

SUMMARY
The pentadecaketide fredericamycin has the longest carbon chain backbone among polycyclic aromatic polyketide antibiotics whose biosynthetic genes have been sequenced. This backbone is synthesized by the bimodular fdm polyketide synthase (PKS). The initiation module is thought to synthesize a C6 intermediate that is then transferred onto the elongation PKS module, which extends it into a C30 poly-β-ketoacyl product. Here we demonstrate that the bimodular fdm PKS as well as its elongation module alone synthesize undecaketides and dodecaketides. Thus, unlike other homologues, the fdm ketosynthase – chain length factor (KS-CLF) heterodimer does not exclusively control the backbone length of its natural product. Using sequence- and structure-based approaches, 48 multiple mutants of the CLF were engineered and analyzed. Unexpectedly, the I134F mutant was unable to turn over, but could initiate and at least partially elongate the polyketide chain. This unprecedented mutant suggests that the KS-CLF heterodimer harbors an as yet uncharacterized chain termination mechanism. Together, our findings reveal fundamental mechanistic differences between the fdm PKS and its well-studied homologues.
INTRODUCTION
Polyketide synthases (PKSs) are a large family of multienzyme assemblies that catalyze the biosynthesis of structurally diverse and medicinally important natural products (O’Hagan, 1991; Rawlings, 1999; Staunton and Weissman, 2001). In analogy with the evolutionarily related fatty acid synthases, they are classified into Type I and Type II PKSs (Hopwood and Sherman, 1990; Smith and Tsai, 2007). The latter sub-class of PKSs, which resemble bacterial and plant fatty acid synthases, play a defining role in the biosynthesis of many polycyclic aromatic antibiotics produced by the actinomycetes (Das and Khosla, 2009; Hertweck et al., 2007). They invariably harbor a minimal PKS module comprised of a ketosynthase (KS), a chain length factor (CLF), an acyl carrier protein (ACP), and a malonyl-CoA:ACP transacylase (MAT, usually from the housekeeping fatty acid synthase in the bacterium). Collectively, these proteins catalyze the formation of a poly-β-ketoacyl-ACP chain of defined length. In addition, some Type II PKSs also have an initiation module that catalyzes the first one or two cycles of polyketide chain growth (reviewed in Das and Khosla, 2009).
The heterodimeric KS-CLF is central to the activity of Type II PKSs, not only because it catalyzes chain elongation but also because it controls product length. In well-studied examples such as the actinorhodin and tetracenomycin PKS, the CLF subunit has been shown to be necessary and sufficient for dictating polyketide chain length (McDaniel et al., 1993; Tang et al., 2003). Because chemical diversity of the resulting natural products correlates with chain length (Shen et al., 1999), we sought to investigate the KS-CLF heterodimer from the fredericamycin A (1, Figure 1) biosynthetic gene cluster (Wendt-Pienkowski et al., 2005). With a C30 backbone, this antitumor aromatic polyketide has the longest known carbon chain length. In earlier work (Das et al., 2010), we showed that the initiation module of the fredericamycin (fdm) PKS catalyzes the synthesis of an ACP-linked C6 intermediate. The goal of the current study was therefore to understand how the fdm KS-CLF catalyzes chain elongation to its full length. To do so, we functionally reconstituted it in a heterologous host as well as in vitro. Using sequence- and structure-based methods, we designed and expressed a set of 48 multiple mutants of the CLF gene, and compared the properties of the resulting KS-CLF heterodimers to those of the wild-type enzyme. Our findings reveal several fundamentally novel aspects of how the fdm KS-CLF controls chain length.
Figure 1.

Polyketides cited in this study.
RESULTS
Reconstitution and analysis of the fdm PKS in S. coelicolor CH999
As a first step in our analysis of the fdm KS-CLF, we exploited the engineered host-vector system Streptomyces coelicolor CH999/pRM5 (McDaniel et al., 1993). Plasmid pAD201 was constructed harboring the KS-CLF (fdmFG) and ACP (fdmH) genes. We also inserted the fdmW phosphopantetheinyl transferase gene on this plasmid, because previous studies had demonstrated that, absent this enzyme, the fdm ACP is inactive in the native host S. griseus (Wendt-Pienkowski et al., 2005) as well as S. coelicolor CH999 (Das et al., 2010). Plasmid pAD201 was introduced via transformation into S. coelicolor CH999 along with a secondary plasmid pBOOST*, which amplifies the copy number of the cloned PKS genes, thereby enhancing protein expression as well as product formation (Hu et al., 2003). Two previously characterized undecaketides (TW94b and TW94c, Fig. 1) and one known dodecaketide (TW94d, Fig. 1) were isolated as the major products from an ethyl acetate: methanol (90:10) extract of this recombinant strain. TW94b (2), TW94c (3) and TW94d (4) were produced in a 2:1:2 ratio. Their identities were verified by 1H and 13C NMR in comparison to authentic standards (Yu et al., 1998). Two conclusions can be drawn from this finding. First, in the absence of the initiation module, the minimal fdm PKS catalyzes chain initiation via decarboxylative priming of a malonyl extender unit. Second, and more importantly, the minimal fdm PKS intrinsically appears to be a dodecaketide synthase. (The concomitant biosynthesis of undecaketides TW94b and TW94c along with the dodecaketide TW94d has precedence in a number of spore pigment PKSs, such as the whiE PKS (Yu et al., 1998).)
To interrogate the influence of the initiation module on the minimal PKS, the fdmS, fdmC, and fdmO genes were inserted onto plasmid pAD201, yielding plasmid pAD210. The resulting transformant, S. coelicolor CH999/pAD210/pBOOST*, synthesized a new product AD210a (5, Fig. 1) with an observed molecular mass of 439 and 441 (in negative and positive ionization modes, respectively) by ESI-MS. High resolution ESI-MS analysis yielded a molecular formula of C24H24O8Na (calcd. 463.1361, observed 463.1369). The structure of AD210a was solved by 1H and 13C NMR spectroscopic analysis as well as HSQC, HMBC and COSY NMR experiments (Table 1), and revealed that, even in the presence of the initiation module, the fdm KS-CLF preferentially synthesizes carbon chain backbones up to 24 carbon atoms in length.
Table 1.
1H and 13C NMR data for AD210a (5). 1H and 13C NMR data were recorded in DMSO-d6 (500 MHz for 1H and 125 MHz for 13C). Chemical shifts are reported in δ (ppm). Coupling constants are reported in Hz. Carbons are labeled according to their number in polyketide backbone.
| AD210a (5)
|
||||
|---|---|---|---|---|
| no. | 13C | 1H | Long-range 13C-1H HMBC correlations | Long-range 1H -1H COSY correlations |
| 1 | 160.5 | |||
| 2 | 88.1 | 5.13 (d, J = 1.7, 1H) | C3, C4, C15 | H4 |
| 3 | 170.2 | |||
| 4 | 100.7 | 5.56 (d, J = 1.7, 1H) | C2, C3, C10, C20 | H2 |
| 5 | 159.0 | |||
| 6 | 36.9 | 3.51 (s, 2H) | C4, C7, C8, C11 | |
| 7 | 136.9 | |||
| 8 | 119.1 | 6.21 (d, J = 1.8, 1H) | C6, C10, C11 | H10 |
| 9 | 160.5 | |||
| 10 | 100.1 | 6.08 (d, J = 1.8, 1H) | C9, C12 | H8 |
| 11 | 164.7 | |||
| 12 | 108.3 | |||
| 13 | 199.7 | |||
| 14 | 110.1 | |||
| 15 | 163.5 | |||
| 16 | 108.4 | 6.07 (d, J = 3.6, 1H) | C15, C18, C21 | H18 |
| 17 | 160.2 | |||
| 18 | 108.3 | 6.16 (d, J = 3.6, 1H) | C8, C16, C17, C20 | H16 |
| 19 | 144.7 | |||
| 20 | 33.1 | 2.19 (t, J = 6.5, 2H) | C12, C19, C21 | H21 |
| 21 | 31.4 | 1.21 (p, J = 5.9, 6.5, 2H) | C20, C22, C23 | H20, H22 |
| 22 | 30.3 | 1.14 (m, 2H) | C20, C21, C23 | H21, H23 |
| 23 | 21.8 | 1.04 (m, 2H) | C21, C22, C24 | H22, H24 |
| 24 | 13.9 | 0.78 (t, J = 5.75, 3H) | C21, C22, C23 | H23 |
Purification and in vitro reconstitution of the fdm minimal PKS
The His6-tagged holo-ACP and KS-CLF heterodimer from the fdm minimal PKS were individually expressed and purified to homogeneity (Fig. 2A, lanes 3 and 6, respectively). (The KS and the His6-CLF have virtually identical molecular masses, and cannot be resolved on SDS-PAGE.) Whereas the holo-ACP was produced in E. coli BAP1, the KS-CLF could only be functionally expressed in S. coelicolor CH999. The MAT from S. coelicolor was expressed in E. coli BL21(DE3). These protein preparations were incubated individually and in selected combinations with [14C]-malonyl-CoA (Fig. 2B). By itself, only the MAT could be labeled (lane 2). When incubated with the MAT and [14C]-malonyl-CoA, the holo-ACP was also labeled (lane 4). Weak labeling of the KS-CLF and the holo-ACP was observed in the absence of the MAT (lane 5); the precise reason for this observation was not investigated. When [14C]-malonyl-CoA was added to the complete minimal PKS, both the KS and the ACP were strongly labeled (lane 6), consistent with the expectation that oligoketides with higher specific radioactivity than malonyl-CoA are attached to their active sites. Taken together, these radio-SDS-PAGE results support the established model for minimal PKS activity, in which the MAT supplies malonyl units to the ACP, whereas polyketide biosynthesis involves back-and-forth transfer of the growing chain between the KS and the ACP.
Figure 2.

(A) SDS-PAGE analysis of purified components of the fdm minimal PKS. Lane 1, protein molecular weight standard; lane 2, S. coelicolor MAT; lane 3, holo-ACP; lane 4, protein molecular weight standard; lane 5, KS-CLF I134F mutant; lane 6, wild-type KS-CLF. The two subunits have nearly identical molecular masses (44 kD and 42 kD, respectively), and therefore cannot be resolved via SDS-PAGE. (B) Activity of the minimal fdm PKS. PKS components were individually or collectively incubated with [14C]-malonyl-CoA, and quenched after 30 min: Lane 1, ACP; Lane 2, MAT; Lane 3, KS-CLF; Lane 4, ACP + MAT; Lane 5, ACP + KS-CLF; Lane 6, ACP + KS-CLF + MAT. (C) Activity of the fdm PKS harboring the I134F CLF mutation. Lane 1, ACP; Lane 2, MAT; Lane 3, KS-CLF; Lane 4, ACP + MAT; Lane 5, ACP + KS-CLF + MAT. The labeled band between the MAT and ACP in lanes 6 (panel A) and 5 (panel B) has not been characterized, but has precedence in the context of other Type II PKSs (Dreier and Khosla, 2000). (D) Activity of purified fdm minimal PKS: Lane 2, Minimal PKS in the presence of [14C]-malonyl-CoA (quenched at 1 h); Lane 1, Control sample pre-incubated with cerulenin for 20 min; Lane 3, Control sample lacking the KS-CLF. (E) Activity of the fdm PKS harboring the I134F CLF mutation: Lane 1, Minimal PKS in the presence of [14C]-malonyl-CoA (quenched at 1 h); Lane 2, the fdm PKS harboring the I134F CLF mutation in the presence of [14C]-malonyl-CoA (quenched at 1 h).
To verify the ability of the reconstituted fdm minimal PKS to turn over, a radio-TLC (thin layer chromatography) assay was performed (Fig. 2D). In lane 2, the minimal PKS is observed to convert malonyl-CoA into polyketide products of unconfirmed structure. Notwithstanding repeated attempts on different types of mass spectrometers, we were unable to verify the identity of the products synthesized in vitro as TW93b, TW93c or TW93d, the expected undeca- or dodeca-ketide products of the PKS (Yu et al., 1998). Nonetheless, two observations support the assumption that the observed radiolabeled products are polyketides synthesized by the fdm minimal PKS. First, omission of the KS-CLF resulted in complete loss of activity (Fig. 2D, lane 3). Second, pre-incubation of the KS-CLF with the irreversible inhibitor, cerulenin, also abolished product formation (Fig. 2D, lane 1). Kinetic measurements showed that the minimal PKS turned over with a Vmax of 0.27 μM/min, as judged by the rate of formation of the polyketide products observed in Fig. 2D, lane 2. This corresponds to a kcat of 0.03 min-1 (referenced to the KS concentration), a comparable value to the turnover number of the act minimal PKS (Carreras and Khosla, 1998).
Given our inability to unambiguously identify the released product(s) of the fdm minimal PKS in vitro, we used electrospray ionization Fourier-Transform Mass Spectrometry (FT-MS) to verify its catalytic activity and to characterize the ACP-bound intermediates. Recently, a 4′-phosphopantetheine (PPant) ejection assay (Fig. 3A) has been developed that allows the masses of ACP-bound substrates and intermediates to be determined (Dorrestein and Kelleher, 2006; Meluzzi et al., 2008). The reaction mixture containing malonyl-CoA, holo-ACP, MAT, and the KS-CLF was incubated at room temperature for 30 min, following which the PPant ejection assay was performed. As seen in Figure 3B (see also MS3 analysis in Figs. S1-S7, Supplementary Information), the predominant PPant ejection peaks corresponded to a tetraketide, a pentaketide, a cyclized tetraketide, and a cyclized pentaketide. Whereas this data does not allow us to verify the longest chain length attainable by the fdm minimal PKS, it confirms that the reconstituted system is catalytically active.
Figure 3.

FT-MS spectra of acyl-pantetheine ejection ions. (A) Overview of PPant ejection assay. Tandem-MS results in characteristic ejection ions bearing polyketide intermediates, which are subjected to further tandem-MS analysis to yield diagnostic fragments. In-source fragmentation of the ACP simultaneously yields multiple pantetheine ejection ions bearing different polyketide intermediates. Results from the following reactions are presented: (B) wild-type fdm PKS; and (C) fdm PKS harboring the I134F mutation in the CLF. The peak labeled (*) is a prominent, unrelated ion. Additional information for each intermediate identified by PPant ejection assay is provided in Figs S1-S7.
Design, synthesis and characterization of an fdm CLF mutant library
A major challenge for structure-function analysis or engineering of the fdm CLF (or any other CLF) is the lack of a high-throughput assay that is adequately sensitive to polyketide product characteristics. To mitigate this limitation, we used a synthetic gene approach to construct a modest library of 48 fdm CLF mutants. Mutants were designed via a combination of sequence and structure-based approaches, as well as available literature data. Specifically, the amino acid sequences of 31 CLF homologues were aligned using ClustalW (Thompson et al., 1994) (Fig. S8). Two types of scores were assigned to each position in the alignment (Minshull et al., 2005). The first score, termed “alternate conservation”, identifies relatively conserved residues whose identity correlates with chain length specificity of the PKS. Namely, PKSs producing shorter polyketide backbones share a conserved residue at this position, while those that synthesize longer chains share an alternative conserved residue. A higher alternate conservation score suggests that an amino acid change at this position has greater potential to influence chain length. The second score, termed “entropy”, reflects the chemical diversity of amino acids at a particular position in the sequence alignment, and is calculated with reference to the Dayhoff substitution matrix. A more conserved residue has a higher entropy score. Twenty residues with the highest alternate conservation scores were targeted in the first set of 24 variants, whereas fifteen substitutions selected on the basis of entropy scores were combined with five substitutions identified from previous analyses of the actinorhodin CLF (Keatinge-Clay et al., 2004; Tang et al., 2003) to design the second set of 24 variants. Because each variant harbored multiple mutations relative to the wild-type fdm CLF, each substitution was programmed to occur at the same frequency (i.e., 5 or 6 times). Moreover, individual mutations in a given variant were randomly combined. Thus, the variants were designed to maximize the number of different pairs of substitutions in the full set of variants. The complete set of variants is defined in Table 2A.
Table 2.
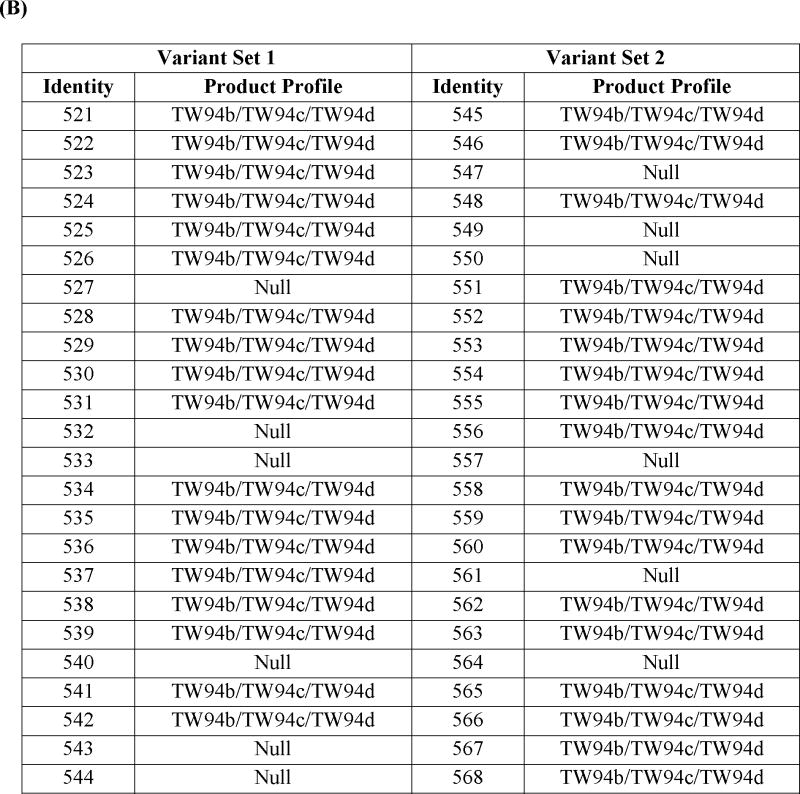
(A)List of the variants with their corresponding mutations. Variant numbers are listed in the 1st column, and the mutations harbored by them are listed the corresponding row. A “1” in a given cell (highlighted in purple for enhanced identification) indicates that the mutation was incorporated into that variant. See also Figure S8. (B) In vivo product profile of fdm CLF variants. Set 1 includes variants whose design was based on high “alternate conservation” scores, whereas Set 2 includes variants designed based on “entropy” scores. For details, see text.

|
|
The product profiles of the 48 variants are summarized in Table 2B. Twelve variants (527, 532, 533, 540, 543, 544, 547, 549, 550, 557, 561, and 564) did not produce TW94b, TW94c, or TW94d. The inactivity of variant 533, which harbors D18G, F77L, Y121W, G129S, and V326A mutations, could not be exclusively ascribed to a single modification, because each mutation was also found in other biosynthetically competent variants. Radio-SDS-PAGE analysis of the corresponding purified KS-CLF confirmed that its inability to be labeled in the presence of MAT, holo-ACP and [14C]-malonyl-CoA (data not shown). Further analysis of variant 533 was not undertaken, as its phenotype was likely due to the combined effect of multiple residues.
Variants 527, 532, 540, 543, and 544, whose designs were based on the alternate conservation score, all harbored the I134F mutation, whereas variants 547, 549, 550, 557, 561, and 564, whose designs were based on the entropy score, shared the G113T mutation. Because both sets of variants exhibited a null phenotype in vivo, the KS-CLF heterodimer from a representative variant of each set was purified. The G113T-harboring mutant was not labeled under any circumstance (data not shown); however, the I134F-harboring mutant showed interesting characteristics. To unambiguously assign the observed properties to the I134 residue, we first constructed and expressed a mutant CLF harboring a single change (I134F). The resulting transformant S. coelicolor CH999/pAD201-I134F/pBOOST* produced no detectable polyketide metabolite.
In vitro analysis of the I134F mutant of the fdm CLF
Using a similar protocol to the wild-type enzyme purification procedure, the KS-CLF heterodimer harboring the I134F mutation in the CLF was purified to homogeneity (Figure 2A, lane 5). As predicted from the in vivo phenotype of this mutant, radio-TLC analysis failed to show evidence of polyketide production in the presence of MAT, holo-ACP, and [14C]-malonyl-CoA (Fig. 2E). However, an unexpected labeling pattern was observed by radio-SDS-PAGE analysis (Fig. 2C). When [14C]-malonyl-CoA was incubated with MAT, holo-ACP and the mutant KS-CLF, both the ACP and the KS were strongly labeled (Fig. 2C, lane 5). The intensity of protein labeling was suggestive of the possibility that polyketide intermediates were covalently attached to the holo-ACP. To test this hypothesis, a reaction mixture containing malonyl-CoA, the holo-ACP, the MAT, and the mutant KS-CLF was incubated at room temperature for 30 min, and analyzed via FT-MS. The same ACP-bound intermediates observed in the wild-type reaction (i.e. a tetraketide, a pentaketide, a cyclized tetraketide, and a cyclized pentaketide) were also observed in the case of the I134F mutant (Fig. 3C), implying that although this mutant was unable to turn over, it could initiate and (at least partially) elongate polyketide chains.
DISCUSSION
The fdm PKS is a bimodular synthase that is believed to synthesize the longest known (C30) polyketide backbone produced by a Type II PKS (Das et al., 2010; Wendt-Pienkowski et al., 2005). Its initiation module synthesizes a C6 primer unit and transfers it to the elongation module (Das et al., 2010), which then presumably incorporates 12 malonyl extender units (Figure 4). In all other Type II PKSs studied thus far, chain length specificity is exclusively programmed in the KS-CLF heterodimer (reviewed in Das and Khosla, 2009). For example, investigations into the actinorhodin and benastatin PKSs have shown that the number of decarboxylative condensation cycles catalyzed by their KS-CLF heterodimers depends on the size of the primer unit (Nicholson et al., 2003; Tang et al., 2004; Xu et al., 2007; Xu et al., 2009). Our findings reported here suggest that the fdm KS-CLF is an exception to this rule in that it only synthesizes C22 – C24 backbones instead of the expected C30 backbone (Figure 4). It is therefore likely that an as yet unidentified auxiliary protein encoded by the fdm gene cluster modifies the inherent chain length specificity of the fdm KS-CLF, in effect enlarging its substrate-binding pocket. Further investigations are warranted to elucidate the mechanism by which this most remarkable synthase controls product length.
Figure 4.

(A) Proposed biosynthesis of TW94b, TW94c and TW94d by the fdm minimal PKS and the act KR in S. coelicolor CH999/pAD201/pBOOST*. The act KR is a ketoreductase from the actinorhodin biosynthetic pathway. (B) Proposed biosynthesis of AD210a by the bimodular fdm PKS in S. coelicolor CH999/pAD210/pBOOST*. The DH (dehydratase) and ER (enoyl reductase) involved in the initiation module are derived from the housekeeping fatty acid synthase in S. coelicolor. (C) Proposed biosynthesis of fredericamycin A in S. griseus.
Besides being of fundamental interest, the ability of the fdm PKS to synthesize exceptionally long poly-β-ketoacyl chains also offers an attractive starting point for the engineering of biosynthetically useful enzymes. Here we have used sequence and structure-based concepts together with machine learning algorithms and high-throughput gene synthesis to identify two important residues, I134 and G116, in the fdm CLF. The I134F mutant is particularly noteworthy because, to our knowledge, it is the first example of a CLF mutant that can initiate and elongate, but not terminate, polyketide chain growth. Earlier efforts to modify the substrate-binding pocket of the act and tcm CLF proteins resulted in mutants that retained the ability to turn over but had altered backbone length specificity (Tang et al., 2003). Alternatively, mutants such as G113T and variant 533 of the fdm CLF (reported here) are completely inactive. In contrast, the partial activity of the I134F mutant suggests that chain termination by the KS-CLF is not simply a default event that occurs when further growth of the polyketide backbone is no longer possible. A homology model of the fdm KS-CLF, based upon the X-ray crystal structure of the act KS-CLF (Keatinge-Clay et al., 2004), is shown in Figure 5. According to this model, I134 is located at the inter-subunit interface. We speculate that the I134F mutation creates a steric block in the substrate-binding channel straddling this interface. However, in contrast to earlier CLF mutants with alterations in the substrate binding channel (Tang et al., 2003), the product of the I134F mutant is not released. Therefore, chain termination must require repositioning of a full-length chain to promote its ejection from the elaborate substrate-binding pocket. Further structural and mechanistic analysis of this fdm PKS mutant will not only shed light on the mechanism of chain length control, but could also provide entirely new insights into the mechanism of chain termination by Type II PKSs.
Figure 5.

Homology model of the fdm KS-CLF (A) along with the structurally characterized act KS-CLF (B). The KS and CLF subunits are shown as green and blue ribbons, respectively. The active site of the KS (C169) and residue 134 of the fdm CLF (F133 is an equivalent residue in the act CLF) are shown in balls and sticks. Polyketide chain growth is initiated via decarboxylation of malonyl-ACP, followed by transfer of the resultant acetyl group onto C169 residue of the KS. The growing polyketide chain is transferred back and forth between and Cys169 residue of the KS and the pantetheinyl thiol of the ACP until it reaches full length. At this point the chain fully occupies the substrate-binding channel (shown in red), and is subsequently released.
SIGNIFICANCE
Type II polyketide synthases (PKSs) catalyze the biosynthesis of highly reactive poly-β-ketoacyl chains of defined length. Understanding and engineering polyketide chain length offers the opportunity to rationally manipulate the structures of many interesting antibiotics. We have harnessed a variety of approaches to study the ketosynthase – chain length factor (KS-CLF) heterodimer from the fredericamycin PKS. Although fredericamycin is derived from a C30 backbone, we show that its KS-CLF synthesizes products in the C22 – C24 range. Thus, this heterodimeric enzyme appears unique in that its chain length specificity is considerably shorter than the backbone length of the final product. We have also identified a prototypical CLF mutant that permits chain initiation and elongation, but not termination. Further characterization of this mutant should yield fundamentally new insights into the mechanisms by which carbon chains of defined length are synthesized and released.
EXPERIMENTAL SECTION
Materials
Radiolabeled coenzyme A (CoA) substrates were from American Radiolabeled Chemicals (St. Louis, MO). All other biochemicals were from Sigma-Aldrich (St. Louis, MO). Deuterated solvents for NMR experiments were from Cambridge Isotope Laboratories (Andover, MA); all other solvents were from Fisher Scientific (Pittsburgh, PA) at highest available grade. Hi-Trap Q anion-exchange column was from GE Healthcare (Madison, WI). Precast SDS-PAGE gels were from Invitrogen (Carlsbad, CA). PCR was performed using PfuTurbo Hotstart polymerase (Stratagene, La Jolla, CA), and amplimers were sequence-verified prior to use. Plasmids used for cloning included pLitmus28i, pUC18 (New England Biolabs, Ipswich, MA) and pET28b (EMD Bioscience, Madison, WI). All cloning steps were performed in E. coli XL1-Blue (Stratagene, La Jolla, CA). Unmethylated DNA was obtained by passaging plasmids through the methylase-deficient strain E. coli ET12567. Protoplast preparation and PEG-assisted transformation were performed as described by Hopwood et al (Kieser et al., 2000). S. coelicolor CH999/pBOOST* (Hu et al., 2003) and E. coli strains BL21(DE3) or BAP1 (Pfeifer et al., 2001) were used for polyketide isolation and protein expression, respectively.
Cloning
The fdm PKS genes were synthesized to introduce suitable restriction sites and ribosomal binding sites. The 5′ and 3′ sequences for fdmC, fdmF, fdmG, fdmH, fdmO, fdmS, and fdmW were engineered as follows:
fdmC: TCTAGAGGAGGAGCCCATATGCTCACGCCCGGCCGG-------
GGCGGGATCGCCCTGTGACTAGTggtaccgagctcGAATTC,
fdmF: TCTAGAttaattaaGGAGGACCCATATGACCGCACGTGCGGTG-------
CCGAGGAGGATCGCATGAACTAGTaagctt,
fdmG: TCTAGAGGAGGACCCATATGACCGGGCCCGTCATC-------
GTCCTCAGATCCGTCTGAACTAGTaagctt,
fdmH: TCTAGAGGAGGAGCGCATATGAGCACCATCAGCTTC-------
CTGTGTGAGGGGGTCTGACTAGTggtaccgagctcGAATTC,
fdmO: TCTAGABGGAGGAGCCCATATGGAACTTCGACTGGTA-------
GTCGACGGGGGTGTCTGACTAGTaagctt,
fdmS: TCTAGAGGAGGAGCCCATATGACTGCGATCCGGGAC-------
GTCGTCCGGCTCCCCTGACTAGTaagctt,
fdmW: TCTAGAGGAGGAGCCCATATGCAACCTGACGACGAG-------
GTCGTCGGTGCGGCCTGACTAGTggtaccgagctcGAATTC.
The fdmG gene was amplified to introduce a His6-tag with the following primers: fdmG_FP: AATTCTAGAGGAGGACCCATATGCATCATCATCATCATCATACCGGGCCCGTCCATCACCGGGGTGGGCG, and fdmG_RP: AGCGAATTCGGGGACTAGTTCAGACGGCTCTGAGGACCA. The underlined sequence encodes the His6-tag. Restriction sites are indicated in italics or in lowercase, and ribosomal binding sites are in bold. The genes fdmF, fdmH, and fdmW were cloned into pLitmus28i vector by XbaI/HindIII digestion to introduce an EcoRI site at the 3′ end. They were then assembled as synthetic operons in pUC18 prior to transfer into a pRM5-derived shuttle vector (McDaniel et al., 1993). The first gene in each operon was inserted between the XbaI and EcoRI sites in pUC18; subsequent genes were inserted downstream following SpeI/EcoRI digestion of the vector and XbaI/EcoRI digestion of the insert. Plasmid pAD201 was constructed by excising the complete operon by PacI/EcoRI digestion, which was inserted between the PacI/EcoRI sites of shuttle vector pRM5. The fdmS, fdmC, and fdmO genes were inserted onto plasmid pAD201 by a similar method described above, yielding plasmid pAD210.
Protein expression and purification
S. coelicolor CH999/pAD201/pBOOST* was the source of the His6-tagged fdm KS-CLF. Spores suspensions of each strain were used to inoculate 4 × 1 L minimal medium (6.25% (w/v) PEG 8000, 25 mM TES, pH 7.2, 0.5% (w/v) casamino acids, 1% (w/v) glucose, 15 mM (NH4)2SO4, 5 mM MgSO4, 1.25 mM NaH2PO4, 1.25 mM K2HPO4, 926 nM FeCl3, 377 nM ZnCl2, 99 nM MnCl2, 73 nM CuCl2, 33 nM Na2B4O7, 10 nM (NH4)6Mo7O24) containing 50 mg/L thiostrepton and 100 mg/L apramycin. Mycelia from the stationary phase cultures (about 2 days) were collected by centrifugation, and resuspended in 140 mL disruption buffer (200 mM NaH2PO4, pH 7.1, 200 mM NaCl, 30% (v/v) glycerol, 2.5 mM EDTA, 2 mM DTT, 1.5 mM benzamidine, 4.3 μM leupeptin, 2.9 μM pepstatin). Cells were disrupted by sonication, and the cell debris was removed by centrifugation (17,000 rpm for 30 min). DNA was precipitated by adding 0.2% polyethyleneimine (PEI), followed by centrifugation (17,000 rpm for 30 min). The KS-CLF protein was precipitated between 30 and 50% (NH4)2SO4. Precipitated proteins were re-dissolved in 100 mM NaH2PO4, pH 7.4, and incubated with 2 mL Ni-NTA resin (Qiagen) for 2 h at 4 °C. The slurry was poured into a capped column, and washed with 20 mL of the first wash buffer (50 mM sodium phosphate, 300 mM NaCl, 20 mM imidazole, pH 8.0), followed by 4 mL of the second wash buffer (50 mM sodium phosphate, 300 mM NaCl, 50 mM imidazole, pH 8.0). The KS-CLF heterodimer was eluted with elution buffer (50 mM sodium phosphate, 100 mM NaCl, 250 mM imidazole, pH 8.0). Fractions containing the KS-CLF were pooled and buffer exchanged into 45 mL of buffer A (50 mM Tris•HCl, pH 8.0, 1 mM TCEP, and 10% glycerol) and loaded onto a Hi-Trap Q column pre-equilibrated with buffer A. The following gradient was applied to this column: 0% buffer B (buffer A with 500 mM NaCl) from 0 to 20 min, 10% buffer B from 20 to 25 min, 80% buffer B from 25 to 85 min, and 100% buffer B from 85 to 100 min. The target protein eluted at 48% buffer B. The eluant was concentrated, buffer exchanged into a storage buffer (100 mM NaH2PO4, pH 7.4, 2 mM TCEP, 2 mM EDTA, and 15% glycerol), flash-frozen with liquid nitrogen, and stored at −80 °C. The typical yield of purified fdm KS-CLF was 1 mg/L.
The fdm holo-ACP was expressed in an engineered E. coli strain BAP1 (Pfeifer et al., 2001) to obtain its 4′-phosphopantetheinylated form. Protein expression was induced at an OD600 of 0.6 with 200 μM IPTG, and allowed to proceed at 18 °C for 18 h. Cells were harvested and lysed by sonication. The protein was initially purified on Ni-NTA resin, followed by anion exchange chromatography on a Hi-Trap Q column (from 0 to 500 mM NaCl in 60 min). The proteins eluted at 200 mM NaCl. Purified protein was buffer exchanged into 100 mM sodium phosphate, 15% glycerol, pH 7.2 and stored at −80 °C. The relative ratio of the holo and apo forms of the ACP was quantified by LC-MS, and was found to be ~50% in the holo form. The typical yield of purified ACP was 12 mg/L.
The S. coelicolor MAT gene was expressed in E. coli BL21(DE3), and the MAT protein was purified as described previously (Carreras and Khosla, 1998). The typical yield of purified MAT was 15 mg/L. All protein concentrations were determined by the Bradford method with the Bio-Rad protein kit.
Detection of covalently bound protein intermediates by radio-SDS-PAGE
To investigate the enzymatic activity of PKS components in vitro, selected protein combinations were incubated with radiolabeled substrates and analyzed via radio-SDS-PAGE. For example, a PKS reaction mixture (12 μL) contained 200 mM [14C]-malonyl-CoA (55 Ci/mol), 10 μM the fdm KS-CLF, 25 μM holo-ACP, and 1 μM MAT in a buffer containing 100 mM sodium phosphate, pH 7.3, 2 mM EDTA, 2 mM TCEP. Following incubation at room temperature for 30 min, the reaction was quenched with 6 μL of non-reducing sample buffer (0.1% bromophenol blue, 1% SDS, 40% glycerol), and loaded directly onto a 4%–12% SDS gel. Following staining, dried gels were subjected to autoradiography.
Radio-TLC analysis of PKS activity in vitro
This assay detects polyketides synthesized by the fdm minimal PKS from radiolabeled malonyl-CoA. Assays were performed at 30 °C in 30 μL of reaction buffer, as described above. Reaction mixtures contained 50 μM holo-ACP, 10 μM KS-CLF, 0.1 μM MAT, and 1 mM [14C]-malonyl-CoA (55 Ci/mol). The reaction was initiated by adding [14C]-malonyl-CoA, and quenched at 2 min, 4 min, 6 min, 10 min, and 20 min by adding 200 μL ethyl acetate. Each quenched mixture was vortexed and extracted with 200 μL ethyl acetate twice. The combined organic phases were evaporated to dryness and re-dissolved in 15 μL ethyl acetate. The reaction products were separated by thin-layer chromatography (TLC) using a solvent containing 99% ethyl acetate and 1% acetic acid, and quantified with a phosphoimager. Cerulenin was used at a final concentration of 1 mM.
Production, isolation, and characterization of polyketide products
For small scale cultivations, Streptomyces strains were grown on R5 agar plates containing 50 mg/L thiostrepton and 100 mg/L of apramycin at 30 °C for 7 days. For HPLC analysis, the pigmented agar medium from 10–12 plates was mashed and extracted with methanol/ethyl acetate (10:90). The solvent was evaporated in vacuo, and the residue was dissolved in methanol. The mixture was analyzed by reverse-phase HPLC outfitted with a UV-Vis detector at 280 and 410 nm, using the Apollo C18 column (250 mm × 4.6 mm). A linear gradient from 30% acetonitrile (MeCN) in water (0.1% TFA) to 55% MeCN in water (0.1% TFA) was used over 40 min with a flow rate of 1 mL/min.
For preparative scale studies, the above fermentation and extraction procedures were scaled up to 3–4 L of R5 agar medium. The solvent was removed in vacuo. The crude extract was flashed through a silica gel column using 1% acetic acid in ethyl acetate. The eluent was dried, re-dissolved in 3–4 mL methanol, and injected onto the preparative reverse-phase HPLC column (250 mm × 22 mm, C18 column, Vydac). Gradients used included 10–40% MeCN in water (0.1% TFA) over 80 min or 20–60% MeCN in water (0.1% TFA) over 100 min, with a flow rate of 5 mL/min. Purified fractions were dried in vacuo or by lyophilization. In cases where further purification was warranted, the relevant HPLC fraction was re-dissolved in 0.5–1 mL acetone and applied to a preparative TLC plate, which was developed in a solvent system consisting of ethyl acetate and hexane with or without 1% acetic acid. 1H and 13C NMR spectra of purified polyketide products were recorded on Varian Inova 500. The 1H NMR spectra were referenced at 2.49 ppm in DMSO-d6 or 3.31 ppm in CD3OD. The 13C NMR spectra were referenced at 39.5 ppm in DMSO-d6.
Synthesis of genes encoding CLF variants
High throughput gene synthesis was carried out to synthesize these variants (Villalobos et al., 2006).
Detection of covalently bound intermediates using Electrospray Ionization (ESI) Fourier-Transform Mass Spectrometry (FT-MS)
A reaction mixture (50 μL) contained 1 mM malonyl-CoA, 6.9 μM KS-CLF heterodimer, 34.7 μM holo-ACP, and 0.7 μM MAT in a buffer containing 100 mM sodium phosphate, pH 7.3, 2 mM EDTA, 2 mM TCEP. Following incubation at room temperature for 30 min, the reaction mixtures were purified using the Phenomenex C4 column (150 mm × 4.6 mm). A linear gradient from 10% acetonitrile (MeCN) in water (0.1% TFA) to 90% MeCN in water (0.1% TFA) was used over 30 min with a flow rate of 1 mL/min. The eluent was collected as 1 mL fractions, lyophilized to dryness, and re-dissolved in an ESI solvent containing 49.5% water, 49.5% methanol, and 1% formic acid for FT-MS analysis. ESI-FT-MS analysis was performed on a Thermo Electron LTQ-FT-ICR hybrid mass spectrometer.
The HPLC fraction containing the fdm ACP was re-dissolved in the ESI solvent and directly introduced into the mass spectrometer with an Advion BioSciences Nanomate 100 Biversa nanospray source. The instrument was operated in positive ion mode, using an electrospray voltage between 1.35 to 1.45 kV and gas pressure of 0.35 psig. Samples were subjected to in-source fragmentation by adjusting the skimmer voltage to 75 V. In-source fragmentation allowed for ejection of the phosphopantetheine arm from the ACP, along with any acyl-intermediate bound to it. The identities of these pantetheine-bound intermediates were determined by intact exact mass analysis followed by MS/MS (MS3) fragmentation. In order to achieve sufficient ion intensity for MS3 identification, pantetheine ejection ions were trapped using an isolation width of 4 m/z centered on the ion of interest. MS3 fragmentation employed the following settings: normalized collision energy of 30%, an activation Q of 0.200 to 0.230, and an activation time of 50 to 100 ms depending on the intensity of the particular pantetheine ejection ion being identified. All spectra were recorded by FT-ICR-MS with a resolution of 100,000.
Supplementary Material
Acknowledgments
We thank Professor Sheryl Tsai and Pouya Javidpour at UC-Irvine for assistance in generating a homology model of the fdm KS-CLF. This research was supported by a grant from the NIH (CA 77248 to CK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Carreras CW, Khosla C. Purification and in vitro reconstitution of the essential protein components of an aromatic polyketide synthase. Biochemistry. 1998;37:2084–2088. doi: 10.1021/bi972919+. [DOI] [PubMed] [Google Scholar]
- Das A, Khosla C. Biosynthesis of aromatic polyketides in bacteria. Acc Chem Res. 2009;42:631–639. doi: 10.1021/ar8002249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Szu PH, Fitzgerald JT, Khosla C. Mechanism and engineering of polyketide chain initiation in fredericamycin biosynthesis. J Am Chem Soc. 2010;132:8831–8833. doi: 10.1021/ja102517q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrestein PC, Kelleher NL. Dissecting non-ribosomal and polyketide biosynthetic machineries using electrospray ionization fourier-transform mass spectrometry. Nat Prod Rep. 2006;23:893–918. doi: 10.1039/b511400b. [DOI] [PubMed] [Google Scholar]
- Dreier J, Khosla C. Mechanistic analysis of a Type II polyketide synthase. Role of conserved residues in the b-ketoacyl synthase-chain length factor heterodimer. Biochemistry. 2000;39:2088–2095. doi: 10.1021/bi992121l. [DOI] [PubMed] [Google Scholar]
- Hertweck C, Luzhetskyy A, Rebets Y, Bechthold A. Type II polyketide synthases: gaining a deeper insight into enzymatic teamwork. Nat Prod Rep. 2007;24:162–190. doi: 10.1039/b507395m. [DOI] [PubMed] [Google Scholar]
- Hopwood DA, Sherman DH. Molecular genetics of polyketides and its comparison to fatty acid biosynthesis. Annu Rev Genet. 1990;24:37–66. doi: 10.1146/annurev.ge.24.120190.000345. [DOI] [PubMed] [Google Scholar]
- Hu Z, Hopwood DA, Hutchinson CR. Enhanced heterologous polyketide production in Streptomyces by exploiting plasmid co-integration. J Ind Microbiol Biotechnol. 2003;30:516–522. doi: 10.1007/s10295-003-0064-y. [DOI] [PubMed] [Google Scholar]
- Keatinge-Clay AT, Maltby DA, Medzihradszky KF, Khosla C, Stroud RM. An antibiotic factory caught in action. Nat Struct Mol Biol. 2004;11:888–893. doi: 10.1038/nsmb808. [DOI] [PubMed] [Google Scholar]
- Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces Genetics. Norwich: The John Innes Foundation; 2000. [Google Scholar]
- McDaniel R, Ebert-Khosla S, Hopwood DA, Khosla C. Engineered biosynthesis of novel polyketides. Science. 1993;262:1546–1550. doi: 10.1126/science.8248802. [DOI] [PubMed] [Google Scholar]
- Meluzzi D, Zheng WH, Hensler M, Nizet V, Dorrestein PC. Top-down mass spectrometry on low-resolution instruments: characterization of phosphopantetheinylated carrier domains in polyketide and non-ribosomal biosynthetic pathways. Bioorg Med Chem Lett. 2008;18:3107–3111. doi: 10.1016/j.bmcl.2007.10.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshull J, Ness JE, Gustafsson C, Govindarajan S. Predicting enzyme function from protein sequence. Curr Opin Chem Biol. 2005;9:202–209. doi: 10.1016/j.cbpa.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Nicholson TP, Winfield C, Westcott J, Crosby J, Simpson TJ, Cox RJ. First in vitro directed biosynthesis of new compounds by a minimal type II polyketide synthase: evidence for the mechanism of chain length determination. Chem Comm. 2003:686–687. doi: 10.1039/b300847a. [DOI] [PubMed] [Google Scholar]
- O’Hagan D. The Polyketide Metabolites. Chichester, UK: Ellis Howard; 1991. [Google Scholar]
- Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C. Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science. 2001;291:1790–1792. doi: 10.1126/science.1058092. [DOI] [PubMed] [Google Scholar]
- Rawlings BJ. Biosynthesis of polyketides (other than actinomycete macrolides) Nat Prod Rep. 1999;16:425–484. doi: 10.1039/a900566h. [DOI] [PubMed] [Google Scholar]
- Shen Y, Yoon P, Yu TW, Floss HG, Hopwood D, Moore BS. Ectopic expression of the minimal whiE polyketide synthase generates a library of aromatic polyketides of diverse sizes and shapes. Proc Natl Acad Sci USA. 1999;96:3622–3627. doi: 10.1073/pnas.96.7.3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S, Tsai SC. The type I fatty acid and polyketide synthases: a tale of two megasynthases. Nat Prod Rep. 2007;24:1041–1072. doi: 10.1039/b603600g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staunton J, Weissman KJ. Polyketide biosynthesis: a millennium review. Nat Prod Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- Tang Y, Tsai SC, Khosla C. Polyketide chain length control by chain length factor. J Am Chem Soc. 2003;125:12708–12709. doi: 10.1021/ja0378759. [DOI] [PubMed] [Google Scholar]
- Tang Y, Lee TS, Khosla C. Engineered biosynthesis of regioselectively modified aromatic polyketides using bimodular polyketide synthases. PLoS Biol. 2004;2:227–238. doi: 10.1371/journal.pbio.0020031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalobos A, Ness JE, Gustafsson C, Minshull J, Govindarajan S. Gene Designer: a synthetic biology tool for constructing artificial DNA segments. BMC Biotechnol. 2006;7:285. doi: 10.1186/1471-2105-7-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt-Pienkowski E, Huang Y, Zhang J, Li B, Jiang H, Kwon H, Hutchinson CR, Shen B. Cloning, sequencing, analysis, and heterologous expression of the fredericamycin biosynthetic gene cluster from Streptomyces griseus. J Am Chem Soc. 2005;127:16442–16452. doi: 10.1021/ja054376u. [DOI] [PubMed] [Google Scholar]
- Xu Z, Metsä-Ketelä M, Hertweck C. Ketosynthase III as a gateway to engineering the biosynthesis of antitumoral benastatin derivatives. J Biothenol. 2009;140:107–113. doi: 10.1016/j.jbiotec.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Xu Z, Schenk A, Hertweck C. Molecular analysis of the benastatin biosynthetic pathway and genetic engineering of altered fatty acid-polyketide hybrids. J Am Chem Soc. 2007;129:6022–6030. doi: 10.1021/ja069045b. [DOI] [PubMed] [Google Scholar]
- Yu TW, Shen Y, McDaniel R, Floss HG, Khosla C, Hopwood DA, Moore BS. Engineered biosynthesis of novel polyketides from Streptomyces spore pigment polyketide synthases. J Am Chem Soc. 1998;120:7749–7759. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.