

Abstract
Clinical studies consistently demonstrate that a single sub-psychomimetic dose of ketamine, an ionotropic glutamatergic n-methyl-d-aspartate receptor (NMDAR) antagonist, produces fast-acting antidepressant responses in patients suffering from major depressive disorder (MDD), although the underlying mechanism is unclear1-3. Depressed patients report alleviation of MDD symptoms within two hours of a single low-dose intravenous infusion of ketamine with effects lasting up to two weeks1-3, unlike traditional antidepressants (i.e. serotonin reuptake inhibitors), which take weeks to reach efficacy. This delay is a major drawback to current MDD therapies, leaving a need for faster acting antidepressants particularly for suicide-risk patients3. Ketamine's ability to produce rapidly acting, long-lasting antidepressant responses in depressed patients provides a unique opportunity to investigate underlying cellular mechanisms. We show that ketamine and other NMDAR antagonists produce fast-acting behavioural antidepressant-like effects in mouse models that depend on rapid synthesis of brain-derived neurotrophic factor (BDNF). We find that ketamine-mediated NMDAR blockade at rest deactivates eukaryotic elongation factor 2 (eEF2) kinase (also called CaMKIII) resulting in reduced eEF2 phosphorylation and desuppression of BDNF translation. Furthermore, we find inhibitors of eEF2 kinase induce fast-acting behavioural antidepressant-like effects. Our findings suggest that protein synthesis regulation by spontaneous neurotransmission may serve as a viable therapeutic target for fast-acting antidepressant development.
Keywords: antidepressant, BDNF, protein translation, animal model, ketamine
We examined ketamine's acute effect in C57BL/6 mice (WTs) and detected significant behavioural responses in antidepressant (AD)-predictive tasks including forced swim (FST), novelty-suppressed feeding (NSF), and learned helplessness (LH) (Supplementary Fig. 1a-e, 2a-c)4. Ketamine also produced such responses in sucrose consumption test (SCT), NSF, and FST after chronic mild stress, an animal model of depression (Supplementary Fig. 1f-i). To elucidate mechanisms underlying ketamine's fast-acting AD action, we focused on FST, a test predictive of non-monoaminergic AD efficacy4. We examined the time-course of behavioural AD effects in WT mice following single, low dose treatment with ketamine, MK801, or CPP (Fig. 1 a-c). After 30 minutes or 3 hours, each NMDAR antagonist significantly reduced immobility in FST compared to vehicle-treated animals, suggesting NMDAR blockade produces fast-acting AD responses. Notably, in our system, acute conventional AD treatment did not produce AD-like FST responses (Supplementary Fig. 3), which may require multiple doses5. Effects of ketamine and CPP, but not MK-801, persisted for 24 hours4. Ketamine's behavioural effect lasted for one week. Acute NMDAR antagonist treatment produced no alterations in hippocampal-dependent learning (Supplementary Fig. 1d) or locomotor activity (Supplementary Fig. 4). These drugs have short half-lives (∼2-3 hours)6-8 suggesting sustained NMDAR antagonist-induced AD responses are due to synaptic plasticity, not persistent receptor blockade.
Figure 1. Time-course of NMDAR antagonist-mediated antidepressant-like behavioural effects.

Mean immobility±SEM of C57BL/6 mice in FST following acute treatment of ketamine, CPP or MK801. Independent groups of mice were used at each time point and drug treatment to avoid behavioural habituation. ANOVA analysis F3,27 =30.31, P<.0001 for treatment groups, F3,27 =19.06, P<.0001 for duration of response, and F9,81=9.32, P<.0001 for treatment-duration interaction; therefore, we examined treatment effects by time-point. a, Ketamine (3.0 mg/kg) significantly reduced immobility, suggestive of an AD-like response, at 30-min., 3-hrs, 24-hrs, and 1-week compared to vehicle treatment (*P<0.05). b, CPP (0.5 mg/kg) significantly reduced immobility at 30-min., 3-hrs, and 24-hrs (*P<0.05) compared to vehicle treatment. c, MK-801 (0.1 mg/kg) produced significant decreases in immobility at 30-min and 3-hrs compared to vehicle treatment (*P<0.05), (n=10/group/time-point). Here and in all figures error bars represent Standard Error of the Mean (SEM).
Brain-derived neurotrophic factor (BDNF) is linked to traditional AD action; BDNF expression is increased in hippocampus (HC) by ADs9 and BDNF deletion in HC attenuates AD behavioural responses10-12. Moreover, intraventricular or intrahippocampal BDNF infusion causes rapid, sustained AD-like effects lasting 3-6 days in FST13,14. To examine whether ketamine's AD-like response is mediated through BDNF, we administered ketamine to inducible BDNF knockout (KO) mice10, then observed FST behaviour. After 30 minutes, ketamine-treated wild-type littermate controls (CTLs) displayed significant reductions in immobility, indicating AD-like responses compared to vehicle-treated CTLs (Fig. 2a). However, ketamine did not produce AD-like effects in BDNF KOs, suggesting fast-acting AD responses require BDNF. After 24 hours, ketamine significantly reduced immobility in CTLs, but not in BDNF KOs (Fig. 2a), indicating ketamine's sustained effects depend on BDNF. To validate this link between NMDAR antagonists and BDNF-mediated AD responses, MK-801 was administered to BDNF KOs or CTLs. After 30 minutes, MK-801 significantly reduced FST immobility in CTLs, but had no effect in BDNF KOs (Supplementary Fig. 6). After 24 hours, MK-801 did not affect FST behaviour (Supplementary Fig. 6), as previously demonstrated (Fig. 1c). We next generated postnatal conditional15 TrkB KOs and found that they are insensitive to ketamine's AD-like effects in FST and NSF (Supplementary Fig. 5a,b). To confirm TrkB engagement, we examined receptor autophosphorylation and found increased TrkB activation after NMDAR antagonist treatment (Supplementary Fig. 5c).
Figure 2. BDNF translation in antidepressant effects of NMDAR antagonists.
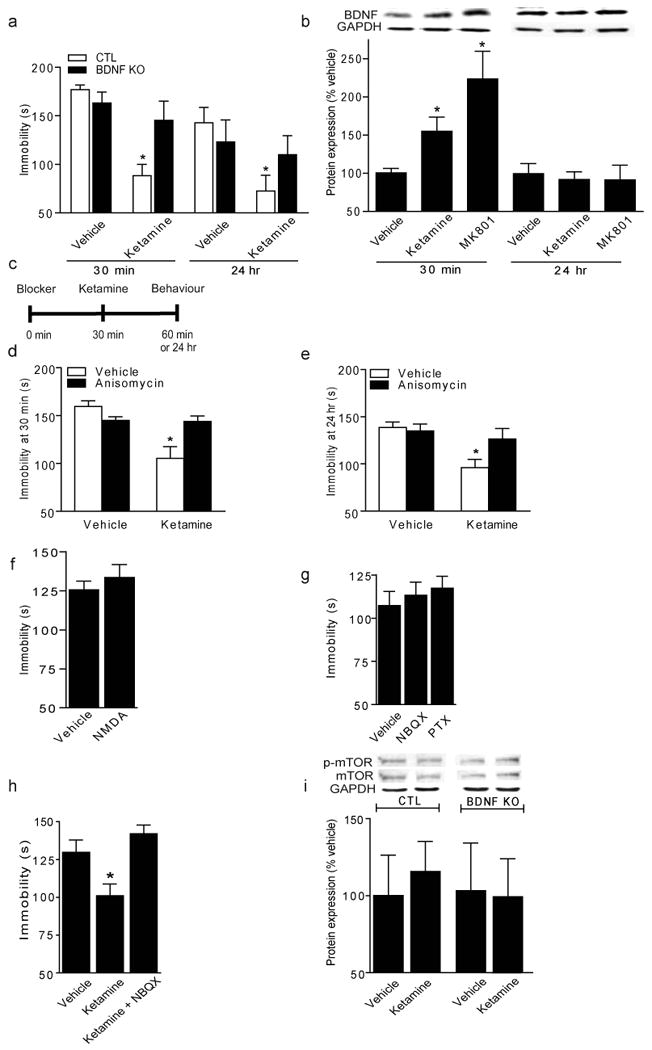
a, Immobility in FST following acute ketamine (3.0 mg/kg). At 30-min, ANOVA F1,35=17.13, P=0.0002 for drug, F1,35=7.57, P=0.0093 for genotype-drug interaction, multiple comparisons with t-test (*P<0.05). At 24-hr, in a separate cohort, ANOVA F1,29=3.77, P=0.0619 for treatment, multiple comparisons (*P<0.05) (n=7-12/group). b, Densitometric analysis of BDNF (normalized-GAPDH) in hippocampus following ketamine (3.0 mg/kg) or MK801 (0.1 mg/kg). 30-min., ANOVA F2,12=6.77, P=0 .0108 for treatment, Bonferroni post-hoc test *P<0.05. 24-hrs, no significance (n=5-6/group). c, Anisomycin and actinomycinD protocol. d, Immobility at 30-min., ANOVA F1,34=11.83, P=0.0016 for treatment and F1, 34 = 10.91, P =0.0023 for treatment-inhibitor interaction, multiple comparisons (*P<0.05)(n=8-10/group). e, Immobility at 24 hrs, ANOVA F1, 31=9.34, P =0.0046 for treatment, multiple comparisons (*P<0.05)(n=8-10/group). f, Immobility of WTs given vehicle or NMDA (75 mg/kg), tested 30-minutes later in FST. g, Immobility of WTs given NBQX (10 mg/kg) or PTX (1.0 mg/kg), tested 30-minutes later in FST. h, Immobility of WTs given vehicle, ketamine (3.0 mg/kg), ketamine+NBQX (10 mg/kg), tested 30-minutes later in FST. ANOVA F2,26=8.226, P<0.0019, Bonferroni post-hoc analysis shows ketamine effect reversed by NBQX, *P<0.05. i, Densitometric analysis of p-mTOR (normalized-mTOR) in hippocampus 30-minutes after vehicle or ketamine.
To determine if NMDAR antagonists alter BDNF expression in HC, WTs were acutely treated with vehicle, ketamine, or MK-801. Quantitative RT-PCR analysis of exon IX coding region demonstrated BDNF mRNA expression was unaltered by ketamine or MK-801 at 30 minutes or 24 hours (Supplementary Fig. 7a). Contrastingly, Western blot analysis and ELISA methodology revealed significantly increased BDNF protein at 30 minutes but not 24 hours after NMDAR antagonist treatment (Fig. 2b, Supplementary Fig. 7b). Moreover, ketamine's acute effects on BDNF extended to precursor proBDNF (Supplementary Fig. 7c). These data suggest rapid increases in BDNF protein translation, not transcription, are necessary for fast-onset AD responses. However, continued BDNF protein up-regulation does not underlie ketamine's long-term behavioural effects.
To study translation or transcription in ketamine's AD-like effects, we examined FST behaviour with the protein synthesis inhibitor anisomycin16 or RNA polymerase inhibitor actinomycin D (ActD)17 which block respective processes by ∼80% within two hours. We pre-treated mice with anisomycin or ActD before ketamine (Fig. 2c). Anisomycin prevented ketamine-induced rapid behavioural responses (30 minutes) in FST and NSF paradigms, suggesting dependence on new protein synthesis (Fig. 2d; Supplementary Fig. 8a,b). Anisomycin also prevented ketamine's long-term FST effect (24 hours), suggesting rapid protein translation was involved in sustained AD-like responses (Fig. 2e). We found both mature and proBDNF synthesis in HC were sensitive to anisomycin treatment (Supplementary Fig. 8c,d). However, ActD did not impact ketamine's AD-like FST behaviour at either time-point, suggesting independence of new gene expression (Supplementary Fig. 9b,c). To confirm that ActD crossed the blood-brain barrier, we examined BDNF mRNA expression in drug-treated animals and found decreased BDNF transcription in HC (Supplementary Fig. 9a). Taken together, these findings suggest rapid, transient BDNF translation is required for ketamine's fast-acting and long-lasting AD-like behavioural effects and that long-term AD responses may be due to synaptic plasticity alterations initiated by transient increases in BDNF translation.
We observed increased BDNF protein in cortex but not nucleus accumbens 30 minutes following actute ketamine or MK801 administration (Supplementary Fig. 10a,b). We further investigated whether NMDAR antagonism affected proteins other than BDNF. We found activity-regulated cytoskeletal (Arc) protein is increased in HC (sensitive to anisomycin treatment; Supplementary Fig. 8e) but not Homer, GluR1, or s6 kinase phosphorylation (Supplementary Fig. 10c-f). Additionally, these proteins remain unaltered in cortex following acute NMDAR antagonist treatment (Supplementary Fig. 11a-e).
Synaptic plasticity and ensuing learning processes are often mediated by NMDAR activation-driven protein translation, but AD-like effects require NMDAR blockade-induced protein translation. To resolve this paradox, we turned to recent evidence that NMDAR blockade by MK-801 or AP5, without neuronal activity, augments protein synthesis through eukaryotic elongation factor 2 (eEF2) dephosphorylation (activation), a critical catalytic factor for ribosomal translocation during protein synthesis18. In this model, resting NMDAR activity causes sustained eEF2 kinase (eEF2K, or CamKIII) activation, which phosphorylates eEF2, effectively halting translation whereas acute NMDAR blockade at rest attenuates eEF2 phosphorylation allowing target transcript translation.
To evaluate this model, we tested whether excess synaptic glutamate possibly elicited by NMDAR blockade was responsible for ketamine's behavioural effects. Acute NMDA administration did not alter FST behaviour (Fig. 2f) as previously demonstrated19 but increased Arc expression (Supplementary Fig. 10i), suggesting excess glutamate does not elicit rapid behavioural AD effects. To define the role of neuronal activity in AD behavioural effects, we tested whether NBQX, an AMPA channel blocker that reduces neuronal activity, or picrotoxin (PTX), a GABA channel blocker that increases activity, impacted FST behaviour4,20. Acute systemic treatment with these drugs did not affect FST behaviour (Fig. 2g), or BDNF synthesis, though PTX enhanced Arc expression in HC (Supplementary Fig. 10 g,h). However, when co-applied with ketamine, NBQX abolished behavioural FST AD-like responses (Fig. 2h) as previously described4. These data suggest behavioural AD effects are not elicited by alterations in evoked neurotransmission, but require ketamine-mediated augmentation of AMPA-receptor activation.
Recent evidence suggests cortical mTOR signalling underlies ketamine-mediated AD responses21. We investigated whether ketamine's rapid behavioural AD effects required mTOR activation, and if this signalling was downstream of BDNF. Regulation of phosphorylated mTOR was not detected after acute ketamine administration in CTL or BDNF KO hippocampal tissue (Fig. 2i) or WT cortex tissue (Supplementary Fig. 11d). In earlier work, rapamycin prevented ketamine-mediated antidepressant responses; however, the link between rapamycin and AD-like effects is equivocal22. We tested whether rapamycin pre-treatment could block acute ketamine-mediated FST behaviour. Thirty minutes after ketamine administration, WTs show AD responses unaffected by rapamycin treatment (Supplementary Fig. 11h). Rapamycin reduced s6 kinase phosphorylation in cortex and HC (Supplementary Fig. 11 f,g) suggesting brain tissue penetration. The earlier study examined molecular effects 2 hours or behavioural effects 24 hours after drug treatment21, therefore mTOR's role in ketamine's AD effect may be maintenance rather than rapid induction.
To determine whether ketamine inhibits spontaneous miniature NMDA-receptor mediated currents (NMDA-mEPSC)23,24 at rest and regulates eEF2 phosphorylation, we tested its impact on hippocampal neurons in vitro. After ketamine perfusion (1, 5, or 50 μM), we recorded NMDA-mEPSCs (Fig. 3c) and within minutes detected a significant decrease in NMDA-mEPSCs similar to AP523. Moreover, protein extracts from ketamine-treated neurons revealed decreased eEF2 phosphorylation (peEF2) compared to vehicle-treated cultures, suggesting ketamine, in the absence of neuronal activity, dose-dependently leads to eEF2 de-phosphorylation, permitting protein synthesis (Fig. 3a,b). Additionally, we evaluated ketamine's effects on hippocampal field potentials. Acute ketamine application (20 uM at rest) potentiated subsequent evoked synaptic responses in hippocampal slices (Fig. 3e), further suggesting increased AMPA-mediated neurotransmission underlies ketamine's AD-like behavioural effects consistent with findings on BDNF- and protein synthesis-dependent synaptic plasticity25.
Figure 3. Ketamine blocks NMDAR spontaneous activity, reduces the level of eEF2 phosphorylation, and strengthens synaptic responses.
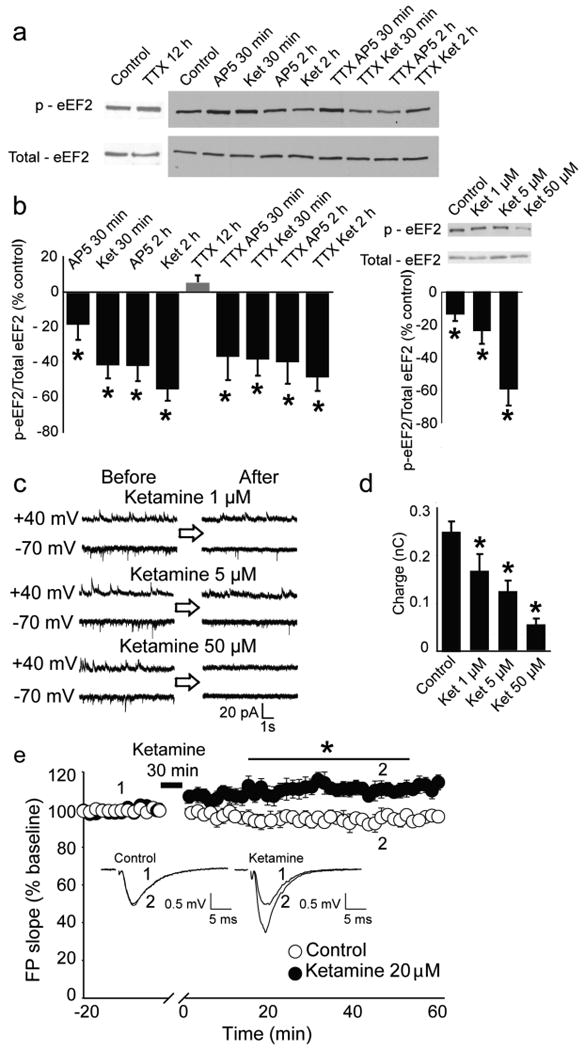
a, Representative western blots from hippocampal primary cultures. b, (left) Densitometric analysis of peEF2 (normalized-total eEF2). Data expressed as mean percentage±SEM. TTX alone does not alter peEF2 while AP5 or ketamine, with or without TTX, significantly decreases peEF2 as assessed by t-test analysis (*P<0.05). (right) Application of 1, 5 and 50 μM of ketamine produces dose-dependent decreases in peEF2 assessed by t-test analysis (*P<0.05). c, Representative traces of NMDAR spontaneous activity after application of 1, 5 and 50 μM. d, Quantification of charge transfer (10 sec) reveals significant effects (*P<0.05) for all ketamine concentrations compared to control (n=6-16) assessed by t-test analysis (*P<0.05). e, Field potential slopes are plotted as a function of time. Representative field potential traces, (average 2-min) are shown during baseline (1) and at 45-min (2). The asterisk refers to significantly different field potentials values (*P<0.05). For statistical analysis we used two-way repeated ANOVA with Bonferroni post-hoc analysis. The drug-time interaction was significant (F143,1430=6.723 P<0.001).
To examine whether fast-acting AD response is mediated via eEF2, we administered ketamine or MK801 to WTs and analyzed eEF2 phosphorylation. Within 30 minutes, ketamine and MK801 lead to rapid decreases in peEF2 in HC (Fig. 4a-c; Supplementary Fig. 12,13), detected by immunostaining and Western blot analysis (Fig. 4d). However, cortical peEF2 levels were unaltered with acute NMDAR antagonist treatment (Supplementary Fig. 11f).
Figure 4. Rapid antidepressant-like behaviour mediated by decreased p-eEF2 and increased BDNF translation.
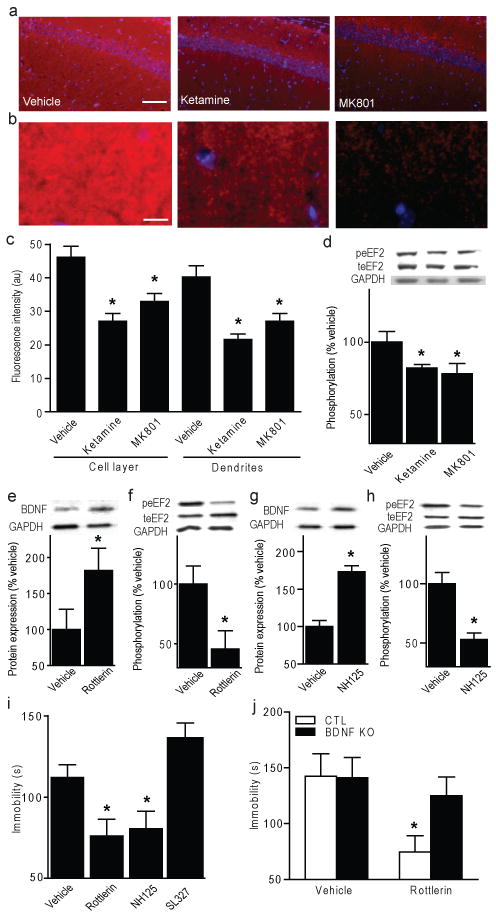
a, Images of CA1 pyramidal and stratum radiatum layers after acute vehicle, ketamine, or MK-801; scale bar=100 μm (red: peEF2, blue: DAPI). b, Stratum radiatum magnification; scale bar=20 μm. c, ImageJ analysis of average fluorescence intensity. ANOVA cell layer F2,23=13.13, P=0.0002 for treatment, dendrites F2,23=14.06, P=0.0001 for treatment (n=4/group). d, Densitometric analysis of peEF2 (normalized-total eEF2) in hippocampus after NMDAR antagonist. ANOVA F2,23=3.183, P=0.03 for treatment (n=8/group). e-h, Densitometric analysis. e, g, Significant increases in hippocampal BDNF protein (normalized-GAPDH) with rottlerin (5.0 mg/kg) versus vehicle (t-test *P<0.05), and NH125 (5.0 mg/kg) versus vehicle (*P<0.05). f, h, Significant decreases in peEF2 (normalized-total eEF2) versus vehicle (*P<0.05) and NH125 versus vehicle (t-test *P<0.05). i, Immobility in FST of WTs given acute rottlerin (5.0 mg/kg) or NH125 (5.0 mg/kg). ANOVA F3, 44= 8.13, P=0.0002 for treatment, Bonferroni post-hoc analysis shows significance with rottlerin or NH125 versus vehicle (*P<0.05), but not ERK inhibitor SL327 (10mg/kg). j, Immobility of BDNF KO or littermate CTLs given acute rottlerin (5.0 mg/kg), tested 30-minutes later in FST. ANOVA F1, 19=5.77, P=0.0267 for treatment, Bonferroni post-hoc analysis for rottlerin versus vehicle CTLs (*P<0.05) (n=5-7/group).
To examine whether eEF2K inhibition alters BDNF protein expression in vivo, WTs were administered eEF2K inhibitors, rottlerin or NH125, then sacrificed 30 minutes later. Rottlerin and NH125 produced significantly increased BDNF protein expression (Fig. 4e, g), with corresponding significant peEF2 decreases in HC (Fig. 4f, h). To directly assess whether eEF2K inhibition is sufficient to mediate fast-acting AD-like responses, WTs were administered rottlerin or NH125 and examined in FST. Both rottlerin and NH125 produced significant decreases in FST immobility at 30-minutes (Fig. 4i), a time-scale similar to NMDAR antagonists' effects, suggesting fast-acting behavioural effects are mediated through eEF2K inhibition. To test whether extracellular-related kinase (ERK), a regulator of protein translation during neural activity, impacts FST behaviour, we treated WTs with inhibitor SL327. This treatment reduced ERK phosphorylation in HC tissue (Supplementary Fig. 10j), but did not affect FST behaviour (Fig. 4i), suggesting AD-like effects are specific to eEF2K inhibition during resting spontaneous glutamatergic signalling. We found that an acute dose of rottlerin or NH125 did not affect locomotor activity, but AD-related behavioural effects were long-lasting (Supplementary Fig. 14a-f). To validate that AD effects following eEF2K inhibition were mediated through BDNF, we administered rottlerin to BDNF KOs and tested FST behaviour. Like NMDAR antagonists, rottlerin is ineffective in BDNF KOs, demonstrating a requirement for increased BDNF expression upon eEF2K inhibition to produce AD-like behavioural responses (Fig. 4j).
In summary, our data support the hypothesis that ketamine produces rapidly acting AD-like behavioural effects through inhibition of spontaneous NMDA-mEPSCs, leading to decreased eEF2 kinase activity, thus permitting rapid increases in BDNF translation (Supplementary Fig. 15) which may in turn exert strong influences on pre- or postsynaptic efficacy26,27. We found that fast-acting AD-like effects cannot be elicited by disinhibition of behavioural circuitry, or by evoked neurotransmission, but must rely on enhanced neurotransmission following NMDAR antagonist-induced plasticity occurring at rest18. The observation of behavioural effects mediated through spontaneous neurotransmission provides the first evidence that tonic resting neurotransmission is involved in behaviour, and supports the notion that spontaneous and evoked forms of glutamatergic signalling are segregated18,23,28,29. These data demonstrate that eEF2K inhibition, resulting in de-suppression of protein translation, is sufficient to produce AD-like effects, implicating eEF2K inhibitors as potential novel MDD treatments with rapid onset. Moreover, our results show that synaptic translational machinery may serve as a viable therapeutic target for development of faster acting antidepressants.
Methods Summary
Behavioural studies were performed using adult male wild type C57BL/6 or mutant strains maintained as previously described10,15. All drugs utilized were administered via intraperitonteal injection. Antidepressant-like behaviour was assessed using the forced swim test as previously described4. Briefly, animals were placed in a cylinder of water (22-24° C) for 6 minutes and immobility was measured during the last 4 minutes of the test. Molecular studies consisted of Western blotting analysis or QPCR performed on whole cell lysates from medial prefrontal cortex or anterior HC. Electrophysiological studies were performed as previously described in cultured neurons (whole cell recordings23) or hippocampal slices (field recordings10).
Methods
Mice
C57BL/6 male mice aged 6-8 weeks old were habituated to animal facilities for one week prior to behavioural testing. Mice were kept on a 12/12 light dark cycle and given access to food and water ad libitium. Inducible BDNF KOs were generated from a trigenic cross of NSE-tTA, TetOp-Cre, and floxed BDNF mice as previously described10. Conditional TrkB knockout mice were made by crossing CamK-cre(93)15 to floxed TrkB mice. For all behavioural testing, male mice were age- (two to four months) and weight-matched, and groups were balanced by genotype. All animal procedures conform to the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at UT Southwestern Medical Center.
Drug
All drugs were injected intraperitoneally (i.p.). Concentrations were the following: ketamine (Fort Dodge Animal Health) 3.0 mg/kg, MK-801 (Sigma) 0.1 mg/kg, and CPP (Sigma) 0.5 mg/kg in 0.9% saline; anisomycin (Sigma) 100 mg/kg (dissolved in HCl/saline final pH 7.4); actinomycin D (Sigma) 0.5 mg/kg in 5% ethanol; rottlerin and NH125 (Sigma) 5mg/kg in 20-100% DMSO; SL327 (Sigma) 10mg/kg in 100% DMSO30; NMDA (Sigma) 75 mg/kg, NBQX (Sigma) 10 mg/kg, and PTX (Sigma) 1 mg/kg in 0.9% saline; Rapamycin (Sigma) 1.0 mg/kg dissolved in 50% DMSO.
Sucrose consumption test
Briefly, group housed mice were habituated to a 1% sucrose/tap water solution for 48 hours. The mice were then habituated to water deprivation periods of 4, 14, and 19 hours, followed by a 1 hour exposure to the sucrose solution for three days with intervening access to regular drinking water. To assess individual sucrose intake, the group-housed mice were water-deprived overnight and then housed temporarily in a new cage. Each test mouse was placed in its home cage for one hour with access to the 1% sucrose solution. The bottle of sucrose solution was weighed before and after the test to determine sucrose intake. A water test was performed in a similar manner the following day. Data are expressed as a percentage of sucrose to total volume consumed in both sucrose and water trials.
Elevated Plus Maze
Mice were placed in the centre of a plus maze (each arm 33 cm × 5 cm) that was elevated 1 meter above the floor with two open arms and two closed arms (25-cm-tall walls on the closed arms) at 40 lux. The exploratory activity was monitored for 5 min with a video tracking system, and the duration in seconds in the closed and open arms was recorded by EthoVision software.
Novelty suppressed feeding
Briefly, group housed animals were food deprived for 24 hours and then placed in a temporary home cage for 30 minutes. For the test, individual mice were placed in a 42 × 42 cm open field arena at 40 lux. A single pellet of the mouse's regular food chow was placed in the centre of the open field arena. Each animal was placed in a corner of the arena and allowed to explore for up to 10 minutes. The trial ended when the mouse chewed a part of the chow. Amount of food consumed in the home cage was taken as weight of chow consumed in 5 minutes as a control measure for appetite.
Context and Cued Fear Conditioning
Fear conditioning was performed as previously described5. Briefly, mice were placed in individual chambers for 2 min followed by a loud tone (90 dB) for 30 s, immediately followed by a 0.5 mA footshock for 2 s. After 1 min, mice received a second pairing of tone and footshock, as described. Mice were placed in home cages until 24 h later, when the mice were placed back in the same boxes without a tone or shock. The amount of time the animal spent freezing was scored by an observer blind to genotype. Freezing behaviour was defined as no movement except for respiration. Four hours later, mice were placed in a novel environment with no tone or shock for 3 min, followed by 3 min of the tone to assess cue-dependent fear conditioning. Again, time spent freezing was recorded as described10.
Learned Helplessness
Mice were trained on one side of a two-chamber shuttlebox (MedAssociates) with the door closed for 1 hour, receiving 120 variable interval (18-44s average 30 sec) shocks (0.35 mA for 2 sec) on two training days. On the test day, the door was raised at the onset of shock and the shock ended either when the mouse stepped through to the other side of the shuttlebox or after 25 sec. Latency to step through the door and number of escape failures were recorded for fifteen trials.
Locomotor activity
Mice were placed in cages and locomotor activity was recorded for one hour under red light by photocell beams linked to computer acquisition software (San Diego Instruments).
Forced swim test
The forced swim test (FST) was performed as previously described12. The FST is an animal model that is sensitive to conventional AD treatment31 as well as non-monoaminergic ADs4. Mice were placed in a 4000-mL Pyrex glass beaker containing 3000 mL of water 24±1°C for six minutes. Water was changed between subjects. All test sessions were recorded by a video camera positioned on the side of the beakers. The videotapes were analyzed and scored by an observer blind to group assignment during the last four minutes of the six minute trial. A decrease in immobility time is suggestive of an AD-like response.
Chronic mild stress
Stressed mice were subjected to 2 randomly selected mild stressors/day of variable duration (1-12 hours) for 28 days. Stressors included water deprivation, 45° cage tilt, food deprivation, exposure to rat faeces, cage overcrowding, wet bedding, overnight illumination, dark exposure during normal light cycle, cold bedding, acoustic disturbance (120 dB), strobe lights, and cagemate rotation. Stressors were not applied within 8 hours of behavioural testing.
Time-course experiments
Separate cohorts of C57BL/6 adult male mice were i.p. injected with vehicle or the NMDAR antagonists ketamine (3.0 mg/kg), MK-801 (0.1 mg/kg), or CPP (0.5 mg/kg) at 30 minutes, 3 hours, 24 hours, or 1 week prior to FST (n=10 per group). The drug doses were based on previous literature demonstrating an AD-like response in mouse models4.
Anisomycin and actinomycin D experiments
Separate cohorts of C57BL/6 adult male mice were i.p. injected with either vehicle or anisomycin (100mg/kg) or saline or actinomycin D (0.5 mg/kg) one hour prior to FST. Thirty minutes prior to testing, mice received either a saline or ketamine injection (3.0 mg/kg) (n=10 per group). For 24 hour experiments, mice were given anisomycin (100 mg/kg) or saline 30 minutes prior to an injection of ketamine and tested in the FST one day later.
Inducible BDNF KO experiments
Separate cohorts of inducible BDNF KO adult male mice, or wild-type littermate controls, were subjected to FST either 30 minutes or 24 hours after injection with saline, ketamine (3.0 mg/kg), or MK-801 (0.1 mg/kg) (n=7-12 per group).
Quantitative RT-PCR
Fresh frozen anterior hippocampal slices (2/mouse, ∼1 mm thick) were dissected and total RNA was extracted using Trizol reagent (Invitrogen) according to manufacturer's instruction. Conditions for cDNA synthesis, amplification, and primer sequences were described previously12. Fold change in BDNF expression of the coding exon IX is normalized to GAPDH.
Protein quantification
Anterior hippocampal slices (2/mouse, ∼1 mm thick) were dissected from C57BL/6 mice receiving saline vehicle, ketamine (3.0 mg/kg), or MK-801 (0.1 mg/kg) i.p. either 30 minutes or 24 hours post-injection and rapidly frozen. Tissue was lysed in buffer containing protease and phosphatase inhibitors, and total protein concentration was quantified by Bradford analysis. BDNF quantification was determined using SDS-PAGE. Primary antibodies for BDNF (Santa Cruz Biotechnology) and GAPDH (Cell Signaling) were used at dilutions of 1:200 and 1:10,000, and anti-rabbit secondary antibodies were used at 1:2,000 and 1:50,000, respectively. For phosphorylated eEF2 (peEF2) (Thr56) and total eEF2 (teEF2), primary antibodies were used at dilutions of 1:1,000, and anti-rabbit secondary antibodies were used at 1:2,000. Mouse anti-Arc (C7) (Cell Signaling) was used at primary dilution 1:1000 and secondary dilution at 1:2,000. Phospho-mTOR and total mTOR (Cell Signaling) were both used at primary dilution 1:500 and secondary dilution 1:10,000. GluR1 (Chemicon) was used at primary dilution 1:5,000 and secondary dilution 1:2,000. Pan-Homer antibody (Cell Signaling) was used at 1:5,000 with 1:2000 dilutions for primary and secondary, respectively. Phospho-S6 kinase and total s6 kinase were used at 1:200 and 1:5,000 for primary dilutions, respectively, and both had secondary dilutions at 1:5,000 (Cell Signaling). Phospho-ERK and total ERK (Cell Signaling) were used at primary dilution 1:10,000 and 1:500 respectively and both had secondary dilutions at 1:2,000. ECL developed bands were exposed to film. Films were analyzed by ImageJ. BDNF was normalized to GAPDH bands, and peEF2 and teEF2 bands were taken as a ratio of GAPDH normalized values.
Immunohistochemistry
C57BL/6 mice were treated i.p. with saline, ketamine (3.0 mg/kg), or MK-801 (0.1 mg/kg) and sacrificed 30 minutes later. Protocol is adapted from a previous study32. Brains were fresh-dissected and post-fixed for 72 hours in ice-cold 4% paraformaldehyde. Brains were cryoprotected for 2 or more hours in 20% glycerol and sectioned on a freezing microtome at 30 μm and preserved in 1X PBS/0.01% sodium azide. Floating sections were washed in 2X SSC followed by antigen unmasking in 50:50 acetone:methanol performed at 4° C. Sections were rinsed and endogenous peroxidase activity was quenched in 1% H2O2 for 30 minutes. Sections were rinsed in 2X SSC/0.05% Tween-20. Tissue was blocked for 30 min in 3% normal goat serum/2X SSC/0.05% Tween followed by primary antibody, rabbit anti-peEF2 (diluted 1:100 in blocking solution; Cell Signaling Technology), incubation for 48 hours at 4° C. After rinsing in 2X SSC, a horseradish peroxidase-labelled secondary antibody at 1:200 was applied and the signal amplified using the tyramide amplification signal (TSA) system (Perkin Elmer). Slides were counterstained with DAPI, mounted on superfrost plus slides and dried for 2 hours, and coverslipped in DPX mountant.
ELISA
High sensitivity enzyme-linked immunosorbent assay to assess BDNF levels was used per manufacturer's instructions (Promega). Briefly, hippocampal lysates were prepared in the recommended buffer, diluted 1:4 in 1X PBS and acid treated as instructed by the manufacturers. A 96-well plate (Nunc) was coated overnight in carbonate coating buffer, blocked in provided sample buffer for 2 hours at RT, and treated with recombinant human BDNF antibody for 2 hours at RT. Acid-treated samples and provided standards were added to the plate in duplicate. Wells were then treated with anti-IgY conjugated to HRP for 1 hour at RT and colour was developed with provided TMB solution for ten minutes stopped with 1N HCl. Absorbance of wells was measured at 450 nm. BDNF concentration was determined by comparing mean absorbance of the duplicate samples to the standards. BDNF concentration was then normalized to total protein content and expressed as pg BDNF/μg total protein.
Cell Culture
Dissociated hippocampal cultures were prepared as previously described33. Briefly, whole hippocampi were dissected from postnatal day 0-3 (P0-3) Sprague-Dawley rats. Tissue was trypsinized (10 mg/ml trypsin) for 10 min at 37°C, mechanically dissociated by pipetting and plated on Matrigel coated coverslips. Cytosine arabinoside (4 μM ARAC, Sigma, St. Louis, MO) was added at day 1 in vitro (DIV), at 4 DIV ARAC concentration was reduced to 2 μM. All experiments were performed on 14-21 DIV cultures.
Cell culture recordings
Whole-cell patch-clamp recordings were performed on hippocampal pyramidal neurons. Data were acquired using a MultiClamp 700B amplifier and Clampex 9.0 software (Molecular Devices). Recordings were filtered at 2 kHz and sampled at 200 μs. A modified Tyrode's solution containing (in mM): 150 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2, 10 glucose, 10 HEPES, pH 7.4, was used as external bath solution. The pipette internal solution contained (in mM): 115 Cs-MeSO3, 10 CsCl, 5 NaCl, 10 HEPES, 0.6 EGTA, 20 Tetraethylammonium-Cl, 4 Mg-ATP, 0.3 Na3GTP, pH 7.35, and 10 QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl)-triethylammonium bromide], 300 mOsm. Series resistance ranged between 10-30 MΩ. To record and isolate NMDAR-mediated miniature EPSCs (NMDA-mEPSCs), MgCl2 concentration was reduced to 0.1 mM and 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX; 10μM, Sigma), picrotoxin (PTX; 50 μM; Sigma) were added to bath solution to block α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptor mediated excitatory currents and γ-Aminobutyric acid (GABA) receptor mediated inhibitory currents respectively. Baseline for the analysis of NMDA-mEPSCs was automatically determined as the average current level of silent episodes during a recording. The events were selected at a minimum threshold of 4 pA and the area under current deflection was calculated to quantify charge transfer18.
Field recordings
Field recordings were made from hippocampal slices. Sprague-Dawley rats were obtained from Charles River Laboratories (Wilmington, MA). Slices (400 μm) were prepared from 15- to 25-d-old rats. Rats were anesthetized with the Euthasol (50 mg/kg) and decapitated soon after the disappearance of corneal reflexes. The brain was removed, dissected and then sliced using a vibratome (1000 Plus) in ice-cold dissection buffer containing the following (in mM): 2.6 KCl, 1.25 NaH2PO4, 26 NaHCO3, 0.5 CaCl2, 5 MgCl2, 212 sucrose, and 10 dextrose. Area CA3 was surgically removed from each slice immediately after sectioning. The slices were transferred into a reservoir chamber filled with ACSF containing the following (in mM): 124 NaCl, 5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 CaCl2, 2 MgCl2, and 10 dextrose. Slices were allowed to recover for 2–3 h at 30°C. ACSF and dissection buffer were equilibrated with 95% O2 and 5% CO2. For recording, slices were transferred to a submerged recording chamber, maintained at 30°C, and perfused continuously with ASCF at a rate of 2–3 ml/min. Field potentials (FPs) were recorded with extracellular recording electrodes (1 MΩ) filled with ACSF and placed in stratum radiatum of area CA1. Field potentials were evoked by monophasic stimulation (duration, 200 μs) of Schaffer collateral/commissural afferents with a concentric bipolar tungsten stimulating electrode (Frederick Haer Company, Bowdoinham, ME). Stable baseline responses were collected every 30 s using a stimulation intensity (10–30 μA), yielding 50–60% of the maximal response. After recording 20 min of stable baseline stimulation was stopped and 20 μM of ketamine was applied for 30 min, after this stimulation was resumed. FPs were filtered at 2 kHz, acquired, and digitized at 10 kHz on a personal computer using custom software (LabVIEW; National Instruments, Austin, TX). Synaptic strength was measured as the initial slope (10–40% of the rising phase) of the FP. The group data were analyzed as follows: (1) the initial slopes of the FP were expressed as percentages of the preconditioning baseline average; (2) the time scale in each experiment was converted to time from the end of ketamine application; and (3) the time-matched, normalized data were averaged across experiments.
Supplementary Material
Acknowledgments
We thank Melissa A. Mahgoub for assistance with the animal experiments, Dr. Shari Birnbaum and Ami Pettersen for assistance with the behavioral testing, and members of the Monteggia and Kavalali laboratories for insightful discussions and comments of the manuscript. This work was supported by grant MH070727 (L.M.M), grant MH066198 (E.T.K.) as well as the Division of Basic Sciences Training Program at UT Southwestern Medical Center T32 MH 76690-02 (A.E.A). E.T.K. is an Established Investigator of the American Heart Association.
Footnotes
Author Contributions A.E.A. performed the behavioral experiments. A.E.A., M.A., and M.F.L. contributed to the molecular experiments. E.N. performed the electrophysiology experiments, E.S.N. performed the TrkB behavioral experiments, and A.E.A. and P-f. C. performed the statistical analyses. A.E.A. also made the figures and wrote the corresponding section of the paper. E.T.K. and L.M.M. designed the study, supervised the experiments and wrote the paper.
References
- 1.Zarate CA, Jr, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63(8):856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 2.Berman RM, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47(4):351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 3.Price RB, Nock MK, Charney DS, Mathew SJ. Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol Psychiatry. 2009;66(5):522–526. doi: 10.1016/j.biopsych.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maeng S, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63(4):349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 5.Detke MJ, Johnson J, Lucki I. Acute and chronic antidepressant drug treatment in the rat forced swimming test model of depression. Exp Clin Psychopharmacol. 1997;5(2):107–112. doi: 10.1037//1064-1297.5.2.107. [DOI] [PubMed] [Google Scholar]
- 6.Sinner B, Graf BM. Ketamine. Handb Exp Pharmacol. 2008;(182):313–333. doi: 10.1007/978-3-540-74806-9_15. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz PH, Wasterlain CG. Cardiac arrest and resuscitation alters the pharmacokinetics of MK-801 in the rat. Res Commun Chem Pathol Pharmacol. 1991;73(2):181–195. [PubMed] [Google Scholar]
- 8.Kristensen JD, Hartvig P, Karlsten R, Gordh T, Halldin M. CSF and plasma pharmacokinetics of the NMDA receptor antagonist CPP after intrathecal, extradural and i.v. administration in anaesthetized pigs. Br J Anaesth. 1995;74(2):193–200. doi: 10.1093/bja/74.2.193. [DOI] [PubMed] [Google Scholar]
- 9.Chen B, Dowlatshahi D, MacQueen GM, Wang JF, Young LT. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry. 2001;50(4):260–265. doi: 10.1016/s0006-3223(01)01083-6. [DOI] [PubMed] [Google Scholar]
- 10.Monteggia LM, et al. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci U S A. 2004;101(29):10827–10832. doi: 10.1073/pnas.0402141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berton O, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311(5762):864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- 12.Adachi M, Barrot M, Autry AE, Theobald D, Monteggia LM. Selective loss of brain-derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. Biol Psychiatry. 2008;63(7):642–649. doi: 10.1016/j.biopsych.2007.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22(8):3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoshaw BA, Malberg JE, Lucki I. Central administration of IGF-I and BDNF leads to long-lasting antidepressant-like effects. Brain Res. 2005;1037(1-2):204–208. doi: 10.1016/j.brainres.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 15.Akbarian S, et al. Brain-derived neurotrophic factor is essential for opiate-induced plasticity of noradrenergic neurons. J Neurosci. 2002;22(10):4153–4162. doi: 10.1523/JNEUROSCI.22-10-04153.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lattal KM, Abel T. Different requirements for protein synthesis in acquisition and extinction of spatial preferences and context-evoked fear. J Neurosci. 2001;21(15):5773–5780. doi: 10.1523/JNEUROSCI.21-15-05773.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Capasso A, Di Giannuario A, Loizzo A, Pieretti S, Sorrentino L. Actinomycin D blocks the reducing effect of dexamethasone on amphetamine and cocaine hypermotility in mice. Gen Pharmacol. 1996;27(4):707–712. doi: 10.1016/0306-3623(95)02077-2. [DOI] [PubMed] [Google Scholar]
- 18.Sutton MA, Taylor AM, Ito HT, Pham A, Schuman EM. Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron. 2007;55(4):648–661. doi: 10.1016/j.neuron.2007.07.030. [DOI] [PubMed] [Google Scholar]
- 19.Poleszak E, et al. NMDA/glutamate mechanism of antidepressant-like action of magnesium in forced swim test in mice. Pharmacol Biochem Behav. 2007;88(2):158–164. doi: 10.1016/j.pbb.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 20.Fernandez F, et al. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat Neurosci. 2007;10(4):411–413. doi: 10.1038/nn1860. [DOI] [PubMed] [Google Scholar]
- 21.Li N, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 329(5994):959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cleary C, et al. Antidepressive-like effects of rapamycin in animal models: Implications for mTOR inhibition as a new target for treatment of affective disorders. Brain Res Bull. 2008;76(5):469–473. doi: 10.1016/j.brainresbull.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 23.Atasoy D, et al. Spontaneous and evoked glutamate release activates two populations of NMDA receptors with limited overlap. J Neurosci. 2008;28(40):10151–10166. doi: 10.1523/JNEUROSCI.2432-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Espinosa F, Kavalali ET. NMDA receptor activation by spontaneous glutamatergic neurotransmission. J Neurophysiol. 2009;101(5):2290–2296. doi: 10.1152/jn.90754.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanaka J, et al. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science. 2008;319(5870):1683–1687. doi: 10.1126/science.1152864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jakawich SK, et al. Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron. 68(6):1143–1158. doi: 10.1016/j.neuron.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindskog M, et al. Postsynaptic GluA1 enables acute retrograde enhancement of presynaptic function to coordinate adaptation to synaptic inactivity. Proc Natl Acad Sci U S A. doi: 10.1073/pnas.1016399107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sutton MA, Schuman EM. Partitioning the synaptic landscape: distinct microdomains for spontaneous and spike-triggered neurotransmission. Sci Signal. 2009;2(65):pe19. doi: 10.1126/scisignal.265pe19. [DOI] [PubMed] [Google Scholar]
- 29.Kavalali ET, et al. Spontaneous neurotransmission: an independent pathway for neuronal signaling? Physiology (Bethesda) 26(1):45–53. doi: 10.1152/physiol.00040.2010. [DOI] [PubMed] [Google Scholar]
- 30.Duman CH, Schlesinger L, Kodama M, Russell DS, Duman RS. A role for MAP kinase signalling in behavioral models of depression and antidepressant treatment. Biol Psychiatry. 2007;61:661–70. doi: 10.1016/j.biopsych.2006.05.047. [DOI] [PubMed] [Google Scholar]
- 31.Porsolt RD, Le Pichon M, Jalfre M. Depression: A new animal model sensitive to antidepressant treatments. Nature. 1977;266:730–732. doi: 10.1038/266730a0. [DOI] [PubMed] [Google Scholar]
- 32.Park S, et al. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron. 2008;59:70–83. doi: 10.1016/j.neuron.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kavalali ET, Klingauf J, Tsien RW. Activity-dependent regulation of synaptic clustering in a hippocampal culture system. Proc Natl Acad Sci U S A. 1999;96:12893–12900. doi: 10.1073/pnas.96.22.12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.