

Abstract
Transthyretin (TTR), a systemic amyloid precursor in the human TTR amyloidoses, interacts with β-amyloid (Aβ) in vitro, inhibits Aβ fibril formation, and suppresses the Alzheimer's disease (AD) phenotype in APP23 mice bearing a human APP gene containing the Swedish autosomal dominant AD mutation. In the present study, we show that TTR is a neuronal product upregulated in AD. Immunohistochemical analysis reveals that, in contrast to brains from non-demented age-matched individuals and control mice, the majority of hippocampal neurons from human AD and all those from the APP23 mouse brains contain TTR. Quantitative PCR for TTR mRNA and Western blot analysis show that primary neurons from APP23 mice transcribe TTR mRNA, and the cells synthesize and secrete TTR protein. TTR mRNA abundance is greatly increased in cultured cortical and hippocampal embryonic neurons and cortical lysates from adult APP23 mice. Antibodies specific for TTR and Aβ pulled down TTR/Aβ complexes from cerebral cortical extracts of APP23 mice and some human AD patients but not from control brains. In complementary tissue culture experiments, recombinant human TTR suppressed the cytotoxicity of soluble Aβ aggregates added to mouse neurons and differentiated human SH-SY5Y neuroblastoma cells. The findings that production of Aβ, its precursor, or its related peptides induces neuronal TTR transcription and synthesis and the presence of Aβ/TTR complexes in vivo suggest that increased TTR production coupled with interaction between TTR and Aβ and/or its related peptides may play a role in natural resistance to human AD.
Introduction
Protein aggregation diseases, such as Alzheimer's disease (AD), involve homotypic interactions between and among similarly amyloidogenic molecules (Querfurth and LaFerla, 2010). Cellular protein homeostasis requires heterotypic interactions between misfolded molecules and components of stress-responsive signaling pathways, chaperones and the degradation apparatus (Balch et al., 2008). It is possible that other heterotypic protein–protein interactions, such as that between transthyretin (TTR) and β-amyloid (Aβ), may also be protective for AD neurons.
Wild-type human TTR is the systemic amyloid precursor in senile systemic amyloidosis and its overexpression produces cardiac and renal deposition in aging mice (Teng et al., 2001). However, in the well validated APP23 transgenic mouse model, rather than amplifying disease, TTR overexpression suppressed both the neuropathologic and behavioral manifestations of AD (Buxbaum et al., 2008). In the same model, silencing the endogenous ttr gene accelerated disease pathogenesis (Buxbaum et al., 2008). We now examine potential mechanisms by which TTR may suppress the AD phenotype in these mice.
Earlier studies in Tg2576 AD model mice showed that ttr transcripts were increased and TTR protein was immunochemically detected in neurons in hippocampal and cerebral cortical slices (Stein and Johnson, 2002; Wu et al., 2006). However, in those studies, it was not clear whether the TTR was synthesized in the neurons or in the choroid plexus followed by neuronal uptake (Stein and Johnson, 2002; Carro et al., 2006).
In human AD, a number of studies have reported reduced TTR levels in the CSF (Serot et al., 1997). This has not been a constant finding and the suggestion that TTR “sequesters” Aβ has had little experimental support. Recent results from the MIRAGE study of AD families indicated that at least one TTR single-nucleotide polymorphism (rs3764479) is associated with MRI-documented hippocampal atrophy in AD patients and are consistent with a role for TTR in AD pathogenesis, although these results have not yet been independently replicated (Cuenco et al., 2009).
In vitro, TTR binds to all forms of soluble Aβ, monomer, oligomer, and fibrils. TTR binds to Aβ better at 37°C than 25°C, binds to Aβ aggregates better than to monomer (Liu and Murphy, 2006; Buxbaum et al., 2008; Du and Murphy, 2010), and to Aβ1–42 better than to Aβ1–40 (Schwarzman et al., 2004; Liu and Murphy, 2006; Buxbaum et al., 2008). The binding is highly dependent on the quaternary structure of TTR (Du and Murphy, 2010). Thus, the findings in human AD, murine models of the disease, and the in vitro studies suggest a relationship between TTR and AD pathogenesis.
The present in vivo and tissue culture studies indicate that the salutary effect of overexpressing TTR in a murine model of Aβ deposition may be the result of the increased neuronal synthesis of TTR and the interaction between TTR and Aβ or one of its related peptides, which reduces Aβ concentration, interferes with its capacity to aggregate, and renders it nontoxic in the context of the neuron and its environment.
Materials and Methods
Transgenic mice.
C57BL/6 [WT (B6)], APP23, mttr−/− (mouse ttr knock-out), hTTR+ (human wild-type TTR transgenic), APP23/mttr−/− (APP23 mice on ttr knock-out background), and APP23/hTTR+ (APP23 mice on hTTR+ transgenic background) were established and maintained as described previously (Teng et al., 2001; Buxbaum et al., 2008). TTR knock-out mice were obtained from V. Episkopou, Columbia University, New York, NY (Episkopou et al., 1993). Mice of either sex were used in the experiments.
Staining of human brains.
Autopsy material was obtained from patients studied neurologically and psychometrically at the Alzheimer Disease Research Center/University of California, San Diego (Table 1). At autopsy, brains were divided sagitally, samples from the left mid-frontal cortex were fixed in 4% PFA and sectioned at 40 μm for immunocytochemical analysis. Frozen samples from the right half were used for immunoblot analysis. For routine neuropathological diagnosis, paraffin sections from neocortical, limbic, and subcortical regions were stained with H&E and thioflavine-S (Masliah et al., 1990; Hansen et al., 1998). The analysis also included Braak stage determination (Braak and Braak, 1997). All cases met the Consortium to Establish a Registry for AD and National Institute of Aging criteria for diagnosis and displayed neuritic plaques and tangle formation in the neocortex and limbic system (McKeith et al., 1996; Jellinger, 1998; Jellinger and Bancher, 1998).
Table 1.
Clinicopathological characteristics of the control and AD cases
| Diagnosis | N | PMT (h) | Age (years) | Gender (M/F) | Duration (years) | Braak stage |
|---|---|---|---|---|---|---|
| Control | 5 | 8 ± 2 | 78 ± 2 | 3/2 | NA | 0–I |
| AD | 10 | 7 ± 2 | 82 ± 3 | 6/4 | 12 ± 1 | V–VI |
PMT, Postmortem time.
As previously described (Buxbaum et al., 2008), vibratome sections from the mid-frontal cortex of control and AD cases were incubated overnight at 4°C with the rabbit antibody against TTR (DAKO; A0002, 1:250) followed by secondary antibody (Vector; 1:75), avidin D horseradish peroxidase (HRP) (ABC Elite; Vector), and reacted with DAB (0.2 mg/ml) in 50 mm Tris, pH 7.4, with 0.001% H2O2. Control experiments consisted of incubation with nonimmune IgG. Sections were then counterstained with cresyl violet to estimate the proportion of pyramidal neurons immunolabeled with the antibody against TTR or with thioflavine S to estimate the proportion of plaques and vessels containing amyloid that were TTR positive (TTR+). Sections were analyzed with a digital Olympus BX41 microscope equipped with epifluorescence and Image Pro Plus program. For each case, three sections and a total of 12 fields at 200× (0.1 mm2) were studied.
Primary neuron cultures.
Primary hippocampal and cortical neuron cultures were established from all the mouse strains of interest [WT (B6), APP23, mttr−/−, hTTR+, APP23/mttr−/−, APP23/hTTR+] following previously established protocols (Kaech and Banker, 2006). Briefly, the neurons were obtained from embryonic day 14 (E14) to E16, with E0 representing the day when the postcoital vaginal plug is observed. Mouse brains were removed and transferred into cold HBSS (Cellgro) with 20 mm d-glucose (Invitrogen). The vessel membrane outside the cortex was torn off, and the hippocampus and cortex were collected and incubated in trypsin-EDTA solution (Invitrogen) with 50 U/ml DNase I (Worthington Biochem) for 10 min at room temperature. Trypsinization was stopped by addition of 10% fetal bovine serum (FBS). The cells were then dissociated with a polished glass Pasteur pipette and centrifuged at 150 × g for 2 min. The pellet was resuspended in Neurobasal medium with B27 supplement, 0.5 mm glutamine, and 1% penicillin/streptomycin (Invitrogen). Cells (200,000/ml) were plated and incubated in Neurobasal medium on poly-d-lysine (Sigma)-coated 100 mm culture dishes, multiwell tissue culture plates (Costar), or coverglasses (VWR). The medium was exchanged the next day and subsequently one-half of the medium was replaced every 3–4 d. For all of the experiments, 7 d in vitro (DIV7) neurons were used, except in Aβ-derived diffusible ligand (ADDL) assays DIV14 neurons were used.
Immunocytochemistry.
Cells were washed with PBS and fixed with 4% formaldehyde (Ted Pella) for 10 min. The cells were then permeabilized with 2% Triton X-100, blocked with 10% goat serum (Vector) for 1 h at room temperature, and incubated with primary antibodies overnight at 4°C. After adding appropriate secondary antibodies, the nuclei were counterstained with Hoechst 33342 (2 μg/ml; Invitrogen). The coverslips were mounted (Shandon Immu-mount; Thermo Scientific), and images were taken using either an inverted fluorescence microscope or a confocal microscope [Bio-Rad (Zeiss) Radiance 2100]. Antibodies used include rabbit anti-human TTR (Dako; A0002; 1:100), a monoclonal (F17E5) anti-TTR (1:200) produced in house, anti-Aβ 6E10 (Signet; 1:100), MAP2 (Abcam; 1:100), anti-NeuN (Millipore Bioscience Research Reagents; 1:200), anti-rabbit IgG-Alexa 488 (Invitrogen; 1:2000), anti-mouse IgG-Alexa 488 (Invitrogen; 1:2000), and anti-mouse IgG-Alexa 546 (Invitrogen; 1:2000).
Real-time PCR (quantitative PCR).
RNA was extracted from cells or tissues using RNeasy kit (Qiagen). Trace genomic DNA was removed by on column digestion (RNase-Free DNase Set; Qiagen). QuantiTect Reverse Transcription Kit (Qiagen) was used to synthesize cDNA, and Fast SYBR Green Master Mix (Roche) was used for quantitative PCR (qPCR) on an Opticon II thermocycler (Bio-Rad). β-Actin was chosen as reference gene to quantify relative expression of the transcripts using the ΔΔCt method. All procedures were performed following the manufacturer's recommendations.
FACS.
Primary cultured embryonic neurons (DIV7) from mttr−/−, WT (B6), APP23, and hTTR+ mouse strains were labeled as described previously (Sergent-Tanguy et al., 2003), using MAP2 antibody as a neuronal marker. The samples were kept in sorting buffer (25 mm HEPES, pH 7.0, and 1% FBS in PBS) on ice and sorted by staff at The Scripps Research Institute Flow Cytometry Core Facility using a FACSAria (BD Biosciences). RT-PCR was performed using an Ambion cells-to-CT kit, and β-actin was used as an internal control.
Recombinant TTR and synthetic Aβ preparation.
Recombinant human and mouse TTR were prepared in an Escherichia coli expression system and purified as described previously (Reixach et al., 2008). Aβ1–40 and Aβ1–42 were synthesized on solid-phase resin using FMOC-based chemistry and purified by HPLC as described previously (Usui et al., 2009). Mass spectrometry was used to confirm their identity by molecular weight.
Mouse brain and cell homogenates.
The primary cultured neurons were lysed (50 mm Tris, pH 7.5, 150 mm NaCl, 3 mm EDTA, 1% Triton X-100) with Complete mini protease inhibitor mixture (Roche). Alternatively, carefully dissected cortex of adult mice (>1 year of age) was homogenized by motor homogenizer (Kontes Glass Company) in cold lysis buffer (above) with protease inhibitors on ice, then sonicated using a microtip probe for 5 s at 80% intensity (Misonix). For Western blots without immunoprecipitation (IP), the lysates were spun at 10,000 × g for 10 min at 4°C, and the amount of protein in supernatant and resupended pellet was quantified by Bradford assay (Bio-Rad).
IP of mouse brain homogenates and cell lysates.
For IP experiments, 1000 μg of total protein from the cell or tissue lysates were precleared by shaking with 15 μl of protein A/G plus agarose beads (Santa Cruz Biotechnology) for 4 h at 4°C. The lysates were then incubated with protein A Dynabeads (Invitrogen) cross-linked with anti-TTR (Dako) per manufacturer's protocol. Last, the complexes were eluted following the manufacturer's recommendations. The samples were analyzed by Western blot probing for Aβ (6E10) as above.
Western blotting.
Recombinant proteins, cell lysates, or IP eluates were boiled in SDS Tricine sample buffer for 10 min. The samples were separated on 15% Tris-Tricine SDS-PAGE, and then transferred onto PVDF membranes. The membranes were blocked with 5% nonfat dry milk in TBS with 0.1% Tween 20 (TBST) for 1 h, washed with TBST, and incubated with 6E10 (1:5000) or anti-TTR (Dako; 1:1000) primary antibodies overnight at 4°C. After washing in TBST, the membranes were incubated with either AP- or HRP-labeled corresponding secondary antibodies, and the blots were developed using standard protocols. To reprobe the blot with different antibodies, the membranes were stripped using stripping buffer (Bio-Rad) for 30 min with rocking.
Aβ fibril formation.
Aβ1–40 and Aβ1–42 were monomerized using previously established protocols (Buxbaum et al., 2008). Briefly, Aβ1–42 was dissolved in HFIP (hexafluoro-2-propanol) (Sigma), evaporated under nitrogen stream, and stored at −80°C, whereas Aβ1–40 was dissolved and sonicated for 10 min in pH 10.5 aqueous NaOH, and then passed through a 10 kDa microcon (Millipore).
Aβ1–40 was diluted to 100 mm with PBS and agitated at 200 rpm for 7 d at 37°C to obtain amyloid fibrils, which were confirmed by thioflavin T fluorescence as described previously (Cohen et al., 2006).
To prepare the Aβ1–42 fibrils, the monomerized peptide was solubilized in DMSO (99.5%; Sigma) at 5 mm by vortexing and sonication for 10 min. The peptide was then diluted in phenol red-free Ham's F12 (Caissonlabs) to a final concentration of 60 μm and incubated at 37°C for 5 d with shaking.
ADDLs.
Aβ1–42 was resuspended in DMSO as above and diluted in ice-cold phenol red-free Ham's F12 to a final concentration of 60 μm and incubated at 4°C overnight. The solutions were centrifuged at 14,000 × g for 10 min at 4°C, and supernatants, consisting of ADDLs, were used for all experiments (Lambert et al., 1998).
Cell culture.
The human hepatoblastoma HepG2 cells (American Type Culture Collection; HB-8065) were grown in DMEM (Invitrogen) supplemented with 10% FBS, 1% penicillin/streptomycin, and 2 mm l-glutamine (Invitrogen). Human cardiac myocyte cells AC16 (Davidson et al., 2005) and human neuroblastoma cells SH-SY5Y (American Type Culture Collection; CRL-2266) were cultured in 1:1 mixture of DMEM:F12 medium (Invitrogen) supplemented with 10% FBS, 1% penicillin/streptomycin, and 2 mm l-glutamine. SH-SY5Y was differentiated using 5 μm all trans-retinoic acid (Sigma-Aldrich) for 7 d. For cell viability and reactive oxygen species (ROS) assays, the cells were cultured using phenol red-free DMEM:F12 media supplemented with B27 (Invitrogen) without FBS.
Cytotoxicity assays and ROS assays.
The primary cultured embryonic neurons were seeded in black-wall clear-bottom 96-well plates (Costar) coated with poly-d-lysine (Sigma-Aldrich) at a density of 20,000 cells/well. For each treatment, at least four replicates were measured and border lanes were not used. The experiments were repeated on neurons from a minimum of two animals. The media were replaced by fresh DMEM:F12 media without phenol red before all cell assays. Cell viability was measured by resazurin reduction assay (O'Brien et al., 2000). Briefly, 10 μl/well resazurin solution (500 μm in PBS) was added and the cells were incubated at 37°C for 4 h. Fluorescence intensity of the formazan generated was measured using a multiwell fluorescent reader (Tecan) with excitation (exc)/emission (em) 530/590 nm. The results are expressed as percentage of cell viability, with 100% viability corresponding to cells treated with TTR (or buffer) only.
For Aβ1–40 cytotoxicity assays, 200 μm Aβ was allowed to form fibrils in PBS (fibril formation protocol, above) and centrifuged at 20,000 × g for 20 min at 4°C. The pellet was resuspended in PBS buffer. Either Aβ1–40 supernatant or resuspended pellet was added to WT (B6), mttr−/−, and hTTR+ neurons to a final concentration of 10 μm for 48 h before viability measurements.
To compare the Aβ1–40 cytotoxicity on neurons cultured from WT (B6), mttr−/−, hTTR+, and APP23 mouse strains, 10 μm Aβ1–40 fibril mixture without centrifugation with or without 2.5 μm human recombinant TTR (hTTR) protein were added to DIV7 neurons. The cells were incubated for 48 h and cell viability was measured as above.
To test the effect of adding human recombinant TTR on Aβ1–40 fibril formation and Aβ cytotoxicity, 100 μm Aβ1–40 solutions with or without 25 μm hTTR, or hTTR alone (control) were incubated under fibril formation conditions. Solutions were diluted to a final concentration of 10 μm Aβ1–40 (and 2.5 μm hTTR) on WT (B6) neurons. Ten micromolar Aβ1–40 with hTTR (2.5 μm) was also tested on the cells. Cells were cultured for 2 d, and viability was measured as above.
For Aβ1–42 oligomer cytotoxicity and ROS assays, 20 μm ADDLs and 5 μm TTR were added to DIV14 neurons or differentiated SH-SY5Y cells for 2 d, and then cell viability was measured. For ROS assays, the treated cells were washed and incubated for 30 min with 20 μm dihydroethidine (DHE) (Invitrogen). The DHE was removed, the cells were washed one more time, and 100 μl of DMEM:F12 media were added to each well. Fluorescence intensity corresponding to the amount of oxidized DHE was measured in at multiwell plate reader (Tecan) with exc/em 518/630 nm (De Felice et al., 2007).
Statistical analysis.
In cell culture assays, most measurements were normalized against a contemporaneous control (buffer or exposure to TTR alone). One-way or two-way ANOVA was used to analyze data. Interaction term was always considered when two-way ANOVA was used. Model assumptions were tested and standardized residuals were studied to test model fit. Post hoc Tukey's pairwise comparisons or Dunnett's comparisons were made only when ANOVAs were statistically significant. Statistical analysis was performed in Minitab 10 (Minitab). Mean ± SD were graphed. For each treatment, the replicates were >4, and each experiment was repeated at least twice. *p < 0.05 and **p < 0.01.
Results
The in vivo observations in human AD and the APP23 mouse model strongly suggest that the presence of Aβ or its related peptides is associated with increased neuronal TTR. In the frontal cortex from the human non-demented controls, the antibody against TTR immunolabeled ∼10% of the cresyl violet-stained pyramidal neurons; in contrast, in the AD cases, almost 70% of the cresyl violet-stained pyramidal neurons displayed TTR immunoreactivity (Tables 1, 2). In sections from the frontal cortex counterstained with thioflavine-S, TTR immunoreactivity was detected in 10% of the mature plaques (Table 2). Approximately 40% of the vessels displayed amyloid deposits in their walls (Table 2). In APP23 mice, using the same methods, all the hippocampal and cortical neurons and the congophilic plaques stained for TTR, a phenomenon also seen in the Tg2576 transgenic model (Stein and Johnson, 2002; Buxbaum et al., 2008).
Table 2.
Image analysis of TTR distribution in the frontal cortex of the control and AD cases
| Neurons |
Plaques |
Amyloid vessels |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Cresyl violet | TTR+ | % TTR+ | Thio-S | TTR+ mature | % TTR+ | Thio-S | TTR+ | % TTR+ | |
| Control | 1230 ± 135 | 119 ± 10 | 10 ± 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| AD | 800 ± 60 | 545 ± 33 | 69 ± 3 | 45 ± 2 | 4.7 ± 0.5 | 10.3 ± 0.5 | 7 ± 1 | 2.9 ± 0.5 | 43 ± 3 |
To further explore the potential role of TTR in neuronal resistance to toxic Aβ aggregation and determine the nature of the synthesizing cell, primary neuronal cultures free of choroid plexus epithelial cells were established from the hippocampi and cerebral cortices of 14–16 d APP23 and control wild-type C57BL/6 [WT (B6)] embryos. APP23-derived neurons were stained for Aβ and its related AβPP precursor (antibody 6E10, specific for Aβ residues 1–17) and TTR. Figure 1C shows apparent colocalization of the two proteins in both cell bodies and dendritic processes, indicating that the molecules were in close proximity. In contrast, neurons from WT (B6) animals showed minimal staining (Fig. 1G). The data indicate that the APP-overexpressing neurons produced increased amounts of TTR in the presence of AβPP and its related peptides well before the appearance of neuropathologic or behavioral evidence of disease. All the cells were positive with antibodies for the neuronal markers NeuN (data not shown) and MAP2 (Fig. 1I), showing that the TTR-positive cells were of bona fide neuronal lineage.
Figure 1.
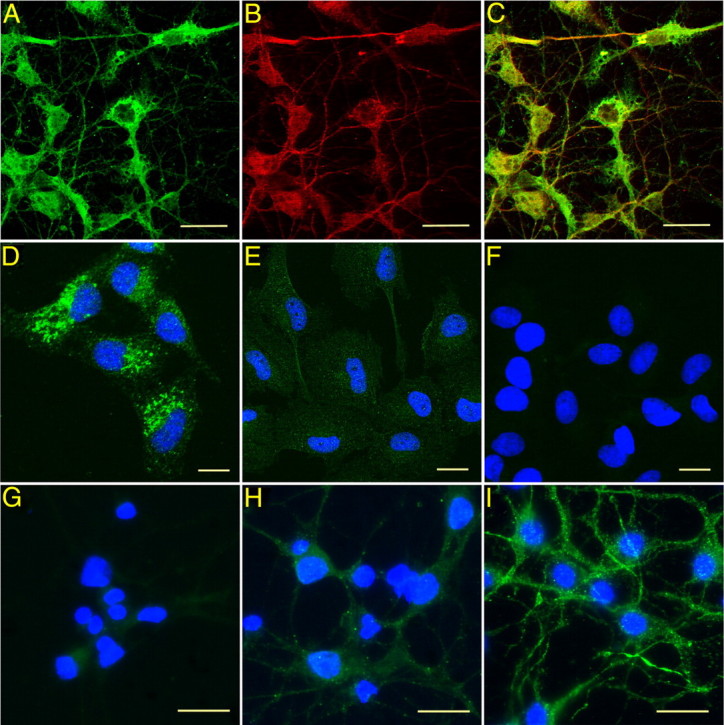
TTR expression in primary cultured embryonic cortical and hippocampal neurons. Choroid plexus-free neuron cultures were established from C57BL/6 [WT (B6)], APP23, mttr−/−, hTTR+, APP23/mttr−/−, APP23/hTTR+ strains. A–C, Immunostaining of APP23 neurons with anti-TTR and anti-Aβ antibodies. A, TTR staining (Dako; green). B, Aβ or related peptide staining (6E10; red). C, Merge of A and B. D, TTR-positive human hepatoblastoma HepG2 cells. E, TTR-negative human cardiac myocyte AC16 cells. F, Second antibody alone with HepG2 cells. G, H, TTR staining in neurons cultured from WT (B6) (G) and APP23 (H) embryos shows increased TTR signal in APP23 cells. I, Embryonic cortical and hippocampal neurons stained with the neuronal marker MAP2 (green) and counterstained by Hoechst 33342 (blue). Scale bars, 10 μm.
The expression of TTR mRNA in neurons was confirmed independently by RT-PCR analysis of FACS-sorted WT (B6), APP23, and human TTR transgenic (hTTR+, overexpressing human TTR in the absence of a human APP gene) hippocampal cells selected for expression of neuronal marker MAP2 (Fig. 2A). No ttr signal was found from mttr−/− control cells (Fig. 2A). Quantitative PCR showed that ttr transcripts were 10-fold more abundant in primary cultured hippocampal and cortical neurons obtained from the APP23 mice than in those from WT (B6) animals (Fig. 2B). The results establish that neuronal synthesis is the source of the TTR seen by immunohistochemistry. No ttr signal was detected in neurons from ttr−/− mice. qPCR of RNA extracted from cerebral cortex (dissected free of choroid plexus) from adult APP23 and WT (B6) mice revealed an even greater relative abundance of ttr mRNA in the APP23 mice than seen in the cultured neurons, further validating the tissue culture findings (data not shown).
Figure 2.

TTR is expressed and upregulated in APP23 embryonic cortical and hippocampal neurons. A, TTR mRNAs were detected by RT-PCR. TTR mRNA was detected in WT (B6), APP23, and hTTR+ neurons collected by FACS for MAP2 positivity, and mttr−/− served as a negative control. B, Quantitation of ttr mRNA in cultured primary neurons. Relative expression was calculated based on qPCR using ΔΔCt method. Error bars indicate SD. *p < 0.05. C, TTR immunoprecipitation of cultured neurons. TTR protein was immunoprecipitated from neuronal lysates with protein A/G plus agarose beads and anti-TTR antibody. Western blots of the immunoprecipitates analyzed by SDS-PAGE were developed with an anti-TTR antibody. D, Immunodetection of TTR in medium of neuronal cultures. Neurons were incubated in B27 free Neurobasal medium overnight. TTR released by the neurons was immunoprecipitated from the medium and processed as in C.
Western blots of lysates made from cultured hippocampal and cortical neurons demonstrated that APP23 neurons produced more TTR than those from control mice and that TTR was secreted into the medium by hTTR+ neurons (Fig. 2C,D).
Analysis of Aβ in cortical extracts from adult APP23 and APP23/hTTR+/mttr−/− mice by Western blot showed that the amount of Aβ1–40/1–42 in the cortex of the AD animals also transgenic for hTTR+ was significantly lower than in the APP23 mice, while the intensities of bands representing AβPP were similar in all the strains carrying the APP23 construct (Fig. 3). These results are consistent with our prior findings that SDS and formic acid-extractable Aβ1–40 and Aβ1–42 (i.e., soluble and insoluble Aβ aggregates) were reduced in the brains of the APP23/hTTR+/mttr−/− mice (Buxbaum et al., 2008). AβPP content appears to be similar in the two strains as does the amount of C99 peptide, although the distribution between the supernatant and pellet fractions is quite different (Fig. 3). The presence of C99 fragments in brain homogenates of APP23/hTTR+/mttr−/− mice suggests that there is substantial β-secretase cleavage of AβPP. If that is the case, the reduction of Aβ species in APP23/hTTR+/mttr−/− mice could result from TTR and Aβ interaction (with sequestration in a form not visible on the gel) or by TTR inhibition of γ-secretase cleavage.
Figure 3.

Brain extracts of APP23 mice transgenic for hTTR have decreased Aβ peptide. Lysates of cortical and hippocampal tissue from APP23/hTTR+/mttr−/− and APP23 (mttr+/+) mice (>1 year) were centrifuged at 10,000 × g for 10 min at 4°C. Protein in supernatant (S) and resuspended pellet (P) were quantified by Bradford assay. Identical amounts of total protein/lane were analyzed in a 15% Tris-Tricine SDS-PAGE. The electrophoresed proteins were transferred onto a PVDF membrane, and the Aβ species were detected with 6E10 antibody.
Intimate interaction between TTR and Aβ in vivo was demonstrated by coimmunoprecipitation of the two molecules from cerebral cortical homogenates of APP23 and APP23/hTTR+ mice using an anti-TTR antibody cross-linked to protein A-coated magnetic beads (Fig. 4). Western blots of identically treated brain homogenates from mice expressing either TTR or AβPP showed no coprecipitation. Similar coimmunoprecipitation results were obtained in some (1/2), but not all, human AD brains, confirming that in vivo TTR was complexed with Aβ (Fig. 4B). These findings approximated those seen when in vitro mixtures of recombinant TTR and synthetic Aβ1–40 or Aβ1–42 were analyzed, showing the same TTR–Aβ interactions in vivo and in vitro (data not shown).
Figure 4.

Coimmunoprecipitation of in vivo Aβ and TTR complexes. A, Mouse brains. Cortices and hippocampi of WT (B6), hTTR+/mttr−/−, APP23/hTTR+/mttr−/−, APP23, and APP23/mttr−/− mice (>1 year) were dissected free of choroid plexus and homogenized in lysis buffer with protease inhibitors, precleared with protein A/G plus agarose beads, and then incubated with anti-TTR antibody cross-linked to protein A magnetic beads overnight at 4°C. Eluted complexes were analyzed in 15% Tris-Tricine SDS-PAGE, and Aβ was detected by Western blot (6E10 antibody). B, Human brains. Human brain homogenates were processed as in A. P indicates brain from AD patient; Ctr indicates age-matched control brain.
The significance of the TTR–Aβ interaction was supported by a series of tissue culture experiments (Fig. 5). Many investigators have shown that Aβ preparations are cytotoxic to a variety of cultured cells, with oligomers being the most likely active component (Lambert et al., 1998; Walsh et al., 2002; Gong et al., 2003; Walsh and Selkoe, 2007). We studied the cytotoxic potential of Aβ1–40 fibrils and the supernatants (i.e., subfibrillar aggregates) of the Aβ1–40 incubations after the fibrils had been removed by centrifugation, on primary cultured neurons from WT (B6), mttr−/−, and hTTR+ mice (Fig. 5A). Compared with the fibril pellet, the supernatant (containing subfibrillar oligomers/protofibrils) was more cytotoxic to the cells (Tukey's pairwise comparisons, p < 0.01), supporting the notion that it is the smaller species of Aβ aggregates rather than mature fibrils that are the potentially toxic molecules in AD.
Figure 5.

Recombinant human TTR suppresses Aβ cytotoxicity and ROS induction. A, Aβ1–40 soluble aggregates are more cytotoxic than resuspended amyloid fibrils. Cell viability was measured by resazurin reduction assay. B, hTTR (2.5 μm) suppressed the cytotoxicity induced by Aβ1–40 (10 μm) aggregates on WT (B6), mttr−/−, hTTR+, and APP23 neurons. C, hTTR (5 μm) reduces Aβ1–42 oligomer (20 μm) cytotoxicity for cultured WT (B6) neurons. D, hTTR suppressed ROS formation in neurons treated with Aβ1–42 oligomers. WT (B6) neurons were treated with Aβ1–42 oligomers with or without TTR, and the fluorescence intensity of oxidized DHE was measured. E, hTTR prevents Aβ1–42 oligomer cytotoxicity for differentiated SH-SY5Y human neuroblastoma cells. F, Adding hTTR to Aβ1–40 fibrilization process suppressed Aβ cytotoxicity on WT (B6) neurons. Aβ1–40 solutions with ([Aβ+TTR]ag) or without (Aβ1–40) TTR, or TTR alone (TTRagctl) was subjected fibril formation condition. Solutions were diluted to final concentration of 10 μm Aβ and 2.5 μm TTR on WT (B6) neurons. Ten micromolar Aβ with 2.5 μm hTTR (Aβ+TTR) or 2.5 μm hTTR (hTTR) were also incubated with cells as control. Cell viability was measured as above. *p < 0.05 and **p < 0.01. Error bars indicate SD.
Although the amount of human TTR produced by hTTR+ neurons was much greater than the quantity of mouse TTR produced by WT (B6) neurons (Fig. 2D), these cells were still susceptible to Aβ-induced cytotoxicity (p < 0.01), suggesting that either the amount or rate of TTR secretion into the media was inadequate to protect the neurons when oligomeric Aβ was added to the culture medium. This hypothesis was tested by quantitative studies in which we found that 10 μm Aβ1–40 fibrils are toxic, while addition of 2.5 μm recombinant human TTR offered significant protection from this challenge to primary neurons derived from WT (B6), mttr−/−, hTTR+, and APP23 strains (Tukey's pairwise comparisons, p < 0.05) (Fig. 5B). Medium harvested from a 22 h incubation of (3 × 106) hTTR+ cells had an approximate TTR concentration of 2 × 10−4 μm (as determined by hTTR-specific ELISA), an amount insufficient to inhibit the cytotoxicity of the added Aβ aggregates.
ADDLs are also toxic to cultured cells producing ROS with subsequent increased oxidative stress (De Felice et al., 2007). Our experiments reveal that incubation with 20 μm ADDLs significantly increased ROS production in WT neurons compared with buffer- or TTR-treated controls (Fig. 5D; Dunnett's comparisons, p < 0.05). Addition of 5 μm TTR reduced ROS production significantly (p < 0.01). Human recombinant TTR also suppressed ADDLs-induced cytotoxicity for primary neurons from WT (B6) mice (p < 0.05) (Fig. 5C) and differentiated SH-SY5Y human neuroblastoma cells (p < 0.01) (Fig. 5E).
To further investigate the relationship between TTR and Aβ cytotoxicity, Aβ1–40 and recombinant human TTR were mixed and incubated under fibril-forming conditions with cytotoxicity measured on WT (B6) neurons (Fig. 5F). Aβ1–40 or TTR alone under the same conditions were used as controls. Preincubation of TTR with Aβ1–40 reduced cytotoxicity (Dunnett's comparisons, p < 0.01) (Fig. 5F).
Discussion
Studies of CSF from AD patients, inhibition of in vitro Aβ fibril formation, suppression of the Aβ-induced phenotype in Caenorhabditis elegans coexpressing TTR with Aβ, and other AD mouse models have all suggested TTR–Aβ interactions in AD (Link, 1995; Serot et al., 1997; Davidsson et al., 2002; Costa et al., 2008a). Some—but not all—studies have shown that homozygous or hemizygous TTR knock-out transgenic mice have accelerated AD disease (Choi et al., 2007; Buxbaum et al., 2008; Wati et al., 2009; Doggui et al., 2010). Our current observations showing both TTR mRNA and protein within cultured hippocampal neurons demonstrate neuronal synthesis of TTR. The increase in ttr mRNA and protein in neurons from the AD model mice and the increase in TTR-stained neurons in human AD brains suggest that neuronal TTR transcription may be substantially induced by Aβ or one of its precursors. Others have suggested that such induction might be mediated by sAPPα or sAPPβ, but before these studies neither of those notions was supported by definitive data in primary neurons (Stein and Johnson, 2002; Li et al., 2010).
The in vivo and cell culture analyses show that TTR interacts with Aβ and reduces its propensity to acquire cytotoxic properties probably by inhibiting its ability to aggregate. Similar phenomena may be responsible for the suppression of the neuropathologic and behavioral AD phenotype seen in the APP23 transgenic mice overexpressing human TTR. Despite convincing evidence of in vitro neuroprotection by TTR (Fig. 5C–E), intrinsic cellular TTR production does not protect neurons from the cytotoxicity produced by extracellular aggregates under these conditions (Fig. 5B). It is likely that Aβ toxicity from the “outside in” overwhelmed the quantitative capacity of the intracellular and/or secreted TTR and the differences in sensitivity between the TTR (+) and TTR (−) cells could not be detected. Measurement of the TTR concentration in the media of the hTTR+ cells, accumulated over 22 h, was <1 nm, far less than that required to inhibit the cytotoxic effect of standard concentrations of Aβ1–40/42 on cultured cells. The experiment does not resolve the question of whether in vivo the cytotoxic molecules are generated intracellularly and the significant interaction between TTR and Aβ-related species occurs within some cellular compartment, or that TTR and Aβ-related peptides form a complex outside the cell, which renders the Aβ oligomers nontoxic. While the relationship of cytotoxicity in tissue culture to AD pathogenesis in vivo is not clear, the fact that TTR protects against exogenous Aβ soluble aggregate toxicity in tissue culture confirms the TTR–Aβ interaction and is consistent with the protective effect in vivo.
The marked reduction in the concentrations of formic acid-extractable Aβ1–40/42 confirmed by the Western blot experiments shown in Figure 3, can be explained by either removal of the Aβ1–40/1–42 peptides from the system via TTR-assisted degradation or TTR inhibition of AβPP cleavage by secretases reducing the amount of Aβ1–40/1–42 that is produced. This question requires further investigation.
Recent discussions of clinical disorders of protein folding have proposed that enhancement of the proteostatic capacity (i.e., that combination of the elements of the stress responses, chaperones, and degradative systems), represents a potential therapeutic approach for these diseases (Balch et al., 2008). Our observations suggest that the structure of TTR may allow it to engage in a protective heterotypic interaction with Aβ or one of its related peptides. If the interaction occurs in the intracellular or pericellular environment of the neuron, it may serve as a neuron-specific proteostatic element. Why should TTR have these properties? Our data suggest that wild-type TTR, and perhaps the wild-type forms of other amyloidogenic proteins (e.g., gelsolin, cystatin) (Kaeser et al., 2007; Mi et al., 2007; Chauhan et al., 2008; Antequera et al., 2009) may have structural properties that allow them to participate in proteostasis to the benefit of the neuron under stress. APP23 animals, even in the presence of increased endogenous ttr expression, still show AD-like pathology in the presence of an overexpressed human AD gene. APP overexpression, which presents an increased Aβ load to the affected cells, is required to overcome the protective capacity of such endogenous mechanisms to produce the transgenic models of the human disease (Ashe and Zahs, 2010). The beneficial effect of increasing the concentration of such a potentially protective molecule by overexpressing the human TTR transgene supports this hypothesis. It is possible that the Aβ–TTR interaction reduces the aggregation-prone protein concentration in a chaperone-like manner supplementing the activity of the endogenous proteostatic apparatus.
Others have suggested proteolysis and/or disaggregation of Aβ by TTR or the generation of large nontoxic complexes as mechanisms of protection (Costa et al., 2008b; Cohen et al., 2009). We have not found evidence for any of these pathways at physiologically relevant concentrations of TTR. One can speculate that the ability of the neuron to marshal a TTR response may be limited genetically or deteriorate in the course of aging and contribute to susceptibility to sporadic human AD (Serot et al., 1997).
The failure of mice overproducing wild-type TTR and patients with unstable TTR variants to develop intraneuronal or intracerebral TTR amyloid deposits, except in the choroid plexus or leptomeninges, suggests that, in the absence of an Aβ-like challenge, as in the WT (B6) mice, there is little or no neuronal ttr transcription. It is also possible that even in the face of intraneuronal aggregates the TTR transcriptional response is limited and the amount of TTR does not exceed some critical concentration required for its aggregation and subsequent fibril formation. This question requires further investigation. The effect of TTR overexpression could be considered to be pharmacologic, while the effect of silencing the ttr gene could be interpreted as compromising a normal physiologic response to molecules involved in the development of AD and more likely to vary from individual to individual (Buxbaum and Reixach, 2009).
In summary, we have shown definitively for the first time that neurons transcribe TTR mRNA, which is subsequently translated to produce TTR protein, and that the neuronal synthesis of TTR may be a natural protective response to the cellular challenge presented by the aggregation of the Aβ fragment occurring in human AD and at least one murine transgenic model. Protection appears to be a function of increased TTR production with binding of Aβ by TTR and subsequent reduction in toxic aggregate formation with a possible role for TTR in reducing Aβ1–40/42 production.
Footnotes
This research was supported by NIH Grants AG R01 030027 (J.N.B.) and AG18440, AG5131, and NS057096 (E.M.), and by the W. M. Keck Foundation (J.N.B., X.L.). We thank Drs. J. W. Kelly, W. E. Balch, and T. Bartfai for constructive advice and Drs. F. Cattaneo and J. Hulleman for helpful discussions. We thank E. Wong for technical support.
References
- Antequera D, Vargas T, Ugalde C, Spuch C, Molina JA, Ferrer I, Bermejo-Pareja F, Carro E. Cytoplasmic gelsolin increases mitochondrial activity and reduces Abeta burden in a mouse model of Alzheimer's disease. Neurobiol Dis. 2009;36:42–50. doi: 10.1016/j.nbd.2009.06.018. [DOI] [PubMed] [Google Scholar]
- Ashe KH, Zahs KR. Probing the biology of Alzheimer's disease in mice. Neuron. 2010;66:631–645. doi: 10.1016/j.neuron.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Diagnostic criteria for neuropathologic assessment of Alzheimer's disease. Neurobiol Aging. 1997;18:S85–S88. doi: 10.1016/s0197-4580(97)00062-6. [DOI] [PubMed] [Google Scholar]
- Buxbaum JN, Reixach N. Transthyretin: the servant of many masters. Cell Mol Life Sci. 2009;66:3095–3101. doi: 10.1007/s00018-009-0109-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T. Transthyretin protects Alzheimer's mice from the behavioral and biochemical effects of Abeta toxicity. Proc Natl Acad Sci U S A. 2008;105:2681–2686. doi: 10.1073/pnas.0712197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro E, Trejo JL, Gerber A, Loetscher H, Torrado J, Metzger F, Torres-Aleman I. Therapeutic actions of insulin-like growth factor I on APP/PS2 mice with severe brain amyloidosis. Neurobiol Aging. 2006;27:1250–1257. doi: 10.1016/j.neurobiolaging.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Chauhan V, Ji L, Chauhan A. Anti-amyloidogenic, anti-oxidant and anti-apoptotic role of gelsolin in Alzheimer's disease. Biogerontology. 2008;9:381–389. doi: 10.1007/s10522-008-9169-z. [DOI] [PubMed] [Google Scholar]
- Choi SH, Leight SN, Lee VM, Li T, Wong PC, Johnson JA, Saraiva MJ, Sisodia SS. Accelerated Aβ deposition in APPswe/PS1ΔE9 mice with hemizygous deletions of TTR (transthyretin) J Neurosci. 2007;27:7006–7010. doi: 10.1523/JNEUROSCI.1919-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Cohen E, Paulsson JF, Blinder P, Burstyn-Cohen T, Du D, Estepa G, Adame A, Pham HM, Holzenberger M, Kelly JW, Masliah E, Dillin A. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell. 2009;139:1157–1169. doi: 10.1016/j.cell.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa R, Gonçalves A, Saraiva MJ, Cardoso I. Transthyretin binding to A-Beta peptide—impact on A-Beta fibrillogenesis and toxicity. FEBS Lett. 2008a;582:936–942. doi: 10.1016/j.febslet.2008.02.034. [DOI] [PubMed] [Google Scholar]
- Costa R, Ferreira-da-Silva F, Saraiva MJ, Cardoso I. Transthyretin protects against A-beta peptide toxicity by proteolytic cleavage of the peptide: a mechanism sensitive to the Kunitz protease inhibitor. PLoS One. 2008b;3:e2899. doi: 10.1371/journal.pone.0002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenco KT, Friedland R, Baldwin CT, Guo J, Vardarajan B, Lunetta KL, Cupples LA, Green RC, DeCarli C, Farrer LA. Association of TTR polymorphisms with hippocampal atrophy in Alzheimer disease families. Neurobiol Aging. 2009;32:249–256. doi: 10.1016/j.neurobiolaging.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson MM, Nesti C, Palenzuela L, Walker WF, Hernandez E, Protas L, Hirano M, Isaac ND. Novel cell lines derived from adult human ventricular cardiomyocytes. J Mol Cell Cardiol. 2005;39:133–147. doi: 10.1016/j.yjmcc.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Davidsson P, Westman-Brinkmalm A, Nilsson CL, Lindbjer M, Paulson L, Andreasen N, Sjögren M, Blennow K. Proteome analysis of cerebrospinal fluid proteins in Alzheimer patients. Neuroreport. 2002;13:611–615. doi: 10.1097/00001756-200204160-00015. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-d-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- Doggui S, Brouillette J, Chabot JG, Farso M, Quirion R. Possible involvement of transthyretin in hippocampal beta-amyloid burden and learning behaviors in a mouse model of Alzheimer's disease (TgCRND8) Neurodegener Dis. 2010;7:88–95. doi: 10.1159/000285513. [DOI] [PubMed] [Google Scholar]
- Du J, Murphy RM. Characterization of the interaction of beta-amyloid with transthyretin monomers and tetramers. Biochemistry. 2010;49:8276–8289. doi: 10.1021/bi101280t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Episkopou V, Maeda S, Nishiguchi S, Shimada K, Gaitanaris GA, Gottesman ME, Robertson EJ. Disruption of the transthyretin gene results in mice with depressed levels of plasma retinol and thyroid hormone. Proc Natl Acad Sci U S A. 1993;90:2375–2379. doi: 10.1073/pnas.90.6.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer's disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen LA, Daniel SE, Wilcock GK, Love S. Frontal cortical synaptophysin in Lewy body diseases: relation to Alzheimer's disease and dementia. J Neurol Neurosurg Psychiatry. 1998;64:653–656. doi: 10.1136/jnnp.64.5.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger KA. The neuropathological diagnosis of Alzheimer disease. J Neural Transm Suppl. 1998;53:97–118. doi: 10.1007/978-3-7091-6467-9_9. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Bancher C. Neuropathology of Alzheimer's disease: a critical update. J Neural Transm Suppl. 1998;54:77–95. doi: 10.1007/978-3-7091-7508-8_8. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kaeser SA, Herzig MC, Coomaraswamy J, Kilger E, Selenica ML, Winkler DT, Staufenbiel M, Levy E, Grubb A, Jucker M. Cystatin C modulates cerebral beta-amyloidosis. Nat Genet. 2007;39:1437–1439. doi: 10.1038/ng.2007.23. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang B, Wang Z, Guo Q, Tabuchi K, Hammer RE, Südhof TC, Zheng H. Soluble amyloid precursor protein (APP) regulates transthyretin and Klotho gene expression without rescuing the essential function of APP. Proc Natl Acad Sci U S A. 2010;107:17362–17367. doi: 10.1073/pnas.1012568107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link CD. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1995;92:9368–9372. doi: 10.1073/pnas.92.20.9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Murphy RM. Kinetics of inhibition of beta-amyloid aggregation by transthyretin. Biochemistry. 2006;45:15702–15709. doi: 10.1021/bi0618520. [DOI] [PubMed] [Google Scholar]
- Masliah E, Terry RD, Mallory M, Alford M, Hansen LA. Diffuse plaques do not accentuate synapse loss in Alzheimer's disease. Am J Pathol. 1990;137:1293–1297. [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47:1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- Mi W, Pawlik M, Sastre M, Jung SS, Radvinsky DS, Klein AM, Sommer J, Schmidt SD, Nixon RA, Mathews PM, Levy E. Cystatin C inhibits amyloid-beta deposition in Alzheimer's disease mouse models. Nat Genet. 2007;39:1440–1442. doi: 10.1038/ng.2007.29. [DOI] [PubMed] [Google Scholar]
- O'Brien J, Wilson I, Orton T, Pognan F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur J Biochem. 2000;267:5421–5426. doi: 10.1046/j.1432-1327.2000.01606.x. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Reixach N, Foss TR, Santelli E, Pascual J, Kelly JW, Buxbaum JN. Human-murine transthyretin heterotetramers are kinetically stable and non-amyloidogenic. A lesson in the generation of transgenic models of diseases involving oligomeric proteins. J Biol Chem. 2008;283:2098–2107. doi: 10.1074/jbc.M708028200. [DOI] [PubMed] [Google Scholar]
- Schwarzman AL, Tsiper M, Wente H, Wang A, Vitek MP, Vasiliev V, Goldgaber D. Amyloidogenic and anti-amyloidogenic properties of recombinant transthyretin variants. Amyloid. 2004;11:1–9. doi: 10.1080/13506120410001667458. [DOI] [PubMed] [Google Scholar]
- Sergent-Tanguy S, Chagneau C, Neveu I, Naveilhan P. Fluorescent activated cell sorting (FACS): a rapid and reliable method to estimate the number of neurons in a mixed population. J Neurosci Methods. 2003;129:73–79. doi: 10.1016/s0165-0270(03)00210-3. [DOI] [PubMed] [Google Scholar]
- Serot JM, Christmann D, Dubost T, Couturier M. Cerebrospinal fluid transthyretin: aging and late-onset Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1997;63:506–508. doi: 10.1136/jnnp.63.4.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein TD, Johnson JA. Lack of neurodegeneration in transgenic mice overexpressing mutant amyloid precursor protein is associated with increased levels of transthyretin and the activation of cell survival pathways. J Neurosci. 2002;22:7380–7388. doi: 10.1523/JNEUROSCI.22-17-07380.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng MH, Yin JY, Vidal R, Ghiso J, Kumar A, Rabenou R, Shah A, Jacobson DR, Tagoe C, Gallo G, Buxbaum J. Amyloid and nonfibrillar deposits in mice transgenic for wild-type human transthyretin: a possible model for senile systemic amyloidosis. Lab Invest. 2001;81:385–396. doi: 10.1038/labinvest.3780246. [DOI] [PubMed] [Google Scholar]
- Usui K, Hulleman JD, Paulsson JF, Siegel SJ, Powers ET, Kelly JW. Site-specific modification of Alzheimer's peptides by cholesterol oxidation products enhances aggregation energetics and neurotoxicity. Proc Natl Acad Sci U S A. 2009;106:18563–18568. doi: 10.1073/pnas.0804758106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. A beta oligomers—a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wati H, Kawarabayashi T, Matsubara E, Kasai A, Hirasawa T, Kubota T, Harigaya Y, Shoji M, Maeda S. Transthyretin accelerates vascular Abeta deposition in a mouse model of Alzheimer's disease. Brain Pathol. 2009;19:48–57. doi: 10.1111/j.1750-3639.2008.00166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZL, Ciallella JR, Flood DG, O'Kane TM, Bozyczko-Coyne D, Savage MJ. Comparative analysis of cortical gene expression in mouse models of Alzheimer's disease. Neurobiol Aging. 2006;27:377–386. doi: 10.1016/j.neurobiolaging.2005.02.010. [DOI] [PubMed] [Google Scholar]