

Abstract
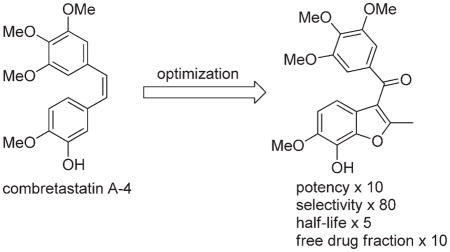
A structure–activity relationship (SAR) guided design of novel tubulin polymerization inhibitors has resulted in a series of benzo[b]furans with exceptional potency toward cancer cells and activated endothelial cells. The potency of early lead compounds has been substantially improved through the synergistic effect of introducing a conformational bias and additional hydrogen bond donor to the pharmacophore. Screening of a focused library of potent tubulin polymerization inhibitors for selectivity against cancer cells and activated endothelial cells over quiescent endothelial cells has afforded 7-hydroxy-6-methoxy-2-methyl-3-(3,4,5-trimethoxybenzoyl)benzo-[b]furan (BNC105, 8) as a potent and selective antiproliferative. Because of poor solubility, 8 is administered as its disodium phosphate ester prodrug 9 (BNC105P), which is rapidly cleaved in vivo to return the active 8. 9 exhibits both superior vascular disrupting and tumor growth inhibitory properties compared with the benchmark agent combretastatin A-4 disodium phosphate 5 (CA4P).
INTRODUCTION
Microtubules are protein biopolymers formed through polymerization of heterodimers of α- and β-tubulin.1 The polymerization is reversible, and the dynamic assembly and disassembly of microtubules are involved in a number of cell functions, including cell division, migration, and shape change. Microtubules are also involved in a host of cell signaling pathways, including those related to apoptosis. Compounds that bind to α,β-tubulin heterodimers and disrupt the dynamics of microtubule assembly and disassembly have emerged as some of the most effective chemotherapeutics for the treatment of cancer. Notable examples include the Catharanthus (or vinca) alkaloids vinblastine and vincristine and the taxoids paclitaxel and docetaxel.2 These drugs are powerful antimitotic agents that inhibit cancer cell proliferation and induce apoptosis. While they have proven to be successful in the treatment of cancer, their narrow therapeutic index and the emergence of resistance have inspired efforts to search for safer and more effective agents that are capable of treating resistant phenotypes.2
The Catharanthus alkaloids and the taxoids bind at separate sites on the α,β-tubulin heterodimer, and another natural product, colchicine 1 (Figure 1), binds to a third distinct site.1 Compounds that bind to the colchicine site have been the subject of intense investigation by researchers seeking to identify new agents capable of addressing the limitations of existing tubulin targeting drugs.3,4 In addition to being potent antimitotics, compounds that bind to the colchicine site also show potent vascular disrupting properties, which result from the effects of these compounds on vascular endothelial cells.5,6 Such vascular disrupting agents (VDAs) take advantage of the significant differences that exist between the vasculature of normal, healthy tissues and that of tumors. The selective shutdown of tumor vasculature starves tumors of oxygen and nutrients, leading to significant cancer cell death through necrosis and apoptotis. While the antimitotic and vascular disrupting properties of colchicine have been known for over 50 years, toxicity issues have prevented its clinical development as an anticancer agent. The significant toxicity of colchicine (LD50 < 5 mg/kg in rodents) is attributed to its very slow dissociation from tubulin (t1/2 = 35 h), which leads to persistence in sensitive tissues for several days.7–9 Compounds that bind more reversibly to the colchicine site often have shorter half-lives in vivo and are much better tolerated.10 N-Acetylcolchicinol 2 and combretastatin A-4 (CA4) 4 are examples of reversible binders to the colchicine site that have short half-lives in vivo and that are much better tolerated than colchicine.5,6,10 Since vascular disruption in tumors peaks after just a few minutes, safe and effective vascular shutdown can be achieved by tubulin binders that have reversible binding kinetics and short half-lives. Both 2 and 4 have entered clinical trials, where they are administered as the soluble disodium phosphate ester prodrugs 3 (ZD6126) and 5 (CA4P), which are rapidly cleaved in vivo to the active agents.10–12 A significant drawback of the short half-lives of 2 and 4 is that a short period of in vivo exposure is often insufficient to elicit a significant cell kill via the antimitotic effect of these compounds.13 As a result, significant tumor growth from the outer rim of the tumor is maintained, since this region is often fed by normal, healthy vasculature and is not susceptible to the vasculature disrupting effects of tubulin binders.14 The more potent tumor growth inhibitory properties of 7 (Oxi4503), a phosphate prodrug of combretastatin A-1 (CA1) 6, are attributed to its capacity to act as both a VDA and a cytotoxic agent.15 After in vivo deesterification of 7, to give the short half-life VDA 6, further metabolism occurs to produce a cytotoxic o-quinone (not shown). Thus 7, represents a dual mode agent capable of targeting the vasculature of the internal regions of the tumor and proliferative cancer cells of the viable rim.15 Since tubulin binding compounds are cytotoxic in their own right, we considered that a moderate half-life agent might act as both a VDA and a cytotoxin in vivo without exhibiting undue toxicity. In order to further offset any increase in toxicity that may result from a moderate increase in half-life, we also sought a selective compound that would exhibit higher potency toward cancer cells and tumor vascular endothelial cells relative to normal, healthy tissues. These efforts have led to the identification of 8 (BNC105), and we have recently described aspects of this compound’s vascular disrupting and tumor growth inhibitory properties in animal models.16 Herein, we describe the SAR-guided discovery process that led to 8 and the key biological data that led to its selection as a candidate for further development, including its potency and selectivity toward various cancer cell lines, effectiveness against multidrug resistant phenotypes, and favorable pharmacokinetics.17
Figure 1.
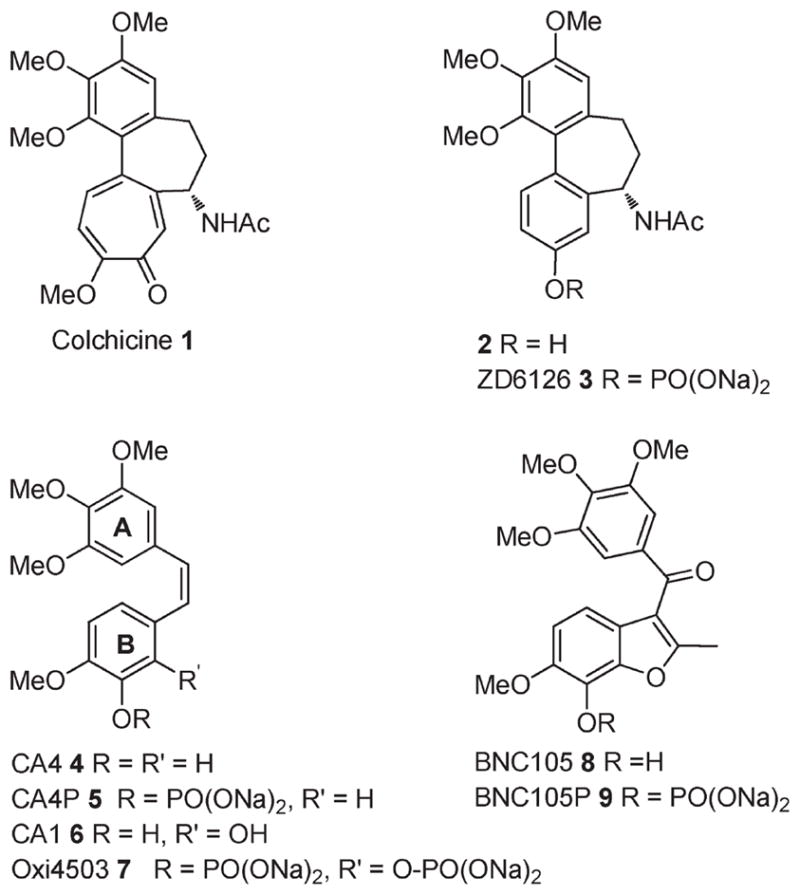
Tubulin binding agents with affinity for the colchicine site.
RESULTS AND DISCUSSION
Background SAR
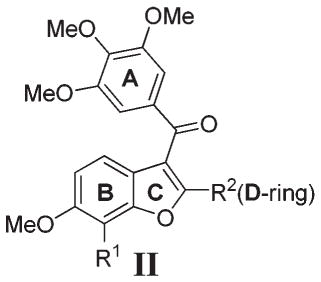
Early studies by Pinney and co-workers identified benzo[b]thiophene 10 as weak inhibitor of tubulin polymerization that binds to the colchicine binding site (entry 2, Table 1).18 X-ray crystal structure analysis of 10 revealed the A-ring to be rotated toward the D-ring in a pseudo-π-stacking arrangement.18b It was concluded that this conformation is likely to be important to binding, and early SAR studies within our group indicated that the A- and D-rings of 10 may coincide with the A- and B-rings of 4.18–20 For example, the introduction of a 3′-hydroxyl to the D-ring of 10 (compound 11) increased structural analogy to 4 and increased potency (entry 3, Table 1). Also, removal of the B-ring in 11 was well tolerated, giving an even more potent compound 14 (entry 4, Table 3). On the other hand, we also found that small changes in the substitution pattern of the B-ring of 10, such as removal or relocation of the C6-methoxy to C5, completely eliminated activity, indicating that the B-ring does contribute to activity when present (data not shown).19 We also identified that the benzo[b]furan 12 and indole 13 analogues of benzo[b]thiophene 11 were much more potent (compare entries 3, 5, and 6, Table 1) and that for all systems I (X = O, S, NH) the carbonyl linker between the A- and C-rings is essential for activity (data not shown).21,22 Later, Hseih and co-workers reported that indole 15 (BPROL075), an analogue of 13 in which the D-ring is absent, is also active.23 On the basis of similarities in the SAR of 15 and 4, they concluded that the A- and B-rings of 15 correlate with the A- and B-rings of 4. These results presented the possibility that the D-ring in compounds I (10–13) may be redundant and that in fact the B-ring in I plays an equivalent role to the B-ring of 4. On the other hand, π-stacking interactions between the A- and D-rings in I may induce a conformational change such that compounds I present a different pose to the tubulin binding site than does 15, that is, where the A- and D-rings in I equate to the A- and B-rings of 4. Therefore, in order to understand the SAR and generate more potent analogues of I and 15, a closer evaluation of the relative contributions of the B- and D-rings in I would be required. Since benzo[b]furans generally gave the most effective leads, based on potency and stability (metabolic and thermal), we focused our investigations on I (X = O).
Table 1.
SAR Studies of Tubulin Polymerization Inhibitors I
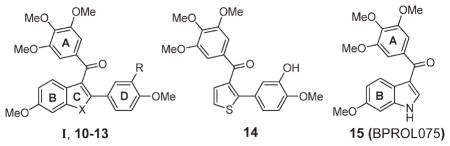
Table 3.
Biological Evaluation of Benzo[b]furans II
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | cmpnd | R1 | R2(D-ring) | tubulina IC50 (μM) | MCF-7b IC50 (nM) | activated HUVECc IC50 (nM) | quiescent HUVECd IC50 (nM) | selectivity ratioe |
| 1 | 4 | - | - | 1.8 ± 0.2 | 2.9 | 3.6 | 3.9 | 1.1 |
| 2 | 12 | H |
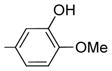
|
1.3 ± 0.2 | 39 ± 18 | 45 | 41 | 0.9 |
| 3 | 21 | H |
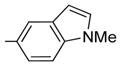
|
1.3 ± 0.04 | 42 ± 10 | 41 | 32 | 0.8 |
| 4 | 25 | H |
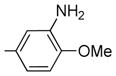
|
1.9 ± 0.4 | 26±5 | 32 | 44 | 1.4 |
| 5 | 26 | H |
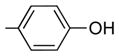
|
2.3 (both runs) | 48 ± 10 | 30 | 34 | 1.1 |
| 6 | 31a | H | H | 1.6 ± 0.2 | 55 ± 5 | 45 | 63 | 1.4 |
| 7 | 31b | OH | H | ND | 45 ± 15 | 83 | 322 | 3.9 |
| 8 | 27 | OH |
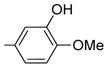
|
0.8 ± 0.2 | 4.0 ± 0.1 | 1.7 | 3.4 | 2.0 |
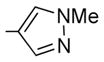
| 9 | 8 | OH | Me | 3.0 ± 0.6 | 2.4 ± 2 | 0.31 | 25 | 81 |
| 10 | 28 | OH |
|
1.3 ± 0.07 | 1.0 ± 0.5 | 0.4 | 0.7 | 1.8 |
| 11 | 24 | OiPr |
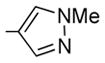
|
ND | 42 ± 6 | 665 | 419 | 0.6 |
| 12 | 33a | H | Br | ND | 495 ± 25 | 510 | 309 | 0.6 |
| 13 | 35 | H |
|
ND | 71 ± 1 | 45 | 27 | 0.6 |
| 14 | 45 | H |
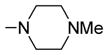
|
ND | 215 ± 15 | 484 | 405 | 0.8 |
| 15 | 36 | OH |
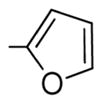
|
8.7 ± 0.8 | 0.6 ± 0.2 | 3.6 | 3.4 | 0.9 |
| 16 | 37 | OH |
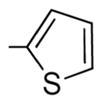
|
ND | 0.5 ± 0.2 | 0.31 | 0.36 | 1.2 |
| 17 | 42 | OH |
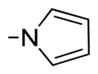
|
ND | ND | 2.6 | 3.3 | 1.3 |
| 18 | 39 | OH |
|
ND | ND | 0.38 | 2.3 | 6.0 |
| 19 | 43 | OH | CN | ND | ND | 1.1 | 3.0 | 2.7 |
| 20 | 40 | OH |
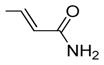
|
ND | ND | 2.6 | 2.3 | 0.9 |
| 21 | 46 | OH |
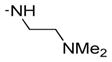
|
ND | ND | 29 | 36 | 1.2 |
| 22 | 47 | OH |
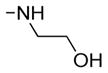
|
ND | ND | 3.1 | 2.9 | 0.9 |
| 23 | 49 | OH | NH2 | ND | ND | 1.9 | 8.8 | 4.6 |
The tubulin concentration was 10 μM. Inhibition of extent of assembly was the parameter measured (n = 2).
Cells were grown for 48 h at 37 °C in a humidified 5%CO2 atmosphere. Cell protein was the parameter measured. At least two independent experiments were performed with each compound.
For activated growth conditions HUVEC cells were seeded at 2500 and 500 cells/well, respectively, and cultured in EGM-2 medium (Lonza) or F12K medium containing 0.03 mg/mL endothelial cell growth supplement.
For quiescent growth conditions HUVEC and HAAE-1 cells were seeded at 15 000 and 5000 cells/well, respectively, in basal medium (EBM-2 or F12K) containing 0.5% fetal calf serum and antibiotics.
Selectivity ratio (IC50 quiescent)/(IC50 activated).
Compound Synthesis
The compounds generated in the course of this work were prepared either by use of a multicomponent coupling (MCC) reaction or by a modified Larocktype coupling (Table 2 and Scheme 1).17,24,25 The one-pot MCC reaction involves deprotonation of an o-iodophenol 16 and a terminal alkyne 17 with methylmagesium chloride to generate a magnesium phenolate and magnesium acetylide that then couple to give 18 in the presence of a catalytic amount of Pd(PPh3)2Cl2 (3 mol %) in THF at reflux (Table 2). The THF is then exchanged for DMSO. The third coupling partner, aryl iodide 19, is introduced and the reaction mixture heated under an atmosphere of CO (g) (balloon). This produces a carbonylative, heteroannulative coupling reaction between 18 and 19, giving the 3-aroylbenzo[b]furans 20–24 in moderate yields (30–55%).17 This step involves initial oxidative insertion of the palladium into the C–I bond of 19, followed by CO insertion to give an electrophilic ArC(O)PdI complex that coordinates to the alkyne bond of 18, promoting cyclization to give a 3-[ArC(O)Pd]-benzo[b]furan complex. Reductive elimination then affords the 3-aroylbenzo[b]furans 20–24. A number of products 20–24 required deprotection in order to afford the desired test compounds. The benzyl protecting groups in 20 and 22 were cleaved by hydrogenation to give 25 (34%) and 26 (63%), respectively.17 The isopropyl ether protecting groups in 23 and 24 were cleaved using AlCl3 in dichloromethane, giving 27 (51%) and 28 (75%), respectively.17
Table 2.
MCC Reaction
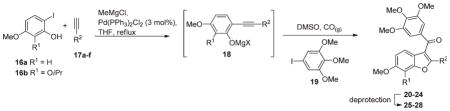 | |||||
|---|---|---|---|---|---|
| entry | 16 R1 |
17 R2 |
product (yield%) | deprotection conditions | product (yield%) R1, R2 |
| 1 |
16a H |
17a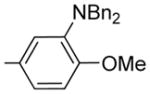
|
20 (31) | Pd/C, H2(g) |
25 (34)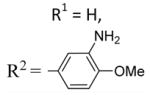
|
| 2 |
16a H |
17b
|
21 (32) | – | – |
| 3 |
16a H |
17c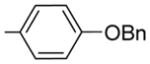
|
22 (30) | Pd/C, H2(g) |
26 (63)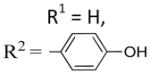
|
| 4 |
16b OiPr |
17d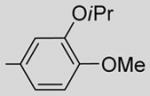
|
23 (46) | AlCl3, CH2Cl2 |
27 (51)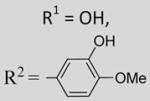
|
| 5 |
16b OiPr |
17f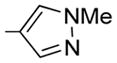
|
24 (55) | AlCl3, CH2Cl2 |
28 (75)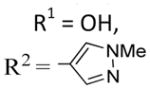
|
Scheme 1.

Modified Larock Coupling
In order to better explore the SAR of the C2 position of the benzo[b]furan lead series (see below) and to aid in the generation of a focused library of active compounds, we devised a concise method for the preparation of 2-substituted benzo[b]-furans (Scheme 1). This approach involves a modified Larocktype coupling between o-iodophenol 16a,b and the 3-silyl-1-arylpropynone 29 to give 2-silylbenzo[b]furans 30a,b.25,26 Silanes 30a,b were subjected to treatment with TBAF in methanol to remove the silyl groups and, in the case of 30b, reaction with AlCl3, to remove the isopropyl group, giving 31a and 31b in good yields (83% and 86%, respectively). Bromo-desilylation of 30a was achieved in reasonable yield to give the 2-bromobenzo-[b]furan 33a (59%).25 In the case of 30b, it was necessary to first exchange an isopropyl for an acetyl group (32) in order to avoid competitive C4-bromination of the benzo[b]furan in the bromo-desilylation step. Subsequent conversion of 32 to bromide 33b proceeded smoothly (69%).25 The bromo substituent in 33a and 33b proved to be a versatile functionality that could be readily substituted by way of palladium mediated coupling or by nucleophilic displacement to produce a host of analogues 35–48 (Methods A–E).17,25 The acetyl group was generally cleaved under the reaction conditions used for C2-substitution or by methanolysis (MeOH, K2CO3) of the crude product. In the case of 48, hydrogenation of the benzyl substituent was required to afford the desired test compound, amine 49.17
Biological Evaluation
The endothelial cells within tumors are constantly exposed to proangiogenic growth factors and, as a consequence, are in a constant state of activation and angiogenesis.27 Since we were primarily interested in identifying a new compound capable of selective inhibition of tumor over normal endothelial cells, all new compounds were screened for their capacity to selectively inhibit human umbilical vein endothelial cells (HUVECs) grown in the presence (activated) or absence (quiescent) of growth factors (Table 3). Key compounds were also evaluated for their capacity to inhibit tubulin polymerization and the proliferation of MCF-7 cancer cells (Table 3).
In our SAR studies of the benzo[b]furan lead structure II (Table 3), we first sought to investigate whether the presence of a C2-aryl group (D-ring) in II influences its mode of binding and which of the two rings B or D in II equates to the B-ring in 4. To do this, we generated three new D-ring analogues of 12, compounds 21, 25, and 26, all of which should be capable of a similar pseudo-π-stacking interaction with the A-ring as seen in the crystal structure of 10 (entries 3–5, Table 3). If the D-ring in II plays the role of the B-ring in 4, then based on previous SAR studies of 4,28–30 compounds 25 and 21 should be of similar potency to 12, whereas 26 should be inactive. However, all new analogues 21, 25, 26 exhibited similar potency to 12 (compare entries 2–5, Table 3). Furthermore, these compounds were all of similar potency to the C2-unsubstituted analogue 31a (a benzo-[b]furan equivalent of 15) (compare entries 2–6, Table 2). These data indicate that while the D-ring in II is tolerated, it does not contribute to the potency of this series. Most likely, all compounds II present a similar pose to tubulin irrespective of whether a C2-aryl substituent (D-ring) is present or not (see also docking studies below). Thus, the B-ring in I/II plays an equivalent role to the B-ring in 4 and the presence of a C2-aryl does not influence this conformation. This is also consistent with our earlier observation that a C6-methoxy is required for the potency of I (X = S), even when a C2-aryl is present.19 An alternative explanation for the increase in potency of 11 relative to 10 (entries 2 and 3, Table 2) may be that the inhibitory effects of 10 are limited by poor solubility at higher assay concentrations and that the presence of a hydroxyl in 11 improves solubility.
Satisfied that the B-ring in II corresponds to the B-ring in 4, irrespective of the presence of a C2-aryl, we next investigated the effect of introducing a hydroxyl group ortho to the C6-methoxy in 31a and 12, giving 31b and 27, respectively (entries 7 and 8, Table 3). We introduced the hydroxyl to the C7-position rather than the C5-position, since superimposing 4, colchicine, and 31b indicated that this would be the most appropriate location (Figure 2). The introduction of a C7-OH to 31a, giving 31b, was tolerated and afforded a compound of similar potency to 31a, which was slightly more selective for activated over quiescent endothelial cells (entry 7, Table 3). This is consistent with the SAR of 4, where removal of the B-ring hydroxyl has little effect on potency.30 Interestingly, the introduction of a C7-OH to indole 15 has recently been shown to reduce potency (~20-fold increase in IC50 values across multiple cell lines), albeit to a lesser degree than a C5-OH.31 While 31a and 31b are of similar potency, the introduction of a C7-OH to 12 afforded a much more potent compound, 27 (compare entries 2 and 8, Table 3). This 10-fold increase in potency is not attributed to any particular pharmacophoric feature of the C2-aryl in 27, as many other C2-substituted systems bearing a C7-OH exhibit similar levels of potency (compare entries 8–23, Table 3). Morevover, the SAR data accumulated in this study indicate a synergistic (>additive) effect for the introduction of both a C7-OHand a C2-substituent. This synergism is apparent for a range of different C2-substitutents where the antiproliferative activity of lead series II exhibits a SAR as follows: [R1 = OH,R2 ≠ H]≫[R1=H, R2=H]≈[R1 = OH,R2 = H] ≈ [R1 = H, R2 ≠ H] (compare entries 2, 6, 7, and 8, Table 3)
Figure 2.
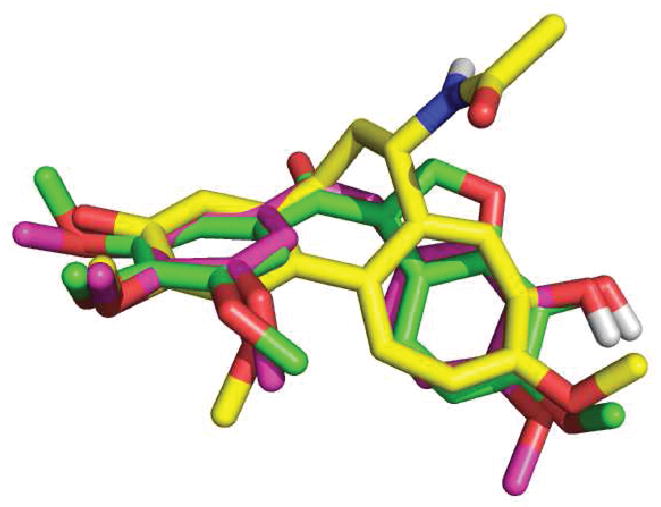
Superpositioning of colchicine 1 (yellow), 4 (pink), and 27a (green). Oxygen atoms are red. Nitrogen atoms are blue.
Presumably, the C2-substituent in II plays a steric role, favoring cisoid-II over transoid-II because of increased steric compression in the latter, forcing the A- and B-rings into the preferred orientation for binding to tubulin (Figure 3). This is supported by quantum chemical calculations on 31b (II, R1 = OH, R2 = H) and 8 (II, R1 = OH, R2 = Me). Calculations using the B3LYP method with the 6-311g** basis set show that although the energy differences are not large, in 31b the transoid conformation is favored by 1.1 kJ/mol while in compound 8 the cisoid conformation is favored by 0.3 kJ/mol.
Figure 3.
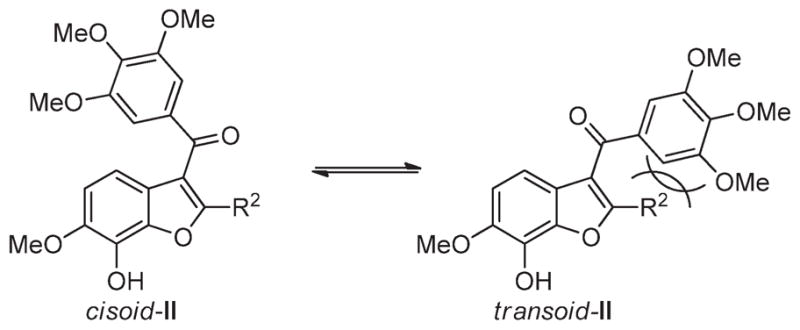
Cisoid and transoid rotamers of II.
Generally speaking, both hydrophobic and hydrophilic substituents are well tolerated in the C2-position of II, giving potent antiproliferative compounds in most cases, particularly for C7-OH analogues (entries 7–23, Table 3).32 Nonetheless, some C2 substituents had significant effects on potency. The presence of a strong base at C2, as in compounds 45 and 46, tended to reduce potency (entries 14 and 21, respectively, Table 3). At this stage, it is uncertain as to whether this is as a result of reduced affinity of these compounds for the target or due to partial sequestration of the active into acidic vesicles in the cell, reducing the concentration in the proximity of the microtubules. Additionally, certain five-membered ring heterocycles in the C2-position, furan 36, thiophene 37, and N-methylpyrazole 28, afforded very potent compounds (entries 10, 15, and 16, respectively, Table 3). Of all the C2-substitutients evaluated,32 only the C2-methyl 8 showed any significant selectivity for activated HUVECs over quiescent HUVECs (81-fold) (entry 9, Table 3). 8 also exhibited good potency against MCF-7 cancer cells (IC50 = 2.4 nM).
Notwithstanding that there is some variation in activity and selectivity as a function of C2-substitution in II, the broader tolerance to structural variation at this position indicates that it is probably oriented away from the binding pocket, directing its substituent into an area of relative “free space” (solvent water).32 This and other features of the SAR were supported by docking studies. Using the X-ray crystal structure of bovine α,β-tubulin dimer complexed to podophylotoxin (PDB entry 1SA1),33 we used Glide34 to dock a variety of benzo[b]furan compounds into the colchicine site (Figure 4), which lies within the β-tubulin subunit adjacent to its interface with α-tubulin.35 The docked structures were oriented so that their 3,4,5-trimethoxyphenyl (A) rings overlapped with that of colchicine and the C6-OMe and C7-OH substituents overlapped with the methoxy and carbonyl groups on the tropone ring of colchicine. The benzo[b]furan C7- OH group makes a hydrogen bond to the side chain of Asn β258, which also forms a hydrogen bond to the backbone amide nitrogen of Val 181 in the adjacent α-tubulin subunit. The C2-position of the benzo[b]furan is oriented toward a gap between the α- and β-tubulin subunits which can accommodate large substituents. The D-ring of compound 27 projects into this gap, lying between residues Thr α179 and Leu β248.
Figure 4.

Comparison between the crystal-bound orientation of colchicine (derived from 3E22) (A) and docked orientations of 31b (B), 8 (C), and 27 (D). The tubulin β subunit is shown with light blue carbon atoms. Residue Thr α179 is shown in dark blue. Note the number scheme used is that described in ref 33.
In light of the favorable potency and selectivity exhibited by 8, we subjected this compound to further in vitro and in vivo biological evaluation. Compound 8 exhibited excellent potency against a panel of different cancer cell lines (Table 4). The selectivity observed for 8 against activated over quiescent HUVECs was also observed in human aortic arterial endothelial cells (HAAECs). Again, this selectivity was not seen with 4. Furthermore, 8 generally exhibited greater potency than 4, up to 10-fold in most cases.
Table 4.
Inhibition of Cell Proliferation by 8 and 4
| entry | cell linea | cell type | 8 IC50 (nM) | 4 IC50 (nM) |
|---|---|---|---|---|
| 1 | activated HAAE-1 | endothelial cell | 0.1 (120)b | 2.2 (1.7)b |
| 2 | quiescent HAAE-1 | endothelial cell | 12 | 1.3 |
| 3 | U87-MG | brain glioblastoma | 0.41 | 2.6 |
| 4 | DU145 | prostate carcinoma | 0.36 | 4 |
| 5 | Calu-6 | lung anaplastic carcinoma | 0.16 | 0.94 |
| 6 | MDA-MB-231 | breast adenocarcinoma | 0.63 | 3.2 |
| 7 | A431 | epidermoid carcinoma | 18.6 | 188 |
| 8 | A375 | malignant skin melanoma | 1.5 | 2.1 |
| 9 | SKOV-3 | ovary adenocarcinoma | 0.59 | 5.5 |
| 10 | LoVo | colorectal adenocarcinoma | 0.24 | 2.9 |
| 11 | AU565 | breast adenocarcinoma | 5.8 | 4.4 |
| 12 | BT549 | breast carcinoma | 0.34 | 3.5 |
HAAE-1 cells were seeded at 500 cells/well in medium containing 0.03 mg/mL endothelial cell growth supplement (activated) or in basal medium containing 0.5% fetal calf serum and antibiotics (quiescent). Cancer cell lines were seeded at an average of 500–2000 cells/well and cultured as recommended by the ATCC.
Selectivity ratio (IC50 quiescent)/(IC50 activated) in parentheses.
We also evaluated 8 for multidrug resistance. Acquired resistance of human cancer cells to chemotherapeutic agents is one of the major causes of treatment failure.36 A key mechanism of acquired resistance is the increased expression of the transporter, multidrug resistance protein MDR-1 (P-glycoprotein). This transporter effluxes drug agents from the cytoplasm of the cell, reducing the drug concentration to ineffective levels. Since more than 80% of current chemotherapeutics are substrates for MDR- 1, it is important to identify new chemotherapeutic agents that are not MDR-1 substrates and that are able to treat multidrug resistant phenotypes. The renal carcinoma cell line 786-0 has high levels of expression ofMDR-1, and the potency of drugs that are MDR-1 substrates is significantly reduced by this transporter. The three key tubulin targeting drugs paclitaxel, vincristine, and vinblastine are all known to be MDR-1 substrates and show enhanced activity against 786-0 when coadministered with the MDR-1 inhibitor verapamil (Table 5). By contrast, 8 was equipotent against 786-0 cells with and without verapamil, indicating that 8 is not a substrate for the MDR-1 transporter (Table 5).
Table 5.
Effect of MDR-1 Inhibition on the Potency of Antitubulin Drugs toward Renal Carcinoma Cell Line 786-0
| conditions | drug IC50 (nM) against 786-0 cells
|
|||
|---|---|---|---|---|
| paclitaxel | vincristine | vinblastine | 8 | |
| drug alone | 31 | 37 | 4.5 | 0.58 |
| drug + verapamil | 4.0 | 1.2 | 0.31 | 0.39 |
MDR-1 overexpression is also a key element of the multidrug resistance acquired by the human ovarian cancer cell line A2780 upon continuous exposure to adriamycin, producing the A2780ADR phenotype.37 Both doxorubicin and cisplatin, which are MDR-1 substrates, showed reduced potency toward this cell line, whereas 8 exhibited similar potency against both A2780 and A2780ADR (Table 6). Compound 8 also exhibited good potency toward the cisplatin resistant cell line A2780cis (Table 6). A2780cis cells exhibit cross-resistance to a number of cytotoxins and radiation in an MDR-1 independent manner.37
Table 6.
Potency of Cytotoxic Drugs toward Resistant Phenotypes of A2780
| cell line | drug IC50 (nM)
|
||
|---|---|---|---|
| doxorubicin | cisplatin | 8 | |
| A2780 | 1.2 | 329 | 0.03 |
| A2780ADR | 194 | 1103 | 0.06 |
| A2780cis | 5.0 | 5021 | 0.01 |
In order to achieve a dualmode agent, capable of acting as a VDA and an antimitotic, we were interested in producing a compound that exhibits increased exposure relative to 4 to improve cell kill levels through the antimitotic effect. Compared with 4, 8 exhibits a longer half-life and larger AUC0–∞ in pharmacokinetic studies in rats (Table 7). It also exhibits a lower level of plasma protein binding (PPB) than 4, resulting in a significantly higher free drug exposure of 8 relative to 4. This increased exposure is expected to produce an increased cell kill through the antimitotic effect of 8, particularly at the viable rim of the tumor that survives vascular disruption.
Table 7.
Pharmacokinetic Properties of 4 and 8
| parameter | compda |
|
|---|---|---|
| 4 | 8 | |
| dose (μmol/kg) | 6 | 6 |
| t1/2 (h) | 0.7 | 3.5 |
| VSS (L/kg) | 7.3 | 7.1 |
| AUC0–∞ | 36 | 48 |
| PPB (% bound) | 98.9 | 81.4 |
Plasma concentration–time profiles are provided in the Supporting Information.
The poor aqueous solubility of 8 necessitated its conversion to the disodium phosphate ester 9, providing a soluble prodrug for in vivo administration (Scheme 2), as has been done for 4 (5) and 2 (3) (Figure 1).38 Prodrug 9 was prepared from 8 by carbon tetrabromide mediated esterification with dibenzyl phosphite to give the dibenzyl phosphate ester 50. Ester 50 was converted to 9 upon sequential treatment with trimethylsilyl bromide and sodium methoxide. Pharmacokinetic evaluation of 9 in rats and mice revealed that it is rapidly converted to 8 and that it achieves similar levels of drug exposure (tumor and other tissues) compared to direct administration of 8.39
Scheme 2.

Synthesis of Prodrug 9
In our previous report on the in vivo efficacy of 9 in murine tumor models, we demonstrated that 9 was more effective at tumor vasculature disruption than the disodium phosphate prodrug of combretastatin A-4 5. Also, in head-to-head studies on tumor growth inhibition, we also observed that 9 is a much more effective inhibitor of tumor growth than 5 (Figure 5). When both compounds were dosed at half their maximum tolerated dose (½MTD) in a murine breast tumor model,40 9 achieved a much greater reduction in tumor growth than did 5 (Figure 5). Furthermore, 9 could achieve a similar reduction in tumor growth at 10 mg/kg (⅛MTD) as was achieved by 5 at 150 mg/kg (½MTD). As previously reported, continued dosing of 9 at 40 mg/kg (½MTD) completely suppresses tumor growth and leads to complete eradication of tumors in 14% of the mice after 70 days.16
Figure 5.
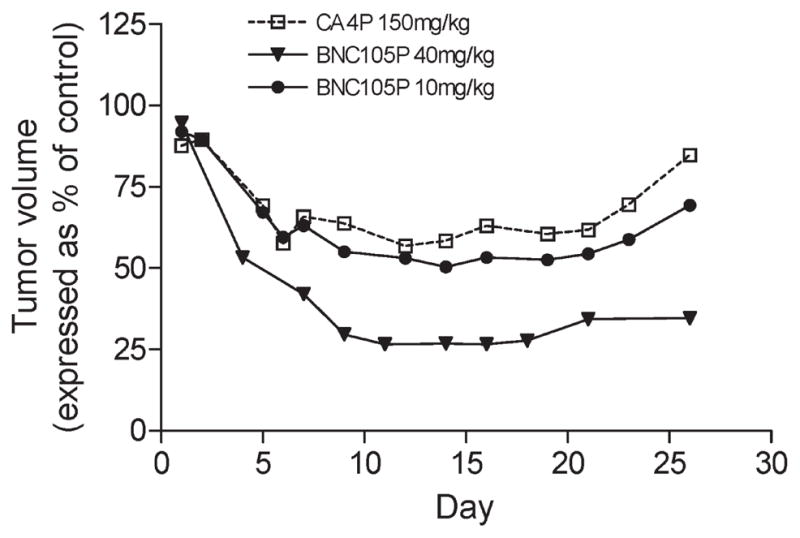
Tumor growth inhibition by 9 and 5 in a MDA-MB-231 breast cancer model: 5 at 150 mg/kg (½MTD), dosed on days 1 and 5 (□); 9 at 10 mg/kg (⅛MTD), dosed days 1 and 5 (●); 9 at 40 mg/kg (½MTD), dosed days 1 and 8 (▼).
The superior efficacy and therapeutic index of 9, relative to 5, can be attributed to a combination of 8’s greater selectivity, potency, and in vivo exposure. The superior vascular disrupting properties of 9, relative to 5, may be derived from 8’s greater potency and higher free drug fraction. The longer half-life exhibited by 8 (3.5 h), relative to 4 (0.7 h), should enable it to achieve a higher cell kill at the viable rim, as a higher proportion of cells will cycle through the most sensitive phase of the cell cycle (G2-M check point) in the presence of this antimitotic agent. Unlike colchicine, though, 8 is completely cleared from healthy tissues over 24 h, which, in combination with its higher selectivity for activated over quiescent phenotypes, minimizes its toxicity.16 As previously reported, 8 is trapped in tumor tissue because of the vascular shutdown effect, such that 64% of drug remains in the tumor after 24 h.16
CONCLUSION
The potency of early leads I has been improved by the introduction of a C7-OH. The presence of both a C7-OH and a C2-substituent are required for optimal potency. The C2-substituent sterically interacts with the trimethoxybenzoyl unit in II, favoring the required cisoid-conformer (cisoid-II, Figure 3). The C7-OH group enables an additional hydrogen bonding interaction with Asn β258 to be achieved. Only when both effects are engendered (C7-OH + C2-substituent) is a significant increase in potency observed (>10 fold). This SAR is maintained across a broad range of C2-subsitutents, and a focused library of potent C2-modified analogues of II has been screened for compounds exhibiting selectivity for activated over quiescent endothelial cells. Compound 8 exhibited good selectivity toward activated endothelial cells and was also more potent than 4 against a panel of cancer cell lines. Studies directed toward the molecular basis of this selectivity are ongoing. Compound 8 also exhibited excellent potency against a range of multidrug resistant phenotypes and does not appear to be a substrate for the efflux pumpMDR-1. The superior in vivo efficacy of 8, relative to 4, is attributed to a combination of its higher potency and greater exposure, enabling it to elicit both vascular disrupting and antimitotic modes of action in vivo. Prodrug 9 is currently undergoing phase II clinical trials for mesothelioma and renal cell carcinoma.41
EXPERIMENTAL SECTION
Chemistry
Melting points were recorded with an Electrothermal melting point apparatus. Proton (1H) and carbon (13C) NMR spectra were recorded at 300 and 75 MHz, respectively. Liquid chromatography–mass spectrometry (LCMS) was conducted with a C8 column (4.6 mm ×150 mm), using an isocratic mobile-phase containing 49% acetonitrile, 50% and 315 water, and a 1% ammonium formate solution (this solution was made up of 1 g of acetic acid and 315 mg of ammonium formate in 1 L of 33% methanol in water) at a constant flow rate of 0.5 mL/min. UV detection was measured at 214 nm, and MS analysis was conducted using an atmospheric pressure ionization (APCI) ion source. High-resolution mass spectrometry (HRMS) was performed on a time-of-flight mass spectrometer fitted with an electrospray (ESI) ion source. Tetrahydrofuran (THF) and diethyl ether were distilled under nitrogen from sodium benzophenone ketyl. Dichloromethane and 1,2-dichloroethane were distilled from calcium hydride under nitrogen. Analytical thin layer chromatography (TLC) was conducted on aluminum sheets coated with silica gel 60 GF254. Flash chromatography was performed on flash grade silica gel. Experimental methods for the following compounds has been previously reported: 8, 16a, 16b, 30a, 30b, 31a, 32, 33a, 33b, 35, 36, 44, 47.24 All test compounds (Table 3) exhibited >95% purity by LCMS except 26, which was 91% pure by LCMS.
General Procedure A: Multicomponent Coupling of 16, 17, and 19
To a solution of 2-iodophenol 16 (1 mmol) and alkyne (1.2 mmol) in dry THF (5 mL) under nitrogen at 0 °C was added MeMgCl (3.0 M solution in THF, 2.5 mmol), and the mixture was allowed to warm to room temperature. Pd(Ph3P)2Cl (3 mol %) was added and the mixture heated to 65 °C for 4–8 h (monitoring by TLC). The reaction mixture was then cooled to room temperature and the THF removed under vacuum. Aryl iodide 19 (1.1 mmol) was added, and the mixture was dissolved in DMSO (12 mL/mmol). The nitrogen atmosphere was replaced with carbon monoxide (balloon). The aryl iodide (1.05 mmol) was added, and the mixture was heated to 80 °C overnight, then quenched with 10% NH4Cl (aq) and extracted with ethyl acetate. The organic layer was washed with brine and the solvent removed under vacuum. The residue was concentrated onto silica gel and purified by flash column chromatography.
2-(3-N,N-Dibenzyl-4-methoxy-phenyl)-6-methoxy-3-(3,4, 5-trimethoxybenzoyl)benzo[b]furan 20
20 was prepared from 16a, 17a, and 19 in accordance with general procedure A. Flash chromatography: silica gel, eluent = hexane/diethyl ether 8:2, yield 31%. 1H NMR (300 MHz, CDCl3) δ 7.50 (d, J = 8.71 Hz, 1H), 7.27–7.05 (m, 16H), 6.88 (dd, J = 8.68, 2.23 Hz, 1H), 4.03 (br s, 4H), 3.87 (s, 6H), 3.80 (s, 3H), 3.58 (s, 6H).
6-Methoxy-2-(N-methyl-5-indolyl)-3-(3,4,5-trimethoxybenzoyl) benzo[b]furan 21
21 was prepared from 16a, 17b, and 19 in accordance with general procedure A. Flash chromatography: silica gel, eluent = hexane/diethyl ether 8:2, yield 32%. 1H NMR (300 MHz, CDCl3) δ 7.91 (d, J = 1.19 Hz, 1H), 7.60 (d, J = 8.65 Hz, 1H), 7.32–7.36 (m, 2H), 7.14 (d, J = 8.63 Hz, 1H), 7.11 (s, 2H), 7.01 (d, J = 3.11 Hz, 1H), 6.92 (dd, J = 9.95, 2.27 Hz, 1H), 6.41 (d, J = 3.08 Hz, 1H) 3.89 (s, 6H), 3.76 (s, 3H), 3.74 (s, 3H), 3.61 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 191.0, 159.1, 158.0, 154.5, 152.4, 141.7, 136.7, 132.6, 129.6, 127.9, 122.0, 121.9, 121.27, 120.8, 113.9, 112.1, 108.7, 107.05, 101.6, 95.4, 60.4, 55.6, 55.4, 32.5. LCMS: tR = 2.71 min, >99%. MS m/z = 472 (M + H)+, 100%. HRMS: calcd for C28H26NO6+ = 472.1760, found = 472.1761.
2-(4-Benzyloxyphenyl)-6-methoxy-3-(3,4,5-trimethoxybenzoyl) benzo[b]furan 22
22 was prepared from 16a, 17c, and 19 in accordance with general procedure A. Flash chromatography: silica gel, eluent = hexane/diethyl ether, 8:2 and then 7:3, yield 29%. 1H NMR (300 MHz, CDCl3) δ 7.54 (d, J = 8.96 Hz, 2H), 7.49 (d, J = 8.67 Hz, 1H), 7.36–7.38 (m, 5H), 7.11 (s, 2H), 7.07 (d, J = 2.19 Hz, 1H), 6.90 (dd, J = 8.60, 2.20 Hz, 1H), 6.86 (d, J = 8.92 Hz, 2H), 5.04 (br s, 2H) 3.87 (s, 3H), 3.86 (s, 3H), 3.67 (s, 6H).
7-Isopropoxy-2-(3-isopropoxy-4-methoxyphenyl)-6-methoxy- 3-(3,4,5-trimethoxybenzoyl)benzo[b]furanbenzo[b]furan 23
23 was prepared from 16b, 17d, and 19 in accordance with general procedure A. Flash chromatography: silica gel, eluent = hexane/diethyl ether, 8:2 and then 7:3, yield 46%. 1H NMR (300 MHz, CDCl3) δ 7.26 (d, J = 8.58 Hz, 1H), 7.18 (dd, J = 8.44, 2.14 Hz, 1H), 7.13 (s, 2H), 7.07 (d, J = 2.02 Hz, 1H), 6.93 (d, J = 8.65 Hz, 1H), 6.81 (d, J = 8.52 Hz, 1H), 4.77 (quintet, 1H), 4.22 (quintet, 1H), 3.91 (s, 3H), 3.85 (s, 3H), 3.83 (s, 3H), 3.68 (s, 6H), 1.42 (d, J = 6.17 Hz, 6H), 1.23 (d, J = 6.17 Hz, 6H).
7-Isopropoxy-6-methoxy-2-(4-N-methylpyrazolyl)-3-(3,4, 5-trimethoxybenzoyl)benzo[b]furan 24
24 was prepared from 16b, 17f, and 19 in accordance with general procedure A. Flash chromatography: silica gel, eluent = hexane/diethyl ether, 8:2 and then 7:3, yield 55%. 1H NMR (300 MHz, CDCl3) δ 8.12 (s, 1H), 7.98 (s, 1H), 7.13 (s, 2H), 6.84 (d, J = 8.63 Hz, 1H), 6.79 (d, J = 8.67 Hz, 1H), 4.73 (quintet, 1H), 3.93 (br s, 6H), 3.88 (s, 3H), 3.77 (s, 6H), 1.41 (d, J = 6.16 Hz, 6H). LCMS: tR = 2.27 min, >99%. MS m/z = 481 (M + H)+, 100%. HRMS: calcd for C24H26NO7 + = 481.1975, found = 481.1974.
2-(3-Amino-4-methoxyphenyl)-6-methoxy-3-(3,4,5-trimethoxybenzoyl) benzo[b]furan 25
Catalyst 10% Pd/C (60 mg) was added to a solution of 20 (60 mg, 0.093 mmol) in ethyl acetate (12 mL), methanol (5 mL), and water (2.5 mL), followed by 1 drop of HCl (aq) (6 M), and the reaction mixture was stirred under an atmosphere of hydrogen (balloon) for 1 h. The mixture was filtered through Celite and washed with dichloromethane (2 × 5 mL) and the solvent removed under vacuum. The crude product was purified by preparative layer chromatography (eluent = hexane/ethyl acetate/triethylamine 6:4:1) to give a yellow paste that was recrystallized from hexane and dichloromethane (vapor diffusion) to afford 25 as a yellow solid (14.7 mg, 34%). 1H NMR (300 MHz, CDCl3) δ 7.47 (d, J = 8.64 Hz, 1H), 7.12 (s, 2H), 7.09–7.05 (m, 2H), 6.99 (dd, J = 8.39, 1.57 Hz, 1H), 6.88 (dd, J = 8.41, 2.18 Hz, 1H), 6.65 (d, J = 8.38 Hz, 1H), 6.31 (br s, 2H), 3.86 (br s, 6H), 3.81 (s, 3H), 3.70 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 190.7, 158.1, 157.0, 154.2, 152.5, 148.4, 142.0, 134.7, 132.4, 122.3, 121.7, 121.3, 119.9, 114.4, 114.3, 112.2, 109.6, 107.0, 95.3, 60.6, 55.8, 55.4, 55.2. LCMS: tR = 2.14 min, >99%. MS m/z = 464 (M + H)+, 100%. HRMS: calcd for C26H26NO7+ = 464.1709, found = 464.1707.
2-(4-Hydroxyphenyl)-6-methoxy-3-(3,4,5-trimethoxyben zoyl)benzo[b]furan 26
A mixture of 30 (45 mg, 0.09 mmol) and Pd/C (10%, 40 mg) in a mixture of ethyl acetate (7 mL) and triethylamine (3 drops) was stirred under an atmosphere of hydrogen for 1 h. The mixture was filtered through Celite and washed with dichloromethane (2 × 5 mL), and the solvent was removed under vacuum. Purification by flash chromatography (silica gel, eluent = hexane/ethyl acetate, 6:4) gave a yellow paste that was recrystallized from hexane and dichloromethane (vapor diffusion) to afford 26 as a yellow solid (23.2 mg, 63%). 1H NMR (300 MHz, CDCl3) δ 7.49 (dd, 2 × J = 8.36 Hz, 3H), 7.11 (s, 2H), 7.07 (d, J = 2.06 Hz, 1H), 6.89 (dd, J = 8.62, 2.18 Hz, 1H), 6.74 (d, J = 8.69 Hz, 2H), 3.87 (s, 3H), 3.86 (s, 3H), 3.69 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 191.0, 158.1, 157.4, 157.2, 154.3, 152.5, 142.2, 132.3, 129.7, 127.1, 121.7, 121.3, 115.2, 114.1, 112.3, 107.3, 95.4, 60.6, 55.8, 55.4. LCMS: tR = 2.38 min, >90%. MS m/z = 435 (M + H)+, 38%; 157, 100%. HRMS: calcd for C25H23NO7+ = 435.1444, found = 435.1442.
7-Hydroxy-2-(3-hydroxy-4-methoxyphenyl)-6-methoxy-3-(3,4,5-trimethoxybenzoyl)benzo[b]furan 27
To a solution of 23 (62 mg, 0.11 mmol) in dry dichloromethane (3 mL) was added AlCl3 (37 mg, 0.28 mmol). The reaction mixture was stirred vigorously at room temperature for 5 h. Another portion of AlCl3 (18 mg, 0.14 mmol) was added, and stirring continued for 20 min (TLC). The reaction was quenched with saturated NH4Cl (aq), and the aqueous layer was extracted with ethyl acetate (20 mL). The organic layer was washed with water (15 mL), dried over MgSO4, and concentrated under vacuum. The crude product was purified by flash chromatography (silica gel, gradient elution = hexane/diethyl ether 2:8, then neat diethyl ether) to afford 27 as a crystalline yellow solid (27mg, 51%). 1HNMR(300 MHz, CDCl3) δ 7.25 (d, J = 2.34 Hz, 1H), 7.12 (d, J = 8.92 Hz, 1H), 7.10 (dd, J = 8.36, 2.14 Hz, 1H), 7.10 (s, 2H), 6.91 (d, J = 8.59Hz, 1H), 6.70 (d, J = 8.45Hz, 1H), 5.71 (br s, 1H), 5.52 (br s, 1H), 3.96 (s, 3H), 3.86 (s, 3H), 3.85 (s, 3H), 3.69 (s, 6H). LCMS: tR = 1.62 min, >99%. MS m/z = 481 (M + H)+, 100%. HRMS: calcd for C26H24O9+ = 481.1499, found = 481.1501.
7-Hydroxy-6-methoxy-2-(4-N-methylpyrazolyl)-3-(3,4,5-trimethoxybenzoyl)benzo[b]furan 28
To a solution of 24 (85 mg, 0.18 mmol) in dry dichloromethane (3 mL) was added AlCl3 (48 mg, 0.36 mmol). The reaction mixture was stirred vigorously at room temperature for 10 min. Another portion of AlCl3 (10 mg, 0.08 mmol) was added, and stirring continued for 20 min (TLC). The reaction was quenched with saturated NH4Cl (aq), and the aqueous layer was extracted with ethyl acetate (20 mL). The organic layer was washed with water (15 mL), dried over MgSO4, and concentrated under vacuum. The crude product was purified by flash chromatography (silica gel, gradient elution = hexane/diethyl ether 2:8, then neat diethyl ether) to afford the title compound as a crystalline yellow solid (58 mg, 75%). 1H NMR (300 MHz, CDCl3) δ 8.15 (s, 1H), 8.03 (s, 1H), 7.13 (s, 2H), 6.79 (d, J = 8.62Hz, 1H), 6.69 (d, J = 8.58Hz, 1H), 3.92 (br s, 6H), 3.91 (s, 3H), 3.77 (s, 6H). LCMS: tR = 1.63min, >99%. MS m/z = 439 (M+ H)+, 100%. HRMS: calcd for C23H22NO7+ = 439.1505, found = 439.1510.
7-Hydroxy-6-methoxy-3-(3,4,5-trimethoxybenzoyl)benzo-[b]furan 31b
Step 1
Tetrabutylammonium fluoride (76.5 μL, 0.076 mmol, 1 M solution in THF) was added to a stirred solution of 30b24 (0.066 mmol) in THF (1 mL). The reaction mixture was stirred at room temperature for 20 min (TLC), diluted with ethyl acetate (10 mL), and washed with 1 M HCl (5 mL). The organic layer was dried over MgSO4, and the solvent was removed under vacuum. The crude product was purified by flash chromatography (silica gel, eluent = hexane/diethyl ether, 7:3) to afford the 3-(3,4,5-trimethoxybenzoyl)-6-methoxy-7-isopropoxybenzo[b]furan as a light yellow paste (23 mg) that was used directly in the next step. 1HNMR (300MHz,CDCl3) δ 8.00 (s, 1H), 7.78 (d, J = 8.60 Hz, 1H), 7.15 (s, 2H), 7.04 (d, J = 8.61Hz, 1H), 4.73 (m, 1H), 3.93 (s, 3H), 3.92 (s, 3H), 3.90 (s, 6H), 1.37 (d, J = 6.14 Hz, 6H).
Step 2
A solution of the product from step 1 (23 mg, 0.058 mmol) in dry dichloromethane (1 mL) was treated with AlCl3 (16 mg, 0.116 mmol). The reaction mixture was stirred for 0.5 h at room temperature, then quenched with saturated NH4Cl (aq) and extracted with ethyl acetate (10 mL). The organic layer was washed with water (5 mL), dried over MgSO4 and concentrated under vacuum. The crude product was purified by flash chromatography (silica gel, eluent = hexane/diethyl ether/ethyl acetate, 80:20:1) to afford 31b as a cream crystalline solid (18 mg, 86%). 1H NMR (300 MHz, CDCl3) δ 8.04 (s, 1H), 7.63 (d, J = 8.53 Hz, 1H), 7.14 (s, 2H), 7.02 (d, J = 8.38 Hz, 1H), 5.69 (s, 1H), 3.97 (s, 3H), 3.93 (s, 3H), 3.89 (s, 6H). 13CNMR(75 MHz, CDCl3) δ 188.7, 152.8, 151.2, 144.5, 143.3, 141.8, 134.0, 130.9, 120.92, 120.6, 112.4, 109.2, 106.1, 60.6, 56.9, 56.0. LCMS: tR = 1.73 min, >99%. MS m/z = 359 (M + H)+, 100%. HRMS: calcd for C19H19O7+ = 359.1131, found = 359.1126.
(E)-3-(7-Hydroxy-6-methoxy-3-(3,4,5-trimethoxybenzoyl)-benzofuran-2-yl)acrylamide 40
Pd(OAc)2 (5.0 mg, 0.02 mmol) was added to a solution of 33b (48 mg, 0.10 mmol) and acrylamide 38b (70 mg, 1 mmol) in a mixture of acetonitrile and triethylamine (2:1, 1.5 mL) and the resulting solution degassed (stirring solution exposed to a partial vacuum and backfilled with N2 gas 3 times). The reaction mixture was stirred at reflux for 1 h. More palladium acetate (5.0 mg, 0.02 mmol) was added, and refluxing continued for 7 h (monitored by TLC). The solvent was removed under vacuum and the residue subjected to flash chromatography (silica gel, eluent = hexane/diethyl ether 1:1) affording the title compound 40 as a yellow crystalline solid (7.3 mg, 17%). 1H NMR (300 MHz, CD3OD) δ: 7.48 (d, J = 15.5 Hz, 1H), 7.18 (s, 2H), 7.00 (d, J = 8.65 Hz, 1H), 6.94 (d, J = 15.5 Hz, 1H), 6.88 (d, J = 8.62 Hz, 1H), 5.46 (s, 1H), 4.55 (br s, 2H), 3.91 (s, 3H), 3.87 (s, 3H), 3.79 (s, 6H). LCMS: tR = 1.49 min, >99%. MS m/z = 424 (M + H)+, 100%. HRMS: calcd for C22H22NO8+ = 428.1345, found = 428.1345.
2-(2,3-Dihydro-1H-pyrrol-1-yl)-7-hydroxy-6-methoxy-3-(3,4,5-trimethoxybenzoyl)benzo[b]furan 42
To a solution of pyrrole (22 mg, 0.32 mmol) in dry THF (2 mL) was added sodium hydride (60%, 24 mg, 0.60 mmol), and the resulting suspension was stirred at room temperature for 24 h. To this solution 33b (50 mg, 0.10 mmol) was added, and the reaction mixture was stirred at room temperature for 18 h (monitored by TLC), quenched with saturated NH4Cl (aq) (10% w/v, 3 mL), extracted with dichloromethane (10 mL), dried over MgSO4, and concentrated under vacuum. The resultant residue was purified by planar chromatography on silica gel (eluent = hexane/ethyl acetate 3:1) to give the title compound 42 as a yellow solid (25 mg, 60%). 1H NMR (300 MHz, CDCl3) δ 8.47 (d, J = 1.21 Hz, 1H), 7.75 (d, J = 1.20 Hz, 1H), 7.27 (d, J = 8.60 Hz, 1H), 7.18 (d, J = 8.66 Hz, 1H), 7.05 (s, 2H), 6.17 (t, J = 2.3 Hz, 2H), 5.27 (s, 1H), 3.94 (s, 3H), 3.74 (s, 6H), 3.73 (s, 3H). LCMS: tR = 1.77 min, >99%. MS m/z = 424 (M +H)+, 100%. HRMS: calcd for C23H22NO7+ = 424.1396, found = 424.1396.
6-Methoxy-2-(4-N-methylpiperazino)-3-(3,4,5-trimethoxybenzoyl) benzo[b]furan 45
N-Methylpiperazine (50 μL, 0.45 mmol) was added to a stirred solution of 33a (17 mg, 0.041 mmol) in acetonitrile/dichloromethane 1:1 (2 mL), and the reaction mixture was stirred at room temperature for 1 h. After this time the solvent was removed under vacuum, and the resultant residue was purified by planar chromatography on silica gel (eluent, hexane/ethyl acetate 4:6 + 1% triethylamine) to give the title compound as a yellow paste (8 mg, 43%). 1H NMR (300 MHz, CDCl3) δ 7.07 (s, 2H), 6.93 (d, J = 8.60 Hz, 1H), 6.84 (d, J = 2.25 Hz, 1H), 6.65 (dd, J = 8.64, 2.30 Hz, 1H), 3.92 (s, 3H), 3.83 (s, 6H), 3.78 (s, 3H), 3.66–3.64 (m, 4H), 2.65–2.63 (m, 4H), 2.40 (s, 3H). 13CNMR(75MHz, CDCl3) δ 188.8, 162.2, 155.9, 152.7, 149.1, 141.1, 135.4, 121.7, 119.64, 110.4, 106.1, 155.9, 95.6, 60.6, 55.9, 55.4, 53.9, 47.1, 45.3. LCMS: tR = 1.78 min, >99%. MS m/z = 441 (M + H)+, 100%. HRMS: calcd for C24H29N2O6+ = 441.2026, found = 441.2030.
2-(2-Dimethylaminoethylamino)-7-hydroxy-6-methoxy-3-(3,4,5-trimethoxybenzoyl)benzo[b]furan 46
N,N-Dimethylethylenediamine (500 μL) was added to a stirred solution of 33b (50 mg, 0.10 mmol) in dry pyridine (2 mL) and the reaction mixture stirred at 80 °C for 1 h. The reaction mixture was cooled to room temperature, concentrated under vacuum and the residue purified by flash chromatography (silica gel, eluent hexane/ethyl acetate 4:1 with 1% triethylamine), affording the title compound as light yellow solid (25 mg, 54%). 1H NMR (300 MHz, CDCl3) δ 9.10 (br s, 1H), 6.95 (s, 2H), 6.60 (d, J = 8.44 Hz, 1H), 6.48 (d, J = 8.48 Hz, 1H), 3.90 (s, 3H), 3.84 (s, 3H), 3.83 (s, 6H), 3.82–3.83 (m, 2H), 2.65 (t, J = 5.63 Hz, 2H), 2.34 (s, 6H). LCMS: tR = 1.65 min, >99%. MS m/z = 445 (M + H)+, 100%. HRMS: calcd for C23H29N2O7+ = 445.1975, found = 445.1978.
2-Benzylamino-7-hydroxy-6-methoxy-3-(3,4,5-trimethoxybenzoyl) benzo[b]furan 48
When the reaction above was performed using benzylamine in the place of N,N-dimethylethylenediamine, product 48 was obtained as a light yellow solid (112 mg, 83%). 1H NMR (300 MHz, CDCl3) δ 9.27 (bs, NH, 1H), 7.39–7.32 (m, 5H), 6.96 (s, 2H), 6.61 (d, J = 8.47 Hz, 1H), 6.50 (d, J = 8.10 Hz, 1H,), 4.83 (d, J = 4.95 Hz, 2H), 3.91 (s, 3H), 3.85 (s, 3H), 3.84 (s, 6H).
2-Amino-7-hydroxy-6-methoxy-3-(3,4,5-trimethoxybenzoyl) benzo[b]furan 49
10% Pd/C (100 mg) was added to a solution of 48 (105 mg, 0.23 mmol) in a mixture of ethyl acetate/THF/water 3:2:1 (6 mL), and 2 drops of HCl (aq) (6 M) were added. The resultant mixture was stirred at room temperature for 2.5 h underH2 (g) (balloon). After this time, the mixture was filtered through Celite, the filtrate diluted with water (10 mL) and extracted with dichloromethane (5 mL × 3). The organic phase was dried over MgSO4 and concentrated under vacuum. The resultant residue was purified by flash chromatography (silica gel, eluent hexane/ethylacetate 4:1) to give 49 as a crystalline yellow solid (79 mg, 93%). 1H NMR (300 MHz, CDCl3) δ 6.98 (s, 2H), 6.93 (br s, 2H), 6.63 (d, J = 8.18 Hz, 1H), 6.52 (d, J = 8.41 Hz, 1H), 5.65 (br s, 1H), 3.92 (s, 3H), 3.86 (s, 3H), 3.84 (s, 6H). LCMS: tR = 1.628 min, >99%. MS m/z = 374 (M + H)+, 100%. HRMS: calcd for C19H20NO7+ = 374.1240, found = 374.1237.
Dibenzyl (6-Methoxy-2-methyl-3-(3,4,5-trimethoxybenzoyl) benzofuran-7-yl)phosphate 50
Triethylamine (0.295 mL, 2.12mmol) was added dropwise with stirring to a suspension of 8 (0.36 g, 0.97 mmol), CBr4 (0.383 g, 1.15 mmol), and dibenzyl phosphite (0.28 mL, 1.27 mmol) in anhydrous acetonitrile (5 mL) at 0 °C under a N2 (g) atmosphere. The resulting homogeneous mixture was stirred for 2 h at room temperature and evaporated to dryness under reduced pressure. The residue was diluted to 40 mL with ethyl acetate, washed with 0.1MHCl (10 mL), water (10 mL), brine (10 mL), and dried over anhydrous MgSO4. Filtration and evaporation of the filtrate under reduced pressure gave 0.68 g of crude product, which was purified by flash column chromatography (silica gel, hexane/ethyl acetate 7:3) to give of 50 as a cream solid (0.455 g, 74%). 1H NMR (300 MHz, CDCl3) δ 7.3–7.43 (m, 10H), 7.21 (d, J = 8.7 Hz, 1H), 7.08 (s, 2H, CH), 6.89 (d, J = 8.7 Hz, 1H), 5.32 (m, 4H,), 3.93 (s, 3H), 3.83 (s, 9H), 2.42 (s, 3H).
Disodium (6-Methoxy-2-methyl-3-(3,4,5-trimethoxybenzoyl) benzofuran-7-yl)phosphate 9
Bromotrimethylsilane (0.3 mL) was added dropwise to a stirred solution of 50 (0.455 g, 0.719 mmol) in anhydrous acetonitrile (5mL) at −5 °C under a N2 (g) atmosphere. The reaction mixture was allowed to warm to 0 °C over 1 h, then evaporated to dryness under reduced pressure. The residue was dissolved in 90% aqueous acetonitrile (15 mL) and evaporated to dryness. The crystalline residue was washed with a 50% hexane/dichloromethane mixture (20 mL) to give the free acid of 9 (0.302 g, 93%). This was suspended in anhydrous methanol (15 mL), and a solution of MeONa (0.073 g, 1.34 mmol) in methanol (1 mL) was added, with stirring, at room temperature. The pH of the resulting mixture was further adjusted to 10 by addition of a small amount of additional MeONa, and the mixture was evaporated to dryness. The residue was dissolved in water (2 mL), and acetonitrile (10 mL) was added. The product crystallized, was removed by filtration, rinsed with cold (0 °C) acetonitrile (5 mL) and diethyl ether (10 mL), and air-dried for 24 h to a constant mass to give pure 9 (tetrahydrate) as a creamy solid (0.33 g, 92%). 1H NMR (300 MHz, D2O) δ 7.10 (s, 2H), 7.04 (d, J = 8.6 Hz, 1H), 6.94 (d, J = 8.6 Hz, 1H), 2.37 (s, 3H), 3.76 (s, 6H), 3.79 (s, 3H), 3.81 (s, 3H), 4.66 (s, hydrate, 8H). LCMS, tR = 1.16, >99%. MS m/z = 452.8 (M – 2Na + 3H), 100%. HRMS: calcd for C20H20O10P− = 451.0794, found = 451.0769.
Docking
Molecular docking was performed with Glide 5.6 (Schrödinger LLC) using XP mode with default settings. Ligands were prepared using LigPrep, version 2.4. Default settings were used, unless stated otherwise. Compounds were docked into the crystal structure of the colchicine binding site of the bovine tubulin/stathmin complex (PDB entry 1SA1) which contains a podophylotoxin ligand.33
Biology
The following assays were run according to previously described methods: tubulin polymerization assay42 and MCF-7 proliferation assay.43
Cell Culture and Cell Lines
All in vitro assays were carried out using endothelial cells derived from human umbilical vein (HUVEC) (Clonetics Lonza, Walkersville, MD, U.S.) or human abdominal aorta (HAAE-1) (ATCC, Manassas, VA, U.S.). HUVEC were routinely cultured in EGM-2 medium (Clonetics), and HAAE-1 cells were cultured in F12K medium (Gibco, Invitrogen, Auckland, NZ) containing 10% fetal calf serum, 0.1 mg/mL heparin (Sigma-Aldrich, St. Louis, MO, U.S.), 0.03 mg/mL endothelial cell growth supplement (Sigma-Aldrich, St. Louis, MO, U.S.), 2 mM penicillin–streptomycin–glutamine (Gibco, Invitrogen, Auckland, NZ), and 10mMHepes buffered solution (Gibco, Invitrogen, Auckland, NZ). Endothelial cell cultures between passages 2 and 6 were used for all assays. Cancer cell lines were obtained from ATCC (Manassas, VA, U.S.) and cultured as recommended by the supplier. All cells were cultured in a humidified incubator at 37 °C with 5% CO2.
Endothelial Cell Proliferation Assay
HUVEC or HAAE-1 cell cultures were exposed to a concentration range of 0.1–1000 nM for each test compound tested. Proliferation assays were carried out in triplicate in 96-well plates. For activated growth conditions HUVEC and HAAE-1 cells were seeded at 2500 and 500 cells/well, respectively, and cultured in EBM-2 or F12K medium containing 0.03 mg/mL endothelial cell growth supplement (Sigma-Aldrich, St. Louis, MO, U.S.) as described above. For quiescent growth conditions HUVEC and HAAE-1 cells were seeded at 15 000 and 5000 cells/well, respectively, in basal medium (EBM-2 or F12K) containing 0.5% fetal calf serum and antibiotics. Cancer cell lines were seeded at an average of 500–2000 cells/well. Cells were allowed to adhere overnight, followed by incubation with compounds under evaluation for 48–72 h. Metabolically active cells were measured using CellTiter 96 Aqueous One Solution (Promega Corp., Madison WI, U.S.) according to the manufacturer’s instructions, and absorbance readings were taken at 492 nm. Absorbance readings for each compound concentration were normalized to corresponding vehicle control cultures. A sigmoidal dose–response curve was fitted to the data, and the concentration at which proliferation decreased by 50% was calculated using Graph Pad Prism 4 software (San Diego, CA, U.S.).
Supplementary Material
ABBREVIATIONS USED
- CA4
combretastatin A-4
- CA4P
combretastatin A-4 disodium phosphate
- HUVEC
human umbilical vein endothelial cell
- HAEEC
human aortic arterial endothelial cell
- IC50
inhibitory concentration 50%
- LD50
lethal dose 50%
- MCC
multicomponent coupling
- MDR
multidrug resistance
- MTD
maximum tolerated dose
- SAR
structure–activity relationship
- VDA
vascular disrupting agent
Footnotes
Supporting Information. Complete listing of benzo-[b]furan compounds tested; synthesis of 17a, 17b, and 17f; results of pharmacokinetic studies of 4, 5, 8, and 9. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]; (b) Lopus M, Yenjerle M, Wilson L. Microtubule Dynamics. In: Begley TP, editor. Wiley Encyclopedia of Chemical Biology. Vol. 3. John Wiley and Sons, Inc; Hoboken, NJ: 2008. pp. 153–160. [Google Scholar]; (c) Etienne-Manneville S. From signaling pathways to microtubule dynamics: the key players. Curr Opin Cell Biol. 2010;22:104–111. doi: 10.1016/j.ceb.2009.11.008. [DOI] [PubMed] [Google Scholar]; (d) Wade RH. On and around microtubules: an overview. Mol Biotechnol. 2009;43:177–191. doi: 10.1007/s12033-009-9193-5. [DOI] [PubMed] [Google Scholar]
- 2.(a) Kavallaris M, Verrills NM, Hill BT. Anticancer therapy with novel tubulin-interacting drugs. Drug Resist Updates. 2001;4:392–401. doi: 10.1054/drup.2002.0230. [DOI] [PubMed] [Google Scholar]; (b) Hearn BR, Shaw SJ, Myles DC. Microtubule Targeting Agents, Compr Med Chem II. 2006;7:81–110. [Google Scholar]; (c) Bedard PL, Di Leo A, Piccart-Gebhart MJ. Taxanes: optimizing adjuvant chemotherapy for early-stage breast cancer. Nat Rev Clin Oncol. 2010;7:22–36. doi: 10.1038/nrclinonc.2009.186. [DOI] [PubMed] [Google Scholar]; (d) Hadfield JA, Ducki S, Hirst N, McGown AT. Tubulin and microtubules as targets for anticancer drugs. Prog Cell Cycle Res. 2003;5:309–325. [PubMed] [Google Scholar]
- 3.Recent reviews: Chen J, Liu T, Dong X, Hu Y. Recent developments and SAR analysis of colchicine binding site inhibitors. Mini-Rev Med Chem. 2009;9:1174–1190. doi: 10.2174/138955709789055234.Chaudhary A, Pandeya SN, Kumar P, Sharma PP, Gupta S, Soni N, Verma KK, Bhardwaj G. Combretastatin A-4 analogs as anticancer agents. Mini-Rev Med Chem. 2007;7:1186–1205. doi: 10.2174/138955707782795647.Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. Medicinal chemistry of combretastatin A4: present and future directions. J Med Chem. 2006;49:3033–3044. doi: 10.1021/jm0512903.Pinney KG, Jelinek C, Edvardsen K, Chaplin DJ, Pettit GR. The Discovery and Development of the Combretastatins. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer Agents from Natural Products. Taylor and Francis; Boca Raton, FL: 2005. pp. 23–46.Bhattacharyya B, Panda D, Gupta S, Banerjee M. Antimitotic activity of colchicine and the structural basis for its interaction with tubulin. Med Res Rev. 2008;28:155–183. doi: 10.1002/med.20097.Hamel E. An Overview of Compounds That Interact with Tubulin and Their Effects on Microtubule Assembly. In: Fojo T, editor. The Role of Microtubules in Cell Biology, Neurobiology and Oncology. Humana Press; Totowa, NJ: 2008. pp. 1–19.
- 4.(a) Romagnoli R, Baraldi PG, Carrion MD, Cruz-Lopez O, Tolomeo M, Grimaudo S, Di Cristina A, Pipitone MR, Balzarini J, Brancale A, Hamel E. Substituted 2-(3′,4′,5′-trimethoxybenzoyl)- benzo[b]thiophene derivatives as potent tubulin polymerization inhibitors. Bioorg Med Chem. 2010;18:5114–5122. doi: 10.1016/j.bmc.2010.05.068. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen J, Wang Z, Lu Y, Dalton JT, Miller DD, Li W. Synthesis and antiproliferative activity of imidazole and imidazoline analogs for melanoma. Bioorg Med Chem Lett. 2008;18:3183–3187. doi: 10.1016/j.bmcl.2008.04.073. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lu Y, Li CM, Wang Z, Ross CR, II, Chen J, Dalton JT, Li W, Miller DD. Discovery of 4-substituted methoxybenzoyl-aryl-thiazoles as novel anticancer agents: synthesis, biological evaluation, and structure–activity relationships. J Med Chem. 2009;52:1701–1711. doi: 10.1021/jm801449a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) La Regina G, Sarkar T, Bai R, Edler MC, Saletti R, Coluccia A, Piscitelli F, Minelli L, Gatti V, Mazzoccoli C, Palermo V, Mazzoni C, Falcone C, Scovassi AI, Giansanti V, Campiglia P, Porta A, Maresca B, Hamel E, Brancale A, Novellino E, Silvestri R. New arylthioindoles and related bioisosteres at the sulfur bridging group. 4. Synthesis, tubulin polymerization, cell growth inhibition, and molecular modeling studies. J Med Chem. 2009;52:7512–7527. doi: 10.1021/jm900016t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Gangjee A, Zhao T, Lin L, Raghavan S, Roberts EG, Risinger AL, Hamel E, Mooberry SL. Synthesis and discovery of water-soluble microtubule targeting agents that bind to the colchicine site on tubulin and circumvent Pgp mediated resistance. J Med Chem. 2010;53:8116–8128. doi: 10.1021/jm101010n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Schobert R, Biersack B, Dietrich A, Effenberger K, Knauer S, Mueller T. 4-(3-Halo/amino-4,5-dimethoxyphenyl)-5-aryloxazoles and -N-methylimidazoles that are cytotoxic against combretastatin A-4 resistant tumor cells and vascular disrupting in a cisplatin resistant germ cell tumor model. J Med Chem. 2010;53:6595–6602. doi: 10.1021/jm100345r. [DOI] [PubMed] [Google Scholar]; (g) Lee J, Kim SJ, Choi H, Kim YH, Lim IT, Yang HM, Lee CS, Kang HR, Ahn SK, Moon SK, Kim DH, Lee S, Choi NM, Lee KJ. Identification of CKD-516: a potent tubulin polymerization inhibitor with marked antitumor activity against murine and human solid tumors. J Med Chem. 2010;53:6337–6354. doi: 10.1021/jm1002414. [DOI] [PubMed] [Google Scholar]; (h) Romagnoli R, Baraldi PG, Cruz-Lopez O, Cara CL, Carrion MD, Brancale A, Hamel E, Chen L, Bortolozzi R, Basso G, Viola G. Synthesis and antitumor activity of 1,5-disubstituted 1,2,4-triazoles as cis-restricted combretastatin analogues. J Med Chem. 2010;53:4248–4258. doi: 10.1021/jm100245q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Jourdan F, Leese MP, Dohle W, Hamel E, Ferrandis E, Newman SP, Purohit A, Reed MJ, Potter BVL. Synthesis, antitubulin, and antiproliferative SAR of analogues of 2-methoxyestradiol-3,17-O,O-bis-sulfamate. J Med Chem. 2010;53:2942–2951. doi: 10.1021/jm9018806. [DOI] [PubMed] [Google Scholar]; (j) Lee L, Robb LM, Lee M, Davis R, Mackay H, Chavda S, Babu BL, O’Brien E, Risinger AL, Mooberry SL, Lee M. Design, synthesis, and biological evaluations of 2,5-diaryl-2,3-dihydro-1,3,4-oxadiazoline analogs of combretastatin-A4. J Med Chem. 2010;53:325–334. doi: 10.1021/jm901268n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Bonezzi K, Taraboletti G, Borsotti P, Bellina F, Rossi R, Giavazz R. Vascular disrupting activity of tubulin-binding 1,5-diaryl-1H-imidazoles. J Med Chem. 2009;52:7906–7910. doi: 10.1021/jm900968s. [DOI] [PubMed] [Google Scholar]; (l) Wu Y-S, Coumar MS, Chang J-Y, Sun H-Y, Kuo F-M, Kuo C-C, CY-J, Chang C-Y, Hsiao C-L, Liou J-P, Chen C-P, Yao H-T, Chiang Y-K, Tan U-K, Chen C-T, Chu C-Y, Wu S-Y, Yeh T-K, Lin C-Y, Hsieh H-P. Synthesis and evaluation of 3-aroylindoles as anticancer agents: metabolite approach. J Med Chem. 2009;52:4941–4945. doi: 10.1021/jm900060s. [DOI] [PubMed] [Google Scholar]; (m) Ty N, Dupeyre G, Chabot GG, Seguin J, Tillequin F, Scherman D, Michel S, Cahcet X. Bioorg Med Chem. 2008;16:7494–7503. doi: 10.1016/j.bmc.2008.06.002. [DOI] [PubMed] [Google Scholar]; (n) Chiang Y-K, Kuo C-C, Wu Y-S, Chen C-T, Coumar MS, Wu J-S, Hsieh H-P, Chang C-Y, Jseng H-Y, Wu M-H, Leou J-S, Song J-S, Chang J-Y, Lyu P-C, Chao Y-S, Wu S-Y. Generation of ligand-based pharmacophore model and virtual screening for identification of novel tubulin inhibitors with potent anticancer activity. J Med Chem. 2009;52:4221–4233. doi: 10.1021/jm801649y. [DOI] [PubMed] [Google Scholar]
- 5.(a) Max MA. Small-molecule, tubulin-binding compounds as vascular targeting agents. Expert Opin Ther Pat. 2002;12:769–776. [Google Scholar]; (b) Tozer GM, Kanthou C, Baguley BC. Disrupting tumour blood vessels. Nat Rev Cancer. 2005;5:423–435. doi: 10.1038/nrc1628. [DOI] [PubMed] [Google Scholar]; (c) Siemann DW, Chaplin DJ, Horsman MR. Vascular targeted therapies for treatment of malignant disease. Cancer. 2004;100:2491–2499. doi: 10.1002/cncr.20299. [DOI] [PubMed] [Google Scholar]; (d) Kanthou C, Tozer GM. The tumor vascular targeting agent combretastatin A-4 phosphate induces reorganization of the actin cytoskeleton and early membrane blebbing in human endothelial cells. Blood. 2002;99:2060–2069. doi: 10.1182/blood.v99.6.2060. [DOI] [PubMed] [Google Scholar]; (e) Young SL, Chaplin DJ. Combretastatin A4 phosphate: background and current clinical status. Expert Opin Invest Drugs. 2004;13:1171–1182. doi: 10.1517/13543784.13.9.1171. [DOI] [PubMed] [Google Scholar]; (f) Nagaiah G, Remick SC. Combretastatin A4 phosphate: a novel vascular disrupting agent. Future Oncol. 2010;6:1219–1228. doi: 10.2217/fon.10.90. [DOI] [PubMed] [Google Scholar]; (g) McKeage MJ, Baguley BC. Disrupting established tumor blood vessels: an emerging therapeutic strategy for cancer. Cancer. 2010;116:1859–1871. doi: 10.1002/cncr.24975. [DOI] [PubMed] [Google Scholar]; (h) Kanthou C, Tozer GM. Tumour targeting by microtubule-depolymerising vascular disrupting agents. Expert Opin Ther Targets. 2007;11:1443–1457. doi: 10.1517/14728222.11.11.1443. [DOI] [PubMed] [Google Scholar]; Schwartz EL. Antivascular actions of microtubule-binding drugs. Clin Cancer Res. 2009;15:2595–2601. doi: 10.1158/1078-0432.CCR-08-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Chaplin DJ, Dougherty GJ, Siemann DW. Vascular disrupting agents. Antiangiogenic Cancer Ther. 2008:329–364. [Google Scholar]; (j) Siemann DW, Chaplin DJ, Walicke PA. A review and update of the current status of the vasculature-disabling agent combretastatin- A4 phosphate (CA4P) Expert Opin Invest Drugs. 2009;18:189–197. doi: 10.1517/13543780802691068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siemann DW, editor. Vascular-Targeted Therapies in Oncology. John Wiley and Sons; New York: 2006. [Google Scholar]
- 7.(a) Patnaik P. A Comprehensive Guide to the Hazadous Properties of Chemical Substances. John Wiley & Sons; Hoboken, NJ: 2007. p. 229. [Google Scholar]; (b) IPCS INCHEM. Colchicine. http://www.inchem.org/documents/pims/pharm/colchic.htm.
- 8.Kang GJ, Getahun Z, Muzaffar A, Brossi A, Hamel E. N-Acetylcolchicinol O-methyl ether and thiocolchicine, potent analogs of colchicine modified in the C ring. J Biol Chem. 1990;256:10255–10259. [PubMed] [Google Scholar]
- 9.Niel E, Scherrmann JM. Colchicine today. Jt, Bone, Spine. 2006;73:672–678. doi: 10.1016/j.jbspin.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 10.(a) Blakey DC, Ashton SE, Westwood FR, Walker M, Anderson JR. ZD6126: a novel small molecule vascular targeting agent. Int J Radiat Oncol, Biol, Phys. 2002;54:1497–1502. doi: 10.1016/s0360-3016(02)03922-6. [DOI] [PubMed] [Google Scholar]; (b) Davis PD, Dougherty GJ, Blakey DC, Galbraith SM, Tozer GM, Holder AL, Naylor MA, Nolan J, Stratford MRL, Chaplin DJ, Hill SA. ZD6126: a novel vascular-targeting agent that causes selective destruction of tumor vasculature. Cancer Res. 2002;62:7247–7253. [PubMed] [Google Scholar]
- 11.LoRusso PM, Gadgeel SM, Wozniak A, Barge AJ, Jones HK, DelProposto ZS, DeLuca PA, Evelhoch JL, Boerner SA, Wheeler C. Phase I clinical evaluation of ZD6126, a novel vascular-targeting agent, in patients with solid tumors. Invest New Drugs. 2008;26:159–167. doi: 10.1007/s10637-008-9112-9.Beerepoot LV, Radema SA, Witteveen EO, Thomas T, Wheeler C, Kempin S, Voest EE. Phase I clinical evaluation of weekly administration of the novel vascular-targeting agent, ZD6126, in patients with solid tumors. J Clin Onocol. 2006;24:1492–1498. doi: 10.1200/JCO.2005.02.7458.Rustin GJ, Shreeves G, Nathan PD, Gaya A, Ganesan TS, Wang D, Boxall J, Poupard L, Chaplin DJ, Stratford MRL, Balkissoon J, Zweifel M. A phase Ib trial of CA4P (combretastatin A-4 phosphate), carboplatin, and paclitaxel in patients with advanced cancer. Br J Cancer. 2010;102:1355–1360. doi: 10.1038/sj.bjc.6605650.and references therein.
- 12.A number of other VDA agents have also entered clinical trials: Lickliter JD, Francesconi AB, Smith G, Burge M, Coulthard A, Rose S, Griffin M, Milne R, McCarron J, Yeadon T, Wilks A, Cubitt A, Wyld DK, Vasey PA. Phase I trial of CYT997, a novel cytotoxic and vascular disrupting agent. Br J Cancer. 2010;103:597–606. doi: 10.1038/sj.bjc.6605841.Mita MM, Spear MA, Yee LK, Mita AC, Heath EI, Papadopoulos KP, Federico KC, Reich SD, Romero O, Malburg L, Pilat M-J, Lloyd GK, Neuteboom STC, Cropp G, Ashton E, LoRusso PM. Phase 1 first-in-human trial of the vascular disrupting agent plinabulin (NPI-2358) in patients with solid tumors or lymphomas. Clin Cancer Res. 2010;16:5892–5899. doi: 10.1158/1078-0432.CCR-10-1096.Baguley BC, McKeage MJ. ASA404: a tumor vascular-disrupting agent with broad potential for cancer therapy. Future Oncol. 2010;6:1537–1543. doi: 10.2217/fon.10.122.Hinnen P, Eskens FALM. Vascular disrupting agents in clinical development. Br J Cancer. 2007;96:1159–1165. doi: 10.1038/sj.bjc.6603694.Lippert JW. Vascular disrupting agents. Bioorg Med Chem. 2007;15:605–615. doi: 10.1016/j.bmc.2006.10.020.
- 13.Rojiani MV, Rojiani AM. Morphologic Manifestations of Vascular-Disrupting Agents in Preclinical Models. In: Siemann DW, editor. Vascular-Targeted Therapies in Oncology. John Wiley and Sons; New York: 2006. pp. 81–94. [Google Scholar]
- 14.Shi W, Horsman MR, Siemann DW. Combined Modality Approaches Using Vasculature-Disrupting Agents. In: Siemann DW, editor. Vascular-Targeted Therapies in Oncology. John Wiley and Sons; New York: 2006. pp. 123–136. [Google Scholar]
- 15.Hua J, Sheng Y, Pinney KG, Garner CM, Kane RR, Prezioso JA, Pettit GR, Chaplin DJ, Edvardsen K. Oxi4503, a novel vascular targeting agent: effects on blood flow and antitumor activity in comparison to combretastatin A-4 phosphate. Anticancer Res. 2003;23:1433–1440. [PubMed] [Google Scholar]
- 16.Kremmidiotis G, Leske AF, Lavranos TC, Beaumont D, Gasic J, Hall A, O’Callaghan M, Matthews CA, Flynn BL. BNC105: a novel tubulin polymerization inhibitor that selectively disrupts tumor vasculature and displays single-agent antitumor efficacy. Mol Cancer Ther. 2010;9:1562–1573. doi: 10.1158/1535-7163.MCT-09-0815. [DOI] [PubMed] [Google Scholar]
- 17.Aspects of the work presented in this report have been drawn from our published patents: Chaplin JH, Gill GS, Grobelny DW, Flynn BL. Preparation of Benzofurans and Related Derivatives as Tubulin Polymerization Inhibitors for Treating Neoplasm and Inflammation. WO2006084338. PCT Int Appl. 2006Chaplin JH, Gill GS, Grobelny DW, Flynn BL, Kremmidiotis G. Substituted Benzofurans, Benzothiophenes, Benzoselenophenes and Indoles and Their Use as Tubulin Polymerization Inhibitors. WO2007087684. PCT Int Appl. 2007
- 18.(a) Pinney KG, Bounds AD, Dingeman KM, Mocharla VP, Pettit GR, Bai R, Hamel E. A new anti-tubulin agent containing the benzo[b]thiophene ring system. Bioorg Med Chem Lett. 1999;9:1081–1086. doi: 10.1016/s0960-894x(99)00143-2. [DOI] [PubMed] [Google Scholar]; (b) Mullica DF, Pinney KG, Mocharla VP, Dingeman KM, Bounds AD, Sappenfield EL. Characterization and structural analyses of trimethoxy and triethoxybenzo[b]thiophene. J Chem Crystallogr. 1998;28:289–295. [Google Scholar]
- 19.Flynn BL, Verdier-Pinard P, Hamel E. A novel palladium-mediated coupling approach to 2,3-disubstituted benzo[b]thiophenes and its application to the synthesis of tubulin binding agents. Org Lett. 2001;3:651–654. doi: 10.1021/ol0067179. [DOI] [PubMed] [Google Scholar]
- 20.Flynn BL, Flynn GP, Hamel E, Jung MK. The synthesis and tubulin binding activity of thiophene-based analogues of combretastatin A-4. Bioorg Med Chem Lett. 2001;11:2341–2343. doi: 10.1016/s0960-894x(01)00436-x. [DOI] [PubMed] [Google Scholar]
- 21.Flynn BL, Hamel E, Jung MK. A one-pot synthesis of benzo[b]furan and indole inhibitors of tubulin polymerization. J Med Chem. 2002;45:2670–2673. doi: 10.1021/jm020077t. [DOI] [PubMed] [Google Scholar]
- 22.Related indenones I (X = carbonyl) are inactive: Kerr DJ, Hamel E, Jung MK, Flynn BL. The concise synthesis of chalcone, indanone and indenone analogues of combretastatin A4. Bioorg Med Chem. 2007;15:3290–3298. doi: 10.1016/j.bmc.2007.02.006.
- 23.Liou J-P, Chang Y-C, Kuo F-M, Chang C-W, Tseng H-Y, Wang C-C, Yang Y-N, Chang J-Y, Lee S-J, Hsieh HP. Concise synthesis and structure–activity relationships of combretastatins A-4 analogues, 1-aroylindoles and 3-aroylindoles, a novel class of potent antitubulin agents. J Med Chem. 2004;47:4247–4257. doi: 10.1021/jm049802l. [DOI] [PubMed] [Google Scholar]
- 24.Chaplin JH, Flynn BL. A multi-component coupling approach to benzo[b]furans and indoles. J Chem Soc, Chem Commun. 2001:1594–1595. doi: 10.1039/b104624c. [DOI] [PubMed] [Google Scholar]
- 25.Gill GS, Grobelny DW, Chaplin JH, Flynn BL. An efficient synthesis and substitution of 3-aroyl-2-bromobenzo[b]furans. J Org Chem. 2008;73:1131–1134. doi: 10.1021/jo701656z. [DOI] [PubMed] [Google Scholar]
- 26.Larock RC, Yum EK, Doty MJ, Sham KC. Synthesis of aromatic heterocycles via palladium-catalyzed annulation of internal alkynes. J Org Chem. 1995;60:3270–3271. [Google Scholar]
- 27.Bussolati B, Deambrosis I, Russo S, Deregibus MC, Camussi G. Altered angiogenesis and survival in human tumor-derived endothelial cells. FASEB J. 2003;17:1159–1161. doi: 10.1096/fj.02-0557fje. [DOI] [PubMed] [Google Scholar]
- 28.Ohsumi K, Nakagawa R, Fukuda Y, Hatanaka T, Morinaga Y, Nihei Y, Ohishi K, Suga Y, Akiyama Y, Tsuji T. Novel combretastatin analogues effective againstmurine solid tumors: design and structure–activity relationships. J Med Chem. 1998;41:3022–3032. doi: 10.1021/jm980101w. [DOI] [PubMed] [Google Scholar]
- 29.Maya ABS, Perez-Melero C, Mateo C, Alonso D, Fernandez JL, Gajate C, Mollinedo F, Pelaez R, Caballero E, Medarde M. Further naphthylcombretastatins. An investigation on the role of the naphthalene moiety. J Med Chem. 2005;48:556–568. doi: 10.1021/jm0310737. [DOI] [PubMed] [Google Scholar]
- 30.Cushman M, Nagarathnam D, Gopal D, Chakraborti AK, Lin CM, Hamel E. Synthesis and evaluation of stilbene and dihydrostilbene derivatives as potential anticancer agents that inhibit tubulin polymerization. J Med Chem. 1991;34:2579–2588. doi: 10.1021/jm00112a036. [DOI] [PubMed] [Google Scholar]
- 31.Subsequent to our published patents on C7-OH benzo-[b]furan, benzo[b]thiophene, and indole analogues of 11 (ref 17), Cachet and Hseih reported that C7-OH analogues of 15 exhibit reduced potency, albeit to a lesser extent than does a C5-OH analogue (refs 4l and 4m). Additionally, in their earlier report (see ref 23), Hseih and co-workers also observed a modest increase in potency for a C2-methyl analogue of indole 15 (~2-fold decrease in IC50 across multiple cell lines). However, consistent with our findings on benzo[b]furans, we have shown that the introduction of both C7-OH and C2-Me to 15 affords a much more potent analogue (ref 17b). A more detailed description of the SAR of indoles and other heterocyclic analogues of 8 will be reported elsewhere.
- 32.See Supporting Information for a complete listing of benzofurans evaluated.
- 33.Ravelli RBG, Gigant G, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature. 2004;428:198–202. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- 34.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein–ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 35.For other docking studies on tubulin see the following: Soulère L. Toward docking-based virtual screening for discovering antitubulin agents by targeting taxane and colchicines binding sites. ChemMedChem. 2009;4:161–163. doi: 10.1002/cmdc.200800319.and references cited therein.
- 36.Goda K, Bacso Z, Szabo G. Multidrug resistance through the spectacle of P-glycoprotein. Curr Cancer Drug Targets. 2009;9:281–297. doi: 10.2174/156800909788166493. [DOI] [PubMed] [Google Scholar]
- 37.(a) Battaglia A, Bernacki RJ, Bertucci C, Bombardelli E, Cimitan S, Ferlini C, Fontana G, Guerrini A, Riva A. Synthesis and biological evaluation of 2′-methyl taxoids derived from baccatin III and 14β-OH-baccatin III 1,14-carbonate. J Med Chem. 2003;46:4822–4825. doi: 10.1021/jm034146v. [DOI] [PubMed] [Google Scholar]; (b) Martirosyan A, Clendening JW, Goard CA, Penn LZ. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: potential therapeutic relevance. BMC Cancer. 2010;10:103. doi: 10.1186/1471-2407-10-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pettit GR, Rhodes MR. Antineoplastic agents 389. New syntheses of the combretastatin A-4 prodrug. Anti-Cancer Drug Des. 1998;13:183–191. [PubMed] [Google Scholar]
- 39.See Supporting Information for pharmacokinetic studies on the in vivo conversion of 9 to 8.
- 40.The maximum tolerated dose (MTD) for 9 and 5 was determined to be 80 and 300 mg/kg, respectively, and is based on a no observable adverse effect level (NOAEL).
- 41.Bionomics. BNC105. http://www.bionomics.com.au/page.php?section=103.
- 42.(a) Hamel E, Lin CM. Synthesis and evaluation of stilbene and dihydrostilbene derivatives as potential anticancer agents that inhibit tubulin polymerization. Biochemistry. 1984;23:4173–4184. doi: 10.1021/jm00112a036. [DOI] [PubMed] [Google Scholar]; (b) Hamel E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem Biophys. 2003;38:1–22. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 43.Verdier-Pinard P, Lai JY, Yoo HD, Yu J, Márquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Structure–activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol Pharmacol. 1998;53:62–76. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.