

Abstract
Cigarette smoking is a significant environmental factor in the human inflammatory bowel diseases, remarkably, conferring protection in ulcerative colitis. We previously demonstrated that a prominent component of cigarette smoke, CO, suppresses Th17-mediated experimental colitis in IL-10−/− mice through a heme oxygenase (HO)-1–dependent pathway. In this study, homeostatic and therapeutic effects of CO and HO-1 were determined in chronic colonic inflammation in TCR-α–deficient (−/−) mice, in which colitis is mediated by Th2 cytokines, similar to the cytokine milieu described in human ulcerative colitis. TCRα−/− mice exposed to CO or treated with the pharmacologic HO-1 inducer cobalt protoporphyrin demonstrated amelioration of active colitis. CO and cobalt protoporphyrin suppressed colonic IL-1β, TNF, and IL-4 production, whereas IL-10 protein secretion was increased. CO induced IL-10 expression in macrophages and in vivo through an HO-1–dependent pathway. Bacterial products regulate HO-1 expression in macrophages through MyD88- and IL-10–dependent pathways. CO exposure and pharmacologic HO-1 induction in vivo resulted in increased expression of HO-1 and IL-10 in CD11b+ lamina propria mononuclear cells. Moreover, induction of the IL-10 family member IL-22 was demonstrated in CD11b− lamina propria mononuclear cells. In conclusion, CO and HO-1 induction ameliorated active colitis in TCRα−/− mice, and therapeutic effects correlated with induction of IL-10. This study provides further evidence that HO-1 mediates an important homeostatic pathway with pleiotropic anti-inflammatory effects in different experimental models of colitis and that targeting HO-1, therefore, is a potential therapeutic strategy in human inflammatory bowel diseases.
Cigarette smoking is one of the most significant environmental risk factors identified in the human inflammatory bowel diseases (IBD): Crohn's disease (CD) and ulcerative colitis (UC). Meta-analyses showed that the risk for developing UC in current smokers is ∼40% that of nonsmokers (1). Former smokers have ∼1.7 times increased risk for developing UC (2). Some studies even suggested a dose response, with heavier smokers having greater protection (1). Based on these compelling epidemiological observations, one of the important unanswered questions in IBD is how does cigarette smoking mechanistically mediate this protective effect?
The gaseous molecule CO is one candidate that may contribute to the beneficial association between smoking and UC. CO is a prominent component of cigarette smoke: blood carboxyhemoglobin levels, a measure of systemic exposure to CO, were reported to range from 1–18% in active smokers (3). CO, best known as a toxic compound, is also produced endogenously during normal physiology by the heme oxygenase (HO) enzymes, which mediate the degradation of heme into equimolar quantities of CO, iron, and biliverdin. In particular, the enzyme HO-1 and its metabolic products regulate immune responses, tissue injury, and repair (4). We previously showed that CO ameliorates active inflammation in an experimental model of chronic IBD, IL-10–deficient (−/−) mice, through induction of HO-1 (5).
CD4+ Th cells play a key role in the regulation of immune responses in the intestine. CD4+ T cells have been divided into functionally important subsets based on the cytokines that they produce (6). Although these subdivisions represent a reduction of complex biology, most applicable to the mouse, they provide a framework to understand mucosal T cell responses in human IBD. Th1 cells produce the cytokines IFN-γ and IL-2. Th1 cells are a hallmark of cell-mediated immunity, necessary for the eradication of intracellular pathogens and the development of long-term immunity against infectious agents (6). Numerous mouse models of IBD are characterized by an overabundance of intestinal Th1 cytokines. CD was initially described as a prototype Th1-mediated chronic inflammatory disorder, characterized by mucosal granulomas (the histological hallmark of a Th1 response), increased expression of IFN-γ, as well as increased IL-12, a cytokine necessary for Th1 development (7). However, the discovery of the cytokine IL-23, which shares a common p40 subunit with IL-12, led to a paradigm shift in our understanding of inflammatory responses in IBD (8). IL-23, unlike IL-12, promotes a distinct CD4+ T cell activation profile, the Th17 cell, characterized by the production of the cytokine IL-17. A pivotal role for IL-23 and Th17 cells was demonstrated in experimental IBD models, such as the IL-10−/− mouse, and recent genetic and immunologic findings highlight the importance of this pathway in human IBD (9).
Th2 cells produce the cytokines IL-4, IL-5, and IL-13. These cytokines provide help for B cell Ab production, and are involved in host defense against extracellular helminthic parasites in the mucosal immune system (10). Inflammation in human UC has been characterized as mediated by Th2 cytokines. Lamina propria T cells from UC patients produce IL-13 and IL-5 and little IFN-γ (11). Although multiple experimental models of chronic Th1/17-driven intestinal inflammation have been elucidated, few have been described in which chronic disease occurs in a Th2 cytokine milieu. Mice with targeted disruption of the TCRα gene (TCRα−/−) perhaps most closely recapitulate the colonic Th2 signature in human UC. IL-4 and IL-1β play important roles in the development of colitis in TCRα−/− mice (12).
Our previous work showed that CO and HO-1 induction ameliorates Th17-mediated colonic inflammation in IL-10−/− mice (5). To model the protective effects of cigarette smoking in human UC, CO and the HO-1 pathway were studied in a murine model with immunologic similarities to UC. We demonstrate the anti-inflammatory effects of CO and HO-1 induction in spontaneous Th2-mediated colitis in TCRα−/− mice.
Materials and Methods
Mice
Wild-type (WT), TCRα−/−, and IL-10−/− mice were obtained from The Jackson Laboratory. Hmox1−/− mice were provided by Leo Otterbein (Harvard Medical School, Boston, MA). All mice used in this study were on the C57BL/6 background and matched for age and sex in all experiments. Animals were housed in accordance with guidelines from the American Association for Laboratory Animal Care and Research Protocols and by the Institutional Animal Care and Use Committee of the University of Pittsburgh and the University of North Carolina Schools of Medicine.
CO exposure
Mice or macrophages were exposed to compressed air or CO at a concentration of 250 ppm, as previously described (5). Briefly, CO at a concentration of 1% (10,000 ppm) was mixed with compressed air before delivery into the exposure chamber. Flow into the animal chamber was maintained at a rate of 12 l/min and into the cell-culture chamber at a rate of 2 l/min. The cell-culture chamber was humidified and maintained at 37°C. A CO analyzer (Interscan) was used to measure CO levels continuously in the chambers. The CO-releasing molecule-186 (ALF186) was a generous gift from Alfama (Porto Salvo, Portugal) and was injected i.p. twice a week for 2 wk. Control mice were injected with an inactive form of ALF186 (iALF186) that does not possess CO-releasing capacity. At the end of CO exposure, cardiac blood samples (0.2 ml) were taken immediately after mice were sacrificed to measure carboxyhemoglobin (HbCO) using a hemoximeter (OSM3; Radiometer Copenhagen). Doses of ALF186 were extensively optimized because hypoxia can become a confounding factor at HbCO levels ≥20% (13). HbCO levels were uniformly <20% in all experiments using CO gas and ALF186.
Pharmacologic inhibition and induction of HO-1
Tin protoporphyrin (SnPP) and cobalt protoporphyrin (CoPP) (Frontier Scientific Porphyrin Products, Logan, UT) were dissolved in sodium hydroxide; a final pH of 7.4 was achieved by adding hydrochloric acid and further dilution with PBS. Because of light sensitivity, SnPP and CoPP were prepared in dim light, light protected, and freshly made before injection.
TLR ligands and cytokines
Ultrapure TLR ligands (Invivogen, San Diego, CA), LPS, synthetic bacterial lipopeptide (sBLP), flagellin, and CpG oligodeoxynucleotides (CpG) were dissolved in endotoxin-free water at concentrations described in the Results. IL-10 (PeproTech, Rocky Hill, NJ) was dissolved in endotoxin-free water.
Cytokine ELISAs
Linco Cytokine-16 plex Mouse ELISA was performed for IL-4, IL-1β, IL-10, TNF, and IL-17 ((Millipore, Billerica, MA), per the manufacturer's instructions. Murine IL-10 was measure with cytokine-specific immuno-assay kits (R&D Systems, Minneapolis, MN).
Real-time RT-PCR analysis
Quantitative real-time PCR, using SYBR Green Master Mix (Applied Biosystems, Bedford, MA), was performed on an HT-7900 (Applied Biosystems, Bedford, MA), as previously published (14). The following primer sequences were used: Hmox1: forward, 5′-CCAGAGTTTCCGCATACAACC-3′, reverse, 5′-TCTCTGGACACCTGACCCTTCG-3′; β-actin: forward, 5′-AGCCATGTACGTAGCCATCCAG-3′, reverse, 5′-TGGCGTGAGGGAGAGCATAG-3′; Il10: forward, 5′-GTCATCGATTTCTCCCCTGTG-3′, reverse, 5′-CCTTGTAGACACCTTGGTCTTGG-3′; and Il22: forward, 5′-GGTGCCTTTCCTGACCAAAC-3′, reverse, 5′-TTTCACTGTCTCCTTCAGCCTTC-3′.
Western immunoblots
Western blots were performed on whole-cell extracts, as described (15). HO-1 Abs were from Stressgen (Plymouth Meeting, PA); NF-E2–related factor 2 (Nrf2) and β-actin Abs were from Abcam (Cambridge, MA).
Bone marrow-derived macrophages
Bone marrow-derived macrophages (BMMs) were cultured as described previously (5).
Colonic tissue explant cultures
Colonic tissue fragments (0.5 g dry weight) were processed, as previously described (14). Tissue-fragment supernatants were collected after 24 h for cytokine ELISAs.
Isolation of colonic macrophages
Lamina propria mononuclear cells (LPMCs) were isolated from mouse colon by an enzymatic method, followed by Percoll (GE Healthcare, Piscataway, NJ) density-gradient centrifugation, as previously described (16). LPMCs were further separated into CD11b+ cells using anti-CD11b microbeads (Miltenyi Biotec, Auburn, CA). Purity was >90% by flow cytometric analysis.
Flow cytometry
Splenic, mesenteric lymph node, and colonic cells were isolated, as previously described (17), and stained with the B cell marker FITC anti-mouse B220 and cell surface marker PE anti-mouse CD1d (1B1) (eBioscience, San Diego, CA). In other experiments, cells were stained extracellularly with FITC-conjugated anti-CD4+ (RM4-5), fixed and permeabilized with Cytofix/Cytoperm solution (BD Pharmingen, San Jose, CA), and stained intracellularly with allophycocyanin-conjugated anti-Foxp3 (FJK-16s). Colonic LPMCs from TCR−/− mice exposed to CO or injected with ALF186, as well as control mice (air-exposed and iALF186-treated mice), were labeled with Abs against macrophage lineage and activation markers (CD14, F4/80, CD86, and CD80; eBioscience, San Diego, CA). Dead cells were excluded with propidium iodide staining. Samples were acquired on a FACSCalibur (Becton Dickinson), and data were analyzed with Cell-Quest Pro software (BD Biosciences, San Jose, CA).
Histology
Colitis scores (0–4) were determined by a staff pathologist who was blinded to the experimental protocol using the criteria reported by Berg et al. (18). Twenty separate microscopic fields (original magnification ×100) were evaluated for each mouse by a pathologist (A.R.S.) blinded to the treatment groups.
Data analysis
Statistical significance for data subsets from experiments performed in cells was assessed by the two-tailed Student t test. Statistical significance for in vivo data subsets was assessed by the Mann–Whitney U test (SPSS, Chicago, IL) with Bonferroni correction.
Results
CO exposure ameliorates active colitis in TCRα−/− mice
TCRα−/− mice were exposed to 250 ppm of CO from 12–16 wk of age (n = 10) and compared with a control group (n = 10) exposed to ambient air. Mice in both treatment groups were matched for age, sex, and initial body weight. CO-exposed mice showed an increase in body weight compared with mice housed in ambient air (Fig. 1A). Assessment of histological improvement was performed by a pathologist blinded to treatment groups. CO-exposed mice demonstrated significantly reduced histologic inflammation (Fig. 1B, Supplemental Fig. 1).
FIGURE 1.
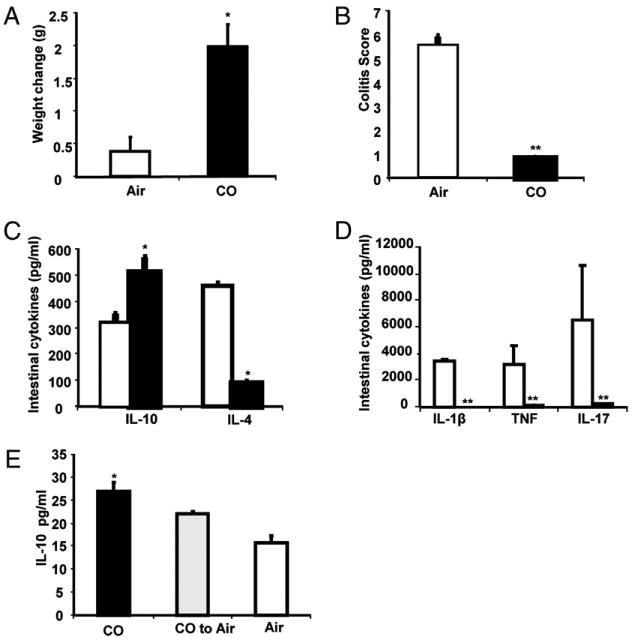
CO ameliorates Th2-mediated colitis in TCRα−/− mice. TCRα−/− mice were housed in ambient air or a chamber maintaining a constant concentration of CO at 250 ppm (n = 10 each) from 12–16 wk of age. A, CO-exposed mice gained more weight than did air-exposed mice. B, Colitis scores were significantly decreased in CO-exposed mice compared with control mice. Results are presented as the sum total of four averaged scores from five regions of the large intestine graded by a pathologist blinded to the groups using a standard scoring system. Bars represent mean ± SEM of 10 mice/group. C, Spontaneous protein secretion determined in 24-h supernatants from colonic explants from CO-exposed (black bars) and air-exposed (white bars) TCRα−/− mice. Spontaneous IL-10 and IL-4 were measured by cytokine-specific ELISA. D, IL-1β, TNF, and IL-17 secretion were assessed by Linco 16-multiplex cytokine assay. Each result represents the mean ± SEM of triplicate assays. E, Ten-week-old TCRα−/− mice were divided into three groups: exposed to CO (250 ppm; black bar) for 4 wk, exposed to air for 4 wk (white bar), or exposed to CO for 2 wk and then transferred to ambient air housing conditions for 2 wk (gray bar). Spontaneous IL-10 secretion was measured in full-length colonic cell-free supernatants using cytokine-specific IL-10 ELISA. Results represent mean ± SEM from three experiments (n = 3 mice/group). *p < 0.05, **p < 0.01 versus air-exposed mice.
We next determined whether CO exposure affects colonic cytokine expression in TCRα−/− mice. Colonic explant cultures from TCRα−/− mice exposed to CO in vivo for 4 wk produced less IL-1β, IL-4, TNF, and IL-17 (Fig. 1C, 1D), correlating with histological improvement. CO treatment in vivo also resulted in increased colonic IL-10 secretion compared with explant cultures from air-exposed TCRα−/− mice (Fig. 1C).
Because IL-10 is an important regulatory cytokine, correlations between CO exposure and colonic IL-10 induction were further explored. Ten-week-old TCRα−/− mice were divided into three groups: group 1 was exposed to CO (250 ppm) for 4 wk, group 2 was exposed to air for 4 wk, and group 3 was exposed to CO for 2 wk and then transferred to ambient air for 2 wk. Mice exposed to CO demonstrated increased secretion of IL-10 in colonic explants. Mice transferred from CO exposure to air after 2 wk showed intermediate IL-10 secretion, with more colonic IL-10 compared with air-exposed mice but less than mice continually exposed to CO (Fig. 1E). These findings suggested that CO may ameliorate inflammation through induction of IL-10. Furthermore, because IL-10 secretion was still increased 2 wk after removing mice from CO, CO may induce a durable change in a cell population that secretes IL-10 in the colon.
CO induces IL-10 and HO-1 in colonic CD11b+ LPMCs
To elucidate potential mechanisms through which CO ameliorates experimental colitis, TCRα−/− mice were treated i.p. with ALF186 (30 mg/kg) and iALF186 (30 mg/kg) twice weekly for 2 wk. Colonic CD11b+ LPMCs, predominantly representing a macrophage cell population (14), were isolated from both groups, and Hmox1 and Il10 expression were analyzed. Hmox1 was induced in CD11b+ and CD11b− LPMCs in ALF186-treated TCRα−/− mice compared with iALF186-treated mice (Fig. 2A), with the most significant induction in CD11b+ cells. Interestingly, markedly increased Il10 expression was demonstrated in CD11b+ LPMCs, but not in CD11b− LPMCs, from ALF186-treated mice compared with iALF186-treated mice (Fig. 2B). No differences were observed in expression of surface markers (F4/80, CD80, CD86, and CD14) in CD11b+ LPMCs from TCRα−/− mice treated with ALF186 or iALF186 (Supplemental Fig. 2A). Similar results were obtained for Hmox1 and Il10 expression in LPMCs from ALF186-treated WT mice, with the exception that Il10 induction was also demonstrated in the CD11b− LPMC population (Supplemental Fig. 3), possibly reflecting increased numbers of CD4+ Foxp3+ lamina propria T regulatory cells in WT mice compared with TCRα−/− mice (see later discussion; Supplemental Fig. 1C).
FIGURE 2.
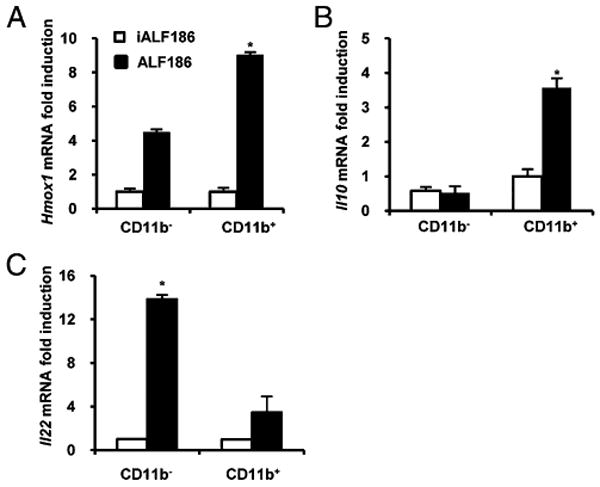
CO upregulates Hmox1 and Il10 expression in colonic CD11b+ LPMCs from TCRα−/− mice. LPMCs were isolated from colons of TCRα−/− mice treated with iALF186 (n = 4) or ALF186 (n = 4). LPMCs were further separated into CD11b− and CD11b+ cells and analyzed for Hmox1 (A), Il10 (B), and Il22 (C) expression by real-time RT-PCR. Results were normalized to β-actin. Bars represent mean ± SEM of triplicate cultures from pooled LPMCs from four mice per group. *p < 0.05 versus iALF186-treated CD11b+ LPMCs.
Another member of the IL-10 family of cytokines, IL-22, was shown to dampen innate mucosal inflammatory responses and attenuate colitis in TCRα−/− mice (19). LPMCs from ALF186-treated TCRα−/− mice demonstrated significantly more IL-22 (Il22) expression compared with iALF186-treated mice, predominantly in the CD11b− LPMC population, but in CD11b+ cells as well (Fig. 2C).
Regulatory CD4+ Foxp3+ T and CD11d+ B cells were demonstrated to be a source of intestinal IL-10 production and have important anti-inflammatory roles in murine IBD (19). However, no differences in the numbers of CD1d+ B cells in LPMCs were found between iALF186- and ALF186-treated TCRα−/− mice (Supplemental Fig. 2B). Interestingly, a marked decrease in the numbers of splenic CD4+ Foxp3+ T cells was observed in TCRα−/− mice compared with WT mice (Supplemental Fig. 2C), and Foxp3+ cells were undetectable in LPMCs from TCRα−/− mice (data not shown). These results suggested that CO may be protective in experimental colitis through induction of IL-10 and HO-1, specifically in CD11b+ LPMCs.
CO induces IL-10 in macrophages through induction of HO-1
To further elucidate regulation of IL-10 by CO in macrophages, WT and TCRα−/− BMMs were stimulated with LPS in CO (250 ppm) or ambient air for 24 h. As previously described (13), CO-augmented LPS stimulated IL-10 secretion in WT BMMs (Fig. 3A) (5). CO also augmented IL-10 secretion from LPS-stimulated TCRα−/− BMMs (Fig. 3B). However, in LPS-activated BMMs from HO-1–deficient (Hmox1−/−) mice, CO failed to induce IL-10 secretion (Fig. 3C), suggesting that CO augments IL-10 secretion through an HO-1–dependent signaling pathway. Moreover, WT BMMs incubated with ALF186 (100 μg/ml) for 3 h demonstrated significantly increased basal Hmox1 and Il10 expression compared with iALF186 BMMs (Fig. 3D, 3E).
FIGURE 3.
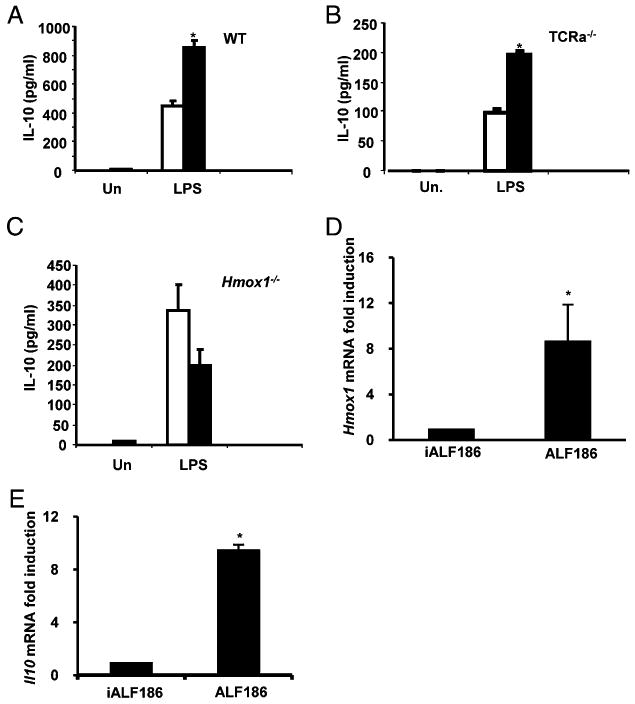
CO induces IL-10 in murine macrophages via the HO-1 pathway. BMMs from WT (A), TCRα−/− (B), or Hmox1−/− (C) mice were cultured in CO (250 ppm, black bars) or ambient air (white bars). Following activation with LPS (1 μg/ml), IL-10 protein secretion was assayed in 24-h supernatants by ELISA. WT BMMs were incubated with iALF186 (100 μg/ml) or ALF186 (100 μg/ml) for 3 h, and Hmox1 (D) and Il10 (E) expression was analyzed by real-time RT-PCR. Results were normalized to β-actin and represent the mean ± SEM of triplicate assays from three independent experiments. *p < 0.05 versus air-exposed or iALF186-treated BMMs.
HO-1 induction recapitulates immunomodulatory effects of CO in vivo
To understand the role of HO-1 in the anti-inflammatory effects of CO in vivo, TCRα−/− mice were treated with a pharmacological inducer of HO-1, CoPP (5 mg/kg i.p.), twice a week for 2 wk and compared with vehicle (DMSO)-treated controls. CoPP treatment resulted in improved histological scores compared with vehicle treatment (Fig. 4A). Moreover, colonic explant cultures revealed decreased IL-4 (Fig. 4B), IL-1β, TNF, and IL-17 (Fig. 4C) secretion in CoPP-treated TCRα−/− mice. CoPP treatment also resulted in robust induction of Hmox1 expression in colonic CD11b− and CD11b+ LPMC populations (Fig. 4D). Colonic CD11b+ LPMCs were the primary source of IL-10 secretion from CoPP-treated TCRα−/− mice, because less IL-10 expression was observed in CD11b− cells (Fig. 4E). These results strongly implicated CD11b+ LPMCs as the primary source of IL-10 in TCRα−/− mice and important targets for the immunomodulatory effects of CO and HO-1.
FIGURE 4.
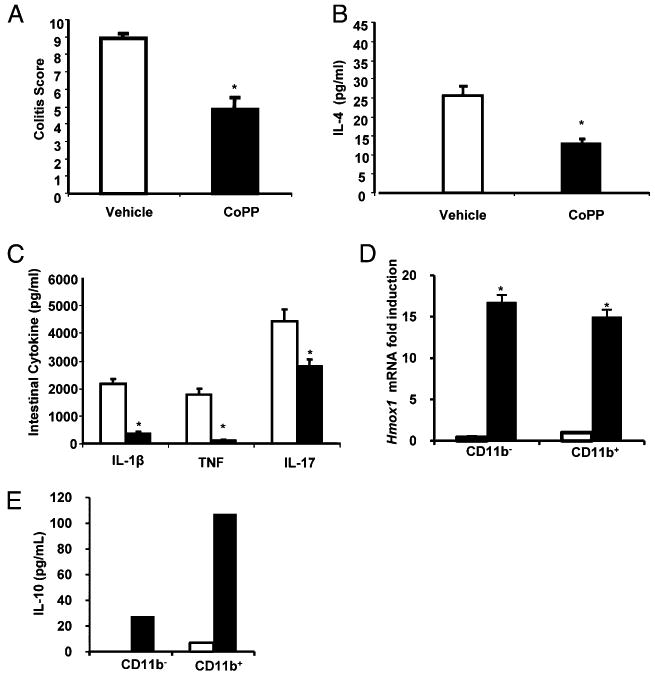
The HO-1 inducer CoPP ameliorates colitis in TCRα−/− mice. Twelve-week-old TCRα−/− mice were treated with i.p. injection of CoPP (5 mg/kg, twice a week for 2 wk) (n = 8), and control mice were treated with DMSO vehicle i.p. (n = 12). A, CoPP-injected mice had less severe colitis. B, Spontaneous protein secretion determined in 24-h supernatants from colonic explants from CoPP-treated (5 mg/kg) and vehicle-treated (DMSO) TCRα−/− mice. C, Spontaneous IL-4 was determined by cytokine-specific ELISA, and IL-1β, TNF, and IL-17 secretion were determined by Linco 16-multiplex cytokine assay. Bars represent mean ± SEM from 12 mice per group. D, LPMCs were isolated from colons of TCRα−/− mice treated with CoPP (black bars) and vehicle (white bars), separated into CD11b− and CD11b+ cells, and analyzed for Hmox1 expression by real-time RT-PCR, with results normalized to β-actin. E, IL-10 secretion was determined by ELISA. Each result represents the mean ± SEM of triplicate assays from four mice/treatment group. *p < 0.05 versus vehicle-treated mice.
Next, to address whether immunomodulatory effects of CO are mediated by HO-1 in vivo, TCRα−/− mice were treated i.p. with ALF186, with or without the HO-1 inhibitor SnPP. ALF186-treated mice demonstrated reduced histologic inflammation compared with ALF186+SnPP-treated mice (Fig. 5A). Importantly, CD11b+ LPMCs from ALF186+SnPP-treated TCRα−/− mice expressed significantly less Il10 than did CD11b+ LPMCs from ALF186-treated mice (Fig. 5B). Likewise, CD11b− and CD11b+ LPMCs from ALF186+SnPP-treated TCRα−/− mice demonstrated lower Il22 expression than did LPMCs from ALF186-treated mice (Fig. 5C). These findings demonstrated that the anti-inflammatory effects of CO are abrogated in the presence of an HO-1 inhibitor.
FIGURE 5.
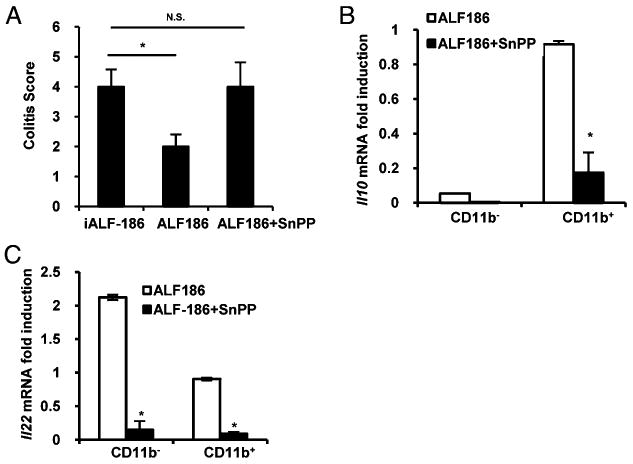
Hmox1 function is required for amelioration of colitis and Il10 and Il22 upregulation by CO in TCRα−/− mice. A, Colitis scores were significantly higher in iALF-186 (n = 6) ALF186+ SnPP (50 μM/kg twice/weekly for 2 wk) (n = 6) treated mice compared with mice treated with ALF186 alone (n = 6). B and C, LPMCs were isolated from colons of TCRα−/− mice treated with ALF186 (n = 6) or ALF186+SnPP (n = 6). They were separated into CD11b− and CD11b+ cells and analyzed for Il10 (B) and Il22 (C) expression by real-time RT-PCR. Results were normalized to β-actin. Bars represent mean ± SEM triplicate cultures from pooled LPMCs from six mice per group. *p < 0.05 versus iALF186-treated TCRα−/− mice and CD11b+ LPMCs.
LPS and IL-10 regulate HO-1 expression in macrophages
Because HO-1 is required for the protective effects of CO, we next studied HO-1 (Hmox1) regulation in macrophages. BMMs from WT and IL-10−/− mice were stimulated with LPS, with or without IL-10, and expression of Hmox1 mRNA and HO-1 protein was determined. IL-10−/− BMMs demonstrated decreased expression of Hmox1 compared with WT BMMs. Addition of rIL-10 restored Hmox1 mRNA and protein expression in LPS-activated IL-10−/− BMMs and augmented Hmox1 expression in WT BMMs (Fig. 6A, 6B). Moreover, incubation of WT BMMs with an IL-10 Ab inhibited LPS-induced expression of HO-1. These results demonstrated that LPS and IL-10 are regulators of HO-1 in macrophages.
FIGURE 6.
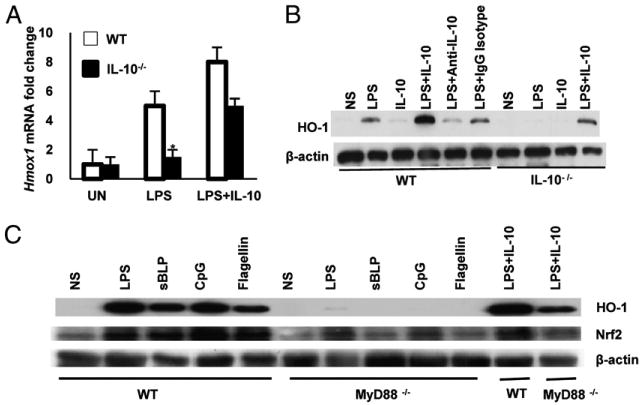
Regulation of HO-1 in macrophages is IL-10 and MyD88 dependent. A, WT and IL-10− BMMs were stimulated with LPS alone (100 ng/ml) or with LPS plus IL-10 (10 ng/ml) for 12 h. Total RNA was isolated and analyzed for Hmox1, and β-actin mRNA expression was detected by real-time RT-PCR. Results are expressed as mean ± SEM from three independent experiments. *p < 0.05 versus LPS-treated WT BMMs. B, WT and IL-10−/− BMMs were stimulated with LPS (100 ng/ml) in the presence of IL-10 (10 ng/ml) for 24 h after initial preincubation with anti–IL-10 Ab (10 μg/ml) for 1 h. HO-1 protein was analyzed by Western blotting. Data are representative of five independent experiments with similar results. C, WT and MYD88−/− BMMs were stimulated with LPS (100 ng/ml), CpG (1 μM), sBLP (100 ng/ml), flagellin (10 ng/ml), or IL-10 (10 ng/ml) for 24 h. HO-1 and Nrf2 protein was analyzed by Western blotting. Data are representative of three independent experiments.
TLRs recognize specific molecular patterns present in a broad range of microbial pathogens. TLR activation uses a common signal-transduction pathway initiated by the adaptor protein MyD88. To further elucidate TLR-mediated induction of HO-1, WT and MyD88−/− BMMs were stimulated with MyD88-dependent (sBLP, LPS, flagellin, and CpG DNA) bacterial ligands. Interestingly, sBLP, LPS, flagellin, and CpG DNA induced HO-1 expression in WT BMMs but not in MyD88−/− BMMs (Fig. 6C). Addition of rIL-10 restored HO-1 expression in MyD88−/− BMMs, suggesting that IL-10–induced expression of HO-1 in macrophages is independent of TLR/MyD88-signaling pathways. As previously reported, TLR-mediated IL-10 expression, another MyD88-dependent gene, was abrogated in MyD88−/− BMMs (Supplemental Fig. 4).
The transcription factor Nrf2 is a critical regulator of HO-1 through binding to antioxidant response elements (20). In the absence of MyD88, Nrf2 protein expression in macrophages was also markedly reduced (Fig. 6C). These results elucidated a novel regulatory circuit, with MyD88-dependent Hmox1 expression by bacterial products, in part through Nrf2, and MyD88-independent regulation by IL-10.
Discussion
In summary, CO exposure ameliorates chronic Th2-mediated colitis in TCRα−/− mice. Immunomodulatory effects of CO were recapitulated by pharmacologic HO-1 induction. Moreover, pharmacologic inhibition of HO-1 blocked the protective effects of CO on colitis, suggesting that in vivo, CO mechanistically requires HO-1 function. CO and HO-1 induction resulted in increased colonic IL-10 expression prominently in CD11b+ LPMCs, with consequent inhibition of inflammatory cytokines. We previously demonstrated that CO ameliorated colitis in IL-10−/− mice (5). IL-10−/− mice exhibit a Th17-mediated immune pathology. The protective effects of CO in IL-10−/− mice were attributed, in part, to inhibition of the common p40 subunit of the inflammatory cytokines IL-12 and IL-23 (5). Our current study elucidated the anti-inflammatory effects of CO and the HO-1 pathway in TCRα−/− mice characterized by a distinctly different immunopathogenesis, with increased colonic Th2 cytokine expression that, to some extent, recapitulates the colonic inflammatory cytokine milieu in human UC (12). This study further elucidated pleiotropic immunomodulatory effects of CO and the HO-1 pathway. There are now illustrations of therapeutic applications of this pathway (5, 21, 22) in multiple experimental models of IBD mediated by divergent immune mechanisms. Given the genetic, immunologic, and clinical heterogeneity of the human IBDs, the therapeutic benefit of CO and HO-1 in numerous preclinical models suggests potentially broad applications in patients.
Notably, in TCRα−/− mice, CO- and HO-1–mediated inhibition of inflammation correlated with increased levels of the anti-inflammatory cytokine IL-10. Cross-talk between CO/HO-1 and IL-10 regulation may underlie the homeostatic function of each modality. Lee and Chau (23) demonstrated that IL-10 induced the expression of HO-1 via a p38 MAPK-dependent pathway. IL-10–induced expression of HO-1 also requires activation of STAT-3 (24). Moreover, HO-1 may be an important downstream mediator of the anti-inflammatory effects of IL-10 in macrophages. HO-1 activity and the generation of endogenous CO were necessary for IL-10–dependent inhibition of TNF expression (25). Likewise, LPS-activated macrophages overexpressing HO-1 or exposed to CO demonstrated reduced TNF production, whereas IL-10 secretion was enhanced (26). Interestingly, we demonstrated that LPS stimulated HO-1 expression in WT BMMs, but not IL-10−/− BMMs, and blocking IL-10 diminished LPS-activated HO-1 expression. Hence, IL-10 is a cofactor for HO-1 induction by TLR ligands. Moreover, Hmox1 induction occurred through MyD88-dependent (bacterial products) and -independent (IL-10) pathways. LPS and inflammatory cytokines (IL-1β and TNF) are well described as potent inducers of HO-1, and several studies linked NF-κB and AP-1 transcription factors in this response (27, 28). However, given the absence of a clearly identified functional NF-κB element, how NF-κB promotes Hmox1 gene transcription is a matter of speculation (29–31). Nrf2, a basic leucine zipper transcription factor, is involved in cellular protection against oxidative stress through antioxidant response element-directed induction of multiple detoxifying and antioxidant enzymes, including HO-1 (20). We demonstrated defective induction of Nrf2 in MyD88−/− BMMs, suggesting a mechanism for how TLR signaling may affect Hmox1 transcription.
HO-1 was also shown to exert its protective effect in experimental asthma through a mechanism mediated by IL-10 expression in CD4+CD25+Foxp3+ T regulatory cells (32). In vivo CO exposure ameliorated intestinal injury induced by LPS or ischemia–reperfusion. Mucosal levels of IL-10 were shown to be increased in CO-exposed mice (33). Similarly, CO-releasing molecules promoted resolution of acute pancreatic inflammation in rats, which correlated with increases in local IL-10 expression (34). CO was also shown to augment local IL-10 and afford protection in other models of inflammation, including sepsis, renal injury, and diabetes (35–37).
Activated macrophages are an abundant source of IL-10 (38). CO augments basal and LPS-induced IL-10 secretion from WT and TCRα−/− BMMs. Specifically, TCRα−/− mice treated with CO and CoPP demonstrated a specific increase in IL-10 secretion exclusively from CD11b+ LPMCs, which include a predominant resident macrophage population. These findings expand upon an important homeostatic role for IL-10–producing colonic macrophages, which acted on T regulatory cells to maintain expression of Foxp3 in a T cell adoptive-transfer model of murine colitis (39).
Several regulatory B cell populations have been characterized in TCRα−/− mice. A subset of regulatory B cells was identified as an important source of IL-10 and was responsible for inhibiting IL-1β and ameliorating colitis (40). However, we could not discern any difference in the numbers of CD1d+ MLNs from ALF186-treated TCRα−/− mice compared with iALF186-treated mice. An IL-12–producing regulatory B cell subset that develops in the presence of IL-10 was also shown to be involved in the regulation of colonic inflammation in this model (41). During CO exposure and pharmacologic HO-1 induction in vivo, IL-10 is almost exclusively detected in the CD11b+ LPMC fraction. These findings suggested that CO and HO-1 induction, mediated in part through IL-10, has anti-inflammatory effects that extend beyond the induction of previously described regulatory B cell populations in this model.
Colonic Foxp3+ cells were not detected in ALF186- or iALF186-treated TCRα−/− mice. Moreover, a significant deficiency of splenic CD4+Foxp3+ T cells was observed in TCRα−/− mice compared with WT mice. The TCR was shown to be involved in the development of CD4+Foxp3+ T cells (42); however, the influence of TCRα-chain repertoire on the development of CD4+ Foxp3+ T cells has not been analyzed. TCRα-chain expression is not essential for CD4+CD25+ T cell development, but its effect on Foxp3 expression remains unknown (43). Although the purpose of our study was not to discern T regulatory cell development in TCRα−/− mice, taken as a whole, our results suggested that the predominant source of IL-10 in LPMCs from TCRα−/− mice, and therefore a target for CO and HO-1 induction, resides in the CD11b+ population and not Foxp3+ T cells.
We unexpectedly detected a Th17-cytokine signature in colonic explants from TCRα−/− mice with abundant levels of IL-17. The IL-17–producing cell population(s) remain(s) to be determined. Given recent reports, it is interesting to speculate that γδ T cells may be a source of IL-17 (44). Interestingly, IL-17 levels decreased following CO exposure or HO-1 induction, correlating with histologic improvement. Recently, the IL-10 family member IL-22, expressed by Th17 cells, was demonstrated to ameliorate colitis in TCRα−/− mice (19). CO-treated TCRα−/− mice demonstrated significant increases in IL-22 mRNA expression in CD11b− LPMCs, consistent with previous studies suggesting that that nonmacrophage-derived IL-22 may also be involved in the protective effects of CO/HO-1. Notably, IL-22 is a potent inducer of IL-10 (19). The description of a Th17 signature in TCRα−/− mice also substantiated this as a model for human UC, in which the same genetic associations within the IL-23/Th17 pathway confer susceptibility to CD and UC (45). Likewise, current biological interventions that inhibit TNF are approved for the treatment of moderate to severe UC (46). CO and pharmacological induction of HO-1 resulted in a significant decrease in TNF secretion in TCRα−/− colons, which may also mediate therapeutic effects.
To our knowledge, these results are the first to characterize anti-inflammatory properties of CO and HO-1 in a Th2-mediated model of chronic colonic inflammation. The anti-inflammatory effects of CO are attributed to the induction of HO-1 and highlight the broad impact of these pathways in intestinal inflammation. HO-1 induction correlated with increased IL-10 and IL-22 expression in vivo, which may be relevant anti-inflammatory mechanisms of this pathway, because both cytokines were previously determined to have a protective role in colonic inflammation in TCRα−/− mice (19, 41). It remains to be determined whether HO-1 induction mediates downstream anti-inflammatory effects in colitis models through increased enzymatic activity and production of endogenous metabolic products, including CO, or through other mechanisms. Nonetheless, these experiments demonstrated that HO-1 is a central regulator of intestinal homeostasis through pleiotropic mechanisms and that understanding these pathways are of mechanistic and therapeutic relevance in human IBD.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant RO1 DK54452 (to S.E.P.), Gastroenterology Research Training Grant T32 DK007737 (to S.Z.S. and J.C.O.), National Research Service Awards F32 DK083186 (to S.Z.S.) and P30 DK034987 (Center for Gastrointestinal Biology and Disease; Immunotechnologies, Gnotobiotic and Histology Cores), and a Crohn's and Colitis Foundation of America Student Research Award (to S.M.R.) and Research Fellowship Award (to R.H., T.K., and K.M.).
Abbreviations used in this article
- ALF186
CO-releasing molecule-186
- BMM
bone marrow-derived macrophage
- CD
Crohn's disease
- CoPP
cobalt protoporphyrin
- CpG
CpG oligodeoxynucleotides
- HbCO
carboxyhemoglobin
- HO
heme oxygenase
- iALF186
inactive form of ALF186
- IBD
inflammatory bowel disease
- LPMC
lamina propria mononuclear cell
- Nrf2
NF-E2–related factor 2
- sBLP
synthetic bacterial lipopeptide
- SnPP
tin protoporphyrin
- UC
ulcerative colitis
- WT
wild-type
Footnotes
The online version of this article contains supplemental material.
Disclosures: L.O. is a scientific consultant for Alfama, Inc., and provided CO-releasing molecules (ALF186) for some experiments. The other authors have no financial conflicts of interest.
References
- 1.Jick H, Walker AM. Cigarette smoking and ulcerative colitis. N Engl J Med. 1983;308:261–263. doi: 10.1056/NEJM198302033080507. [DOI] [PubMed] [Google Scholar]
- 2.Boyko EJ, Koepsell TD, Perera DR, Inui TS. Risk of ulcerative colitis among former and current cigarette smokers. N Engl J Med. 1987;316:707–710. doi: 10.1056/NEJM198703193161202. [DOI] [PubMed] [Google Scholar]
- 3.Smith CJ, Guy TD, Stiles MF, Morton MJ, Collie BB, Ingebrethsen BJ, Robinson JH. A repeatable method for determination of carboxyhemoglobin levels in smokers. Hum Exp Toxicol. 1998;17:29–34. doi: 10.1177/096032719801700105. [DOI] [PubMed] [Google Scholar]
- 4.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 5.Hegazi RA, Rao KN, Mayle A, Sepulveda AR, Otterbein LE, Plevy SE. Carbon monoxide ameliorates chronic murine colitis through a heme oxygenase 1-dependent pathway. J Exp Med. 2005;202:1703–1713. doi: 10.1084/jem.20051047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maynard CL, Weaver CT. Intestinal effector T cells in health and disease. Immunity. 2009;31:389–400. doi: 10.1016/j.immuni.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okazawa A, Kanai T, Watanabe M, Yamazaki M, Inoue N, Ikeda M, Kurimoto M, Ishii H, Hibi T. Th1-mediated intestinal inflammation in Crohn's disease may be induced by activation of lamina propria lymphocytes through synergistic stimulation of interleukin-12 and interleukin-18 without T cell receptor engagement. Am J Gastroenterol. 2002;97:3108–3117. doi: 10.1111/j.1572-0241.2002.07107.x. [DOI] [PubMed] [Google Scholar]
- 8.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 9.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finkelman FD, Shea-Donohue T, Morris SC, Gildea L, Strait R, Madden KB, Schopf L, Urban JF., Jr Interleukin-4- and interleukin-13-mediated host protection against intestinal nematode parasites. Immunol Rev. 2004;201:139–155. doi: 10.1111/j.0105-2896.2004.00192.x. [DOI] [PubMed] [Google Scholar]
- 11.Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, Fiocchi C, Strober W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn's disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–1270. [PubMed] [Google Scholar]
- 12.Mizoguchi A, Mizoguchi E, Chiba C, Spiekermann GM, Tonegawa S, Nagler-Anderson C, Bhan AK. Cytokine imbalance and autoantibody production in T cell receptor-alpha mutant mice with inflammatory bowel disease. J Exp Med. 1996;183:847–856. doi: 10.1084/jem.183.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6:422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 14.Sheikh SZ, Matsuoka K, Kobayashi T, Li F, Rubinas T, Plevy SE. Cutting edge: IFN-gamma is a negative regulator of IL-23 in murine macrophages and experimental colitis. J Immunol. 2010;184:4069–4073. doi: 10.4049/jimmunol.0903600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiong H, Zhu C, Li F, Hegazi R, He K, Babyatsky M, Bauer AJ, Plevy SE. Inhibition of interleukin-12 p40 transcription and NF-kappaB activation by nitric oxide in murine macrophages and dendritic cells. J Biol Chem. 2004;279:10776–10783. doi: 10.1074/jbc.M313416200. [DOI] [PubMed] [Google Scholar]
- 16.Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, Sakuraba A, Kitazume MT, Sugita A, Koganei K, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest. 2008;118:2269–2280. doi: 10.1172/JCI34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murali-Krishna K, Altman JD, Suresh M, Sourdive DJ, Zajac AJ, Miller JD, Slansky J, Ahmed R. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 18.Berg DJ, Davidson N, Kühn R, Müller W, Menon S, Holland G, Thompson-Snipes L, Leach MW, Rennick D. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98:1010–1020. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 2003;24:449–455. doi: 10.1016/s1471-4906(03)00181-9. [DOI] [PubMed] [Google Scholar]
- 21.Sun X, Suzuki K, Nagata M, Kawauchi Y, Yano M, Ohkoshi S, Matsuda Y, Kawachi H, Watanabe K, Asakura H, Aoyagi Y. Rectal administration of tranilast ameliorated acute colitis in mice through increased expression of heme oxygenase-1. Pathol Int. 2010;60:93–101. doi: 10.1111/j.1440-1827.2009.02490.x. [DOI] [PubMed] [Google Scholar]
- 22.Varga C, Laszlo F, Fritz P, Cavicchi M, Lamarque D, Horvath K, Posa A, Berko A, Whittle BJ. Modulation by heme and zinc protoporphyrin of colonic heme oxygenase-1 and experimental inflammatory bowel disease in the rat. Eur J Pharmacol. 2007;561:164–171. doi: 10.1016/j.ejphar.2006.12.022. [DOI] [PubMed] [Google Scholar]
- 23.Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med. 2002;8:240–246. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- 24.Ricchetti GA, Williams LM, Foxwell BM. Heme oxygenase 1 expression induced by IL-10 requires STAT-3 and phosphoinositol-3 kinase and is inhibited by lipopolysaccharide. J Leukoc Biol. 2004;76:719–726. doi: 10.1189/jlb.0104046. [DOI] [PubMed] [Google Scholar]
- 25.Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, Ohta K, Kasahara Y, Koizumi S. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Invest. 1999;103:129–135. doi: 10.1172/JCI4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Minamino T, Christou H, Hsieh CM, Liu Y, Dhawan V, Abraham NG, Perrella MA, Mitsialis SA, Kourembanas S. Targeted expression of heme oxygenase-1 prevents the pulmonary inflammatory and vascular responses to hypoxia. Proc Natl Acad Sci USA. 2001;98:8798–8803. doi: 10.1073/pnas.161272598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Camhi SL, Alam J, Otterbein L, Sylvester SL, Choi AM. Induction of heme oxygenase-1 gene expression by lipopolysaccharide is mediated by AP-1 activation. Am J Respir Cell Mol Biol. 1995;13:387–398. doi: 10.1165/ajrcmb.13.4.7546768. [DOI] [PubMed] [Google Scholar]
- 28.Camhi SL, Alam J, Wiegand GW, Chin BY, Choi AM. Transcriptional activation of the HO-1 gene by lipopolysaccharide is mediated by 5′ distal enhancers: role of reactive oxygen intermediates and AP-1. Am J Respir Cell Mol Biol. 1998;18:226–234. doi: 10.1165/ajrcmb.18.2.2910. [DOI] [PubMed] [Google Scholar]
- 29.Xiao W. Advances in NF-kappaB signaling transduction and transcription. Cell Mol Immunol. 2004;1:425–435. [PubMed] [Google Scholar]
- 30.Wijayanti N, Huber S, Samoylenko A, Kietzmann T, Immenschuh S. Role of NF-kappaB and p38 MAP kinase signaling pathways in the lipopolysaccharide-dependent activation of heme oxygenase-1 gene expression. Antioxid Redox Signal. 2004;6:802–810. doi: 10.1089/ars.2004.6.802. [DOI] [PubMed] [Google Scholar]
- 31.Hill-Kapturczak N, Thamilselvan V, Liu F, Nick HS, Agarwal A. Mechanism of heme oxygenase-1 gene induction by curcumin in human renal proximal tubule cells. Am J Physiol Renal Physiol. 2001;281:F851–F859. doi: 10.1152/ajprenal.2001.281.5.F851. [DOI] [PubMed] [Google Scholar]
- 32.Xia ZW, Xu LQ, Zhong WW, Wei JJ, Li NL, Shao J, Li YZ, Yu SC, Zhang ZL. Heme oxygenase-1 attenuates ovalbumin-induced airway inflammation by up-regulation of foxp3 T-regulatory cells, interleukin-10, and membrane-bound transforming growth factor- 1. Am J Pathol. 2007;171:1904–1914. doi: 10.2353/ajpath.2007.070096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu SH, Ma K, Xu XR, Xu B. A single dose of carbon monoxide intraperitoneal administration protects rat intestine from injury induced by lipopolysaccharide. Cell Stress Chaperones. 2010;15:717–727. doi: 10.1007/s12192-010-0183-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen P, Sun B, Chen H, Wang G, Pan S, Kong R, Bai X, Wang S. Effects of carbon monoxide releasing molecule-liberated CO on severe acute pancreatitis in rats. Cytokine. 2010;49:15–23. doi: 10.1016/j.cyto.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 35.Lancel S, Hassoun SM, Favory R, Decoster B, Motterlini R, Neviere R. Carbon monoxide rescues mice from lethal sepsis by supporting mitochondrial energetic metabolism and activating mitochondrial biogenesis. J Pharmacol Exp Ther. 2009;329:641–648. doi: 10.1124/jpet.108.148049. [DOI] [PubMed] [Google Scholar]
- 36.Goebel U, Mecklenburg A, Siepe M, Roesslein M, Schwer CI, Pahl HL, Priebe HJ, Schlensak C, Loop T. Protective effects of inhaled carbon monoxide in pig lungs during cardiopulmonary bypass are mediated via an induction of the heat shock response. Br J Anaesth. 2009;103:173–184. doi: 10.1093/bja/aep087. [DOI] [PubMed] [Google Scholar]
- 37.Rémy S, Blancou P, Tesson L, Tardif V, Brion R, Royer PJ, Motterlini R, Foresti R, Painchaut M, Pogu S, et al. Carbon monoxide inhibits TLR-induced dendritic cell immunogenicity. J Immunol. 2009;182:1877–1884. doi: 10.4049/jimmunol.0802436. [DOI] [PubMed] [Google Scholar]
- 38.O'Garra A, McEvoy LM, Zlotnik A. T-cell subsets: chemokine receptors guide the way. Curr Biol. 1998;8:R646–R649. doi: 10.1016/s0960-9822(07)00413-7. [DOI] [PubMed] [Google Scholar]
- 39.Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, Kronenberg M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. 2009;10:1178–1184. doi: 10.1038/ni.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, Bhan AK. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity. 2002;16:219–230. doi: 10.1016/s1074-7613(02)00274-1. [DOI] [PubMed] [Google Scholar]
- 41.Sugimoto K, Ogawa A, Shimomura Y, Nagahama K, Mizoguchi A, Bhan AK. Inducible IL-12-producing B cells regulate Th2-mediated intestinal inflammation. Gastroenterology. 2007;133:124–136. doi: 10.1053/j.gastro.2007.03.112. [DOI] [PubMed] [Google Scholar]
- 42.Piccirillo CA, Shevach EM. Naturally-occurring CD4+CD25+ immunoregulatory T cells: central players in the arena of peripheral tolerance. Semin Immunol. 2004;16:81–88. doi: 10.1016/j.smim.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 43.Bosco N, Agenes F, Rolink AG, Ceredig R. Peripheral T cell lymphopenia and concomitant enrichment in naturally arising regulatory T cells: the case of the pre-Talpha gene-deleted mouse. J Immunol. 2006;177:5014–5023. doi: 10.4049/jimmunol.177.8.5014. [DOI] [PubMed] [Google Scholar]
- 44.Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. 2006;177:4662–4669. doi: 10.4049/jimmunol.177.7.4662. [DOI] [PubMed] [Google Scholar]
- 45.Silverberg MS, Cho JH, Rioux JD, McGovern DP, Wu J, Annese V, Achkar JP, Goyette P, Scott R, Xu W, et al. Ulcerative colitis-risk loci on chromosomes 1p36 and 12q15 found by genome-wide association study. Nat Genet. 2009;41:216–220. doi: 10.1038/ng.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, Johanns J, Travers S, Rachmilewitz D, Hanauer SB, Lichtenstein GR, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353:2462–2476. doi: 10.1056/NEJMoa050516. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.