance is not very high) PT is most unlikely to occur via several water molecules. In fact we are not aware of any case where a proposal of PT trough several water molecules in catalyzing phosporyl transfer reaction have been shown (by proper sampling to have a lower barrier than a PT through a single water molecule.
II. 2 Initial structures preparation for EVB calculations and simulations protocol
The preparation of the initial structure for the simulations is described in the SI (IV. 1). Furthermore, the preparation of the system configurations for the conformational study using renormalization approach described below are also described in the SI (IV. 2).
The simulation protocol is also described in the SI (V) but we like to clarify that the EVB and FEP calculations were done with the LRF long rang treatment (which is equivalent to having no cutoff for the electrostatic interactions). All the simulations were performed using the MOLARIS simulation package.66 The nucleic acid bases were represented by AMBER charges67 and ENZYMIX vdW parameters (after validation that these parameters give reliable solvation energies and base-pairing energies and structures). A single center model with the vdW parameters r* = 1.30 and ε = 0.06 kcal/mol12 was used for the Mg2+ ions.
The effect of ionizable groups was taken into account as follows: (i) by performing EVB calculations where only the ionizable residues within the 10 Å region from the geometric center of the EVB atoms are ionized, and then (ii) evaluating the effect of the remaining ionizable groups beyond the 10 Å from the geometric center of the EVB atoms by using a macroscopic treatment with a large dielectric constant for charge-charge interaction.68 The ionization states were evaluated by using our Monte Carlo (MC) approach. The overall effect of the ionizable groups beyond the 10 Å region was found to be quite small (i.e., a change of ± 1.5 kcal/mol of the activation free energy).69,70
II.3 The renormalization approach
Although the EVB mapping approach explores the conformational landscape around the starting configuration, it does not explore regions that are separated from the starting configurations by large barriers (excluding of course these changes that are captured by the EVB mapping procedure). Since we are also interested here in the effect of large conformational changes, we have use the renormalization approach.71-73 In this approach one tries to get the best correspondence between the free energy and dynamics of a full explicit model (all-atom molecular dynamics) and an implicit Langevin Dynamics (LD) model of reduced dimensions. The correspondence is achieved by using constraints of varying strengths in both the implicit and the explicit models and then adjusting the friction in LD treatment to maximize the agreement between the time dependence responses of both models. The external constraint allows us to run all-atom molecular dynamics simulation in a reasonable computer time and then to obtain key information for LD simulations of long time processes. This approach had already been tested and used very effectively in studies of the selectivity of ion channels,74 in proton transport simulations,62 in protein dynamics,71 and in evaluating PMFs.72
III. Results and Analysis
The central aspect in modeling enzyme catalysis is the comparison of the free energy profiles of enzymatic reactions to the corresponding reference reactions in water, using the same computational methodology. Such a strategy, which is the main point of EVB studies, allows one to elucidate the factors responsible for the rate enhancement in the active site of polymerases and to quantify the effects that determine the fidelity. Thus we determine in the current work the free energy surfaces of nucleotide insertion reaction of Pol β with correct (R) and incorrect (W) nucleotide pairs (as well as the reference solution reaction), and explore different key structure-function aspects of the DNA pol β fidelity problem.
III. 1 Exploring the Reaction Surface in the protein
One of our main tasks has been to obtain free energy surfaces that are not only sufficiently reliable to capture the correct reaction path, but also able to reproduce the proper free energy surfaces for possible prechemistry events. This is crucial since previous fidelity studies,17,20,27,44,47,48,75 with the exception of (our PROTEINS 200816), have used rigid protein model with what amounts to problematic energy minimization studies. In particular it is unlikely that such studies can capture the free energy relaxation associated with the protein response to the motion from the W to R system.
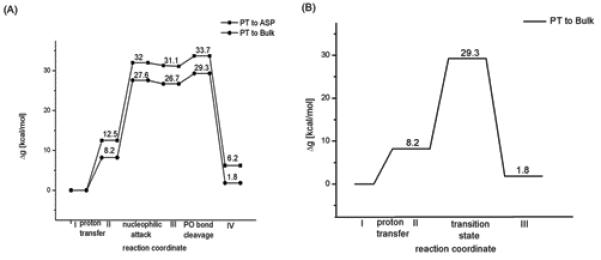
After calibrating the reference reaction (section II), we considered the reaction in Pol β using the simulation protocol described in section II and SI, and the results of these simulations are depicted in Figure 6 and Table 1. More specifically, the reaction free energy for PT from the 3′-O-ribosyl group to OH− in the bulk water was estimated by calculating the difference in salvation free energy of the 3′-O-ribosyl group in aqueous and protein environments. This approach does not give us the Δg≠. However, the calculated magnitude of ΔG0 for both the R (2.9 kcal/mol) and W (2.4 kcal/mol) systems suggests that PT step is not the rate limiting step for the overall nucleotidyl transfer reaction. Interestingly, the corresponding reaction free energy associated with the PT to Asp256 in the R system of Pol β is 8.2 kcal/mol, which is approximately 2 kcal/mol higher than the corresponding free energy in the active site of T7 pol.12 The calculated activation free energy barrier associated with this PT step is 10.0 kcal/mol.
Figure 6.
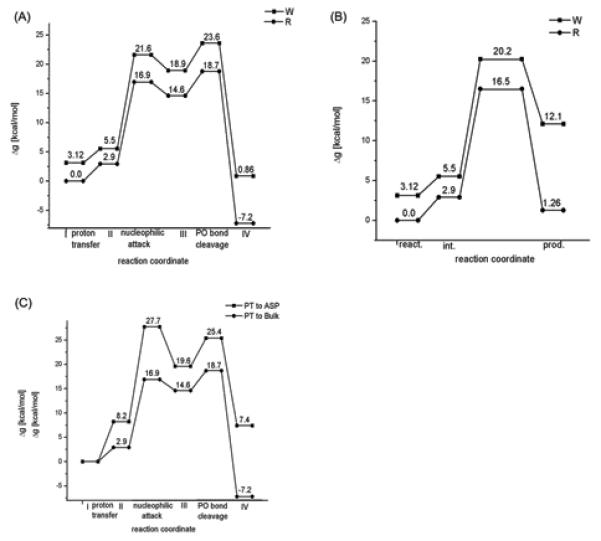
The energetics of the catalytic reaction for different systems and different mechanism. (A) and (B) describe, respectively, the energetics of the associative and concerted mechanisms for the R and W systems with an initial PT to the bulk solvent. (C) provides a comparison of the activation and reaction free energies of the R system following associative mechanism with the initial PT to both the bulk solvent and to Asp256. Note that both in (A) and (B), the reference reactant state energy of the W system is at 3.12 kcal/mol, which is the free energy associated in moving 3′-primer from its x-ray state to the catalytically more favorable state as shown and described in Figure 8.
Table 1.
Calculated activation and reaction free energies for individual steps for the different reaction mechanisms for pol β.
| System | Reaction Step |
Reaction Typec |
Reaction Mechanism |
ΔG0 (kcal/mol) |
Δg≠ (kcal/mol) |
|---|---|---|---|---|---|
| R | I → IIa | PT | Bulk | 2.9 | |
| II → IIIa | NucA | Bulk | 11.7 | 14.0 | |
| III →IVa | DepLg | Bulk | −21.8 | 4.1 | |
| I → Va | PT | Asp | 8.2 | ||
| V →VIa | NucA | Asp | 11.4 | 19.5 | |
| VI→VIIa | DepLg | Asp | −12.2 | 5.8 | |
| I → IIb | PT | Bulk | 2.9 | ||
| II → IIIb | Bulk | −1.64 | 13.61 | ||
| W | I → IIa | PT | Bulk | 2.4 | |
| II → IIIa | NucA | Bulk | 13.4 | 16.1 | |
| III → IVa | DepLg | Bulk | −18.04 | 4.7 | |
| I → IIb | PT | Bulk | 2.4 | ||
| II → IIIb | Bulk | 6.61 | 14.75 |
The nucleophilic attack of the 3′-O− group of the deoxyribose on the Pα phosphorous atom ( step II → III in Figure 4), following a PT to the bulk water, was evaluated for R and W systems of Pol β. The calculated energies indicate that in the case of R, the reaction proceeds via a transition state (TS) with activation free energy of about 14.0 kcal/mol relative to the deprotonated state and leads to the formation of penta-coordinated intermediate state which is 11.7 kcal/mol higher than the deprotonated state (similar barrier was found with the concerted path). However, in the case of the W system, a direct nucleophilic attack from r(ES(W) (the configuration of the reactant enzyme-substrate complex where the position of the template O3′ primer corresponds to its x-ray position in W system) the energy continued to rise and the transition state was found to lie above 50 kcal/mol above the corresponding initial deprotonated state. This clearly demonstrated that in the unrelaxed ground state geometry of the W system, the nucleotide transfer reaction involves a very large barrier. Obviously, we have to allow a proper relaxation and to generate the proper least energy path for the nucleophilic attack step (see below). At any rate, generating a relaxed prechemistry state for W (where the configuration of the W system is similar to that found in the x-ray structure of pol β with the R system42) leads to the formation of a penta coordinated intermediate state with an energy 13.42 kcal/mol higher than the corresponding deprotonated RS state. The activation free energy barrier associated with this process is 16.1 kcal/mol, which is higher by 2.1 kcal/mol from its corresponding matched nucleotide pair (R) reaction. Here, it is important to realize that while carrying out these protein simulations in both R and W cases, no positional or distance constraints were imposed on the systems. In other words, unlike the previous studies,17,20,44,47 the results obtained here were not biased or influenced by any constraints and they were obtained by proper exploration of the phase space. These results reproduced the observed trend in kpol where adding the difference in the binding energy (see below) reproduces the observed fidelity (Table 1).
In the associative stepwise mechanism we have to consider, in addition to the nucleophilic attack step, the departure of the leaving group from the pentavalent phosphorane intermediate (the step that corresponds to the III → IV transition in Figure 4A). The barrier associated with this step increases the overall activation barrier by 4.1 and 4.7 kcal/mol for R and W systems respectively.
In addition to the PT to bulk water mechanism, we also explored the mechanism with Asp256 as a general base in R system. This corresponds to the V → VI transition in Figure 4A. In this mechanism, Asp256 is neutral in the reactant state. The resulting activation barriers and the reaction free energies are shown in Figure 6C. The activation barrier for this mechanism appears to be higher than the corresponding barrier for the path with PT to the bulk water. Thus the barriers for the mechanism with Asp256 as a general base , for the W system, would be even higher. With this consideration in mind we did not explore the Asp256 as a general base mechanism for the W system .
As stated above the true transition state probably involves a concerted mechanism but the catalytic effect of the protein can be assessed by considering the stepwise mechanism. To establish this point we calculated the free energy surface , for the concerted path of the nucleophilic attack of the 3′-O− group of the deoxyribose on the Pα of the incoming nucleotide. The resulted free energy surface is depicted in Figure 6B. As seen from the Figure the activation barriers for the concerted and associative paths are comparable suggesting that the exact nucleotidyl transfer mechanism (concerted or stepwise associative) have very little impact on our conclusions about fidelity and prechemistry as long as we model both systems with the same EVB.
It is important to address here previous QM/MM studies,17,44 which obtained concerted reaction path in pol β. The calculated path is probably quite reasonable. However, as a general note for workers in the field we would like to point out that the surface is energy minimized potential surface rather than a free energy surface (this clearly leads to problems in determining the prechemistry barrier) and there are other potential problems in using energy minimization approaches (see ref.49). However, our main concern which is clearly not limited to the work of ref,17,44 is the fact that it has not been validated by reproducing the observed barriers for the solution reaction. The tendency to assume that just performing ab initio QM/MM calculations in proteins should give the exact barrier is quite risky in our view. Thus having a calibrated EVB surface that exactly reproduces the best information on the solution reaction is clearly a powerful way of getting reliable results. However, unfortunately ref17,44 overlooked the existence of proper QM/MM-EVB free energy calculations (e.g., ref.12) that could have helped at least in validating the prechemistry idea.
It is also instructive to comment here on the recently proposed nucleotide transfer mechanism in pol β and other polymerases,47,48 where the reaction proceeds in a concerted fashion with the simultaneous transfer of the proton to the O1γ of pyrophosphate through several water molecules (following Grotthuss like mechanism) and the attack of the deprotonated O3′ primer on the Pα atom of the incoming nucleotide. Though there will be a pool of water molecules in the vicinity of the enzyme active site, the actual PT pathway depends on the electrostatic environment and the pKa values of the donor and acceptor (this can also be a water molecule). This point has been repeatedly argued and shown several times in the literature.59,62 Hence PT to a water molecule is possible only if its pKa and the surrounding electrostatic environment favor this transfer. Unfortunately, in the above mentioned study, the PT process has not been calibrated or validated by any pKa or related calculations in solution. Furthermore, the evaluation of proton transfer barriers and paths by energy minimization has been show by us (e.g., refs.59,62) to be extremely problematic. Therefore, one needs to critically assess all the possible pathways before adopting it. Here it is important to realize that the reported attempt48 to explore this path obtained a high barrier only because it involves large distance between the donor and acceptor, which is most probably entirely due to the use of energy minimization without proper relaxation of the protein (see the related prechemistry examination below).
Similarly, a related proposal of concerted PT between several acidic groups47 is problematic i.e., any proposal of PT between acidic groups is as reliable (or less) than the corresponding ability to calculate its pKa of the bridging groups. Here the challenge of obtaining pKa by QM/MM calculations has only been accomplished by only few groups in particular in our study.60 Finally, any proposal that suggest a PT trough several water molecules must be evaluated by considering a path without any bridging water molecule.
III. 2 Examining the Proposals of Major Prechemistry Barriers
After reproducing the observed fidelity it is possible to explore the origin of the corresponding difference in barriers. In doing so we started by examining the idea that this effect involves differences in the so called prechemistry barriers. More specifically, a recent study,44 estimated the relative barriers in moving the primer terminal O3′ atom of the W system from r(ES(W)) to its prechemistry state, using a quantum mechanical cluster model. This study suggested that the prechemistry state corresponds to a local minimum at 4.4 kcal/mol above the corresponding initial state (the reactant state). It was also concluded that the system has to surmount a potential barrier of 14.1 kcal/mol to form the prechemistry state, starting from r(ES(W)). These findings led to the suggestion that the free energy required for formation of the prechemistry state is a major factor contributing to the decrease in the rate of incorrect nucleotide incorporation as compared to the correct insertion and therefore to their observed fidelity. Although estimating activation barriers in enzyme catalysis using quantum mechanical cluster models or QM/MM methods and energy minimization approaches is a common practice in the scientific community,17,27,44,47,48,75-77 it has been argued and demonstrated several times in the literature that these models cannot estimate the free energy barriers accurately and consistently (e.g., ref.49).
The main problem with the conclusions from the above studies is the fact that energy minimization approaches involve major problems in estimating the free energy barriers in multidimensional systems. Thus we focused here on using the proper free energy calculations. That is, in order to estimate the relative free energy and the free energy barrier associated with the formation of prechemistry state, we evaluated the PMF of moving the template primer terminal O3′ oxygen in the W system from its ground state position to its position in R (prechemistry state). To achieve this we generated 5 intermediate protein conformations between the ground and prechemistry states by gradually decreasing the distance between O3′-Mg2+. Each of these intermediate structures were equilibrated for 20 ps with a constraint of 10 kcal/mol to obtain the minimized protein structures at that particular O3′-Mg2+ distance. These structures were further subjected to another 20 ps equilibration, this time by only imposing the 10 kcal/mol distance constraint on O3′-Mg2+ distance. Utilizing these structures we evaluated the free energy barrier between the reactant and prechemistry state by estimating the free energy difference between the successive intermediate states. This was done using the creation and annihilation transformations by following the thermodynamic cycle shown in Figure 7. The resulting PMF for this motion is shown in Figure 8 (profile A). The barrier obtained by this procedure appeared to be very small (3.58 kcal/mol) relative to the barrier deduced by the energy minimization treatment (14.1 and 4.0 to 9.0 kcal/mol in refs.44,48 respectively). The reason for the low barrier is the fact that we actually calculated the PMF for the process of moving the O3′ to its catalytic position without any constraints on the protein environment. Apparently the energetics of this process is drastically overestimated in calculations that keep the protein fix beyond the immediate reaction region.44 To illustrate this important point we repeated the above PMF calculation by restricting the protein motions within 10 Å distance from the geometric center of template primer residue (reactive region) using force constants 0.5, 1.0 and 3.0 Kcal/mol. The corresponding PMFs are depicted in Figure 8 (profiles B-D), where we reproduced artificially high PMF barriers (5.61, 5.93 and 7.6 kcal/mol) with the application of varying constraint forces on the immediate reaction region. These results clearly demonstrate that the prechemistry barrier cannot be evaluated reliably without allowing the protein to relax. Thus, in addition to the sampling issues, and the problem with inappropriate energy minimizations, it appears that fixing of the protein environment leads to a drastic overestimation of the free energy barriers associated in moving the primer O3′ from reactant state to the prechemistry state. Apparently the barrier in W does not involve any rate determining prechemistry step and the 3.58 kcal/mol contribution to the chemistry barrier is simply of the penalty of moving to the correct preorganization (see section III. 3).
Figure 7.
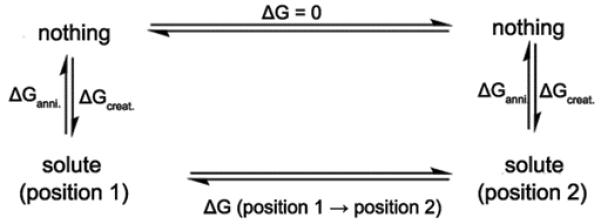
The thermodynamic cycle utilized for evaluating the free energy profile associated with the movement of O3′-primer of W from its x-ray position to the catalytically more favorable position. The template primer residue was considered as the solute which undergoes the corresponding changes as shown by the thermodynamic cycle.
Figure 8.
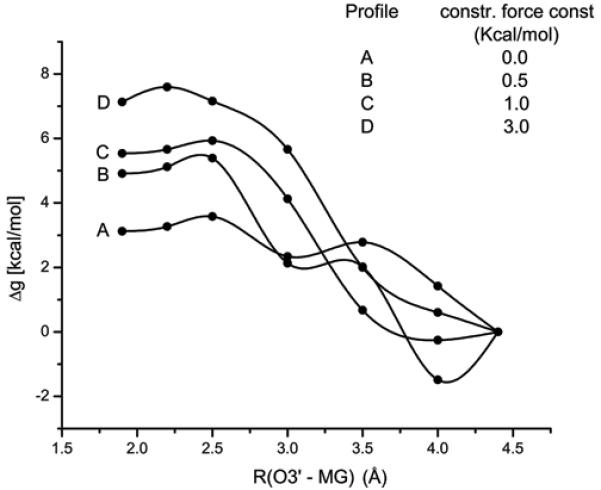
The PMFs associated with the movement of the O3′-primer of the W system from its x-ray position r(ES(W)) to the catalytically more favorable position (where the O3′-P position is the same as in r(ES(R)) using the thermodynamic cycle of Figure 7. Note the Figure contains four PMF profiles (A – D) with varying constraint force constants acting on the surrounding protein environment. Profile A corresponds to the PMF obtained with zero constraints acting on the surrounding protein environment (this is the correct way of obtaining the free energy since all the degrees of freedom of the protein are allowed to relax, system reaches its equilibrium configuration). Whereas the profiles B – D correspond to the PMFs obtained with the incorrect treatment (where the fluctuations of the surrounding protein environment are restricted which might correspond to the non equilibrium exploration of the phase space and leads to the artificially high reaction free energies and high barriers as shown in the Figure). This clearly indicate that the artificially high barrier (14 kcal/mol) observed in moving the O3′ primer from r(ES(W)) in the W system reported in ref44 is due to the unrelaxed treatment of the protein environment during the evaluation of energy profile .
To gain further insight about the catalytic landscape of R and W systems, we generated two dimensional (2D) surfaces for the free energy in the space generated by the chemical coordinate (the coordinate that describes the nucleotide transfer reaction) and the coordinate that describes the movement of the template 3′ primer. Figure 9 describes the calculated surfaces for R (Figure 9A) and W (Figure 9B). In both cases we depict the conformational coordinate along the path from (r(ES(R)) (configuration of the reactant enzyme-substrate complex where the position of the template O3′ primer corresponds to its x-ray position in R) to r(ES(W)). The movement of the template primer along the conformational coordinate was achieved, in both the R and W systems, by generating 9 intermediate structures between these states by gradually changing O3′–Mg2+ distance. Each of these intermediate structures were equilibrated for 20 ps with a constraint of 50 kcal/mol acting upon the 24 Å region of the protein from the simulation center defined earlier. These structures were further subjected to another 20 ps equilibration, using 10 kcal/mol on the 24 Å region of the protein. Then the nucleophilic attack of the O3′ primer onto the corresponding Pα atom of the substrate nucleotide was carried on all these conformational states. The observed results are depicted in Figure 9A and 9B for both R and W systems respectively. Both the EVB surfaces of R and W systems show the common trend of a TS of more than 35 kcal/mol when the O3′-Mg2+ distance is larger than 3.0 Å. The calculated activation barriers from initial to the target state through the intermediate states in both the R and W systems clearly indicate that the nucleotide transfer is more favorable from r(ES(R)) where the primer (nucleophile) strongly interacts with the catalytic Mg2+ ion. Furthermore, the chemical barrier from the r(ES(R)) structure is lower for R than the W thus reflecting the fact that the system R is better preorganized at r(ES(R)).
Figure 9.
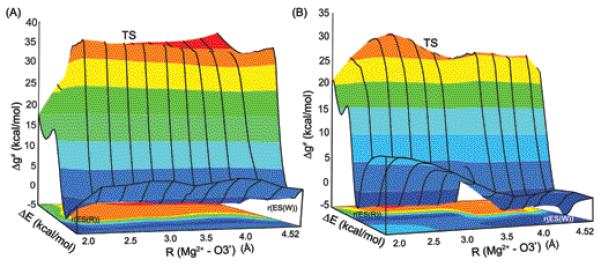
The 2-D free energy surfaces for R (A) and W (B) systems in the space defined by chemical coordinate (the coordinate that describes the nucleotide transfer reaction) and the coordinate that describes the movement of the template 3′ primer (i.e., moving from r(ES(R)) to r(ES(W)). r(ES(R)) and r(ES(W)) designate the configuration of the reactant enzyme-substrate complex where the position of the template O3′ primer corresponds to its x-ray position in R and W systems respectively. The evaluated activation barriers from the initial ( r(ES(R)) ) to the target ( r(ES(W)) ) state through the intermediate states indicate that chemical reaction is more favorable from r(ES(R)) position in both R and W systems. Also, the calculated barriers (14.0 (R) vs 16.1 (W) kcal/mol) indicate that the R system is better preorganized at r(ES(R)). Note that the energy gap ΔE is used here as the reaction coordinate.
III. 3 Examining the Nature of the Contributions to Fidelity
As stated above we reproduced the observed fidelity and also illustrated that it is not due to prechemistry effects. Thus it is interesting to elucidate the origin of the calculated (and observed) fidelity. To address this issue we evaluated the binding free energy of the incoming nucleotide at r(ES(R)) and r(ES(W)) both in the R and W systems. The corresponding binding energies are obtained by following the same strategy used in our recent study where we reproduced the observed change in transition state binding free energy in different mutants and upon change of the bases.45 In this approach we evaluated the binding energy of the chemical (triphosphate) and the base sites separately, by running independent protein dipole Langevin dipole simulations in the linear response approximation version (PDLD/S-LRA),66,78 using dielectric constants of 40 and 2 for the chemical (triphosphate) and base sites, respectively (this approach is justified in ref.45). The corresponding calculated binding free energies are summarized in Figure 10. As seen from the Figure 10, the interactions with the base binding site (Figure 10A) favors the W system at r(ES(W)), whereas the R system is favored at r(ES(R)). However, the energetics of the chemical site (Figure 10B) follows exactly the opposite trend i.e., the R system prefers r(ES(W)) and the W system prefers r(ES(R)). Therefore, the balance between the binding of the base and the chemical sites is the essence of the allosteric effect that controls the fidelity of DNA polymerases (see also the discussion section IV).
Figure 10.
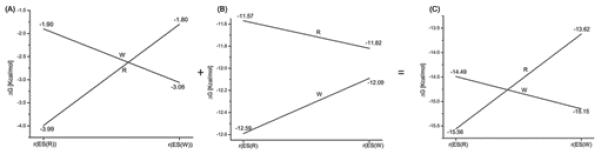
A PDLD/S-LRA decomposition of the binding free energies of the incoming dNTP nucleotide. The calculations estimate the energy contributions to the binding free energy at r(ES(R)) and r(ES(W)) (where r(ES(R)) and r(ES(W)) designate the configuration of the reactant enzyme-substrate complex where the position of the template O3′ primer corresponds to its x-ray position in R and W systems respectively). The figure presents the different contributions, where (A) provides the binding free energy contribution of the base part of the incoming nucleotide, evaluated using εp = 2, (B) provides the binding energy contribution of the triphosphate part of the incoming nucleotide evaluated using the εp = 40, whereas (C) provides the total binding free energy of the nucleotide ( obtained as a sum of the individual binding free energies of the base (A) and the triphosphate (B) parts of the nucleotide ) . This use of different dielectric constants was introduced and successfully employed in what appears to be a reliable evaluation of the TS binding free energies for different mutants of pol β.45
III. 4 Exploring the Idea of Coupled Motions and Checkpoints
Recent ideas that the fidelity of DNA polymerases may involve “gate keeping” dynamical effects46 and related earlier ideas79 are of interest and should be examined in a well defined way. Time-resolved fluorescence80 and NMR81,82 studies have demonstrated that the motions (which have often been referred to as dynamics) of α-helix N are reduced when Watson-Crick hydrogen bonding occurs (i.e., correct nucleotide binding), but not when hydrogen bonding is absent. In contrast, protein side-chain motions are increased in the vicinity of the active site aspartates when the closed complex is formed.81,82 However, these motional effects are not likely to represent true dynamical effects but merely equilibrium thermal fluctuations that follow the Boltzmann populations in different landscapes. In this case we simply have well defined entropic effects; an analysis and interpretation of these types of dynamical effects is discussed in refs.34,83 We have not found convincing theoretical or experimental evidence that dynamical effects play a significant role in catalysis.34
Here we would like to point out that we have already provided clear illustration of the problem with the dynamical proposals35,71 and that none of argument of dynamical effect has ever include a full consistent study of the time dependence of such effect.35 In the present work we do not like to repeat our previous examination of the dynamical proposal but rather to relate it to the fidelity problem. That is, as outlined in section II.3 we try to get the best correspondence between the free energy and dynamics of a full explicit model (all-atom molecular dynamics) and an implicit Langevin Dynamics (LD) model of reduced dimensions. Since we have a powerful way of obtaining the EVB profile for the chemical coordinate at each conformational state, we focused on evaluating the energetics of the conformational transition. That is, in both R and W systems, we studied the energetics of the conformational transition from the open to closed structure (with the bound nucleotide) , through three intermediate states (in the sequence open → Int1, Int1→ Int2, Int2 → Int3, and Int3 → close). This was done sequentially using targeted molecular dynamics (TMD) simulations applying various force constants, ranging from 0.0002 to 0.001 kcal/mol, on all the heavy atoms. In each case we selected the lowest force constant that was sufficient to lead to a complete transfer of the initial state to the target state. The corresponding time dependence of the conformational changes in each segment where evaluated and the PMF of the simplified model (as well as the friction) was then adjusted so that the LD simulation with this model would produce a similar time dependence (see ref.72 for a more systematic description). The resulting refined conformational free energies combined with the EVB chemical profiles were used to construct the free energy landscape described in Figure 11.
Figure 11.
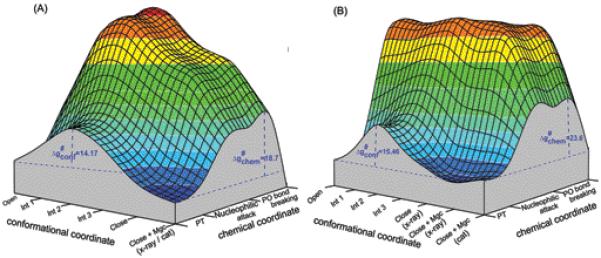
The calculated catalytic landscapes for (A) matched nucleotide pair (R) and (B) mismatched nucleotide pair (W) in the space defined by the protein conformational coordinate (with the bound nucleotide) and the chemical reaction coordinate. The chemical reaction profiles for open, closed, and for three of the intermediates were evaluated and then connected by a simple interpolation. The conformational change along the open → close path was studied using the renormalization approach whereas the chemical reaction pathway was explored using the stepwise associative mechanism. The conformational barriers obtained in this study are 14.17 and 15.46 kcal/mol for R and W respectively. These barriers correspond to the open → close transition of pol β bound with DNA, dNTP and only the Mg2+ ion assisting the dNTP binding was bound. In addition, the figure also contains, the state after the binding of catalytic Mg2+ ion (close + Mgc (x-ray)). In case of mismatched nucleotide pair (B), a prechemistry state (close + Mgc (cat)) is also included.
In considering the idea of checkpoints it is important to try to estimate the rate determining conformational barrier. Here we start by noting that a recent stopped flow fluorescence analysis84 suggested that, in pol β, the rate of conformational step for G:dCTP pair is faster than the nucleotide insertion step (64.4 s−1 vs 12.5 s−1). This suggests that after binding of the nucleotide to the open conformation of pol β, the complex has to surmount a free energy barrier of ~15.0 kcal/mol to reach to the catalytically favorable closed state. In this respect, it is important to realize that the second Mg2+ ion only binds to the catalytic site after the N-subdomain undergoes open → close conformational change. Thus the barrier for the open → closed transition should be evaluated with only one bound Mg2+ ion which assists the incoming dNTP. With this point in mind we evaluated the conformational barriers associated with the open → close transition of pol β ternary complex in the presence of DNA, dNTP and only one Mg2+ ion, using the renormalization approach, and obtained the values of 14.17 and 15.46 kcal/mol for the R and W systems, respectively.
It is important to realize that while evaluating the time dependence of the conformational changes in every segment, the protonation states of all the ionizable residues were evaluated using Monte Carlo (MC) approach and set to the corresponding values. At any rate, it is encouraging to note that the agreement between the calculated and observed values of the conformational barriers demonstrated the reliability of the renormalization approach in particularly in cases when the associated changes are multidimensional where a particular reaction coordinate is extremely difficult to define.
After gaining clear insight on the conformational barrier we further explored the conformational and chemical coupling by constructing the free energy landscape in the space of the open → close coordinate and the chemical coordinate. It is interesting to note that in the case of W, the energy of nucleophilic attack initiated from the open, intermediate and closed states, continued to rise and the transition state was found to lie above 50 kcal/mol above the corresponding initial deprotonated state. Similar trend was observed when the nucleophilic attack was initiated from r(ES(W)) as described in III.1. This clearly indicates that in W system, after forming the complete active site ground state geometry, starting from open configuration, a prechemistry state must needs to be formed before the chemical step and the proper PMF calculations (section III. 2) without any artificial constraints acting on the protein environment suggested that the prechemistry state is 3.12 kcal/mol higher in energy relative to the complete active site ground state geometry.
Here, we mainly want to emphasize that we can capture the main features of the catalytic landscape and that as long as the conformational barrier is lower than the chemical barrier (which is the case in pol β44,85) we will not find any effect of the motion on the conformational coordinate on the overall rate constant (see below). This point can be proven by running LD on the surfaces of Figure 11 without any constraints but the general results have already been established in ref.71
At this stage we can return to the discussion of the idea that dynamical effects help in the controlling fidelity. First as pointed out above we cannot obtaining any significant dynamical coupling between the conformational motions (moving from open to close) to the chemical step. Furthermore even if there was some dynamical coupling it would be difficult to see how it would be advantageous to use these as a mechanism to control replication fidelity. Fidelity is determined by the ratio between kpol/Kd of R and W. If the rate limiting step is the conformational transition between the open and closed conformations and the barrier for these states are much larger for W than for R, then there could, in principal, be conformational control of fidelity. However, this mechanism is in conflict with chemistry being rate limiting (see refs.14,15,39). If the chemical step is rate limiting, then it seems that the only way for conformational changes to control fidelity is that the TS for both W and R must occur in a different conformation than the RS, and the barrier along the conformational axis in Figure 11 is higher than the chemical barrier, and that this barrier will be higher in W than in R.
Finally, it may be useful to comment here on the implication of ref.19 that prechemistry steps can have a major effect on the fidelity. Apparently as long as the barriers for the pre-chemistry steps (e.g., the open to closed conformational change) are significantly lower than the chemical barrier they cannot change the kinetics and the corresponding fidelity (except in some particular saturation conditions). That is, the reaction rate is determined by the difference between the energy of the TS and the E+S state for the chemical step. Having many barriers between the open and closed configurations in Figure 11 is not going to change the kinetics as long as these barriers are smaller than the TS barrier. An additional insight is obtained for example from inspection of Figure 1 of ref.20 This figure describes the calculated barriers for the pre-chemistry steps. Since the barriers for W are smaller than that for R it is very hard to see how these barriers could account for the observed fidelity. The obvious answer is that the real difference is in the chemical step.
Some workers (e.g., ref.86) have argued that the rate of substrate release from the active site plays a major role in determining the fidelity. However, in our view, having the free energy surfaces (and barriers) does provide the complete picture about the fidelity issue, and talking on forward and backward rate does not provide any additional insight. Of course, in some cases when the barrier heighths (e.g. for prechemistry and chemistry) are similar or when we have very deep traps one has to convert the energy diagram to rate diagram and solve the rather trivial kinetic problem for the relevant concentrations. However, it is not useful to basically confuse the issues and to talk on dynamical like effect when the energetics determines all the rate constants and when simple kinetic treatment of these rate constants gives the fidelity. As a case in point it is useful to look at the kinetic diagram of Figure 12, This type of surface, suggested by Johnson86, presents the interesting situation when the chemical barrier in T7 may be lower than the prechemistry barrier. Although it is unlikely that this is the situation in T7 we cannot exclude it without careful calculations. However, the only insight provided by the diagram of Figure 12, is that the overall rate in the case of R is controlled by the presumed prechemistry barrier. There is no need to address this feature by talking on the fact that there will be a larger population in FDnN of R than of W, since the bound substrate will go more frequently to EDn in W. This a completely obvious consequence of the corresponding free energy differences and not a dynamical time dependent issue.The key real question is simply whether or not the prechemistry is rate determining. If this is the case, then in fact we have a bad use of evolution, since with lower prechemistry barrier we will have an even better fidelity, since the rate in R will be then determined by the chemical barrier, (which is presumably lower than the original prechemistry barrier).
Figure 12.
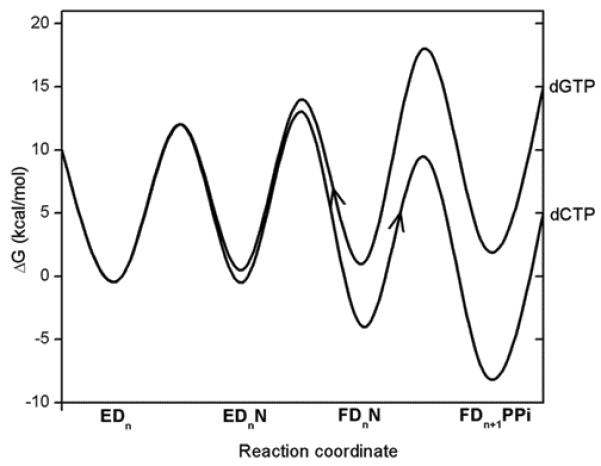
A schematic free energy diagram for correct (green: dCTP) and incorrect (red: dGTP) nucleotide incorporation reactions for the T7 DNA polymerase (adopted from ref.86). Where EDn represents the enzyme-DNA complex with a primer strand n residues in length, N: the incoming dNTP, and FDnN: the closed state of the enzyme with bound DNA and nucleotide.
IV. Discussion
At this point it is useful to discuss our main findings and their relationship to the different concepts about the control of replication fidelity. This will be done below.
IV. 1 Landscape and fidelity
As demonstrated in the previous sections, the fidelity of DNA polymerase reflects an allosteric competition between the binding of the base of the incoming dNTP to the base-binding site and the binding of the TS to the catalytic site. In the case of an incorrect dNTP, poor preorganization in the base-binding site and poor binding of the base lead to structural rearrangements that are propagated to the catalytic site and destroy its preorganization.28 This allosteric control of fidelity, which is described in Figure 13, has been established here at least in a qualitative way by the calculations summarized in Figure 10. This compensating allosteric effect reflects a rather simple interplay of free energy contributions and should not be described by such terms and “population shifts” since the population shift is a simple result of the change in the free energy of being in different configuration and not a reason for any well defined physical effect.
Figure 13.
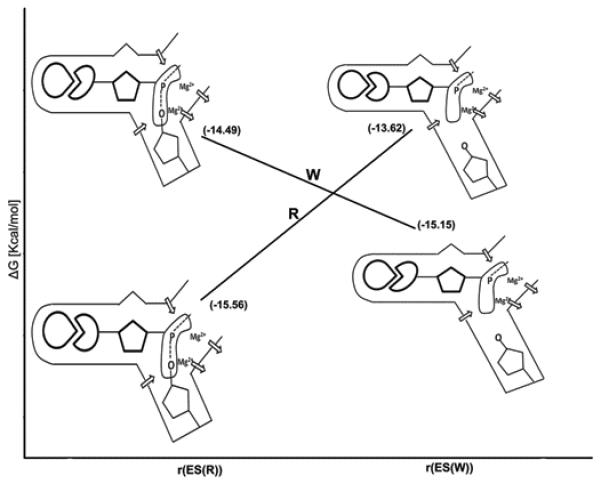
Illustrating the origin of the allosteric effect. The figure uses the calculations from Fig 10 (C) and illustrates at each point what are the relevant changes in the R and W systems. The protein rearrangement upon binding of R and W at r(ES(R)) and r(ES(W)). The protein provides optimal sites for both the chemical and base-pairing parts of R. On the other hand, in the case of W, the protein has to relax in the base-pairing site (to provide good binding) and this relaxation destroys the preorganization in the chemical part. The diagrams that represent the different configurations illustrate this allosteric effect where the arrows indicate the protein dipoles. In the R state these dipoles are reorganized in an optimal way and thus provide maximum TS stabilization. On the other hand in the W state the dipoles are forced to assume less effective prorganization and thus yield less TS stabilization.
At this point it might be useful to discuss a recent study87 that used a conformationally sensitive fluorophore attached to the recognition domain of T7 DNA polymerase to probe structural changes. The kinetics analyses performed at that work suggested that the conformational changes associated with the florescence kinetics are those involved in the alignment of the catalytic site for R and for the misalignment of the active site for W. It was then suggested that conformational changes that occur after the binding step and before the chemical step play a major role in controlling the fidelity of DNA polymerases. It was suggested that this step is used to align the active site for the correct nucleotide. Now, while these findings are potentially important, it is important to realize that there are many possible structural changes and only some of them have an impact on the chemical step. It is likely that the fluorescence probe reflects changes in the base-binding site and does not necessarily tell us about the preorganization in the catalytic site. That is, the most important catalytic preorganization occurs upon binding of the charged part of the incoming nucleotide, and this preorganization is expected to be similar for R and W. Then the base binding process is used, in the case of W, to destroy the preorganization (see Figure 13). The florescence changes observed with the current probe may reflect mainly the structural changes in the base binding site rather than the relevant part of the induced fit. It seems that only direct structural studies and/or simulation approaches can tell us about the nature of the preorganization effect that follows the binding step. Using fluorescence labeling can be very useful in estimating the time scale of these changes, but even this should be done with great care, with two or more probes, trying to correlate the position of the given probe with the two allosteric regions.
Regardless of the above uncertainty, it is important to consider the argument (e.g., ref.86) that the rate of substrate release from the active site plays a major role in determining the fidelity. The problems with this concept are considered in section III.3.
IV.2 What is the relationship between induced fit and fidelity?
Since our work explored the role of conformational changes in pol β, it is important to discuss the idea that the fidelity of DNA polymerase is related to the induced-fit proposal.36,88-91 The fact that the substrate binding induces conformational changes that bring Pol β to its catalytic configuration can be described as an induced fit effect. However, this fact does not provide any information about the nature of the catalytic effect in the preorganized active site, nor does it tell us how the structure of the ES (or in our case the E-DNA-dNTP) complex determines the activation barrier of the chemical step. In other words, the chemical catalysis does not depend on the structure of the enzyme before it binds the substrate. Thus, the induced fit concept does not describe the origin of the ES structure. Enzyme catalysis can be considered on the same level as other parts of the folding to ES state (in general the folding energy is invested in reorganizing the active site92) i.e., the catalytic power of enzymes is largely due to the preorganized electrostatic environment of their active sites and the enzymes inherent folding energy is invested in reorganizing its active site.
The use of the term “induced fit” to describe the origin of the polymerase fidelity is also quite problematic. In fact, the use of kinetic diagrams of the type considered by Post and Ray93 does not provide a unique thermodynamic analysis because they are not based on well defined free energy surfaces (surfaces of the type presented in Figure 11). With actual surfaces we can define the relationship between structural changes upon binding and the corresponding energetics, including the energy change of the enzyme upon moving from r(E) to r(ES) (this energy is defined as the reorganization energy upon binding94), the change in the enzyme substrate interaction upon moving from r(E) to r(ES) and the change in the activation barrier for the chemical step as the result of the binding induced structural change (this is defined in terms of the change in the reorganization energy of the chemical step and the work term.95). Without such estimates it is likely that the discussion will become circular.
As discussed and established by the logical analysis of ref.16 it is almost impossible to conduct a logical analysis of the induced fit effect without describing the relevant free energy surface. However even with well-defined surface we must define what is the reference state for the induced fit effect. When this is done16 we get different results for different definitions. However, none of these results tell us how the induced fit determined the fidelity. This point is further discussed below.
Since the induced fit upon binding of R leads to an improved catalytic configuration and since some of the binding energy is being invested in the reorganization process, it might seem reasonable to suggest that W will induce less reorganization (less induced fit) because its active site is less preorganized. Unfortunately, such a suggestion, which is equivalent to the assumption of a direct relationship between induced fit and fidelity, is unjustified. That is, W and R are different molecules and there is no way to predict a priori which of them will involve larger reorganization upon binding. In fact, if we assume that the binding of W and R is similar without the induced fit effect and then follow the above idea we will conclude that W binds better than R (since it has presumably smaller reorganization effect). However, in reality W binds almost always less strongly than R. Apparently there is no way to tell if the binding of W will lead to smaller or larger reorganization than the binding of R since we only know that the binding of R improves the preorganization of the catalytic site. The binding of W may involve small reorganization or a very large reorganization with a very large cost. For example, the binding of W may be used to move the active site to an anti catalytic configuration.
IV. 3 Can the binding energy be used to control the fidelity ?
The idea that the enzyme uses the “intrinsic” substrate binding energy to achieve its optimal catalytic effect (e.g.,96) has also been invoked in analyzing the induced fit effect.87,97 In particular it has been implied that a portion of the binding energy must be consumed by reorganizing the active site and thus will not be available to stabilize the TS.87 Unfortunately this line of thought is problematic. First, the whole assumption that the enzyme uses the binding energy of distant groups lead to destabilization of the ground state of the chemical part (this is the essence of Jencks’ idea96 about the usage of binding energy) has not been confirmed by careful computational and conceptual studies of specific cases.92 One of the simplest ways to realize this point is to see that most mutations that reduce catalysis also reduce binding rather than increase it. Second, the binding energy in the case of induced fit is invested in part in pushing the TS to the preorganized structure and not “consumed” in destabilizing the ground state. In other words, the binding energy that leads to structural rearrangement increases rather than decrease catalysis. Now, as much as the fidelity is concerned we may try to view the base as the distant part of the substrate (the part that whose energy should be used for inducing catalysis). Doing so we face with the problem that a good binding of the base (a good base pairing) is used to stabilize the TS rather than destabilize the RS. This point can be seen by noting that mutations that increase kpol significantly for R usually decrease KD (lower KD means stronger binding), or leave it unchanged, rather than increase it,28 as would be expected if the reduction of Δg‡cat is due to ground state destabilization. Interestingly the distant part is quite effective in destroying rather than increasing catalysis, as is obviously the case with W.
IV. 4 Catalytic landscape and dynamical effects
The present work considered in a preliminary way the effect of the complexity of the landscape in enzyme catalysis. As illustrated in Figure 11, we may be faced with a complex situation in assessing the probability of reaching the TS region in the multidimensional surface of the enzyme-substrate complex. However, in contrast to some recent proposals31 we do not see any fundamental problem with this complexity, nor do we find a catalytic advantage from this complexity. The possibility that the TS region has more configurations than the RS is a simple entropic effect that does not appear to be supported by the fact that the observed values of the activation entropies in enzymes are not large.98 The possible dynamical effects associated with transitions between different conformational subspaces during the fluctuations that lead to the TS are not expected to be large although simulation studies of this issue are clearly needed. Of course, it is clear that different conformations can be associated with different activation barriers as indicated by recent single molecule studies of enzymes,29 but this point has been taken into account (at least in part) already in our approach of averaging over different initial configurations (e.g.,99,100). That is, in our simulations we frequently run series of FEP/US calculations starting with different initial conditions and take the average of the calculated activation barriers. This approach accomplishes more or less what is done in the so called replica exchange studies (see discussion in ref.95) and provides an estimate of the effective activation barriers.
V. Concluding Remarks
This work explored the origin for DNA replication fidelity using the actual structures of W and R in Pol β. It was found that the difference in fidelity can be completely accounted for by the difference in the corresponding activation free energies and binding free energies, without the need to invoke exotic factors. Furthermore, specific proposals of prechemistry barriers were shown to be problematic and not supported by careful calculations that allowed the protein to relax.
In addressing the prechemistry idea we also considered the appealing concept of checkpoints.46,50 Obviously, if the prechemistry barrier is rate determining it may determine the fidelity. However, regardless of the height of this barrier one may suggest that it helps fidelity by changing the open to close dynamics and allows more time to distinguish between correct and incorrect nucleotide. Unfortunately, this idea suffers from the same problems as the related dynamical idea. That is, as long as the prechemistry barrier is smaller than the chemical barrier, the system will “forget” any information about the situation in intermediate configurations and the overall rate and fidelity will not remember much about the intermediate barriers.35,71 The only checkpoint in our case are the high energy barriers. Intermediates and low energy barriers cannot affect the kinetics unless we have extremely deep low energy intermediates ( which can also be treated by simple kinetics in the Michalis-Menton type treatment.
The consideration of the nucleotide incorporation reaction of DNA polymerases, has led to several mechanisms proposed especially for the initial proton transfer step.12,16,17,27,47,48,75,77,101,102 It is proposed that the general base accepting the proton from 3′-OH primer could be either an aspartate residue, OH−, O atom of Pα, or it could be transferred to the triphosphate moiety of the incoming dNTP through either a single or several water molecules. It is important to clarify here that computational proposal of PT process in proteins has to be calibrated or validated by pKa or related calculations in solution and also validated by a demonstrated ability to evaluate pKas in proteins.60 Furthermore, the evaluation of proton transfer barriers and paths by energy minimization approaches has been show by us to be extremely problematic.59,62 Therefore, one needs to critically assess all the possible pathways ( comparing the relative heights ) before adopting any of them. Proposals that are based on a PT through a single or several water molecules27,47,48 are questionable since they have neither been validated by any pKa calculations nor based on proper free energy simulations , extensive relaxation and careful comparison of alternative paths .
Some have wondered (e.g., ref.86) how can the weak binding of the substrate generate large conformational changes leading to tighter binding and alignment of the catalytic residues. It was also assumed that special theoretical approaches are needed for exploring this issue. However, once one views allosteric effects as they actually are, namely a balance between opposing free energy contributions (e.g., Figure 10) it is not so much an issue of the strength of the interaction, but simply of having two states with a relatively small free energy difference, so that a modest increase in the base-protein interaction in the binding site will change the equilibrium to favor a state with smaller TS binding in the catalytic site. Here there is no need to fully understand (the otherwise intriguing) path of conformational changes but simply to evaluate the free energy contributions in the initial and final states (as is done in Figure 10).
The surfaces obtained in this work allowed us to discuss the induced fit effect in a consistent way. Previous discussions of this issue have been incomplete because they were not related to well defined structure-energy relationships. Although our discussion of the induced fit effect seems complex, we view it as an important step in illustrating that there is no way to predict the trend in fidelity by using the typical induced fit concept since it is ambiguous as to whether or not the fidelity might decrease or increase. In contrast, the calculations described in Figure 8 probe the actual effect of the structural change. These calculations can predict the consequence of the induced fit. Thus accepting that the induced fit concept does not explain fidelity, we can still investigate the consequence of induced fit on fidelity in specific cases using computer simulation or experimental approaches.
We also consider the relationship between the landscape and dynamic effects and concluded that it is unlikely that there are major dynamical effects involved in controlling the fidelity of DNA polymerases. Similarly we find that the barriers along the path from the open to closed structures in the pre-chemistry step are not likely to contribute to the overall fidelity, unless the corresponding barriers are higher than that for the chemical step.
Supplementary Material
Figure 5.
The calibrated EVB free energy surfaces for the reference solution reactions corresponding to the mechanisms proposed (stepwise associative (A) and concerted (B)) for pol β.
Acknowledgments
This work was supported by National Institutes of Health Grants 5U19CA105010. We acknowledge the University of Southern California’s High Performance Computing and Communications Center for computer time. BRP would like to thank Dr. Suman Chakrabarty, Anatoly Dryga, and Nikolay Plotnikov for useful help.
Abbrevations
- EVB
empirical valance bond
- Pol β
DNA polymerase β
- PMF
potential of mean force
- QM/MM
quantum mechanics/molecular mechanics
- LD
langevin dynamics
References
- 1.Kornberg A, Baker TA. DNA Replication. W. H. Freeman; New York: 1992. [Google Scholar]
- 2.Wolfenden R, Snider MJ. The Depth of Chemical Time and the Power of Enzymes as Catalysts. Acc Chem Res. 2001;34:938–945. doi: 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- 3.Echols H, Goodman MF. Fidelity mechanisms in DNA replication. Annual Review Of Biochemistry. 1991;60(1991):477–511. doi: 10.1146/annurev.bi.60.070191.002401. [DOI] [PubMed] [Google Scholar]
- 4.Johnson KA. Conformational coupling in DNA polymerase fidelity. Annu Rev Biochem. 1993;62:685–713. doi: 10.1146/annurev.bi.62.070193.003345. [DOI] [PubMed] [Google Scholar]
- 5.Joyce CM, Benkovic SJ. DNA polymerase fidelity: Kinetics, structure, and checkpoints. Biochemistry. 2004;43(45):14317–14324. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- 6.Beard WA, Wilson SH. Structural insights into the origins of DNA polymerase fidelity. Structure. 2003;11(5):489–496. doi: 10.1016/s0969-2126(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 7.Goodman MF, Creighton S, Bloom LB, Petruska J. Biochemical Basis of DNA Replication Fidelity. Crit Reviews in Biochem and Mol Biol. 1993;28:83–126. doi: 10.3109/10409239309086792. [DOI] [PubMed] [Google Scholar]
- 8.Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- 9.Goodman MF. Error-prone repair DNA polymerases in prokaryotes and eukaryotes. Annu Rev Biochem. 2002;71:17–50. doi: 10.1146/annurev.biochem.71.083101.124707. [DOI] [PubMed] [Google Scholar]
- 10.Showalter AK, Lamarche BJ, Bakhtina M, Su MI, Tang KH, Tsai MD. Mechanistic comparison of high-fidelity and error-prone DNA polymerases and ligases involved in DNA repair. Chem Rev. 2006;106(2):340–360. doi: 10.1021/cr040487k. [DOI] [PubMed] [Google Scholar]
- 11.Florian J, Goodman MF, Warshel A. Theoretical Investigation of the Binding Free Energies and Key Substrate-Recognition Components of the Replication Fidelity of Human DNA Polymerase β. J Phys Chem B. 2002;106:5739–5753. [Google Scholar]
- 12.Florian J, Goodman MF, Warshel A. Computer simulation of the chemical catalysis of DNA polymerases: Discriminating between alternative nucleotide insertion mechanisms for T7 DNA polymerase. J Am Chem Soc. 2003;125:8163–8177. doi: 10.1021/ja028997o. [DOI] [PubMed] [Google Scholar]
- 13.Florian J, Warshel A, Goodman MF. Molecular dynamics free-energy simulations of the binding contribution to the fidelity of T7 DNA polymerase. J Phys Chem B. 2002;106(22):5754–5760. [Google Scholar]
- 14.Florian J, Goodman MF, Warshel A. Computer simulation studies of the fidelity of DNA polymerases. Biopolymers. 2003;68(3):286–299. doi: 10.1002/bip.10244. [DOI] [PubMed] [Google Scholar]
- 15.Florian J, Goodman MF, Warshel A. Computer simulations of protein functions: Searching for the molecular origin of the replication fidelity of DNA polymerases. Proc Natl Acad Sci U S A. 2005;102(19):6819–6824. doi: 10.1073/pnas.0408173102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiang Y, Goodman MF, Beard WA, Wilson SH, Warshel A. Exploring the role of large conformational changes in the fidelity of DNA polymerase β. Proteins: Struct, Funct, Genet. 2008;70:231–247. doi: 10.1002/prot.21668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin P, pedersen LC, Batra VK, Beard WA, Wilson SH, Pedersen LG. Energy analysis of chemistry for correct insertion by DNA polymerase β. Proc Natl Acad Sci U S A. 2006;103:13294–13299. doi: 10.1073/pnas.0606006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sucato CA, Upton TG, Kashemirov BA, Batra VK, Martinek V, Xiang Y, Beard WA, Pedersen LC, Wilson SH, McKenna CE, Florian J, Warshel A, Goodman MF. Modifying the beta-gamma leaving-group bridging oxygen alters nucleotide incorporation efficiency, fidelity and catalytic mechanism of DNA polymerase beta. Biochemistry. 2007;46:461–471. doi: 10.1021/bi061517b. [DOI] [PubMed] [Google Scholar]
- 19.Radhakrishnan R, Schlick T. Orchestration of cooperative events in DNA synthesis and repair mechanism unraveled by transition path sampling of DNA polymerase beta’s closing. Proc Natl Acad Sci U S A. 2004;101:5970–5975. doi: 10.1073/pnas.0308585101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Radhakrishnan R, Schlick T. Fidelity Discrimination in DNA polymerase β: Differening closing profiles for a mismatched (G:A) versus matched (G:C) base pair. J Am Chem Soc. 2005;127:13245–13252. doi: 10.1021/ja052623o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang LJ, Beard WA, Wilson SH, Broyde S, Schlick T. Highly organized but pliant active site of DNA polymerase beta: Compensatory mechanisms in mutant enzymes revealed by dynamics simulations and energy analyses. Biophys J. 2004;86:3392–3408. doi: 10.1529/biophysj.103.036012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feig M, Zacharias M, Pettitt BM. Conformations of an adenine bulge in a DNA octamer and its influence on DNA structure from molecular dynamics simulations. Biophys J. 2001;81(1):352–370. doi: 10.1016/S0006-3495(01)75705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orozco M, Perez A, Noy A, Luque FJ. Theoretical methods for the simulation of nucleic acids. Chem Soc Rev. 2003;32(6):350–364. doi: 10.1039/b207226m. [DOI] [PubMed] [Google Scholar]
- 24.Seibert E, Ross JB, Osman R. Contribution of opening and bending dynamics to specific recognition of DNA damage. J Mol Biol. 2003;330(4):687–703. doi: 10.1016/s0022-2836(03)00598-9. [DOI] [PubMed] [Google Scholar]
- 25.Oostenbrink C, van Gunsteren WF. Efficient calculation of many stacking and pairing free energies in DNA from a few molecular dynamics simulations. Chemistry. 2005;11(15):4340–4348. doi: 10.1002/chem.200401120. [DOI] [PubMed] [Google Scholar]
- 26.Arora K, Beard WA, Wilson SH, Schlick T. Mismatch-induced conformational distortions in polymerase support an induced-fit mechanism for fidelity. Biochemistry. 2005;44(40):13328–13341. doi: 10.1021/bi0507682. [DOI] [PubMed] [Google Scholar]
- 27.Alberts IL, Wang Y, Schlick T. DNA Polymerase β catalysis: Are Different Mechanisms Possilbe? J Am Chem Soc. 2007;129:11100–11110. doi: 10.1021/ja071533b. [DOI] [PubMed] [Google Scholar]
- 28.Xiang Y, Oelschlaeger P, Florian J, Goodman MF, Warshel A. Simulating the Effect of DNA Polymerase Mutations on Transition State Energetics and Fidelity: Evaluating Amino Acid Group Contribution and Allosteric Coupling for Ionized Residues in Human Pol β. Biochemistry. 2006;45(23):7036–7048. doi: 10.1021/bi060147o. [DOI] [PubMed] [Google Scholar]
- 29.Min W, English BP, Luo GB, Cherayil BJ, Kou SC, Xie XS. Fluctuating enzymes: Lessons from single-molecule studies. Acc Chem Res. 2005;38(12):923–931. doi: 10.1021/ar040133f. [DOI] [PubMed] [Google Scholar]
- 30.Leulliot N, Varani G. Current topics in RNA-protein recognition: Control of specificity and biological function through induced fit and conformational capture. Biochemistry. 2001;40(27):7947–7956. doi: 10.1021/bi010680y. [DOI] [PubMed] [Google Scholar]
- 31.Kumar S, Ma BY, Tsai CJ, Sinha N, Nussinov R. Folding and binding cascades: Dynamic landscapes and population shifts. Protein: Struct, Funct, Genet. 2000;9:10–19. doi: 10.1110/ps.9.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hammes-Schiffer S, Benkovic SJ. Relating protein motion to catalysis. Annu Rev Biochem. 2006;75:519–541. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]
- 33.Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313(5793):1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 34.Olsson MHM, Parson WW, Warshel A. Dynamical contributions to enzyme catalysis: Critical tests of a popular hypothesis. Chem Rev. 2006;106(5):1737–1756. doi: 10.1021/cr040427e. [DOI] [PubMed] [Google Scholar]
- 35.Kamerlin SCL, Warshel A. At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis? Proteins: Struct, Funct, Genet. 2010;78:1339–1137. doi: 10.1002/prot.22654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koshland DE. The Key-Lock Theory And The Induced Fit Theory. Angewandte Chemie-International Edition. 1995;33(23-24):2375–2378. [Google Scholar]
- 37.Yang LJ, Beard WA, Wilson SH, Broyde S, Schlick T. Polymerase beta simulations suggest that Arg258 rotation is a slow step rather than large subdomain motions per se. J Mol Biol. 2002;317(5):651–671. doi: 10.1006/jmbi.2002.5450. [DOI] [PubMed] [Google Scholar]
- 38.Berg BJV, Beard WA, Wilson SH. DNA structure and aspartate 276 influence nucleotide binding to human DNA polymerase beta - Implication for the identity of the rate-limiting conformational change. J Biol Chem. 2001;276(5):3408–3416. doi: 10.1074/jbc.M002884200. [DOI] [PubMed] [Google Scholar]
- 39.Bakhtina M, Lee S, Wang Y, Dunlap C, Lamarche B, Tsai MD. Use of viscogens, dNTP alpha S, and rhodium(III) as probes in stopped-flow, experiments to obtain new evidence for the mechanism of catalysis by DNA polymerase beta. Biochemistry. 2005;44(13):5177–5187. doi: 10.1021/bi047664w. [DOI] [PubMed] [Google Scholar]
- 40.Arndt JW, Gong WM, Zhong XJ, Showalter AK, Liu J, Dunlap CA, Lin Z, Paxson C, Tsai MD, Chan MK. Insight into the catalytic mechanism of DNA polymerase beta: Structures of intermediate complexes. Biochemistry. 2001;40(18):5368–5375. doi: 10.1021/bi002176j. [DOI] [PubMed] [Google Scholar]
- 41.Rothwell PJ, Mitaksov V, Waksman G. Motions of the fingers subdomain of klentaq1 are fast and not rate limiting: Implications for the molecular basis of fidelity in DNA polymerases. Mol Cell. 2005;19(3):345–355. doi: 10.1016/j.molcel.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 42.Batra VK, Beard WA, Shock DD, Krahn JM, Pedersen LC, Wilson SH. Magnesium-induced assembly ofa complete DNA polymerase catalytic complex. Structure. 2006;14:757–766. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Batra VK, Beard WA, Shock DD, Pedersen LC, Wilson SH. Structures of DNA Polymerase β with active-site mismatches suggest a transient abasic site intermediate during misincorporation. Cell. 2008;30:315–324. doi: 10.1016/j.molcel.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin P, Batra VK, Pedersen LC, Beard WA, Wilson SH, Pedersen LG. Incorrect nucleotide insertion at the active site of a G:A mismatch catalyzed by DNA polymerase β. Proc Natl Acad Sci U S A. 2007;105:5670–5674. doi: 10.1073/pnas.0801257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rucker R, Oelschlaeger P, Warshel A. A binding free energy decomposition approach for accurate calculations of the fidelity of DNA polymerases. Proteins: Struct, Funct, Genet. 2009;78:671–680. doi: 10.1002/prot.22596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Radhakrishnan R, Arora K, Wang Y, Beard WA, Wilson SH, Schlick T. Regulation of DNA Repair Fidelity by Molecular Checkpoints: “Gates” in DNA Polymerase β’s Substrate Selection. Biochemistry. 2006;45:15142–15156. doi: 10.1021/bi061353z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Radhakrishnan R, Schlick T. Correct and incorrect nucleotide incorporation pathways in DNA polymerase β. Biochem Biophys Res Commun. 2006;350:521–529. doi: 10.1016/j.bbrc.2006.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y, Schlick T. Quantum mechanics/Molecular mechanics investigation of the chemical reacion in Dpo4 reveals water-dependent pathways and requirements for active site reorganizaation. J Am Chem Soc. 2008;130:13240–13250. doi: 10.1021/ja802215c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klähn M, Braun-Sand S, Rosta E, Warshel A. On Possible Pitfalls in ab Initio Quantum Mechanics/Molecular Mechanics Minimization Approaches for Studies of Enzymatic Reactions. J Phys Chem B. 2005;109:15645–15650. doi: 10.1021/jp0521757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Joyce CM, Benkovic SJ. DNA polymerase fidelity: Kinetics, structure, and checkpoints. Biochemistry. 2004;43:14317–14324. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- 51.Hwang J-K, King G, Creighton S, Warshel A. Simulation of Free Energy Relationships and Dynamics of SN2 Reactions in Aqueous Solution. J Am Chem Soc. 1988;110(16):5297–5311. [Google Scholar]
- 52.Warshel A, Chu ZT, Hwang J-K. The Dynamics of the Primary Event in Rhodopsins Revisited. Chem Phys. 1991;158:303–314. [Google Scholar]
- 53.Warshel A. Computer Modeling of Chemical Reactions in Enzymes and Solutions. John Wiley & Sons; 1997. [Google Scholar]
- 54.Florián J, Warshel A. Phosphate Ester Hydrolysis in Aqueous Solution: Associative Versus Dissociative Mechanisms. J Phys Chem B. 1998;102:719–734. [Google Scholar]
- 55.Klahn M, Rosta E, Warshel A. On the mechanism of hydrolysis of phosphate monoester dianions in solution and proteins. J Am Chem Soc. 2006;128:15310–15323. doi: 10.1021/ja065470t. [DOI] [PubMed] [Google Scholar]
- 56.Rosta E, Kamerlin SCL, Warshel A. On the interpretation of the observed linear free energy relationship in phosphate hydrolysis: A thorough computational study of phosphate diester hydrolysis in solution. Biochemistry. 2008;47:3725–3735. doi: 10.1021/bi702106m. [DOI] [PubMed] [Google Scholar]
- 57.Kamerlin SCL, Mckenna CE, Goodman MF, Warshel A. A Computational study of the Hydrolysis of dGTP analogues with halomethylene-Modified leaving groups in solution: Implications for the mechanism of DNA polymerases. Biochemistry. 2009;48:5963–5971. doi: 10.1021/bi900140c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kamelin SCL, Florian J, Warshel A. Associative versus dissociative mechanisms of phosphate monoester hydrolysis: On the interpretation of activation entropies. ChemPhysChem. 2008;9:1767–1773. doi: 10.1002/cphc.200800356. [DOI] [PubMed] [Google Scholar]
- 59.Braun-Sand S, Sharma PK, TChu Z, Pisliakov AV, Warshel A. The energetics of the primary proton transfer in bacteriorhodopsin revisited: it is a sequential light-induced charge separation after all. Biochim Biophys Acta. 2008;1777:441–452. doi: 10.1016/j.bbabio.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kamerlin SCL, Haranczyk M, Warshel A. Progress in Ab Initio QM/MM Free-Energy Simulations of Electrostatic Energies in Proteins: Accelerated QM/MM Studies of pKa, Redox Reactions and Solvation Free Energies. J Phys Chem B. 2009;113:1253–1272. doi: 10.1021/jp8071712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schweins T, Langen R, Warshel A. Why Have Mutagenesis Studies Not Located the General Base in ras p21. Nature Struct Biol. 1994;1(7):476–484. doi: 10.1038/nsb0794-476. [DOI] [PubMed] [Google Scholar]
- 62.Braun-Sand S, Strajbl M, Warshel A. Studies of Proton Translocations in Biological Systems: Simulating Proton Transport in Carbonic Anhydrase by EVB Based Models. Biophys J. 2004;87(4):2221–2239. doi: 10.1529/biophysj.104.043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bondar AN, Fischer S, Smith JC, Elstner M, Suhai S. Key role of electrostatic interactions in bacteriorhodopsin proton transfer. J Am Chem Soc. 2004;126:14668–14677. doi: 10.1021/ja047982i. [DOI] [PubMed] [Google Scholar]
- 64.Bondar AN, Elstner M, Suhai S, Smith JC, Fischer S. Mechanism of primary proton transfer in bacteriorhodopsin. Structure. 2004;12:1281–1288. doi: 10.1016/j.str.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 65.Riccardi D, Koenig P, Guo H, Cui Q. Proton transfer in Carbonic Anhydrase is controlled by electrostatics rather than the orientation of the acceptor group. Biochemistry. 2008;47:2369–2378. doi: 10.1021/bi701950j. [DOI] [PubMed] [Google Scholar]
- 66.Lee FS, Chu ZT, Warshel A. Microscopic and Semimicroscopic Calculations of Electrostatic Energies in Proteins by the POLARIS and ENZYMIX Programs. J Comp Chem. 1993;14:161–185. [Google Scholar]
- 67.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Jr., Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. A Second Generation Force Field for the Simulation Of Proteins, Nucleic-Acids, And Organic-Molecules. J Am Chem Soc. 1995;117(19):5179–5197. [Google Scholar]
- 68.Schutz CN, Warshel A. What are the dielectric “constants” of proteins and how to validate electrostatic models. Proteins: Struct, Funct, Genet. 2001;44:400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- 69.Messer BM, Roca M, Chu ZT, Vicatos S, Kilshtain AV, Warshel A. Multiscale simulations of protein landscapes: Using coarse-grained models as reference potentials to full explicit models. Proteins: Struct, Funct, Genet. 2010;78:1212–1227. doi: 10.1002/prot.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rychkova A, Vicatos S, Warshel A. On the energetics of translocon-assisted insertion of charged transmembrane helices into membranes. Proc Natl Acad Sci U S A. 2010;107:17598–17603. doi: 10.1073/pnas.1012207107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pisliakov AV, Cao J, Kamerlin SCL, Warshel A. Enzyme millisecond conformational dynamics do not catalyze the chemical step. Proc Natl Acad Sci U S A. 2009;106:17359–17364. doi: 10.1073/pnas.0909150106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dryga A, Warshel A. Renormalizing SMD: The Renormalization approach and its use in long time simulations and accelerated PMF calculations of macromolecules. J Phys Chem B. 2010;114:12720–12728. doi: 10.1021/jp1056122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kamerlin SCL, Vicatos S, Dryga A, Warshel A. Coarse-Grained (Multiscale) Simulations in Studies of Biophysical and Chemical Systems. Annu Rev Phys Chem. 2010;62:41–64. doi: 10.1146/annurev-physchem-032210-103335. [DOI] [PubMed] [Google Scholar]
- 74.Burykin A, Kato M, Warshel A. Exploring the origin of the ion selectivity of the KcsA potassium channel. Proteins: Struct, Funct, Genet. 2003;52:412–426. doi: 10.1002/prot.10455. [DOI] [PubMed] [Google Scholar]
- 75.Bojin MD, Schlick T. A Quantum mechanial investigation of possible mechanisms for the nucleotidyl transfer reaction catalyzed by DNA polymerase β. J Phys Chem B. 2007;111:11244–11252. doi: 10.1021/jp071838c. [DOI] [PubMed] [Google Scholar]
- 76.Tittenhouse RC, Apostoluk WK, Miller JH, Straatsma TP. Characterization of the active site of DNA polymerase β by molecular dynamics and quantum chemical calculation. Proteins: Struct, Funct, Genet. 2003;53:667–682. doi: 10.1002/prot.10451. [DOI] [PubMed] [Google Scholar]
- 77.Abashkin YG, Erickson JW, Burt SK. Quantum Chemical Investigation of Enzymatic Activity in DNA Polymerase β. A Mechanistic Study. J Phys Chem B. 2001;105:287–292. [Google Scholar]
- 78.Sham YY, Chu ZT, Tao H, Warshel A. Examining Methods for Calculations of Binding Free Energies: LRA, LIE, PDLD-LRA, and PDLD/S-LRA Calculations of Ligands Binding to an HIV Protease. Proteins: Struct, Funct, Genet. 2000;39:393–407. [PubMed] [Google Scholar]
- 79.Patel SS, Wong I, Johnson KA. Pre-Steady-State Kinetic-Analysis Of Processive Dna-Replication Including Complete Characterization Of An Exonuclease-Deficient Mutant. Biochemistry. 1991;30(2):511–525. doi: 10.1021/bi00216a029. [DOI] [PubMed] [Google Scholar]
- 80.Kim SJ, Beard WA, Harvey J, Shock DD, Knutson JR, Wilson SH. Rapid segmental and subdomain motions of DNA polymerase beta. J Biol Chem. 2003;278(7):5072–5081. doi: 10.1074/jbc.M208472200. [DOI] [PubMed] [Google Scholar]
- 81.Kirby TW, DeRose EF, Beard WA, Wilson SH, London RE. A thymine isostere in the templating position disrupts assembly of the closed DNA polymerase beta ternary complex. Biochemistry. 2005;44(46):15230–15237. doi: 10.1021/bi0511742. [DOI] [PubMed] [Google Scholar]
- 82.Bose-Basu B, DeRose EF, Kirby TW, Mueller GA, Beard WA, Wilson SH, London RE. Dynamic characterization of a DNA repair enzyme: NMR studies of [methyl-C-13]methionine-labeled DNA polymerase beta. Biochemistry. 2004;43(28):8911–8922. doi: 10.1021/bi049641n. [DOI] [PubMed] [Google Scholar]
- 83.Warshel A, Sharma PK, Kato M, Parson WW. Modeling electrostatic effects in proteins. Biochimica Et Biophysica Acta-Proteins And Proteomics. 2006;1764(11):1647–1676. doi: 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 84.Bakhtina M, Lee S, Wang Y, Dunlap C, Lamarche B, Tsai M-D. Use of Viscogens, dNTPαS, and Rhodium(III) as Probes in Stopped-Flow Experiments To Obtain New Evidence for the Mechanism of Catalysis by DNA Polymerase β. Biochemistry. 2005;44:5177–5187. doi: 10.1021/bi047664w. [DOI] [PubMed] [Google Scholar]
- 85.Ahn J, Kraynov VS, Zhong X, Werneburg BG, Tsai MD. DNA polymerase beta: effects of gapped DNA substrates on dNTP specificity, fidelity, processivity and conformational changes. Biochem J. 1998;331:79–87. doi: 10.1042/bj3310079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Johnson KA. Role of induced fit in enzyme specificity: A molecular forward/reverse switch. J Biol Chem. 2008;283:26297–26301. doi: 10.1074/jbc.R800034200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tsai YC, Johnson KA. A New Paradigm for DNA Polymerases Specificity. Biochemistry. 2006;45:9675–9687. doi: 10.1021/bi060993z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Benitez BAS, Arora K, Schlick T. In Silico Studies of the African Swine Fever Virus DNA Polymerase X Support an Induced-Fit Mechanism. Biophys J. 2006;90:42–56. doi: 10.1529/biophysj.105.071944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Post CB, Jr., WJR Reexamination of induced fit as a determinant of substrate specificity in enzymatic reactions. Biochemistry. 1995;34:15881–15885. doi: 10.1021/bi00049a001. [DOI] [PubMed] [Google Scholar]
- 90.Sawaya MR, Prasad R, Wilson SH, Kraut J, Pelletier H. Crystal structures of human DNA polymerase beta complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry. 1997;36(37):11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- 91.Arora K, Schlick T. In silico evidence for DNA polymerase b’s substrate-induced conformational change. Biophys J. 2004;87:3088–3099. doi: 10.1529/biophysj.104.040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Warshel A, Sharma PK, Kato M, Xiang Y, Liu H, Olsson MH. Electrostatic Basis for Enzyme Catalysis. Chem Rev. 2006;106(8):3210–3235. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- 93.Post CB, Ray WJ. Reexamination of induced fit as a determinant of substrate specificity in enzymatic reactions. Biochemistry. 1995;34(49):15881–15885. doi: 10.1021/bi00049a001. [DOI] [PubMed] [Google Scholar]
- 94.Muegge I, Qi PX, Wand AJ, Chu ZT, Warshel A. The Reorganization Energy of Cytochrome c Revisited. J Phys Chem B. 1997;101:825–836. [Google Scholar]
- 95.Liu H, Warshel A. The Catalytic Effect of Dihydrofolate Reductase and Its Mutants Is Determined by Reorganization Energies. Biochemistry. 2007;46:6011–6025. doi: 10.1021/bi700201w. [DOI] [PubMed] [Google Scholar]
- 96.Jencks WP. Catalysis in Chemistry and Enzymology. Dover Publication; New York: 1986. [Google Scholar]
- 97.Herschlag D. The Role Of Induced Fit And Conformational-Changes Of Enzymes In Specificity And Catalysis. Bioorganic Chemistry. 1988;16(1):62–96. [Google Scholar]
- 98.Warshel A, Parson WW. Dynamics of biochemical and biophysical reactions: insight from computer simluations. Q Rev Biophys. 2001;34:563–670. doi: 10.1017/s0033583501003730. [DOI] [PubMed] [Google Scholar]
- 99.Glennon TM, Villa J, Warshel A. How does GAP catalyze the GTPase reaction of Ras? A computer simulation study. Biochemistry. 2000;39:9641–9651. doi: 10.1021/bi000640e. [DOI] [PubMed] [Google Scholar]
- 100.Rosta E, Klahn M, Warshel A. Towards accurate ab initio QM/MM calculations of free-energy profiles of enzymatic reactions. J Phys Chem B. 2006;110(6):2934–2941. doi: 10.1021/jp057109j. [DOI] [PubMed] [Google Scholar]
- 101.Cisneros GA, Perera L, Garcia-Diaz M, Bebenek K, Kunke TA, Pedersen LG. Catalytic mechanism of human DNA polymerase λ with Mg2+ and Mn2+ from ab initio quantum mechanical/molecular mechanical studies. DNA Rep. 2008;7:1824–1834. doi: 10.1016/j.dnarep.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang L, Yu X, Hu P, Broyde S, Zhang Y. A Water-Mediated and Substrate-Assisted Catalytic Mechanism for Sulfolobus solfataricus DNA Polymerase IV. J Am Chem Soc. 2007;129:4731–4737. doi: 10.1021/ja068821c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kirby AJ, Younas MJ. The reactivity of phosphate esters. Diester hydrolysis. J Chem Soc B. 1970:510–513. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.