

Abstract
The field of Alzheimer’s disease (AD) research eagerly awaits the results of a large number of Phase III clinical trials that are underway to investigate the effectiveness of anti-amyloid-β (Aβ) immunotherapy for AD. In this case report, we review the pertinent clinical history, examine the neuropathology, and characterize the Aβ profile of an AD patient who received bapineuzumab immunotherapy. The patient received four bapineuzumab infusions over a 39 week period. During the course of this treatment, there was no remarkable change in cognitive impairment as determined by MMSE scores. Forty-eight days after the fourth bapineuzumab infusion was given, MRI revealed that the patient had developed lacunar infarcts and possible vasogenic edema, probably related to immunotherapy, but a subsequent MRI scan 38 days later demonstrated resolution of vasogenic edema. The patient expired due to acute congestive heart failure complicated by progressive AD and cerebrovascular accident 378 days after the first bapineuzumab infusion and 107 days after the end of therapy. Neuropathological and biochemical analysis did not produce evidence of lasting plaque regression or clearance of Aβ due to immunotherapy. The Aβ species profile of this case was compared with non-immunized AD cases and non-demented controls and found to be similar to non-immunized AD cases. SELDI-TOF mass spectrometric analysis revealed the presence of full-length Aβ1-42 and truncated Aβ peptides demonstrating species with and without bapineuzumab specific epitopes. These results suggest that, in this particular case, bapineuzumab immunotherapy neither resulted in detectable clearance of amyloid plaques nor prevented further cognitive impairment.
Keywords: active immunotherapy, Alzheimer’s disease, amyloid-β, amyloid plaques, bapineuzumab, cerebral amyloid angiopathy, neurofibrillary tangles, passive immunotherapy
Introduction
Alzheimer’s disease (AD) is the most common form of dementia affecting the elderly population worldwide. At present there is no effective treatment to modify the course of this disease. It is widely believed that central to the pathogenesis of AD is the profuse accumulation of amyloid-β (Aβ) peptides in the gray matter as amyloid plaques and in the walls of cerebral blood vessels. A promising potential therapy to stop the progression of AD is the administration of passive or active immunizations against the Aβ peptides. In this case report, we examined the clinical history and neuropathology as well as characterized the Aβ species extant after immunotherapy in a patient who received infusions of bapineuzumab, a humanized antibody (3D6) raised against the N-terminus (residues 1-5) of the Aβ peptide [1;2]. We compared the bapineuzumab-treated patient with 6 non-immunized AD cases, and 5 non-demented controls (NDC).
Clinical and neuropathological reports and treatment synopsis
The patient had a past medical history notable for depression, hypercholesterolemia, hypertension, coronary atherosclerosis, coronary stenting, asthma, and gastrointestinal reflux. The patient’s mother had died of late onset AD. The patient started to experience memory problems in 1999. At that time the patient had a Mini Mental State Examination (MMSE) score of 29 while EEG and cranial CT imaging demonstrated no abnormalities. In 2000, the patient was diagnosed as having mild dementia and Aricept was prescribed then later changed to Exelon in 2001. In 2002, the patient had an MMSE score of 26 and was still capable of playing golf. By 2004 the patient was forgetful, sleeping excessively, with decreased interest in daily activities; the MMSE score was 22. The patient was enrolled in a bapineuzumab double blind study during which the patient received 6 doses of placebo between June 2005 and September 2006. In February 2007, the patient entered the open-label extension study and was placed on active bapineuzumab immunotherapy. A total of 4 doses of bapineuzumab (0.5 mg/kg per dose) were administered over the course of 39 weeks, with the last dose given in the first week of November 2007. Along the course of immunotherapy, four MMSE scores were obtained which fluctuated between 11 and 8. During this time, the patient had an acute myocardial infarction in August 2007. In the last week of December 2007, the patient presented with an ataxic gait. An MRI scan was interpreted as showing an acute lacunar infarct in the left head of the caudate nucleus and putamen, with signs of vasogenic edema in the same region. In early January 2008, the patient was admitted to the hospital with ataxia, gait disturbance, and a decreased level of consciousness. The CSF analysis revealed a protein concentration of 61 mg/dl and 5 mononuclear white blood cells with no signs of infection. Imaging with CT revealed no acute changes. Intravenous Dexamethasone was administered. Three days after the CT scan, an MRI scan was interpreted as showing acute infarction and/or edema in the left frontal periventricular white matter and also involving the basal ganglia. An EEG demonstrated epileptiform waves with severely disorganized rhythm. The patient was started on Keppra. In subsequent days, the patient began to improve in terms of following commands, orientation, and ambulatory and speech capacities. The patient was discharged to a nursing home in mid-January, with noticeable clinical improvement. A follow-up MRI scan a week later reported a lacunar infarct in the left caudate with increased white matter hyperintensities in the right temporal tip and right frontal lobe, with worsening of the mental status, presumably due to an evolving stroke. The patient was hospitalized in a non-ambulatory condition with severe cognitive and verbal impairments and was re-started on Decadron and Keppra. An MRI scan demonstrated apparent progression of the white matter and basal ganglia abnormalities although this was not considered to represent vasogenic edema. The patient expired in the third week of February 2008 at age 79. The direct cause of death was attributed to acute congestive heart failure complicated by progressive AD and stroke.
The brain weighed 1000 grams. The dura was normal and the leptomeninges showed mild fibrosis. There was mild to moderate gyral atrophy. The base of the brain showed severe patchy atherosclerosis of the vessels of the circle of Willis. The mammillary bodies were normal while the unci showed moderate atrophy without signs of herniation. The brainstem and cerebellum were externally unremarkable. Cerebral slices showed moderate and marked enlargement of the frontal horns and of the body of the lateral ventricles, respectively. The basal ganglia showed a reddish cavity in the head of the left caudate nucleus and putamen that also focally involved the internal capsule, measuring 2.0 × 2.0 × 1.2 cm. Similar cavities but yellowish in color rather than red and smaller, each about 1 cc in total, were present in the right head of the caudate nucleus and in the right posterior putamen at the level of the lentiform nucleus. The thalamus and subthalamic regions were unremarkable. The amygdala showed moderate atrophy and there was moderate atrophy of the head and body of the hippocampi and the parahippocampal gyri. The substantia nigra showed mild to moderate depigmentation. Axial slices of the brainstem were unremarkable, as were parasagittal slices of the cerebellum.
Paraffin-embedded, H & E-stained sections showed moderate to marked gliosis in the upper neocortical layers, with focal full-depth gliosis and frequent hypertrophic astrocytes. Frequent senile plaques were evident. There were no chronic inflammatory cell infiltrates in gray or white matter and no evidence of edema. There was an old cortical microinfarct in the middle frontal gyrus. The amygdala and entorhinal cortex showed very marked gliosis and moderate tissue rarefaction. The hippocampal formation showed marked gliosis and tissue rarefaction in the outer part of the dentate gyrus molecular layer and moderate gliosis in area CA1, with the latter region also containing neurofibrillary tangles, granulovacuolar degeneration and Hirano bodies but showing no obvious neuronal loss. The grossly-described cavitating lesion in the left head of the caudate nucleus and putamen was seen microscopically to be consistent with a resolving, subacute old infarction. The cavity was filled with foamy macrophages and persisting islands of acellular neuropil and was bordered by frequent hypertrophic astrocytes. Uninvolved striatal areas showed rare large neurons with neurofibrillary tangles. There were multiple mineralized blood vessels in the globus pallidus and other blood vessels in the basal ganglia and thalamus showed thickened collagenous walls. There was an old microinfarct in the left dorsal putamen at the level of the full development of the lentiform nucleus. The nucleus basalis of Meynert was depleted of neurons and multiple remaining neurons contained neurofibrillary tangles. The medial thalamus showed marked gliosis and moderate tissue rarefaction. The substantia nigra was mildly depleted of pigmented neurons without evidence of Lewy bodies. The superior vermis and hemispheric cerebellar cortex showed mild patchy depletion of Purkinje cells but otherwise the cerebellum was unremarkable as were sections of the remainder of the brainstem and major levels of the spinal cord. Large, 3 × 5 cm H & E-stained sections showed marked and extensive white matter rarefaction in the frontal lobe, with mild rarefaction in the temporal and occipital lobes. There were two old microscopic cortical infarctions in the inferior parietal lobule and eight of these in the lateral occipital lobe. There was an old cystic microinfarct in the cerebellar hemispheric cortex, as well as a single microscopic focus of cortical sclerosis. Large, 3 × 5 cm sections stained with Gallyas, Campbell-Switzer and thioflavine-S methods showed, in neocortical areas, very frequent senile plaques, with frequent diffuse as well as classical (neuritic and cored) plaques. These plaques were evenly distributed in all sections, with no evidence of focal or patchy depletion. There were very frequent neurofibrillary tangles in all cortical areas as well as in the amygdala, hippocampus, entorhinal, and transentorhinal areas. Amyloid angiopathy was present at sparse densities in the frontal and temporal lobes, with moderate to frequent densities in the parietal and occipital lobes. Immunohistochemical staining for phosphorylated alpha-synuclein showed frequent Lewy bodies and Lewy-related neurites in the amygdala and superior cervical sympathetic ganglion, with sparse densities of Lewy-related pathology in the olfactory bulb and dorsal medulla. No synuclein-immunoreactive histopathology was present in frontal, temporal or parietal neocortex.
As the neuropathological findings met diagnostic criteria for both AD (NIA-Reagan “high” probability that dementia was due to AD [3]) as well as the Binswanger’s type of vascular dementia (NINDS-AIREN criteria [4]), both diagnoses were assigned. The acute infarct of the left head of the caudate nucleus and putamen described on MR imaging during the final months of life correlated well with the postmortem histological appearance of a resolving, subacute to old infarct in the same region. There was no neuropathological evidence, however, that corresponded to the imaging findings suggestive of a more extensive infarction involving the frontal lobe white matter. Rather, it is likely that this imaging finding corresponded to the extensive chronic white matter rarefaction seen on large sections of the frontal lobe, consistent with leukoaraiosis. The presence of Lewy bodies restricted to the brainstem and limbic regions was insufficient for the diagnosis of dementia with Lewy bodies (DLB Consortium criteria [5]), and therefore the diagnosis of dementia with Lewy bodies was not assigned. Analysis of DNA isolated from postmortem cerebellum indicated an apolipoprotein E (ApoE) genotype of ε4/ε4.
In addition to the immunized individual, 5 neuropathologically-confirmed NDC and 6 neuropathologically-confirmed non-immunized AD cases were provided by our own Brain Bank at Banner Sun Health Research Institute (BSHRI) for comparison. The NDC included 2 females and 3 males with a mean age of death of 82 years. The AD group was composed of 1 male and 5 females with mean disease duration of 9 years and average age at the time of death of 82 years. Detailed neuropathological and biochemical data on these NDC and AD individuals has been recently published [6].
Materials and Methods
Neuropathological evaluation
All cases, including the bapineuzumab case presented here as well as the non-immunized AD and NDC individuals, were evaluated for total plaque score, plaque density, total neurofibrillary tangle score, white matter rarefaction (WMR) score, CERAD criteria, neuritic plaque score [7] and Braak stage [8]. These neuropathological assessments are detailed in a previous publication [9]. Histopathological appraisals of the AD lesions were obtained by thioflavine-S staining, Switzer-Campbell silver staining and Gallyas staining [9]. In addition, the leptomeningeal membranes from the bapineuzumab patient from both cerebral hemispheres were separated from the surface of the brain prior to its coronal sectioning, washed 12 times with 4 L each of cold distilled water to remove blood, spread on the surface of 5 inch plastic Petri dishes, allowed to dry in an oven at 60°C overnight, fixed for 2 h with absolute ethanol and stained by thioflavine-S. Based on experience, the degree of vascular leptomeningeal amyloidosis was semi-quantitatively classified as severe.
Quantification of soluble and insoluble Aβ by ELISA
Gray matter tissue (100 mg) was homogenized in 6 volumes of 20 mM Tris-HCl, 5 mM EDTA, pH 7.8 with a protease inhibitor cocktail (PIC, Roche Diagnostics, Mannheim, Germany), centrifuged at 435,000 × g for 20 min (Beckman Optima TLA ultracentrifuge, 120.2 rotor; Beckman, Fullerton, CA) and the total protein concentration in the soluble fraction quantified using the Pierce Micro BCA protein assay (Rockford, IL). In addition, 400 mg samples of gray and white matter tissue were homogenized in 3 ml of 90% glass distilled formic acid (GDFA) and centrifuged at 250,000 × g for 1 h (Beckman LE-80 ultracentrifuge, SW41 rotor). The de-lipidated supernatants were collected and the top fat layer discarded. This procedure solubilized fibrillar, diffuse, membrane-bound, and intra- and extra-cellular oligomeric Aβ species. Aliquots of the GDFA-soluble fraction were submitted to fast protein liquid chromatography (FPLC) size-exclusion chromatography in 80% GDFA mobile phase (see below). The fractions containing Aβ peptides were pooled, reduced to 2 ml by vacuum centrifugation (SpeedVac; Savant/Thermo, Waltham, MA) and dialyzed (1000 MW cutoff tubing) against two changes of water, 2 changes of 0.1 M ammonium bicarbonate followed by lyophilization. The samples were dissolved in 500 μl of 5 M guanidine hydrochloride (GHCl), 50 mM Tris-HCl, pH 8.0 and shaken overnight at 4°C. Total protein was determined by the Pierce Micro BCA protein assay. The ELISA kits to quantify Aβ40 and Aβ42 were obtained from Invitrogen (Carlsbad, CA) and Innogenetics (Gent, Belgium), respectively, and performed following the manufacturer’s instructions.
Quantification of tumor necrosis factor-α (TNF-α) by ELISA
From each of the specimens, gray matter (100 mg) was homogenized in 20 volumes of 20 mM HEPES, 1.5 mM EDTA, pH 7.4, PIC (Roche) with an Omni TH electric tissue grinder and centrifuged at 3000 × g for 15 min in an IEC Centra CL3R centrifuge (Thermo, Waltham, MA). The supernatants were collected, centrifuged at 40,000 × g for 1 h (Optima LE-80K ultracentrifuge, 50.4 Ti rotor; Beckman). Again, the supernatants were collected and total protein concentrations determined (BCA protein assay, Pierce). Human TNF-α levels were evaluated using an ELISA kit (PromoKine, Heidelberg, Germany) following the manufacturer’s directions.
Fast protein liquid chromatography (FPLC)
Cerebral cortex was homogenized in 90% GDFA, and centrifuged and the supernatant was submitted to size-exclusion FPLC using a Superose 12 column (10 × 300 mm, General Electric, Uppsala, Sweden) as described in detail elsewhere [10]. The fraction containing the Aβ peptides was reduced by vacuum centrifugation (SpeedVac, Savant Instruments Inc. Farmingdale NY), and stored at -80°C.
High performance liquid chromatography (HPLC)
The FPLC fractions were further purified by HPLC using a reverse-phase C8 column (4.6 × 250 mm, Zorbax SB, Mac Mod) maintained at 80°C. A linear gradient was developed from 0-60% water/acetonitrile containing 0.1% trifluoroacetic acid (TFA) at a flow rate of 1 ml per min over a period of 120 min and the chromatography monitored at 214 nm. A total of 9 fractions were collected and the volume reduced by vacuum centrifugation (SpeedVac). The fractions were washed 3 times with water (200 μl each) and the volume reduced by vacuum centrifugation to neutralize the acid. Each fraction was then reconstituted in 2xLDS sample loading buffer (Invitrogen, Carlsbad, CA) containing 50 mM dithiothreitol. Western blots were performed as previously reported [11] using anti-Aβ40 and anti-Aβ42 (Invitrogen) and CT9APP (Millipore, Billerica, MA) as primary antibodies.
Surface enhanced laser desorption/ionization-time of flight mass spectrometry (SELDI-TOF) mass spectrometry (MS). Aβ40/42 Method
Following HPLC fractionation samples were analyzed using Western blots and the amyloid-containing peaks subjected to SELDI-TOF mass spectrometry as described in a previous publication [10]. Polyclonal anti-Aβ40 and anti-Aβ42 antibodies (Invitrogen) were used at a concentration of 0.38 mg/ml on PS20 chips as capture antibodies (Bio-Rad, Hercules, CA). The molecular mass assignments resulted from 100 averaged shots in a Bio-Rad SELDI Protein Biology System II with external calibration acquired with the ProteinChip Peptide Mass Calibration Kit (Bio-Rad).
6E10 Method ProteinChip β-Amyloid MPD Kit
The HPLC fractions were pooled to yield one combined sample per case. All experimental steps were carried out at room temperature. The 6E10 protein chips, calibration standards and experimental instructions were provided by the manufacturer (Bio-Rad). Fifty μl/ml of the internal standard (Aβ Cys1-24 Mr = 2,979.3) was added to each calibration standard (Aβ1-16, 1-38, 1-40 and 1-42) as well as to each of the specimens (~200-400 μl) under investigation. The ProteinChip arrays were equilibrated with 5 μl of PBS per spot for 5 min. The arrays were then loaded with 5 μl of sample or calibrant, placed in a humidified chamber and incubated for 1 h. Following removal of the sample, each array was placed in a 15 ml Falcon tube and rinsed 3 times (5 min each) with wash buffer (PBS, 0.5% Triton X-100) and 3 times (5 min each) with PBS. Finally, to desalt the arrays, 10 ml of 0.1 M HEPES was added to each tube, incubated for 5 min and air dried. Acetonitrile (200 μl) and 1.0% TFA (200 μl) were added to 5 mg of α-cyano-4-hydroxycinnamic acid (CHCA) and the mix vortexed for 2 min. After centrifugation at 1000 × g for ~1 min to remove particles, 200 μl of the CHCA solution was combined with 400 μl acetonitrile and 400 μl 1.0% TFA and vortexed 1 min. Finally, 1 μl of the prepared CHCA solution was applied to each spot and air dried. The molecular mass assignments resulted from 100 averaged shots in a Bio-Rad SELDI Protein Biology System II with external calibration attained using the ProteinChip Peptide Mass Calibration Kit (Bio-Rad).
Results
Neuropathological examination of the bapineuzumab immunized patient revealed brain atrophy (1000 g), total plaque score of 15 (range 0-15), plaque density as frequent, total neurofibrillary tangles score of 3 (range 0-15), Braak stage of VI (range I-VI), total cerebral cortex amyloid angiopathy of 7 (range 0-12) and total WMR score of 5 (range 0-12). The CERAD NP score classified this brain as definite AD. Figures 1A and B show illustrations of the amyloid core plaques and diffuse amyloid densities and Figure 1C demonstrates neurofibrillary tangle pathology. In general, there were no areas of apparent amyloid plaque clearance (Figure 1). In addition, this patient exhibited severe leptomeningeal cerebral amyloid angiopathy (Figure 1D) and moderate areas of WMR (demyelination).
Figure 1.
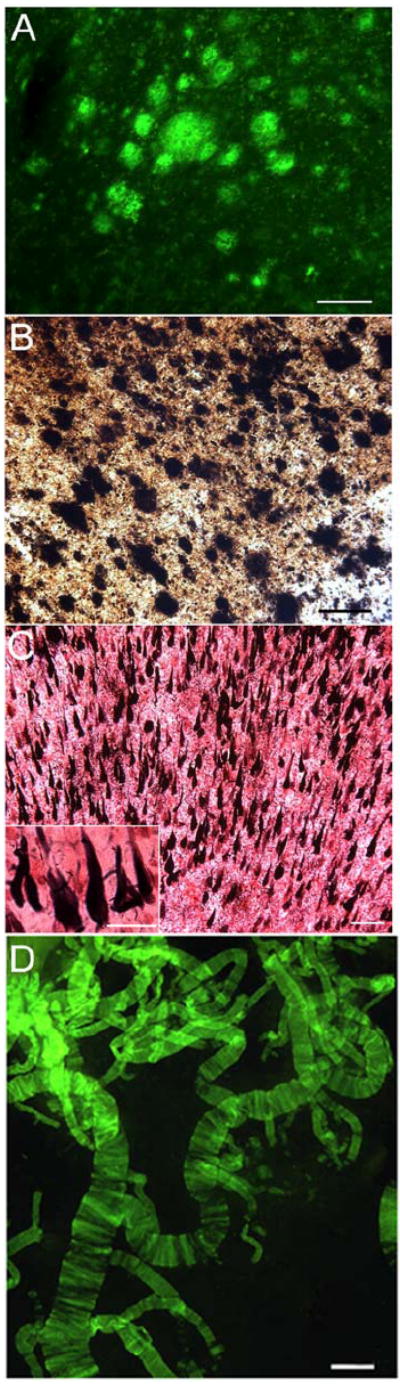
Frontal cortex and leptomeningeal blood vessels histopathology from the Bapineuzumab patient. Amyloid plaques stained by: A) thioflavin-S and B) Campbell-Switzer. C) Gallyas staining demonstrating numerous neurofibrillary tangles. D) Blood vessels stained by thioflavin-S showing severe cerebrovascular amyloidosis. Scale bars for: A and B = 100 μm; C = 100 μm (inset = 25 μm); D = 250 μm.
Immunoassay analysis of the bapineuzumab patient demonstrated high levels of GDFA/GHCl-soluble Aβ in both the gray and white matter, similar to mean levels present in the non-immunized AD group (Figure 2A and 2B). The total gray matter Tris-soluble Aβ level was similar to the mean of the NDC and non-immunized AD groups (Figure 2C). The scatter plots (Figure 2) of the Aβ ELISA show the wide individual variation among the AD group. In the bapineuzumab case, the level of gray matter TNF-α was higher than the mean observed in the non-immunized AD and NDC individuals (Figure 2D). Interestingly, there were no significant differences between AD and NDC TNF-α levels. The levels of TNF-α are in agreement with a previous study using the same technique on control and demented individuals [12].
Figure 2.

Scatter plots of ELISA data derived from Aβ peptides isolated from Superose 12 size-exclusion FPLC fraction 3 (MW range 2-10 kDa), tris-buffer homogenates and HEPES-buffer homogenates. A) Total gray matter GDFA/GHCl-soluble Aβ peptides in ng per mg of total protein. B) Total white matter GDFA/GHCl-soluble Aβ peptides in pg per mg of total protein. C) Total gray matter tris-soluble Aβ peptides in pg per mg of total protein. D) Total gray matter TNF-α in pg per mg of total protein. NDC, non-demented controls; AD, Alzheimer’s disease; Bapi, bapineuzumab immunized AD patient; GM, gray matter; WM, white matter; GDFA, glass distilled formic acid; GHCl, 5 M guanidine hydrochloride (GHCl), 50 mM Tris-HCl, pH 8.0 buffer; tris, 20 mM Tris-HCl, 5 mM EDTA, pH 7.8 buffer; TNF-α, tumor necrosis factor-alpha.
After sequential FPLC size exclusion and HPLC reverse-phase chromatographic separations (Figure 3), Western blot analyses demonstrated that the bapineuzumab-treated individual harbored monomers, dimers and larger oligomeric Aβ forms (Figure 3). A broad array of amyloid-β protein precursor (AβPP) C-terminal fragments were observed comprising the CT99/83 (~13 kDa) and some C-terminal peptides with lower molecular weight as well as a longer AβPP C-terminal fragment at ~40 kDa, which are currently under investigation.
Figure 3.
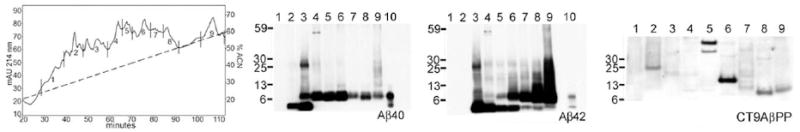
Reverse-phase HPLC and related Western blots derived from FPLC fraction 3 from the Bapineuzumab patient. The Western blots were developed with antibodies against Aβ40, Aβ42 and CT9APP. Lanes # 10 contain the corresponding Aβ40 or Aβ42 peptides standards. Notice that in both Aβ cases there were a preponderance of Aβ dimers over monomers as well as the presence of multiple oligomeric forms. Aside from revealing the presence of the CT99 and CT83 AβPP C-terminal peptides at ~ 13 kDa, which are related to the β- and α-secretase AβPP cleavages, the CT9AβPP antibody also demonstrated the presence of ~40 kDa band, suggesting a longer AβPP C-terminal peptide.
SELDI-TOF mass spectrometry, using anti-Aβ40, anti-Aβ42 and 6E10 capture antibodies, revealed a heterogeneous mixture of monomeric Aβ-related peptides starting at positions: 1, 3, 4, 11, and 17 with C-termini at positions 40, 42, 43, 46, 49, and 50. Mass spectrometry also demonstrated elongated Aβ related peptides with C-termini at positions: 68, 73, 96, and 99 (sequence numbering relative to the Aβ peptide starting at position 1 Asp). Dimeric forms of Aβ peptides ending at positions 40, 42, 43, and 46 as well as peptides with N-termini at position 3 with glutamyl modified to pyroglutamyl were also detected.
Discussion
Despite administration of 4 doses of bapineuzumab immunotherapy, thorough examination of large coronal sections covering the 4 cerebral lobes and the hippocampus in this patient revealed extensive numbers of plaques throughout the cerebral cortex. In addition, the finding of severe cerebral amyloid angiopathy in this case generally coincided with increased neuropathology and clinical evolution of AD as previously reported for ApoE4 carriers [13;14]. This subject died with dementia after disease duration of 5 years.
In a recent study, investigators at the University of Pittsburgh reported the neuropathological findings observed in an AD individual who received four 1 mg/kg doses of bapineuzumab. This individual did not complete the clinical trial because of progression of clinical symptoms and died 11 months after the last infusion of bapineuzumab [15]. Immunochemical and histochemical observations demonstrated frequent amyloid deposits in neocortex mainly composed of neuritic plaques with rare diffuse deposits, and scattered diffuse Aβ accumulation in the neostriatum. A comparison between the magnitude of neuropathological lesions and a 11C-PiB PET study taken 36 months prior to the patient’s death, suggested a ‘mismatch’ between pretreatment radiological and postmortem pathology especially in the neostriatum. In this case, however, it cannot be ruled out that the lack of a significantly noticeable plaque removal may be a consequence of the time interval between the last immunization (11 months) and the time of death. During this period, while amyloid concentrations remain elevated, antibody levels inevitably declined, perhaps diminishing to levels inadequate to prevent plaque reconstitution. Furthermore, a group comparison between 7 placebo and 19 bapineuzumab-treated patients evaluated with 11C-PiB PET showed an estimated decreased mean dye retention of ~25%, interpreted as revealing a reduction in fibrillar amyloid over 78 weeks relative to baseline, while an increased retention was observed in the placebo control group [16]. However, these observations would have been more informative if the differences between baseline and the last available 11C-PiB PET were individually defined and their respective ApoE genotypes specified.
In the present investigation, the bapineuzumab-treated patient exhibited no apparent areas of amyloid plaque clearance in contrast to those observed in the actively immunized AN1792 individuals [17-20]. Moreover, in our bapineuzumab-treated patient, the total gray matter and white matter GDFA/GHCl-soluble and the tris-soluble Aβ levels were similar to the mean observed in the non-immunized AD cases and higher than the NDC.
In general, a remarkable range of variations in Aβ values existed among the individuals in the NDC and AD groups. Similar disparities were also observed in a previous comparison of two AN1742 immunized cases studied in our laboratory [20]. In a more recent study of 7 AN1792 immunized AD individuals the range of Aβ peptide levels among these cases as measured by ELISA was extensive which supports the contention of a wide spectrum of human responses to amyloid removal [6]. The wide variability of amyloid plaque removal by AN1792 in individual patients has also been recently reviewed by Boche et al. [21].
In bapineuzumab-treated individuals, vasogenic edema in the presence of the ApoE4 allele may be complicated by the abundant vascular amyloidosis that probably exacerbated the inflammatory response. Both the classical and alternative complement cascades are preferentially activated by fibrillar Aβ42 which is more plentiful in AD ApoE4 individuals [22-24]. As part of the humoral branch of the immune system, complement responses promote inflammation, opsonization and cytolysis. These inflammatory reactions are compounded by the activation of microglia [25;26], and the secretion of proinflammatory cytokines and chemokines that result in the breaching of the blood-brain barrier and promotes diapedesis of vascular monocytes/macrophages, as observed in an in vitro model [27]. Interestingly, the bapineuzumab-treated individual had a level of TNF-α about twice the average observed in AD and NDC, suggesting an ongoing inflammatory reaction.
The interaction of antibodies with vascular Aβ may also be hampered by the tight enmeshing of the amyloid fibrils with the extracellular matrix molecules. In support of this assumption, it is necessary to employ collagenase to release the Aβ filaments from the extracellular vascular matrix during the amyloid purification procedures [28]. However in Tg mice, the 3D6 antibody appears to remove or prevent the deposition of vascular amyloid [29].
The antigen-antibody recognition sites for the N-terminal sequence of Aβ have been investigated using X-ray crystallography for 3 potentially therapeutic monoclonal antibodies 12A11, 10D5, and 12B4 [30]. These antibodies are capable of recognizing Aβ in amyloid plaques as well as soluble forms and therefore should, in principle, be efficacious anti-amyloid therapeutic interventions. The bapineuzumab antibody was very efficient in removing plaque amyloid in AβPP transgenic mice [31]. This efficacy may reflect the fact that the N-terminal degradation, interfering modifications and ancillary molecular steric hindrance prevalent in humans are absent or present in negligible quantities in transgenic mice [32;33]. We have observed that the Aβ peptides in humans with sporadic AD and familial AD have substantially degraded N-termini with Aβ molecules starting at positions 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 14, 15, 16, 17, and 19 [20;34-36]. N-terminal truncated Aβ peptides starting at positions 3, 4, 5, 8, and 9 have also been detected in CSF of living individuals with MCI [37], and in amyloid deposits of NDC individuals [38]. In human AD cases, the Aβ in amyloid plaque cores and capillaries contains a high proportion of isoaspartyl modifications at position 1 and 7 [34] that drastically changes the progression of the peptide bonds and secondary structure of Aβ. In addition, there are significant amounts of Aβ N-terminally degraded peptides at positions 3 and 11 with modified glutamyl to pyroglutamyl residues [39-41] that strongly alter the secondary and tertiary structures of these molecules. Therefore, key antigenic determinants in human amyloid plaques that the bapineuzumab antibody specifically target could either be chemically modified to yield alterations in N-terminal primary and secondary structures or masked due to binding to ancillary molecules or absent entirely. SELDI-TOF mass spectrometry of the bapineuzumab patient revealed the persistence of full-length Aβ40/42 as well as a spectrum of N-terminally truncated forms, at least two of which (starting at position Aβ11 and at position Aβ17) have been recognized as potentially toxic [42;43] as well as several oligomeric species.
Further complications for hopes to intervene against amyloid deposition pathology arise from the fact that in humans a large amount of Aβ deposits are of the diffuse type which are mainly composed of P3 peptides corresponding to the sequence of Aβ17-42 [44], although diffuse amyloid deposits also contain a variable amount of Aβ1-42. P3 is a highly insoluble and potentially toxic Aβ-related species that entirely lacks the N-terminal region epitopes which the bapineuzumab IgG therapy specifically target. It will be of interest to see whether or not Lilly’s solanezumab or Pfizer’s ponezumab immunization protocols elicit the severe inflammatory response which has been interpreted as the result of T-cell Aβ epitopes in the central and C-terminal domains of the Aβ molecule.
Although senile plaques have been substantially disrupted by bapineuzumab immunotherapy [16], in a Phase II clinical trial, beneficial effects on cognition were not apparent [2], although this trial was primarily designed as a safety and pharmacokinetic study and not powered for clinical endpoints. An additional explanation for this observation is that subsequent neuropathological examinations have revealed that clinical accuracy for AD diagnosis is about 54-97% [45]. Furthermore, about two-thirds of AD patients show mixed pathologies [45] that are also likely to complicate the effectiveness of therapeutic interventions. It is also important to bear in mind that although senile plaques have been disrupted, the data unequivocally reveal that all potentially toxic amyloid species are not eliminated completely or exported from the brain tissue [6;20;21]. This may explain why in spite of amyloid plaque removal, the immunized patients still died of complications related to AD. Moreover, morphological examinations have consistently revealed that disruption therapy leaves a legacy of plaque skeletal remnants [17;18;20;21] of unknown toxicity in the parenchyma that may harbor a residuum of deleterious amyloid species as well as other molecules recalcitrant to disruption by immunotherapy. Thus far, even for cases in which substantial clearance of plaque amyloid was evident, immunotherapy apparently did not commensurately alter the course of AD, although the much greater statistical power of incoming studies will help decide this issue unequivocally. Regardless of the ultimate capacity of Aβ immunotherapy to ameliorate soluble Aβ toxicity and amyloid deposition, this intervention may prove to be a component of wider and more comprehensive strategies designed to mitigate a broader range of molecular pathologies and cognitive dysfunctions.
Acknowledgments
This study was supported by the National Institute on Aging (NIA) grants: R01 AG-19795, the NIA Arizona Alzheimer’s Disease Core Center P30 AG-19610 and by grants from the State of Arizona to the Arizona Alzheimer’s Research Consortium. We express our gratitude to Dr. Douglas Walker for performing ApoE genotyping and to Drs. Walter M. Kalback and Dean C. Luehrs for critical review of the manuscript.
Footnotes
Authors’ disclosures available online (http://www.j-alz.com/disclosures/view.php?id=695).
Reference List
- 1.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, Sabbagh M, Honig LS, Doody R, van Dyck CH, Mulnard R, Barakos J, Gregg KM, Liu E, Lieberburg I, Schenk D, Black R, Grundman M. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73:2061–70. doi: 10.1212/WNL.0b013e3181c67808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging. 1997;18:S1–S2. [PubMed] [Google Scholar]
- 4.Roman GC, Tatemichi TK, Erkinjuntti T, Cummings JL, Masdeu JC, Garcia JH, Amaducci L, Orgogozo JM, Brun A, Hofman A. Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology. 1993;43:250–260. doi: 10.1212/wnl.43.2.250. [DOI] [PubMed] [Google Scholar]
- 5.McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VM, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–72. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 6.Maarouf CL, Daugs ID, Kokjohn TA, Kalback WM, Patton RL, Luehrs DC, Masliah E, Nicoll JA, Sabbagh MN, Beach TG, Castano EM, Roher AE. The biochemical aftermath of anti-amyloid immunotherapy. Mol Neurodegener. 2010;5:39. doi: 10.1186/1750-1326-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mirra SS. The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer’s disease: a commentary. Neurobiol Aging. 1997;18:S91–S94. doi: 10.1016/s0197-4580(97)00058-4. [DOI] [PubMed] [Google Scholar]
- 8.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 9.Beach TG, Sue LI, Walker DG, Roher AE, Lue L, Vedders L, Connor DJ, Sabbagh MN, Rogers J. The Sun Health Research Institute Brain Donation Program: description and experience, 1987-2007. Cell Tissue Bank. 2008;9:229–45. doi: 10.1007/s10561-008-9067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esh C, Patton L, Kalback W, Kokjohn TA, Lopez J, Brune D, Newell AJ, Beach T, Schenk D, Games D, Paul S, Bales K, Ghetti B, Castano EM, Roher AE. Altered APP processing in PDAPP (Val717 --> Phe) transgenic mice yields extended-length Abeta peptides. Biochemistry. 2005;44:13807–19. doi: 10.1021/bi051213+. [DOI] [PubMed] [Google Scholar]
- 11.Maarouf CL, Daugs ID, Spina S, Vidal R, Kokjohn TA, Patton RL, Kalback WM, Luehrs DC, Walker DG, Castano EM, Beach TG, Ghetti B, Roher AE. Histopathological and molecular heterogeneity among individuals with dementia associated with Presenilin mutations. Mol Neurodegener. 2008;3:20. doi: 10.1186/1750-1326-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mulugeta E, Molina-Holgado F, Elliott MS, Hortobagyi T, Perry R, Kalaria RN, Ballard CG, Francis PT. Inflammatory mediators in the frontal lobe of patients with mixed and vascular dementia. Dement Geriatr Cogn Disord. 2008;25:278–86. doi: 10.1159/000118633. [DOI] [PubMed] [Google Scholar]
- 13.Premkumar DR, Cohen DL, Hedera P, Friedland RP, Kalaria RN. Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer’s disease. Am J Pathol. 1996;148:2083–95. [PMC free article] [PubMed] [Google Scholar]
- 14.Wolk DA, Dickerson BC. Apolipoprotein E (APOE) genotype has dissociable effects on memory and attentional-executive network function in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2010;107:10256–61. doi: 10.1073/pnas.1001412107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez OL, Hamilton R, Ikonomovic M, Mathis CA, Price JA, Becker JT, Saxton JA, Dekosky ST, Klunk WE. In vivo amyloid deposition and neuropathological findings after humanized amyloid β-specific monoclonal antibodies therapy in a patient with Alzheimer’s disease. Alzheimers Dement. 2009;5:P64–P65. [Google Scholar]
- 16.Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, Mathis CA, Blennow K, Barakos J, Okello AA, Rodriguez Martinez de Liano S, Liu E, Koller M, Gregg KM, Schenk D, Black R, Grundman M. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9:363–72. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 17.Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, Vlachouli C, Wilkinson D, Bayer A, Games D, Seubert P, Schenk D, Holmes C. Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol. 2006;65:1040–1048. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- 18.Ferrer I, Boada RM, Sanchez Guerra ML, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathol. 2004;14:11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64:129–31. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- 20.Patton RL, Kalback WM, Esh CL, Kokjohn TA, Van Vickle GD, Luehrs DC, Kuo YM, Lopez J, Brune D, Ferrer I, Masliah E, Newel AJ, Beach TG, Castano EM, Roher AE. Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer’s disease patients: a biochemical analysis. Am J Pathol. 2006;169:1048–63. doi: 10.2353/ajpath.2006.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boche D, Denham N, Holmes C, Nicoll JA. Neuropathology after active Abeta42 immunotherapy: implications for Alzheimer’s disease pathogenesis. Acta Neuropathol. 2010;120:369–84. doi: 10.1007/s00401-010-0719-5. [DOI] [PubMed] [Google Scholar]
- 22.Webster S, Bonnell B, Rogers J. Charge-based binding of complement component C1q to the Alzheimer amyloid beta-peptide. Am J Pathol. 1997;150:1531–36. [PMC free article] [PubMed] [Google Scholar]
- 23.Emmerling MR, Morganti-Kossmann MC, Kossmann T, Stahel PF, Watson MD, Evans LM, Mehta PD, Spiegel K, Kuo YM, Roher AE, Raby CA. Traumatic brain injury elevates the Alzheimer’s amyloid peptide A beta 42 in human CSF. A possible role for nerve cell injury. Ann N Y Acad Sci. 2000;903:118–22. doi: 10.1111/j.1749-6632.2000.tb06357.x. [DOI] [PubMed] [Google Scholar]
- 24.Morishima-Kawashima M, Oshima N, Ogata H, Yamaguchi H, Yoshimura M, Sugihara S, Ihara Y. Effect of apolipoprotein E allele epsilon4 on the initial phase of amyloid beta-protein accumulation in the human brain. Am J Pathol. 2000;157:2093–99. doi: 10.1016/s0002-9440(10)64847-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giulian D, Haverkamp LJ, Yu JH, Karshin W, Tom D, Li J, Kirkpatrick J, Kuo YM, Roher AE. Specific domains of beta-amyloid from Alzheimer plaque elicit neuron killing in human microglia. J Neurosci. 1996;16:6021–37. doi: 10.1523/JNEUROSCI.16-19-06021.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giulian D, Haverkamp LJ, Yu J, Karshin W, Tom D, Li J, Kazanskaia A, Kirkpatrick J, Roher AE. The HHQK domain of beta-amyloid provides a structural basis for the immunopathology of Alzheimer’s disease. J Biol Chem. 1998;273:29719–26. doi: 10.1074/jbc.273.45.29719. [DOI] [PubMed] [Google Scholar]
- 27.Fiala M, Zhang L, Gan X, Sherry B, Taub D, Graves MC, Hama S, Way D, Weinand M, Witte M, Lorton D, Kuo YM, Roher AE. Amyloid-beta induces chemokine secretion and monocyte migration across a human blood--brain barrier model. Mol Med. 1998;4:480–489. [PMC free article] [PubMed] [Google Scholar]
- 28.Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, Gowing E, Ball MJ. beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:10836–40. doi: 10.1073/pnas.90.22.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schroeter S, Khan K, Barbour R, Doan M, Chen M, Guido T, Gill D, Basi G, Schenk D, Seubert P, Games D. Immunotherapy reduces vascular amyloid-beta in PDAPP mice. J Neurosci. 2008;28:6787–93. doi: 10.1523/JNEUROSCI.2377-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Basi GS, Feinberg H, Oshidari F, Anderson J, Barbour R, Baker J, Comery TA, Diep L, Gill D, Johnson-Wood K, Goel A, Grantcharova K, Lee M, Li J, Partridge A, Griswold-Prenner I, Piot N, Walker D, Widom A, Pangalos MN, Seubert P, Jacobsen JS, Schenk D, Weis WI. Structural correlates of antibodies associated with acute reversal of amyloid beta-related behavioral deficits in a mouse model of Alzheimer disease. J Biol Chem. 2010;285:3417–27. doi: 10.1074/jbc.M109.045187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seubert P, Barbour R, Khan K, Motter R, Tang P, Kholodenko D, Kling K, Schenk D, Johnson-Wood K, Schroeter S, Gill D, Jacobsen JS, Pangalos M, Basi G, Games D. Antibody capture of soluble Abeta does not reduce cortical Abeta amyloidosis in the PDAPP mouse. Neurodegener Dis. 2008;5:65–71. doi: 10.1159/000112834. [DOI] [PubMed] [Google Scholar]
- 32.Kalback W, Watson MD, Kokjohn TA, Kuo YM, Weiss N, Luehrs DC, Lopez J, Brune D, Sisodia SS, Staufenbiel M, Emmerling M, Roher AE. APP transgenic mice Tg2576 accumulate Abeta peptides that are distinct from the chemically modified and insoluble peptides deposited in Alzheimer’s disease senile plaques. Biochemistry. 2002;41:922–28. doi: 10.1021/bi015685+. [DOI] [PubMed] [Google Scholar]
- 33.Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21:372–81. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roher AE, Lowenson JD, Clarke S, Wolkow C, Wang R, Cotter RJ, Reardon IM, Zurcher-Neely HA, Heinrikson RL, Ball MJ. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J Biol Chem. 1993;268:3072–83. [PubMed] [Google Scholar]
- 35.Miravalle L, Calero M, Takao M, Roher AE, Ghetti B, Vidal R. Amino-terminally truncated Abeta peptide species are the main component of cotton wool plaques. Biochemistry. 2005;44:10810–10821. doi: 10.1021/bi0508237. [DOI] [PubMed] [Google Scholar]
- 36.Roher AE, Kokjohn TA, Esh C, Weiss N, Childress J, Kalback W, Luehrs DC, Lopez J, Brune D, Kuo YM, Farlow M, Murrell J, Vidal R, Ghetti B. The human amyloid-beta precursor protein770 mutation V717F generates peptides longer than amyloid-beta-(40-42) and flocculent amyloid aggregates. J Biol Chem. 2004;279:5829–36. doi: 10.1074/jbc.M311380200. [DOI] [PubMed] [Google Scholar]
- 37.Vanderstichele H, De Meyer G, Andreasen N, Kostanjevecki V, Wallin A, Olsson A, Blennow K, Vanmechelen E. Amino-truncated beta-amyloid42 peptides in cerebrospinal fluid and prediction of progression of mild cognitive impairment. Clin Chem. 2005;51:1650–1660. doi: 10.1373/clinchem.2005.051201. [DOI] [PubMed] [Google Scholar]
- 38.Sergeant N, Bombois S, Ghestem A, Drobecq H, Kostanjevecki V, Missiaen C, Wattez A, David JP, Vanmechelen E, Sergheraert C, Delacourte A. Truncated beta-amyloid peptide species in pre-clinical Alzheimer’s disease as new targets for the vaccination approach. J Neurochem. 2003;85:1581–91. doi: 10.1046/j.1471-4159.2003.01818.x. [DOI] [PubMed] [Google Scholar]
- 39.Harigaya Y, Shoji M, Kawarabayashi T, Kanai M, Nakamura T, Iizuka T, Igeta Y, Saido TC, Sahara N, Mori H, Hirai S. Modified amyloid beta protein ending at 42 or 40 with different solubility accumulates in the brain of Alzheimer’s disease. Biochem Biophys Res Commun. 1995;211:1015–22. doi: 10.1006/bbrc.1995.1912. [DOI] [PubMed] [Google Scholar]
- 40.Russo C, Saido TC, DeBusk LM, Tabaton M, Gambetti P, Teller JK. Heterogeneity of water-soluble amyloid beta-peptide in Alzheimer’s disease and Down’s syndrome brains. FEBS Lett. 1997;409:411–16. doi: 10.1016/s0014-5793(97)00564-4. [DOI] [PubMed] [Google Scholar]
- 41.Kuo YM, Emmerling MR, Woods AS, Cotter RJ, Roher AE. Isolation, chemical characterization, and quantitation of A beta 3-pyroglutamyl peptide from neuritic plaques and vascular amyloid deposits. Biochem Biophys Res Commun. 1997;237:188–91. doi: 10.1006/bbrc.1997.7083. [DOI] [PubMed] [Google Scholar]
- 42.Jang H, Arce FT, Ramachandran S, Capone R, Azimova R, Kagan BL, Nussinov R, Lal R. Truncated beta-amyloid peptide channels provide an alternative mechanism for Alzheimer’s Disease and Down syndrome. Proc Natl Acad Sci U S A. 2010;107:6538–43. doi: 10.1073/pnas.0914251107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei W, Norton DD, Wang X, Kusiak JW. Abeta 17-42 in Alzheimer’s disease activates JNK and caspase-8 leading to neuronal apoptosis. Brain. 2002;125:2036–43. doi: 10.1093/brain/awf205. [DOI] [PubMed] [Google Scholar]
- 44.Gowing E, Roher AE, Woods AS, Cotter RJ, Chaney M, Little SP, Ball MJ. Chemical characterization of A beta 17-42 peptide, a component of diffuse amyloid deposits of Alzheimer disease. J Biol Chem. 1994;269:10987–90. [PubMed] [Google Scholar]
- 45.Jellinger KA. Con: Can neuropathology really confirm the exact diagnosis? Alzheimers Res Ther. 2010;2:11. doi: 10.1186/alzrt34. [DOI] [PMC free article] [PubMed] [Google Scholar]