

Abstract
Most SOS mutagenesis in Escherichia coli is dependent on the UmuD and UmuC proteins. Perhaps as a consequence, the activity of these proteins is exquisitely regulated. The intracellular level of UmuD and UmuC is normally quite low but increases dramatically in lon− strains, suggesting that both proteins are substrates of the Lon protease. We report here that the highly purified UmuD protein is specifically degraded in vitro by Lon in an ATP-dependent manner. To identify the regions of UmuD necessary for Lon-mediated proteolysis, we performed ‘alanine-stretch’ mutagenesis on umuD and followed the stability of the mutant protein in vivo. Such an approach allowed us to localize the site(s) within UmuD responsible for Lon-mediated proteolysis. The primary signal is located between residues 15 and 18 (FPLF), with an auxiliary site between residues 26 and 29 (FPSP), of the amino terminus of UmuD. Transfer of the amino terminus of UmuD (residues 1–40) to an otherwise stable protein imparts Lon-mediated proteolysis, thereby indicating that the amino terminus of UmuD is sufficient for Lon recognition and the ensuing degradation of the protein.
Keywords: UmuD′, ATP-dependent protease, SOS mutagenesis, degradation signal
Exposure of Escherichia coli to DNA-damaging agents, whether they be natural or man-made, invokes the induction of a number of unlinked genes required for DNA repair, cell division, and damage tolerance. This inducible pathway is often referred to as the SOS response (for review, see Friedberg et al. 1995; Koch and Woodgate 1998). One of the hallmarks of the SOS response is that it is graded in such a way that error-free pathways of DNA repair are induced early (so that DNA fidelity remains high), whereas pathways that may ensure survival under more severe conditions (but are error-prone) are induced much later (Sommer et al. 1998). Key participants in the latter pathway, which is often called SOS mutagenesis, are the UmuD′ and UmuC proteins (Woodgate and Levine 1996; Smith and Walker 1998). Together with RecA and Ssb proteins, the Umu proteins allow DNA polymerase III holoenzyme to traverse otherwise replication-blocking lesions (Rajagopalan et al. 1992; Reuven et al. 1998; Tang et al. 1998), but with a concomitant reduction in replication fidelity. As a consequence, it is believed that the regulatory mechanisms that determine the expression and levels of these proteins have evolved so that UmuD′ and UmuC are used only as a last resort (Woodgate and Levine 1996).
To achieve such regulation, the cell utilizes transcriptional control (the umu operon is one of the most tightly regulated in the SOS regulon), together with a variety of post-translational mechanisms to keep the activity of the Umu proteins to a minimum. The pivotal step in controlling the activity of the Umu proteins is the RecA-mediated cleavage of UmuD. This cleavage reaction is primarily intermolecular in nature (McDonald et al. 1998a) and results in the removal of the 24 amino-terminal residues from UmuD to generate UmuD′ (Burchhardt et al. 1988; Shinagawa et al. 1988). Not only does cleavage activate UmuD′ for its mutagenesis functions (Nohmi et al. 1988), it also converts UmuD from a substrate that is sensitive to the Lon protease to one that is relatively insensitive to Lon (Frank et al. 1996a). The increased stability of UmuD′ is problematic in that it is likely to result in an increase in aberrant mutagenesis. The cell generally avoids such problems, however, by targeting UmuD′ for degradation by the ClpXP protease (Frank et al. 1996a). Both UmuD and UmuD′ form homodimers (Burckhardt et al. 1988; Woodgate et al. 1989; Battista et al. 1990); but under conditions of limited cleavage (as might be expected in cells exposed to mild DNA damage), UmuD′ preferentially interacts with the more abundant UmuD protein to form a UmuD/D′ heterodimer and this complex directs ClpXP degradation of UmuD′ (Frank et al. 1996a). All three dimer species of UmuD and UmuD′ (the individual homodimers and the heterodimer) are capable of interacting with UmuC but with varying affinities (Woodgate et al. 1989; Frank et al. 1996a,b; Jonczyk and Nowicka 1996). The UmuC protein, in the absence of interacting partners, is also highly labile and has also been shown to be a substrate of the Lon protease in vivo (Frank et al. 1996a).
It is evident that the extent of SOS mutagenesis is contingent on the proper protein–protein interactions and the resulting stability of these complexes. The regulated proteolysis of the Umu proteins therefore provides a critical mechanism by which the cell maintains the correct levels of the Umu proteins, both during and after exposure to DNA damage (Gonzalez et al. 1998; Sommer et al. 1998). The question remains as to what features these proteins possess that specifically target them for degradation. There are numerous reports implicating the carboxyl terminus in protease recognition. For example, mutants of the P22 Arc repressor or the λcI repressor were found to be unstable, and this instability is dependent on the carboxy-terminal 5 amino acids (Bowie and Sauer 1989; Parsell et al. 1990; Keiler et al. 1995). These 5 amino acids are nonpolar residues, and any change in them to a polar or charged carboxyl terminus make the protein stable (Parsell et al. 1990; Keiler et al. 1995). Similarly, recent work has demonstrated that addition of a nonpolar 11-amino-acid carboxy-terminal sequence, encoded by E. coli ssrA, to peptides results in their rapid degradation (Tu et al. 1995; Keiler et al. 1996). The carboxyl terminus of the Mu transposase, MuA, is known to have a role in ClpX recognition and disassembly of the transposase–DNA complex as well as in its degradation by ClpXP (Levchenko et al. 1995; Levchenko et al. 1997). In contrast to the nonpolar nature necessary for recognition of the SsrA-tagged fusions, the MuA carboxyl terminus is positively charged (Levchenko et al. 1995, 1997).
Comparison of Lon substrates yields no identifiable features that might function in substrate recognition. The carboxyl terminus of the Lon substrate, SulA, was demonstrated to be sufficient for recognition, but not for degradation, of a fusion of β-galactosidase with the carboxy-terminal 20 amino acids of SulA (Higashitani et al. 1997).
In an attempt to identify the signal in UmuD that targets it for degradation by Lon, we have used a dual approach; first, we demonstrate that as predicted from our previous in vivo studies (Frank et al. 1996a), highly purified UmuD protein is a substrate of the Lon protease in vitro; second, we use the in vivo assay to monitor the susceptibility of various mutant UmuD proteins to proteolysis by Lon. Our results suggest that certain regions within the amino-terminal tail of UmuD are necessary and sufficient for Lon-mediated degradation of UmuD and that these signals can be transferred to an otherwise stable protein to impart Lon-mediated proteolysis.
Results
In vitro degradation of UmuD
Previously, we have demonstrated that the UmuD protein is highly labile in vivo and is stabilized in lon− cells (Frank et al. 1996a). To determine whether UmuD stability is directly related to the Lon protease, we incubated highly purified UmuD and Lon proteins in the presence or absence of exogenous ATP (Fig. 1A). To maximize the in vitro activity of Lon (Van Melderen et al. 1996), the reaction also utilized an ATP regeneration system. As demonstrated clearly in Figure 1A, the Lon protease degraded UmuD in the presence of ATP. No detectable degradation products were observed, indicating that degradation proceeds in a processive manner (data not shown). In contrast, no degradation was evident in the absence of ATP. On the basis of these experiments, we calculate that the in vitro half-life of UmuD is ∼30 min (Fig. 1B). These results clearly demonstrate, in concordance with our in vivo observations (Frank et al. 1996a), that UmuD is specifically recognized and degraded by the Lon protease in vitro.
Figure 1.
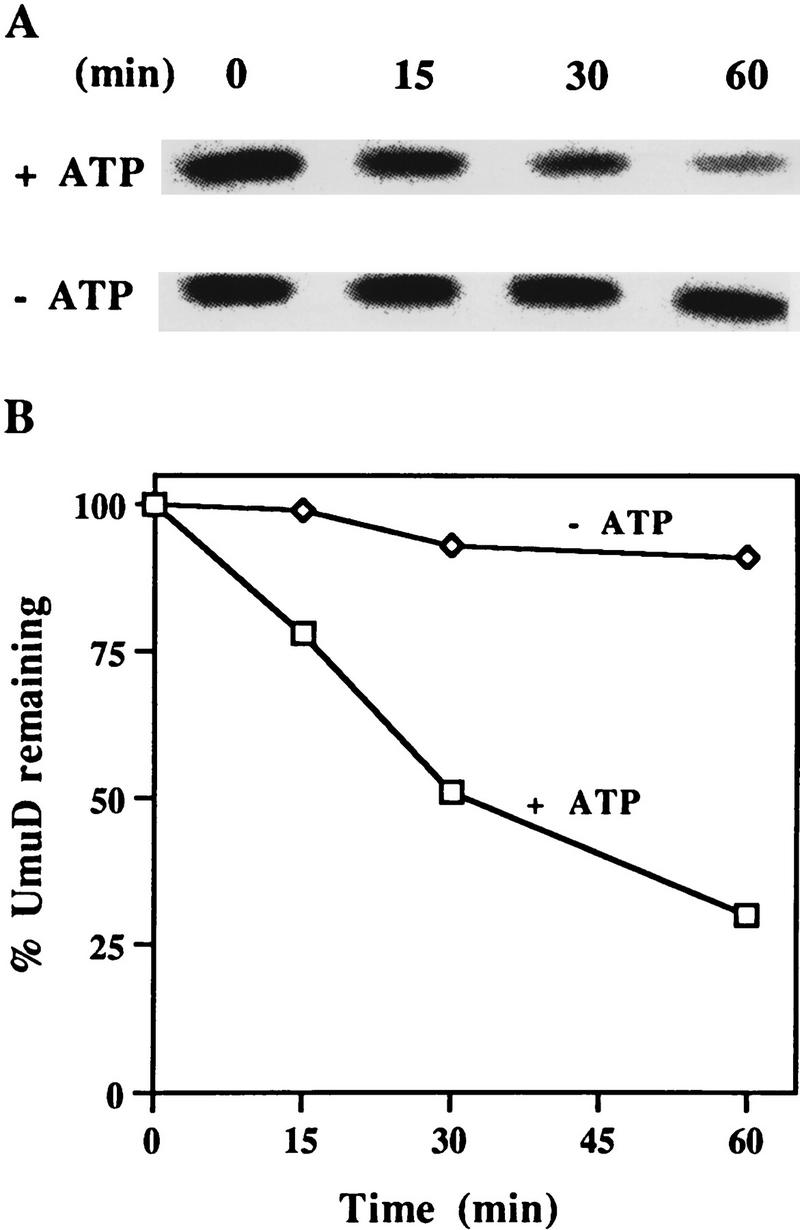
In vitro degradation of UmuD by Lon. (A) Highly purified UmuD and Lon proteins were incubated at 37°C in the presence or absence of ATP, as described in Materials and Methods. An aliquot was removed at the indicated time, electrophoresed in 17% SDS–polyacrylamide gels, and visualized after staining with Coomassie brilliant blue. (B) Kinetics of UmuD degradation. These data were generated by quantifying the gels described in A, on a ChemiImager 4000 low-light imaging system.
Specificity of Lon-mediated degradation of UmuD
The Lon protease has long been known for the housekeeping role it has in the removal of abnormal or damaged cellular proteins (for review, see Gottesman 1996). Lon also functions in the selective degradation of proteins such as SulA (Huisman and D’Ari 1981; Mizusawa and Gottesman 1983), λN (Maurizi 1987), CcdA (Van Melderen et al. 1996), RcsA (Torres-Cabassa and Gottesman 1987; Stout et al. 1991), and UmuC (Frank et al. 1996a). To date, little is known as to what ‘signature’ identifies a protein as a substrate of Lon. As noted above (Fig. 1), UmuD is degraded by Lon in the presence of ATP. To determine the extent of Lon specificity, we purified an engineered version of the post-translationally processed UmuD protein, UmuD′, and examined the effects of Lon on the stability of UmuD′. In vivo, UmuD′ appears relatively insensitive to Lon degradation (Frank et al. 1996a). We reasoned, therefore, that assaying the stability of UmuD′ would be a rigorous test of the in vitro specificity of the Lon protease. As shown in Figure 2A, the UmuD′ protein is relatively stable after a 1-hr incubation in the presence of Lon, thus verifying our previous observations in vivo (Frank et al. 1996a).
Figure 2.
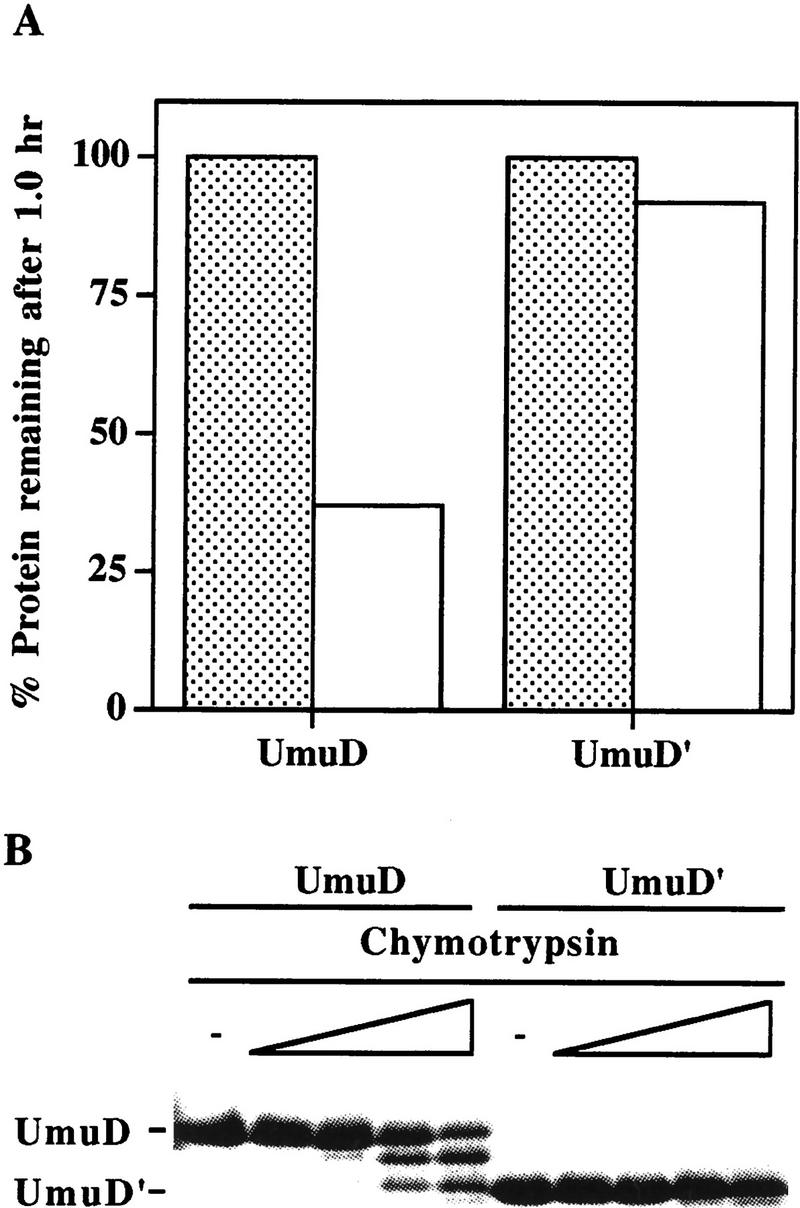
In vitro specificity of Lon. (A) Highly purified UmuD and UmuD′ proteins were individually incubated with ATP in the presence (open bars) or absence (stippled bars) of Lon at 37°C for 1 hr. Reactions were processed, visualized, and quantitated as described in Fig. 1. (B) Limited α-chymotrypsin digest of either purified UmuD or UmuD′ was performed at 25°C for 20 min with increasing concentrations of α-chymotrypsin. The concentrations of α-chymotrypsin used were 0 ng (lanes 1,6), 5 ng (lanes 2,7), 50 ng (lanes 3,8), 250 ng (lanes 4,9), and 500 ng (lanes 5,10). Reactions were electrophoresed and visualized as described in Materials and Methods. The positions of intact UmuD and UmuD′ are indicated at left.
One possible explanation for these results is that UmuD and UmuD′ possess very different tertiary structures (Guzzo et al. 1996). To test this hypothesis, we examined the effect of chymotrypsin digestion on both UmuD and UmuD′. Under limiting protease conditions, UmuD was efficiently processed to a species of similar electrophoretic mobility as UmuD′ (Fig. 2B). In contrast, however, no significant degradation of UmuD′ was observed. This suggests that the globular structures of UmuD and UmuD′ (Peat et al. 1996a,b; Ferentz et al. 1997) are essentially similar and that the 24-amino-acid amino terminus of UmuD does not destabilize UmuD by imparting major structural changes not evident in UmuD′. On the basis of these observations, the amino-terminal 24 amino acids of UmuD appear to be all that is efficiently cleaved by chymotrypsin during the 20-min incubation time. Similar results were obtained after tryptic digestion of UmuD (D. Wall and W. Hendrickson, pers. comm.).
Identification of the regions in UmuD necessary for Lon degradation
The fact that UmuD, but not UmuD′, is a substrate of Lon suggests that the signal conferring Lon-mediated proteolysis is most likely located in the very amino terminus of UmuD that is naturally absent in UmuD′. To identify these regions, we have taken advantage of the fact that the orthologous Salmonella typhimurium UmuD protein also appears to be a substrate of Lon (Gonzalez et al. 1998). Therefore, we hypothesized that the signal for Lon-mediated degradation would be located within the amino-terminal residues of UmuD that are identical in the two proteins. Overall, the two UmuD proteins share 73% identity, but as seen in Figure 3A, the amino termini are more diverged with only 54% identical residues in the region from the amino terminus to the RecA-mediated post-translational cleavage site. To test our hypothesis, we constructed plasmid-encoded mutants of E. coli UmuD in which the wild-type residues (identical or highly conserved in S. typhimurium UmuD) were substituted with 4 alanine residues (Fig. 3B). Transformed lon+ cells expressing the individual mutant UmuD proteins were grown to early logarithmic phase at which time chloramphenicol was added to block further protein synthesis. Samples were taken at subsequent time intervals and UmuD visualized by standard Western blotting techniques. Figure 4A shows time course experiments for the individual UmuD ‘alanine-stretch’ mutants. The half-life of wild-type UmuD was calculated to be ∼11 min and was similar to that of the UmuD9-4 and UmuD26-4 mutants. In striking contrast, the UmuD15-4 alanine mutant was appreciably stabilized with a half-life of ∼38 min. In parallel experiments, UmuD and all the UmuD alanine-stretch mutants were further stabilized (half-life >60 min) in a lon− background (data not shown). The marked stabilization of UmuD154 indicates, therefore, that amino acids 15–18 (FPLF) of UmuD are important for efficient Lon-mediated degradation, but the increased stability noted in a lon− background suggests that other regions of UmuD are also important for efficient Lon recognition.
Figure 3.


Proteins used to identify the Lon recognition signal of UmuD. (A) E. coli and S. typhimurium UmuD proteins are both substrates of Lon (Gonzalez et al. 1998). Their primary amino acid sequences were aligned using the program Geneworks, and areas that are boxed indicate regions that are 100% conserved between the two proteins. Overall, the proteins are 73% identical, but most divergence occurs in the amino- and carboxy-terminal tails. (B) The amino terminus of the UmuD alanine-stretch mutants. Areas boxed indicate the multiple alanine substitutions made within the UmuD amino terminus for each mutant protein. The normal post-translational cleavage site is located between Cys-24 and Gly-25. The positions of the genetically engineered silent restriction sites for NdeI and SacII in the umuD gene are indicated at their approximate position in the corresponding UmuD protein.
Figure 4.


Stability of the UmuD mutants in a lon+ strain. Cultures were grown in LB medium at 37°C to logarithmic phase at which time chloramphenicol was added to block protein synthesis. An aliquot was removed at the indicated times thereafter. Whole cell protein extracts from an equivalent number of cells were electrophoresed in 17% SDS–polyacrylamide gels, blotted onto an Immobilon P membrane, probed with anti-UmuD antibody, and quantitated as described in Materials and Methods. (A) Single region (4-alanine stretch) mutants of UmuD. (○) Wild-type UmuD; (▵) UmuD9-4; (□) UmuD15-4; (⋄) UmuD26-4. (B) The double region (4 plus 4 alanine stretch) mutants of UmuD. (○) Wild-type UmuD; (▴) UmuD15/9; (♦) UmuD26/9; (█) UmuD26/15. All experiments were performed two to three times with no significant variation in results.
To determine the region of UmuD that might be required for complete stabilization, we created plasmids expressing UmuD mutants, each consisting of four alanine stretches within two regions of the amino terminus (see Fig. 3B). The kinetics of degradation of the double alanine-stretch mutant, UmuD15/9 were very similar to that of UmuD15-4 (Fig. 4, cf. A and B), and therefore no additional role in Lon-mediated degradation could be assigned to UmuD9-4. Likewise, UmuD26/9 behaved identically to the individual alanine-stretch mutants UmuD9-4 and UmuD26-4, resulting in a half-life of ∼12 min (Fig. 4B). However, substitution of four alanine amino acids at residues 26–29 in the relatively stable UmuD15-4 mutant resulted in a dramatic increase in stability. The half-life of UmuD26/15 is >1 hr (Fig. 4B), and the decay kinetics for UmuD26/15 in a lon+ background are similar to those of the wild-type UmuD protein in a lon− background (data not shown).
These data support a model in which the Lon protease recognizes two specific regions within the amino terminus of UmuD, the primary site being located between residues 15 and 18 and the auxiliary site between residues 26 and 29. Note that the auxiliary site alone is insufficient to target UmuD degradation (Fig. 4A), explaining why UmuD′ (which lacks the primary Lon recognition signal but possesses the secondary site) is insensitive to proteolysis by Lon (Fig. 2A; Frank et al. 1996a).
Functional activity of the UmuD alanine-stretch mutants
One possible explanation for our finding that certain mutant UmuD proteins are rendered insensitive to Lon is that we have grossly altered the conformation of the UmuD protein and, as a consequence, made it less susceptible to Lon proteolysis. A critical step in the mutational process is the RecA-mediated self-cleavage of mutagenically active UmuD to mutagenically active UmuD′ (Burckhardt et al. 1988; Nohmi et al. 1988; Shinagawa et al. 1988). UmuD′ then associates with UmuC, RecA, and DNA polymerase III to promote error-prone trans-lesion DNA synthesis (Rajagopalan et al. 1992; Reuven et al. 1998; Tang et al. 1998). A simple, but indirect, assay of the conformational structure of the various UmuD mutants is, therefore, an ability to promote damage-inducible SOS mutagenesis in vivo. Presumably strains that are mutable express a UmuD protein that can undergo cleavage as well as the subsequent protein–protein interactions necessary for trans-lesion DNA synthesis. Such analysis revealed, however, that the majority of the UmuD alanine-stretch mutants do not promote significant levels of SOS mutagenesis (Fig. 5A). The lone exception is UmuD9-4, which displays a slightly greater mutation frequency than wild-type UmuD.
Figure 5.
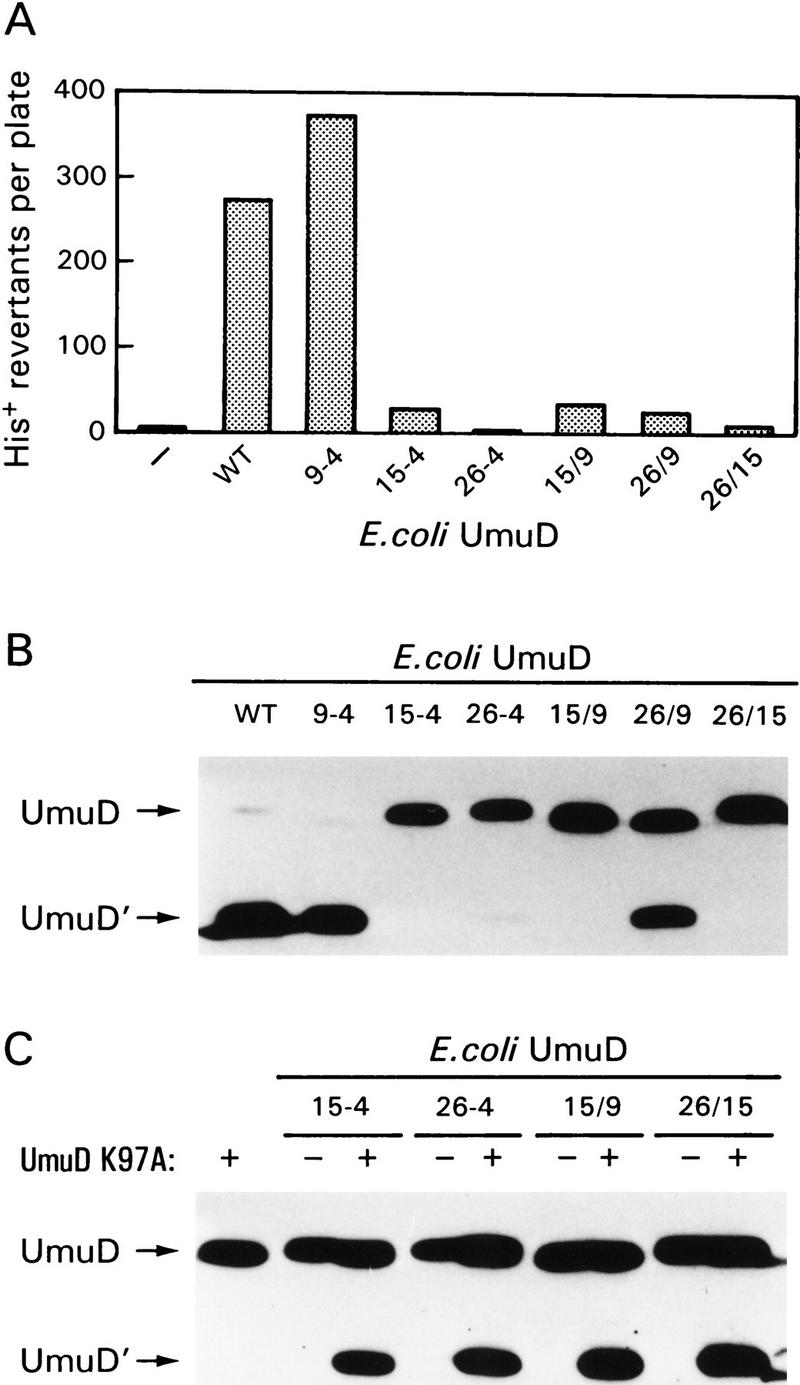
Functional activity of the UmuD alanine-stretch mutants. (A) The ability of the alanine-stretch mutants to function in SOS mutagenesis was assayed using strain RW126 harboring the low-copy-number UmuC-expressing plasmid pRW274 and a compatible plasmid expressing each of the individual UmuD alanine mutants. Strain RW126 carries an ocher mutation in hisG [hisG4(Oc)] that renders it auxotrophic for histidine. In the presence of functional Umu proteins it can, however, revert to His+. The number of His+ revertants per plate after exposure to MMS represents the mean number from a minimum of three cultures. (B) Western blot analysis of the RecA-mediated self cleavage reaction in vivo. The ability of the alanine-stretch mutants to undergo post-translational cleavage was assayed in the lexA51(Def) recA730 strain RW244. This strain constitutively expresses all LexA-regulated proteins (including the Umu proteins) and promotes constitutive cleavage of UmuD in the absence of exogenous DNA damage. The positions of UmuD and UmuD′ are indicated by arrows at left. (C) The ability of the UmuD alanine mutants to act as an enzyme in the intermolecular UmuD cleavage reaction. The UmuDK97A protein cannot undergo intramolecular cleavage because it carries a mutation at the active site of UmuD. It can, however, serve as a substrate in the intermolecular cleavage reaction if a UmuD enzyme is provided in trans (in our case, the various alanine-stretch mutants). Cleavage of the UmuDK97A protein was monitored in strain RW244 in the absence or presence of the individual UmuD alanine mutants and visualized by Western blot analysis as described in Materials and Methods. The positions of UmuD and UmuD′ are indicated by arrows at left.
Such findings prompted us to directly assay the ability of the mutant UmuD protein to be processed to UmuD′ (Fig. 5B). In a recA730 background, wild-type UmuD is converted to UmuD′ with >90% efficiency (Fig. 5B; Shinagawa et al. 1988; Woodgate and Ennis 1991). In concordance with the in vivo mutagenesis assay, UmuD9-4 was converted to UmuD′ with efficiency similar to that of the wild-type protein. Interestingly, despite its inability to promote mutagenesis, ∼50% of UmuD26/9 was converted to UmuD′, indicating that the mutant protein probably retains the same overall structure as the wild-type UmuD protein. Given that the resultant UmuD′ protein (UmuD′26-4) remains functionally inactive for mutagenesis (Fig. 5A), it would argue that residues 26–29 are important for the subsequent activity of UmuD′. These residues are absolutely conserved in all of the bona fide UmuD(D′) homologs identified to date (Woodgate and Levine 1996).
We have recently demonstrated that UmuD cleavage predominantly occurs via an intermolecular reaction in vivo (McDonald et al. 1998a). That is, one monomer of UmuD (or UmuD′) can act as an enzyme to facilitate the cleavage of another substrate UmuD monomer (McDonald et al. 1998a). Given the fact that the alanine-stretch mutants are in close proximity to (or actually span) the cleavage site, we hypothesized that such changes might simply alter the ability of the protein to act as a substrate molecule. However, if the mutant protein retains the same overall globular structure of the wild-type protein, it should still be capable of acting as an enzyme in the intermolecular cleavage reaction (McDonald et al. 1998a). To test this hypothesis, the mutant plasmids were cotransformed with pKSD10 (a compatible low-copy-number plasmid expressing UmuDK97A) into a recA730 strain. The UmuDK97A protein expressed by pKSD10 is unable to undergo intramolecular self-cleavage because it has a mutation at the catalytic active site but is able to undergo intermolecular cleavage as a substrate molecule if a UmuD enzyme is provided in trans (McDonald et al. 1998a). As shown in Figure 5C, all of the alanine-stretch mutants (15-4, 26-4, 15/9, and 26/15) are able to act as enzymes in the intermolecular cleavage reaction.
The assays described above not only allow us to conclude that the globular bodies of the mutant 15-4, 26-4, 15/9, and 26/15 UmuD proteins are similar to that of the wild-type protein, they also reveal the multiple types of interactions that are mediated by the amino terminus of UmuD (see Discussion).
Transfer of the UmuD degradation signal
Our finding that the mutant UmuD proteins appear to retain the same overall structure as the wild-type protein supports our notion that the region of UmuD identified in our mutational studies is a bona fide Lon recognition/degradation signal. One obvious test of this hypothesis is an ability of these sequences to impart instability on an otherwise stable protein. To test this hypothesis, we constructed a plasmid (pKSD–PRP) expressing a chimeric gene encoding the first 40 amino acids of UmuD fused to a 7-amino-acid linker joined to the entire Streptoalloteichus hindustanus Ble protein. The S. hindustanus ble gene encodes a small, stable protein that provides resistance to the antibiotics of the phleomycin family (Dumas et al. 1994; Gatignol et al. 1988). For convenience, the S. hindustanus ble gene product will be referred to as the phleomycin resistance protein (or PRP). The wild-type PRP is itself quite stable when assayed in our wild-type background (data not shown). In contrast, the UmuD–PRP fusion is unstable in the lon+ background and displays a half-life of ∼9 min (Fig. 6). Interestingly, UmuD–PRP demonstrates a marked increase in stability when assayed in a lon− strain (half-life of ∼38 min), indicating that the UmuD–PRP fusion is degraded in a Lon-dependent fashion (Fig. 6). Furthermore, subsequent experimentation demonstrated that residues 1–29 of UmuD are sufficient to impart Lon recognition and degradation of the PRP similar to that seen with the 40-amino-acid fusion (data not shown). By comparison, a UmuD–PRP fusion protein containing the two alanine-stretch mutants located between residues 15 and 18 and 26 and 29, was stable, and the decay kinetics of the UmuD26/15–PRP fusion in a lon+ background (half-life of ∼30 min) were similar to those of the wild-type UmuD–PRP fusion in the lon− background (Fig. 6). It is important to note, however, that the UmuD–PRP fusion, as well as the UmuD26/15–PRP fusion, is obviously degraded by at least one additional protease other than Lon. These results therefore confirm our hypothesis that residues 15–19 and 26–29 of UmuD are recognized by Lon and that these sequences confer Lon-mediated instability on an otherwise stable protein.
Figure 6.

The amino-terminal 40 amino acids of UmuD are sufficient to target the PRP for degradation by Lon in vivo. The amino-terminal 40 amino acids of UmuD were fused to the amino terminus of PRP to create a UmuD–PRP fusion protein. Western blot analysis of UmuD–PRP stability was determined in lon+ (EC10) and lon− (EC18) cells. Stability of the plasmid expressed UmuD26/15-PRP fusion protein was also assessed in the lon+ strain (EC10). UmuD26/15-PRP describes a similar fusion construct except that the first 40 amino acids of UmuD contain the UmuD26/15 alanine-stretch mutations described in Figure 3B. Cultures were grown in LB medium at 37°C to logarithmic phase at which time chloramphenicol was added to block protein synthesis. An aliquot was removed at the indicated times thereafter. Whole cell protein extracts from an equivalent number of cells were electrophoresed in 17% SDS–polyacrylamide gels, blotted onto an Immobilon P membrane, and probed with antibodies directed against the amino-terminal 24 amino acids of UmuD. This antibody demonstrated limited cross-reactivity with the 8-alanine-substituted UmuD26/15-PRP, and as a consequence, the UmuD26/15-PRP mutant protein was visualized using anti-PRP antibodies.
Discussion
Trans-lesion DNA synthesis in E. coli involves numerous protein–protein interactions, all of which are vital for ensuring continued survival of the organism when challenged with various stressful conditions. Many of the protein–protein interactions function in maintaining the proper equilibrium of the Umu proteins so that SOS mutagenesis occurs only as a last resort. The E. coli Lon protease contributes to this process by regulating the levels of the UmuD and UmuC proteins in vivo (Frank et al. 1996a). Our studies demonstrate that the degradation of UmuD is directly related to the proteolytic action of Lon and occurs in an ATP-dependent fashion. Furthermore, we have shown that the Lon protease displays striking specificity for UmuD in vitro, thereby mirroring our previous observations in vivo (Frank et al. 1996a). The relative rate of degradation of UmuD, however, is much more rapid in vivo (Fig. 4) than in vitro (Fig. 1A), but such findings are also characteristic of other Lon substrates such as λN (Maurizi 1987), CcdA (Van Melderen et al. 1996), and a SulA fusion protein (Sonezaki et al. 1995).
There is an emerging consensus that the termini of labile proteins, both amino and carboxyl, have a vital role in the targeting of many proteins for rapid degradation. For example, a carboxy-terminal tagging system in E. coli is dedicated to identifying stalled ribosomes at the 3′ end of a truncated mRNA and tagging the carboxyl terminus of the respective truncated protein with a nonpolar 11-amino-acid (AANDENYALAA) peptide (Tu et al. 1995; Keiler et al. 1996). This carboxy-terminal addition, a function of the SsrA RNA, results in the rapid degradation of the fusion protein (Keiler et al. 1996). Moreover, Gottesman et al. (1998) identified the proteases responsible for the degradation of the SsrA-tagged peptides as the ClpAP and ClpXP proteases. In addition, the membrane-bound HflB protease of E. coli is also known to degrade SsrA–tagged peptides (Herman et al. 1998). Because HflB is membrane-bound, Herman et al. (1998) postulate that the primary role of HflB is to act upon abnormal membrane proteins. The SsrA tag is composed primarily of nonpolar amino acids that are normally found buried within the protein. The nonpolar nature of the SsrA tag is therefore likely to resemble exposed areas of damaged and/or denatured proteins that are known to be rapidly degraded by many of the E. coli cytoplasmic proteases, possibly including ClpAP and ClpXP (for review, see Gottesman 1996).
The bacterial N-end rule reflects a proteolytic pathway whose specificity is dictated by the amino-terminal amino acid (Tobias et al. 1991). The pathway was identified by assessing the stability of engineered β-galactosidase fusions, each beginning with a different amino acid. The protease in E. coli responsible for the degradation of the unstable N-end rule substrates was identified as ClpAP (Tobias et al. 1991). This protease was also shown to be responsible for the degradation of a fusion protein consisting of the first 40 amino acids of ClpA fused to the amino terminus of β-galactosidase (Gottesman et al. 1990). When wild-type ClpA stability was assessed in vivo, it was found to be unstable, albeit at a much reduced degradation rate when compared to the rate of degradation of the ClpA–β-galactosidase fusion (Gottesman et al. 1990). Much like the SsrA tag described above, the ClpA–β-galactosidase fusion presumably places the ClpAP recognition signal in a context allowing greater accessibility.
The amino- and carboxy-terminal recognition pathways described above most likely function because terminal recognition signals provide easier access for the protease. The UmuD′ crystal structure was determined recently and the amino-terminal region (starting at residue 32) of UmuD′ was described as extended and unstructured (Peat et al. 1996a,b). Therefore, if one assumes that UmuD and UmuD′ share the same globular structure (Fig 2A; Ferentz et al. 1997), the entire amino terminus (residues 1–40) is likely to be extended and possibly unstructured. The experiments presented here suggest that this extended amino terminus of UmuD is essential for efficient Lon-mediated degradation. Using alanine-stretch mutagenesis, we have localized the primary Lon recognition signal in UmuD to residues Phe-15, Pro-16, Leu-17, and Phe-18. Multiple alanine substitutions at these residues result in significant stabilization of UmuD, strongly suggesting that this specific region of the UmuD protein is recognized by the Lon protease. Interestingly, multiple alanine mutations at residues 15–18 in unison with alanine substitutions at residues 26–29 result in almost complete stabilization of UmuD. In contrast, multiple alanine mutations spanning residues 26–29 had no apparent affect on UmuD degradation. This suggests that residues 26–29 can function in either stabilizing the Lon–UmuD interaction at the putative Lon recognition site (residues 15–18) or in maintaining the accessibility of the amino terminus for Lon recognition. The 26-4 site is therefore considered an auxiliary site in that it only functions in Lon-mediated proteolysis in concert with the primary site.
As noted earlier, the S. typhimurium UmuD protein is also a substrate of Lon (Gonzalez et al. 1998). The auxiliary site (residues 26–29) in the S. typhimurium UmuD protein is identical to that of the E. coli protein. By comparison, the primary site is not identical but is highly conserved (FPLF(Ec) → LPFF(St)). Moreover, the UmuD homolog RumA, which is found on the incJ plasmid R391 as part of the RumAB operon, also has an identical auxiliary site and a highly conserved primary site (FPLF(Ec) → IPLF(RumA)) (Kulaeva et al. 1995). RumA, however, is not degraded by the Lon protease (M. Gonzalez, unpubl.). A possible explanation for this finding is that the amino acid sequences immediately surrounding the primary site of RumA are highly charged in comparison to the same region in the E. coli and S. typhimurium UmuD proteins (Woodgate and Levine 1996). Such findings suggest, therefore, that the major determinants leading to Lon-mediated proteolysis are not primary amino acid sequences per se but, rather, an exposed stretch of nonpolar amino acid residues and the surrounding protein environment. Site-directed mutagenesis of the primary and auxiliary sites, as well as mutagenesis of the surrounding residues, should further elaborate the Lon–UmuD interaction necessary for degradation.
The amino terminus of UmuD assumes no identifiable secondary structure, and no sequence similarity is evident between UmuD and other known substrates of Lon. Higashitani et al. (1997) identified a region of similarity between two Lon substrates, the λ N protein and the E. coli SulA protein. However, whereas fusion of the carboxy-terminal 20 amino acids of SulA to the carboxyl terminus of β-galactosidase was sufficient for Lon binding, it was not enough to promote Lon-mediated proteolysis (Higashitani et al. 1997). In contrast, our results demonstrate that fusion of the amino terminus of UmuD (residues 1–40) to an otherwise stable PRP is all that is necessary to target the protein for Lon-mediated proteolysis (Fig. 5). Furthermore, mutations in the amino terminus of the UmuD–PRP fusion, identical to those found in the stable UmuD26/15 mutant, inhibited Lon-mediated degradation (Fig. 5). The ability to transfer the Lon recognition signal of UmuD and thereby provoke Lon-mediated degradation signifies that the amino terminus of UmuD not only comprises the information necessary for binding but also provides the foundation for degradation of the substrate.
Our finding that the two regions of the UmuD amino terminus necessary for Lon-mediated degradation span the UmuD cleavage site (Cys-24 → Gly-25) raises the intriguing possibility that Lon recognition and degradation of UmuD is in direct competition with the RecA-mediated self-cleavage of UmuD that occurs during the SOS response (Fig. 7). Such competition allows exquisite post-translational regulation of UmuD and provides yet another mechanism by which Umu-dependent error-prone trans-lesion DNA synthesis is regulated (Woodgate and Levine 1996; Sommer et al. 1998). Clearly, recognition of protein termini is a common mechanism employed by many proteases. The targeting of the Lon protease to the UmuD amino terminus is one of the critical mechanisms that ensures cell survival in the face of irreparable DNA damage at minimal mutational cost.
Figure 7.

Lon degradation vs. RecA processing of the UmuD SOS mutagenesis protein. Competition for UmuD by Lon and RecA provides one mechanism whereby the level of mutagenically active UmuD′ protein is kept to a minimum. In the absence of exogenous DNA damage, the equilibrium of the reaction favors Lon degradation and most of the UmuD protein is rapidly degraded before it is converted to UmuD′ (Gonzalez et al. 1998). After DNA damage, when the need for functional Umu proteins increases, the equilibrium begins to shift toward the RecA-mediated cleavage of UmuD to UmuD′. This cleavage reaction most likely occurs as a consequence of RecA altering the conformation of UmuD′s amino-terminal tail so as to bring the cleavage site of UmuD into close proximity with the active site (McDonald et al. 1998a). Initially, most of the UmuD′ reassociates with the more abundant UmuD protein to form a UmuD/D′ heterodimer that targets UmuD′ for proteolysis by ClpXP (Frank et al. 1996a) (not shown). If cellular damage is severe, more UmuD′ is generated than is degraded, and these will associate to form a stable UmuD′ homodimer (Frank et al. 1996b).
Materials and methods
Bacterial strains and plasmids
Four E. coli K-12 strains were utilized to investigate Lon-mediated degradation of UmuD in vivo. All four strains carry a lexA(Def) mutation (either the mis-sense lexA51(Def) allele or the Tn5 insertion allele, lexA71(Def)::Tn5), which results in constitutive expression of LexA-regulated genes (including the umu operon) in the absence of exogenous DNA damage. They also carry a deletion of the chromosomal umu operon (either ΔumuDC595::cat or ΔumuDC596::ermGT) that allows us to assay the stability of our various plasmid-encoded umuD mutants in the absence of contaminating chromosomally encoded UmuD. Strains EC10 [relevant genotype: recA+ lexA51(Def) ΔumuDC596::ermGT] and EC18 [recA+ lexA51(Def) ΔumuDC596::ermGT lon146::Tn10] (Frank et al. 1996a) were used to characterize the stability of the UmuD alanine-stretch mutants and the UmuD–PRP fusions. RW126 [recA718 srlC300::Tn10 lexA71(Def)::Tn5 ΔumuDC595::cat hisG4(Oc)] (Ho et al. 1993), harboring pRW274 (see below), was used to measure damage-induced Umu-dependent mutagenesis. To determine the ability of the various UmuD alanine-stretch mutants to undergo RecA-mediated cleavage to UmuD′, we utilized strain RW244 [recA730 srlC300::Tn10 lexA51(Def) ΔumuDC595::cat]. In a wild-type background, UmuD cleavage is normally damage inducible, but in certain coprotease-proficient recA mutants, like recA730 (Shinagawa et al. 1988; Woodgate and Ennis 1991), it is rendered constitutive.
All of the plasmid constructs described below express their respective protein from the natural LexA-regulated umu operon promoter–operator. Normally, expression of the Umu proteins is damage inducible, but as noted above, all of the strains used in this study carry lexA(Def) mutations, thereby resulting in damage-independent constitutive expression of the various Umu proteins.
Construction of all of the alanine-stretch mutants, except pKSD15/9, was accomplished by digesting the pBR322-derived UmuD-expressing plasmid pJM125 (McDonald et al. 1998b) with NdeI and SacII, and the resulting ∼90 bp fragment containing the 5′ end of the umuD gene was replaced with four annealed oligonucleotides (of ∼90 bp) corresponding to the multiple desired alanine changes (Fig. 3B). For the ease of identification, the DNA sequence coding for the 4 alanine substitutions also created a novel NotI restriction site in the umuD gene. pKSD15/9 was created by digesting pKSD15-4 with NotI and NdeI, and the ∼42 bp fragment was replaced with two annealed oligonucleotides (of ∼42 bp) that encoded the UmuD15/9 mutations. All of the UmuD alanine-stretch mutant plasmids were sequenced (Lark Technologies Inc., Houston, TX) to ensure that they did not contain any additional nucleotide changes.
The UmuD–PRP-expressing plasmid (pKSD–PRP) was constructed by cloning the appropriately modified ∼617-bp BclI–NotI fragment from pUT649 (Cayla, Toulouse, France) into the BsaBI-digested plasmid pJM125 (McDonald et al. 1998b). The resultant plasmid pKSD-PRP is a pBR322-based vector expressing the amino-terminal 40 amino acids of UmuD fused to a 7-amino-acid linker followed by the entire PRP. pKSD26/15–PRP, which creates the alanine-stretch mutations in pKSD–PRP identical to those mutations in pKSD26/15, was constructed by ligating a ∼790-bp SacII–PvuI fragment from pKSD26/15 into the similarly digested pKSD–PRP vector.
pKSD10 (UmuDK97A) was constructed by cloning the ∼1 kb BglII fragment from pRW414 (McDonald et al. 1998a) into the BglII–BamHI-digested vector pRW362 (Frank et al. 1996b). pRW274 (umuC+) was constructed by digesting pRW134 (umuD′C+) (Ennis et al. 1995) with NcoI and the site blunt-ended with Pol I (Kf) so as to introduce a frameshift mutation in the religated umuD′ gene. Both pKSD10 and pRW274 are derived from the low-copy-number spectinomycin-resistance plasmid pGB2 (Churchward et al. 1984) and are compatible with the pBR322-derived plasmids described above.
Proteins
Purified Lon protease was a gift from Michael Maurizi (National Institutes of Health, Bethesda, MD). UmuD and UmuD′ were purified as described (Frank et al. 1993). Creatine kinase was purchased from Sigma (St. Louis, MO), and the α-chymotrypsin from bovine pancreas was purchased from Boehringer Mannheim (Indianapolis, IN).
In vitro protein degradation
Purified UmuD (12.5 μm) was incubated with Lon (0.5 μm) in buffer containing 50 mm Tris-HCl (pH 8.0), 10 mm MgCl2, 1 mm DTT, 50 mm creatine phosphate, and 80 μg/ml creatine kinase. In the reactions containing ATP the concentration of ATP was 4 mm. The reaction mixture was incubated at 37°C and a 25-μl aliquot was removed at the specified times and added to 4× SDS sample buffer (Sambrook et al. 1989). Samples were subsequently visualized by 17% SDS-PAGE, stained with Coomassie Blue R-250. Identical reaction conditions (UmuD or UmuD′ at 12.5 μm ± Lon 0.5 μm were employed when assaying Lon specificity except that only a 1 hr time point was assayed.
Limited α-chymotrypsin digests (10 μl) contained 65 μm protein substrate (either UmuD or UmuD′) and increasing concentrations of α-chymotrypsin (0, 5, 50, 250, and 500 ng final concentration) in a buffer containing 20 mm Tris-HCl (pH 7.5), 50 mm NaCl, 1 mm DTT, and 0.1 mm EDTA. The reactions were incubated at 25°C for 20 min and visualized by a 17% SDS–polyacrylamide gel stained with Coomassie blue.
Measurement of UmuD mutant stability
The stability of the UmuD alanine-stretch mutants, as well as UmuD fusions, was assayed as described previously (Frank et al. 1996b). Briefly, cells were grown in Luria–Bertani medium at 37°C until they reached early exponential phase. At time zero, 100 μg/ml chloramphenicol was added to the medium to block protein synthesis and a 1.5-ml aliquot was removed at the indicated times. Cells were harvested by centrifugation and the resulting cell pellet resuspended in 4× SDS sample buffer. Aliquots representing equal cell numbers were electrophoresed on 17% SDS–polyacrylamide gels. Proteins were transferred to an Immobilon P membrane (Millipore) and subsequently probed with polyclonal antibodies raised against UmuD/UmuD′ (Frank et al. 1996b). Visualization of the UmuD–PRP protein was performed using polyclonal antibodies to the extreme 24-amino-acid N-terminus of UmuD raised in rabbits by Covance Laboratories (Vienna, VA). Because of limited cross-reactivity of the UmuD antibodies with UmuD26/15-PRP, UmuD26/15-PRP was visualized using polyclonal anti-PRP antibodies (Cayla, Toulouse, France). The transferred proteins were visualized on Kodak Bio-MaxMR film using the Western light chemiluminescent assay (Tropix, Bedford, MA). Quantitation was performed on a ChemiImager 4000, low-light imaging system (Alpha Innotech Corporation, San Leandro, CA). All experiments were performed two to three times with no significant variation in results.
Ability of UmuD mutants to undergo RecA-mediated intermolecular cleavage
RecA-mediated cleavage of various UmuD alanine-stretch mutants was followed in strain RW244 essentially as described above. However, because this assay only determines the steady-state level of the UmuD (or UmuD′) protein, no chloramphenicol was added to the reaction and only one time point was taken at time zero.
The ability of the various alanine-stretch mutants to act as an enzyme and promote intermolecular UmuD cleavage was assayed in strain RW244/pKSD10 (UmuDK97A). UmuDK97A has a mutation at the active site of UmuD that inactivates its ability to function as an enzyme in the intermolecular cleavage reaction. It does, however, still posses a functional cleavage site so that it can serve as a substrate if an active UmuD (or UmuD′) enzyme is provided in trans (McDonald et al. 1998a).
Ability of the UmuD mutants to promote damage-inducible mutagenesis
The ability of the various plasmid-encoded UmuD mutants to function in SOS mutagenesis was assayed in strain RW126/pRW274. This strain carries a chromosomal deletion of the entire umu operon, but cellular mutagenesis can be restored in trans by introducing compatible plasmids expressing UmuC (pRW274) and UmuD (UmuD′). Briefly, bacterial cultures were grown overnight in LB medium containing the appropriate antibiotics. A 1.0-ml aliquot was centrifuged and resuspended in an equal volume of SM buffer (Sambrook et al. 1989). The ability of particular plasmid bearing strains to promote Umu-dependent SOS mutator activity was judged by plating a 100-μl aliquot on Davis and Mingioli minimal agar plates containing a trace amount of histidine (1 μg/ml) (Ho et al. 1993). A small sterile disk was placed in the center of the plate, and 5 μl of a 1:5 dilution of methylmethane sulfonate (MMS) (Sigma, St Louis, MO) in dimethylsulfoxide (Sigma, St Louis, MO) was added to the disk. MMS-induced His+ mutants were scored after 4 days of incubation at 37°C. The results represent the average number of His+ colonies from at least three cultures from each strain, with six plates per culture.
Acknowledgments
M.G. dedicates this paper to the memory of Roy Scott. We thank Michael Maurizi for the generous gift of purified Lon protein and helpful advice. We also thank the members of the laboratory for helpful comments and discussion throughout the course of this work and Agnès Tissier for designing Figure 7. This work was performed while the M.G. held an National Institutes of Health/National Research Council Section on DNA Replication, Repair, and Mutagenesis Research Associateship.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL woodgate@helix.nih.gov; FAX (301) 594-1135.
References
- Battista JR, Ohta T, Nohmi T, Sun W, Walker GC. Dominant negative umuD mutations decreasing RecA-mediated cleavage suggest roles for intact UmuD in modulation of SOS mutagenesis. Proc Natl Acad Sci. 1990;87:7190–7194. doi: 10.1073/pnas.87.18.7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie JU, Sauer RT. Identification of C-terminal extensions that protect proteins from intracellular proteolysis. J Biol Chem. 1989;264:7596–7602. [PubMed] [Google Scholar]
- Burckhardt SE, Woodgate R, Scheuermann RH, Echols H. UmuD mutagenesis protein of Escherichia coli: Overproduction, purification and cleavage by RecA. Proc Natl Acad Sci. 1988;85:1811–1815. doi: 10.1073/pnas.85.6.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchward G, Belin D, Nagamine Y. A pSC101-derived plasmid which shows no sequence homology to other commonly used cloning vectors. Gene. 1984;31:165–171. doi: 10.1016/0378-1119(84)90207-5. [DOI] [PubMed] [Google Scholar]
- Dumas P, Bergdoll M, Cagnon C, Masson JM. Crystal structure and site-directed mutagenesis of a bleomycin resistance protein and their significance for drug sequestering. EMBO J. 1994;13:2483–2492. doi: 10.1002/j.1460-2075.1994.tb06535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennis DG, Levine AS, Koch WH, Woodgate R. Analysis of recA mutants with altered SOS functions. Mutat Res. 1995;336:39–48. doi: 10.1016/0921-8777(94)00045-8. [DOI] [PubMed] [Google Scholar]
- Ferentz AE, Opperman T, Walker GC, Wagner G. Dimerization of the UmuD′ protein in solution and its implications for regulation of SOS mutagenesis. Nat Struct Biol. 1997;4:979–983. doi: 10.1038/nsb1297-979. [DOI] [PubMed] [Google Scholar]
- Frank EG, Hauser J, Levine AS, Woodgate R. Targeting of the UmuD, UmuD′ and MucA′ mutagenesis proteins to DNA by RecA protein. Proc Natl Acad Sci. 1993;90:8169–8173. doi: 10.1073/pnas.90.17.8169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank EG, Ennis DG, Gonzalez M, Levine AS, Woodgate R. Regulation of SOS mutagenesis by proteolysis. Proc Natl Acad Sci. 1996a;93:10291–10296. doi: 10.1073/pnas.93.19.10291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank EG, Gonzalez M, Ennis DG, Levine AS, Woodgate R. In vivo stability of the Umu mutagenesis proteins: A major role for RecA. J Bacteriol. 1996b;178:3550–3556. doi: 10.1128/jb.178.12.3550-3556.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W. DNA repair and mutagenesis. Washington, D.C.: American Society for Microbiology; 1995. [Google Scholar]
- Gatignol A, Durand H, Tiraby G. Bleomycin resistance conferred by a drug-binding protein. FEMS Lett. 1988;230:171–175. doi: 10.1016/0014-5793(88)80665-3. [DOI] [PubMed] [Google Scholar]
- Gonzalez M, Frank EG, McDonald JP, Levine AS, Woodgate R. Structural insights into the regulation of SOS mutagenesis. Acta Biochim Polon. 1998;45:163–172. [PubMed] [Google Scholar]
- Gottesman S. Proteases and their targets in Escherichia coli. Annu Rev Genet. 1996;30:465–506. doi: 10.1146/annurev.genet.30.1.465. [DOI] [PubMed] [Google Scholar]
- Gottesman S, Clark WP, Maurizi MR. The ATP-dependent Clp protease of Escherichia coli. Sequence of clpA and identification of a Clp-specific substrate. J Biol Chem. 1990;265:7886–7893. [PubMed] [Google Scholar]
- Gottesman S, Roche E, Zhou Y, Sauer RT. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes & Dev. 1998;12:1338–1347. doi: 10.1101/gad.12.9.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzzo A, Lee MH, Oda K, Walker GC. Analysis of the region between amino acids 30 and 42 of intact UmuD by a monocysteine approach. J Bacteriol. 1996;178:7295–7303. doi: 10.1128/jb.178.24.7295-7303.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman C, Thevenet D, Bouloc P, Walker GC, D’Ari R. Degradation of carboxy-terminal-tagged cytoplasmic proteins by the Escherichia coli protease HflB (FtsH) Genes & Dev. 1998;12:1348–1355. doi: 10.1101/gad.12.9.1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashitani A, Ishii Y, Kato Y, Koriuchi K. Functional dissection of a cell-division inhibitor, SulA, of Escherichia coli and its negative regulation by Lon. Mol & Gen Genet. 1997;254:351–357. doi: 10.1007/s004380050426. [DOI] [PubMed] [Google Scholar]
- Ho C, Kulaeva OI, Levine AS, Woodgate R. A rapid method for cloning mutagenic DNA repair genes: Isolation of umu-complementing genes from multidrug resistance plasmids R391, R446b, and R471a. J Bacteriol. 1993;175:5411–5419. doi: 10.1128/jb.175.17.5411-5419.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisman O, D’Ari R. An inducible DNA replication-cell division coupling mechanism in E. coli. Nature. 1981;290:797–799. doi: 10.1038/290797a0. [DOI] [PubMed] [Google Scholar]
- Jonczyk P, Nowicka A. Specific in vivo protein-protein interactions between Escherichia coli SOS mutageneis proteins. J Bacteriol. 1996;178:2580–2585. doi: 10.1128/jb.178.9.2580-2585.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiler KC, Silber KR, Downard KM, Papayannopoulos IA, Biemann K, Sauer RT. C-terminal specific protein degradation: Activity and substrate specificity of the Tsp protease. Protein Sci. 1995;4:1507–1515. doi: 10.1002/pro.5560040808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keiler KC, Waller PR, Sauer RT. Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science. 1996;271:990–993. doi: 10.1126/science.271.5251.990. [DOI] [PubMed] [Google Scholar]
- Koch WH, Woodgate R. The SOS response. In: Nickoloff JA, Hoekstra MF, editors. DNA damage and repair: DNA repair in prokaryotes and lower eukaryotes. Totowa, NJ: Humana Press; 1998. pp. 107–134. [Google Scholar]
- Kulaeva OI, Wootton JC, Levine AS, Woodgate R. Characterization of the umu-complementing operon from R391. J Bacteriol. 1995;177:2737–2743. doi: 10.1128/jb.177.10.2737-2743.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levchenko I, Luo L, Baker TA. Disassembly of the Mu transposase tetramer by the ClpX chaperone. Genes & Dev. 1995;9:2399–2408. doi: 10.1101/gad.9.19.2399. [DOI] [PubMed] [Google Scholar]
- Levchenko I, Yamauchi M, Baker TA. ClpX and MuB interact with overlapping regions of Mu transposase: Implications for control of the transposition pathway. Genes & Dev. 1997;11:1561–1572. doi: 10.1101/gad.11.12.1561. [DOI] [PubMed] [Google Scholar]
- Maurizi MR. Degradation in vitro of bacteriophage lambda N protein by Lon protease from Escherichia coli. J Biol Chem. 1987;262:2696–2703. [PubMed] [Google Scholar]
- McDonald JP, Frank EG, Levine AS, Woodgate R. Intermolecular cleavage of the UmuD-like mutagenesis proteins. Proc Natl Acad Sci. 1998a;95:1478–1483. doi: 10.1073/pnas.95.4.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JP, Maury EE, Levine AS, Woodgate R. Regulation of UmuD cleavage: Role of the amino-terminal tail. J Mol Biol. 1998b;282:721–730. doi: 10.1006/jmbi.1998.2044. [DOI] [PubMed] [Google Scholar]
- Mizusawa S, Gottesman S. Protein degradation in Escherichia coli: The lon gene controls the stability of sulA protein. Proc Natl Acad Sci. 1983;80:358–362. doi: 10.1073/pnas.80.2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohmi T, Battista JR, Dodson LA, Walker GC. RecA-mediated cleavage activates UmuD for mutagenesis: Mechanistic relationship between transcriptional derepression and posttranslational activation. Proc Natl Acad Sci. 1988;85:1816–1820. doi: 10.1073/pnas.85.6.1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsell DA, Silber KR, Sauer RT. Carboxy-terminal determinants of intracellular protein degradation. Genes & Dev. 1990;4:277–286. doi: 10.1101/gad.4.2.277. [DOI] [PubMed] [Google Scholar]
- Peat TS, Frank EG, McDonald JP, Levine AS, Woodgate R, Hendrickson WA. Structure of the UmuD′ protein and its regulation in response to DNA damage. Nature. 1996a;380:727–730. doi: 10.1038/380727a0. [DOI] [PubMed] [Google Scholar]
- Peat TS, Frank EG, McDonald JP, Levine AS, Woodgate R, Hendrickson WA. The UmuD′ protein filament and its potential role in damage induced mutagenesis. Structure. 1996b;4:1401–1412. doi: 10.1016/s0969-2126(96)00148-7. [DOI] [PubMed] [Google Scholar]
- Rajagopalan M, Lu C, Woodgate R, O’Donnell M, Goodman MF, Echols H. Activity of the purified mutagenesis proteins UmuC, UmuD′ and RecA in replicative bypass of an abasic DNA lesion by DNA polymerase III. Proc Natl Acad Sci. 1992;89:10777–10781. doi: 10.1073/pnas.89.22.10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuven NB, Tomer G, Livneh Z. The mutagenesis proteins UmuD′ and UmuC prevent lethal frameshifts while increasing base substitution mutations. Mol Cell. 1998;2:191–199. doi: 10.1016/s1097-2765(00)80129-x. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- Shinagawa H, Iwasaki H, Kato T, Nakata A. RecA protein-dependent cleavage of UmuD protein and SOS mutagenesis. Proc Natl Acad Sci. 1988;85:1806–1810. doi: 10.1073/pnas.85.6.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BT, Walker GC. Mutagenesis and more: umuDC and the Escherichia coli SOS response. Genetics. 1998;148:1599–1610. doi: 10.1093/genetics/148.4.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer S, Boudsocq F, Devoret R, Bailone A. Specific RecA amino acid changes affect RecA-UmuD′C interaction. Mol Microbiol. 1998;28:281–291. doi: 10.1046/j.1365-2958.1998.00803.x. [DOI] [PubMed] [Google Scholar]
- Sonezaki S, Ishii Y, Okita K, Sugino T, Kondo A, Kato Y. Overproduction and purification of SulA fusion protein in Escherichia coli and its degradation by Lon protease in vitro. Appl Microbiol Biotechnol. 1995;43:304–309. doi: 10.1007/BF00172829. [DOI] [PubMed] [Google Scholar]
- Stout V, Torres-Cabassa A, Maurizi MR, Gutnick D, Gottesman S. RcsA, an unstable positive regulator of capsular polysaccharide synthesis. J Bacteriol. 1991;173:1738–1747. doi: 10.1128/jb.173.5.1738-1747.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M, Bruck I, Eritja R, Turner J, Frank EG, Woodgate R, O’Donnell M, Goodman MF. Biochemical basis of SOS-induced mutagenesis in Escherichia coli: Reconstitution of in vitro lesion bypass dependent on the UmuD′2C mutagenic complex and RecA. Proc Natl Acad Sci. 1998;95:9755–9760. doi: 10.1073/pnas.95.17.9755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobias JW, Shrader TE, Rocap G, Varshavsky A. The N-end rule in bacteria. Science. 1991;254:1374–1377. doi: 10.1126/science.1962196. [DOI] [PubMed] [Google Scholar]
- Torres-Cabassa AS, Gottesman S. Capsule synthesis in Escherichia coli K-12 is regulated by proteolysis. J Bacteriol. 1987;169:981–989. doi: 10.1128/jb.169.3.981-989.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu GF, Reid GE, Zhang JG, Moritz RL, Simpson RJ. C-terminal extension of truncated recombinant proteins in Escherichia coli with a 10Sa RNA decapeptide. J Biol Chem. 1995;270:9322–9326. doi: 10.1074/jbc.270.16.9322. [DOI] [PubMed] [Google Scholar]
- Van Melderen L, Thi MHD, Lecchi P, Gottesman S, Couturier M, Maurizi MR. ATP-dependent degradation of CcdA by Lon protease. Effects of secondary structure and heterologous subunit interactions. J Biol Chem. 1996;271:27730–27738. doi: 10.1074/jbc.271.44.27730. [DOI] [PubMed] [Google Scholar]
- Woodgate R, Ennis DG. Levels of chromosomally encoded Umu proteins and requirements for in vivo UmuD cleavage. Mol & Gen Genet. 1991;229:10–16. doi: 10.1007/BF00264207. [DOI] [PubMed] [Google Scholar]
- Woodgate R, Levine AS. Damage inducible mutagenesis: Recent insights into the activities of the Umu family of mutagenesis proteins. In: Lindahl T, editor. Cancer surveys: Genetic instability in cancer. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1996. pp. 117–140. [PubMed] [Google Scholar]
- Woodgate R, Rajagopalan M, Lu C, Echols H. UmuC mutagenesis protein of Escherichia coli: Purification and interaction with UmuD and UmuD′. Proc Natl Acad Sci. 1989;86:7301–7305. doi: 10.1073/pnas.86.19.7301. [DOI] [PMC free article] [PubMed] [Google Scholar]