

Abstract
Previous studies have shown that the change of cerebral metabolic rate of glucose (CMRglc) in response to traumatic brain injury (TBI) is different in young (PND35) and adult rats (PND70), and that prolonged ketogenic diet treatment results in histological and behavioral neuroprotection only in younger rat brains. However, the mechanism(s) through which ketones act in the injured brain and the biochemical markers of their action remain unknown. Therefore, the current study was initiated to: 1) determine the effect of injury on the neurochemical profile in PND35 compared to PND70 rats; and 2) test the effect of early post-injury administration of ketogenic diet on brain metabolism in PND35 versus PND70 rats. The data show that alterations in energy metabolites, amino acid, and membrane metabolites were not evident in PND35 rats on standard diet until 24 h after injury, when the concentration of most metabolites was reduced from sham-injured values. In contrast, acute, but transient deficits in energy metabolism were measured at 6 h in PND70 rats, together with deficits in N-acetylaspartate that endured until 24 h. Administration of a ketogenic diet resulted in significant increases in plasma β-hydroxybutyrate (βOHB) levels. Similarly, brain βOHB levels were significantly elevated in all injured rats, but were elevated by 43% more in PND35 rats compared to PND70 rats. As a result, ATP, creatine, and phosphocreatine levels at 24 h after injury were significantly improved in the ketogenic PND35 rats, but not in the PND70 group. The improvement in energy metabolism in the PND35 brains was accompanied by the recovery of NAA and reduction of lactate levels, as well as amelioration of the deficits of other amino acids and membrane metabolites. These results indicate that the PND35 brains are more resistant to the injury, indicated by a delayed deficit in energy metabolism. Moreover, the younger brains revert to ketones metabolism more quickly than do the adult brains, resulting in better neurochemical and cerebral metabolic recovery after injury.
Key words: ketogenic diet, nuclear magnetic resonance spectroscopy, phosphorus, proton, TBI
Introduction
Traumatic brain injury (TBI) results in excessive glutamate efflux and loss of ionic balance across neuronal membranes (Faden et al., 1989; Katayama et al., 1995; Palmer et al., 1993; Rose et al., 2002). Ionic imbalance induces a transient increase in the cerebral metabolism of glucose (CMRglc) followed by a prolonged phase of depressed CMRglc, as well as an increased anaerobic glycolysis reflected by tissue and extracellular accumulation of lactate (Alessandri et al., 2000; Goodman et al., 1999; Sunami et al., 1989; Sutton et al., 1994). It is generally thought that the immediate post-injury increase in CMRglc is caused by increased energy demand to maintain cellular membrane ionic balance (Andersen and Marmarou, 1992; Hovda et al., 1990; Kawamata et al., 1992; Kocher, 1990; Vink et al., 1994). The reason for the post-injury period of depressed CMRglc is currently unknown, but it has been consistently observed in different experimental injury models as well as clinically (Bergsneider et al., 1997; O'Connell et al., 2005; Sutton et al., 1994; Yoshino et al., 1991). The injury-induced alterations in glucose uptake are age-dependent after fluid percussion injury (FPI) in the rat. The duration of CMRglc depression persists for 10 days before returning to sham levels in adult rats, whereas recovery is observed within 3 days post-injury in pre-weanling (PND17) rats (Thomas et al., 2000). Whereas cortical CMRglc remains depressed for 10 days in both adult and PND35 rats after cortical controlled impact (CCI) injury, (Sutton et al., 1994), PND35 rats show earlier recovery of CMRglc in sub-cortical regions (Prins and Hovda, 2009).
Multiple lines of evidence indicate that cellular and biochemical alterations may impact the metabolic fate of glucose during the acute phase of depressed glucose uptake. For example, there is an increased flux of glucose through the pentose phosphate pathway (PPP) during this post-injury time, which is possibly in response to oxidative stress induced by TBI (Bartnik et al., 2005). Increased consumption of poly ADP-ribose polymerases (PARP) caused by DNA oxidative damage that occurs after injury can deplete cytosolic nicotinamide adenine dinucleotide (NAD+), resulting in glyceraldehyde-3-phosphate (GAPDH) inhibition and glycolytic dysfunction (Satchell et al. 2003; Sheline et al., 2000; Suh et al., 2000). Under these circumstances, glucose may not provide sufficient cerebral energy and use of alternative fuel types during this critical period may improve cell viability and enhance functional outcome.
Administration of ketones during the period of CMRglc depression has shown to be neuroprotective after TBI (Prins, 2008). More importantly, a week-long ketogenic diet was shown to confer greater neuroprotection in post-weanling rats (PND 35 and 45) compared to both suckling (PND17) and adult rats (Prins et al., 2005). Using the same therapeutic paradigm, motor and cognitive behavioral scores improved in the younger group, but not in adults (Appelberg et al., 2009). Whereas the age-related neuroprotective differences are likely attributed to age-related differences in ketone uptake after injury (Prins and Giza, 2006), the mechanism for the neuroprotection is unknown. Cerebral metabolism of ketones has been shown to decrease oxidative damage (Haces et al. 2008; Maalouf et al., 2007), decrease inflammation (Ruskin et al., 2009), and improve ATP levels (Haces et al., 2008). In order to determine the potential mechanism(s) for the beneficial effects of ketone metabolism after TBI, the current study was designed to address the effects of ketone metabolism on cerebral bioenergetics and metabolism in CCI injured young (PND35) versus adults (PND70) rats using quantitative, high resolution proton (1H) and phosphorus (31P) nuclear magnetic resonance spectroscopy (NMRS) of brain extracts. We hypothesize that the mechanism of ketogenic neuroprotection after TBI is directly related to improved cellular bioenergetics.
Methods
Experimental procedures and groups
Sexual maturity in rats is achieved at PND60. Based on metabolic developmental profile studies (i.e., glucose metabolism), PND35 rats reach ∼90% of adult values (Nehlig et al., 1988). Therefore, we believe that PND35 versus PND70 rats mirror pre-adolescent versus adult age groups in humans. Following CCI or sham injury, PND35 and PND70 male Sprague-Dawley rats (Charles River, Wilmington, MA) were placed on either standard (SD) or ketogenic (KG) diet. The brain tissue was harvested at either 6 or 24 h after sham injury or CCI injury (n=6–7 per group and per time point).
Surgery
Anesthesia was induced with 3% isoflurane vaporized in 100% O2 and then maintained with 2% isofluorane during surgery. The head was positioned in a stereotaxic frame, a midline incision was made, and a 6-mm craniotomy was drilled centered at −4 mm anterior-posterior (AP), 5 mm midlateral (ML) relative to bregma. A CCI injury was produced on the exposed left cortex using an electronically controlled pneumatic piston cylinder (Hydraulics Control, Inc., Emeryville, CA) as previously described (Dixon et al., 1991; Sutton et al., 1993). In the present study, the 5 mm diameter flat rod tip was angled at 22.5° away from vertical and compressed the cortex at 1.9 m/sec to a depth of 2 mm. After injury, the bone flap was replaced and a dental acrylic cement cap was made to seal the craniotomy before suturing the skin closed. Body temperature was monitored throughout the surgery and recovery using a homeothermic heated pad (37.0±0.5°C). Sham injured rats received only a craniotomy but no cortical injury. All procedures were approved by the UCLA Chancellor's Committee for Animal Research.
Diet
Animals were provided with free access to water and either standard rodent chow (Teklad no. 7013) or KG diet (no. F3666; Bioserv, Frenchtown, NJ) immediately after they regained consciousness. Previous studies have shown that providing rodents with a KG diet results in an increase in the blood concentration of ketone bodies within hours (Prins and Hovda, 2009; Prins et al., 2005; Rho et al. 1999). The KG diet consists of 8.4% protein, 78.8% fat, 0.8% carbohydrates, and 5% fiber. The SD consists of 18.6% protein, 6.2% fat, 59.8% carbohydrates, and 4.5% fiber.
Arterial blood measurement and tissues fixation
Tail arterial blood samples were collected in polyethylene tubes (PE-50) immediately before the microwaving procedure. Blood-gas measurements were taken (Instrumentation Laboratory, Stockholm, Sweden) and the remaining blood samples were centrifuged and plasma glucose and lactate levels were measured (YSI 2700 Biochemistry Analyzer, Yellow Springs, OH). Plasma levels of β-hydroxybutyrate (βOHB) were determined with Ketosite test cards (Stanbiochem, Boerne, TX) on a GDS STATSite Analyzer (Elkhart, IN).
Tissue was fixed using microwave irradiation in order to effectively reduce postmortem changes of energy-related metabolites/enzymes in the nervous tissues (Sharpless and Brown, 1978). All animals were anesthetized with isoflurane before a beam of microwave irradiation at 2450 MHz and nominal output of 3.5 kW was focused for 2.45–2.90 sec directly on the head (Thermex-Thermatron Model 4104 Microwave Fixation System, Louisville, KY). Individual irradiation time was adjusted based on the minimal time necessary for the temperature of the core to reach 80°C, which is sufficient to denature enzymes (Guidotti et al., 1974). Rats were quickly decapitated and the brains were dissected hemispherically, placed into 1.5 mL Eppendorf tubes, and submerged into methylbutane on dry ice before storing at −70°C prior to metabolite extraction.
Chloroform–methanol metabolic extraction
The frozen ipsilateral hemispheric brain samples were weighed and grossly pulverized with a dry ice-chilled pulverizer. The powder was removed and transferred to a steel mortar contained within a liquid nitrogen bath for further grinding. The fine powder was mixed with 1:2 (vol/vol) chloroform/methanol at a ratio of 3 mL solvent to 1 g wet weight. Additional chloroform/de-ionized water was added at a 1:1 ratio to achieve the final ratio of chloroform/methanol/water at 2:2:1 (Harris et al. 1996; Miccheli et al. 1988). The resulting emulsion was centrifuged at 14,000g for 20 min at 4°C to produce a three phase solution: aqueous metabolites, proteins, and hydrophobic lipids. All three phases were collected into separate glass vials and air dried at room temperature under N2 gas flow. The dried aqueous, protein, and lipid pellets were weighed and stored at −70°C until use. The dried aqueous extracts were reconstituted in 0.7 mL deuterium oxide containing 2.86 mM trimethylsilyl propionate (TSP) and 22.7 mM methylenediphosphonic acid (MDPA) immediately before spectroscopic analysis.
NMRS
Spectra were obtained on a 14.1-Tesla Bruker Avance 600 spectrometer at a proton frequency of 600 MHz and at a phosphorus frequency of 242 MHz. Tissue extract samples were shimmed at 4°C until the line width of the TSP peak at half-height was <2 Hz (non-spinning). One-dimensional proton spectra were acquired with a spectral width of 7184 Hz and an acquisition time of 9 sec, resulting in a digital resolution of 0.11 Hz/point. The residual water peak in the reconstituted sample was pre-saturated with a 1-sec soft pulse prior to the excitation pulse. Spectra were acquired in 256 transitions using a 70° flip angle and an overall repetition time of 11 sec. This resulted in fully T1-relaxed spectra at this field strength (Harris et al., 1997). All spectra were processed with 0.5 Hz line broadening before Fourier transformation. Peak assignments were made using published chemical shift information relative to TSP at 0.0 ppm (Harris et al., 1997).
Proton-decoupled phosphorus data were acquired using a 12,019-Hz spectra window and an acquisition time of 5.45 sec, resulting in a digital resolution of 0.18 Hz/point. Spectra were acquired for 1024 transients using a 45° flip angle and an overall repetition time of 5.54 sec for fully T1-relaxed spectra. All spectra were processed with 5-Hz line broadening before Fourier transformation. Peak assignments were made using published chemical shift information relative to glycerophosphorylcholine (GroPChol) at −0.13 ppm (Harris et al., 1996).
Figure 1 demonstrates both 1H and 31P high resolution NMR spectra of a PND35 rat brain extract from the ipsilateral hemisphere. The peak assignments were made according to Glonek and associates (1982), Harris and associates (1997), and Trehout and associates (2002). The following peaks were integrated and normalized to the protein concentration of the dried pellet resulting from the tissue extraction (Fig. 1 A): myo-inositol (Inos), glycine (Gly), Taurine (Tau), Choline (Chol), phosphocreatine (PCr), creatine (Cr), aspartate (Asp), glutamine (Gln), glutamate (Glu), γ-aminobutyric acid (GABA), N-acetyl aspartate (NAA), alanine (Ala), lactate (Lac) (Fig. 1B) phosphorylethanolamine (PEtn), phosphorylcholine (PChol), glycerophosphorylethanolamine (GroPEtn), glycerophosphorylcholine (GroPChol), PCr, (γ, α, β)-ATP, β-ADP, and nicotinamide adenine dinucleotides (NAD).
FIG. 1.

Representative (A) 1H-NMRS and (B) 31P-NMRS spectra of chloroform–methanol extract of ipsilateral hemisphere from PND35 rats. (A) Peak assignment with the chemical shift scale set relative to trimethylsilyl propionate (TSP) at 0 ppm; (B) peak assignment with the chemical shift scale set relative to glycerophosphorylcholine (GroPChol) at −0.13 ppm. The following peaks assignments were made from the NMR spectrum: (A) myo-inositol (Inos), 4.06 ppm; glycine (Gly), 3.56 ppm; taurine (Tau), 3.44 ppm; choline (Chol), 3.20 ppm; phosphocreatine (PCr), 3.05 ppm; creatine (Cr), 3.04 ppm; aspartate (Asp), 2.82 ppm; glutamine (Gln), 2.46 ppm; glutamate (Glu), 2.36 ppm; γ-aminobutyric acid (GABA), 2.31 ppm; N-acetyl aspartate (NAA), 2.02 ppm; alanine (Ala), 1.48 ppm; lactate (Lac), 1.33 ppm; (B) phosphorylethanolamine (PEtn), 3.86 ppm; phosphorylcholine (PChol), 2.63 ppm; glycerophosphorylethanolamine (GroPEtn), 0.81 ppm; glycerophosphorylcholine (GroPChol), −0.13 ppm; PCr, −3.12 ppm; (γ, α, β)-ATP, (-5.80, −10.92, −21.45) ppm; β-ADP, −6.11 ppm; nicotinamide adenine dinucleotides (NAD), −11.37 ppm.
The absolute metabolite amounts were determined using:
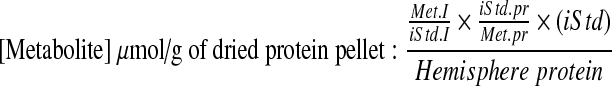 |
Where Met.I and iStd.I are the integrals of the metabolite and internal standard respectively; iStd.pr and Met.pr are the number of protons per resonance of the internal standard and metabolite; and (iStd) is the concentration of the internal standard in moles.
Statistical analysis
A two-way analysis of variance was performed for each metabolite to determine any overall significant difference between experimental groups and between different time points of each group, and any significant interaction of group by time. As there was no significant effect of time among the sham groups, data were pooled. A Tukey HSD post-hoc analysis was used to determine individual group and time difference. A p-value of <0.05 was considered significant.
Results
Contusion injury in PND35 rats results in delayed deficits in brain energy metabolism
Energy metabolism
We first determined the effect of CCI injury on cerebral metabolites in PND35 on SD using 31P and 1H NMRS (Fig. 2). Whereas ipsilateral ATP levels remained unaltered at 6 h after injury, they declined by up to 30% at 24 h compared to sham-injured PND35 rats (p<0.05). Similarly, although Cr levels were lower by 12% compared to sham-injured values at 6 h, this was not significant (p>0.05). But these levels were significantly reduced to 31% by 24 h (p<0.05). Although PCr levels trended above sham values by 14% at 6 h, this was then followed by a significant decrease to values 12% lower than sham at 24 h (p<0.05). As a result of the different direction and degree of change in these two metabolites, the total Cr (PCr+Cr) did not differ at 6 h, whereas at 24 h it was significantly lower than sham values by up to 24%(p<0.05). The PCr/Cr ratio, relevant to in vivo spectroscopy, was significantly increased by 16% and 25% at 6 h and 24 h, respectively (p<0.05). Considering the prior data, this reflects a greater effect of injury on Cr levels than on PCr. Phosphorus was unaffected at 6 h but was significantly reduced at 24 h by up to 25%. Although NAD and β-ADP followed similar trends to PCr and ATP, respectively, there were no significant changes in these metabolites throughout the first 24-h post-injury period.
FIG. 2.

Cerebral energy metabolite concentrations in PND35 rats on standard diet at 6 and 24 h after CCI injury compared to sham-injured (6 and 24 h sham data were pooled because there was no significant effect of time among the sham groups) rats on standard diet. All values are expressed as μmol per gram of sample dried protein pellet. Box plots: the horizontal line inside the box represents the median; the area above this line represents the 75th percentile of a variable; the area below this line represents the 25th percentile of a variable; the small horizontal markers above and below the box represent the maximal and minimal values respectively. *p<0.05, compared to sham-injured values with standard diet (control); #p<0.05, compared to CCI-injured values with standard diet at 6 hr post-injury.
Amino acids and membrane metabolites
We used 1H NMRS to determine the effect of injury on cerebral amino acids and other proton-containing metabolites (Fig. 3A). NAA remained largely unaffected by injury in PND35 rats until 24 h, when it was significantly reduced to 26% of sham-injured values. However, brain Lac levels were significantly elevated by twofold over sham levels at both time points. Glu levels were variable but overall were not significantly different at any time. However, Gln was significantly increased by 26% at 6 h but normalized by 24 h. In contrast, GABA, Gly, and Asp levels were all significantly reduced at 24 h compared to 6 h (p<0.05), although higher within-group variability did not result in any detectable difference compared to the sham-injured group at any time point. No significant changes were measured for Tau and Ala at any time-point.
FIG. 3.

Cerebral amino acids (A) and membrane metabolite concentrations (B) in PND35 rats on standard diet at 6 and 24 h after CCI injury compared to sham-injured rats (6 and 24 h sham data were pooled because there was no significant effect of time among the sham groups) on standard diet. All values are expressed as μmol per gram of sample dried protein pellet. Box plots: the horizontal line inside the box represents the median; the area above this line represents the 75th percentile of a variable; the area below this line represents the 25th percentile of a variable; the small horizontal markers above and below the box represent the maximal and minimal values respectively. *p<0.05, compared to sham-injured values with standard diet; #p<0.05, compared to CCI-injured values with standard diet at 6 h post-injury.
There were no significant changes in membrane metabolites until 24 h, when Inos, Chol, and PEtn were significantly lower than sham values by 23–26%. Inos, Chol, and PChol levels were significantly reduced at 24 h compared to values at 6 h. GroPChol and GroPEtn remained unaffected by injury at all time points (Fig. 3B).
Contusion injury in PND70 rats results in early reductions in brain metabolism
We next determined the effect of contusion injury in adult (PND70) rats on SD. Although some data exist that describe the cerebral metabolic profile after adult TBI (Marklund et al., 2006; Mautes et al., 2001; Thomale et al., 2007), none has used microwave fixation, a method that more efficiently preserves brain metabolites post mortem (Medina et al. 1975; Swaab 1971). ATP levels were reduced by 13% at 6 h after injury, but this was not significant (p>0.05, Table 1), in agreement with previous work that showed amelioration of earlier ATP deficits by 6 h (Lee et al., 1999; Prins et al., 2004; Thomale, et al. 2007). There were no further differences in ATP levels at 24 h in the injured adult brain, in contrast to the PND35 data where levels were significantly reduced at 24 h. NAA levels in PND70 injured rats decreased by 15% below sham-injured values at both 6 and 24 h (p<0.05). Total Cr concentration was not significantly different from sham values at both time points, although there was a trend toward 9–15% lower levels. Lac levels were significantly increased by approximately twofold at 6 h after injury, similar to in PND35 rats. However, unlike in the younger group, Lac levels in PND70 injured rats began to normalize thereafter, so that at 24 h they were not significantly raised above sham-injured levels. Glu and Gln levels remained unchanged at both time points examined (data not shown). PEtn levels were significantly decreased at 24 h post-injury, similar to the PND35 rats (data not shown), but there was no significant effect of brain injury on other membrane-containing metabolites in the PND70 at the time points examined.
Table 1.
Comparison of the Effect of Injury on ATP, Total Creatine, NAA, and Lactate Levels in PND35 and PND70 Rats at 6hrs and 24hrs after CCI Injury
| |
PND35 |
PND70 |
||||
|---|---|---|---|---|---|---|
| Group | Sham-SD | 6hrs CCI-SD | 24hrs CCI-SD | Sham-SD | 6hrs CCI-SD | 24hrs CCI-SD |
| ATP | 17.23±2.67 | 17.40±1.46 | 12.09±1.30* | 15.15±1.89 | 12.86±1.44 | 15.07±2.01 |
| Tol Cr | 39.14±5.81 | 39.23±3.60 | 29.79±3.85* | 42.49±3.77 | 38.50±5.66 | 36.14±8.52 |
| NAA | 33.66±4.85 | 31.25±5.10 | 24.79±3.01* | 36.08±4.12 | 30.83±4.96* | 30.26±4.87* |
| Lac | 2.55±0.34 | 5.12±1.28* | 5.30±1.60* | 3.42±1.08 | 6.79±2.80* | 4.37±1.53 |
Data are expressed as mean±STD (μmol per gram of dry protein pellet). *p<0.05 significantly different compared to age-matched sham animal with standard diet (SD) (highlighted in grey).
Early KG diet decreases metabolic defects in PND35 but not in PND70 rats
Next we investigated whether administration of KG diet would ameliorate the effects of injury on brain metabolism in both young and adult rats. All rats were placed on designated diets immediately following recovery after injury. There were no significant differences in blood gas values in any group. Plasma levels of βOHB, Glu, and Lac in SD-fed PND35 injured rats were maintained at sham-injured levels at 6 and 24 h after injury (Table 2). As expected, the KG diet significantly elevated plasma βOHB levels in all PND35 rats by two- to- fourfold at both 6 and 24 h after injury. The KG diet also resulted in reductions in plasma glucose and Lac levels at 6 h, but this was only significant in the sham group. By 24 h both glucose and Lac were similarly reduced in sham and CCI-injured PND35 rats (p<0.05). Similar changes in plasma metabolites were observed in PND70 and PND35 rats (data not shown) and were very similar to those reported previously (Prins and Giza, 2006; Prins and Hovda, 2009). More importantly, after 24 h on the KG diet, a βOHB doublet of peaks was detectable at 1.2 ppm on the 1H NMRS spectra, reflecting the accumulation of metabolized ketones into the CNS (Fig. 4). No peaks were visible at 6 h post-injury or at any time in spectra from rats on SD. Brain βOHB levels were 43% higher in the injured PND35 brains compared to PND70.
Table 2.
Physiological and Plasma Metabolic Data (Means±STD) in PND35 Rats
| |
6hrs |
24hrs |
||||||
|---|---|---|---|---|---|---|---|---|
| |
Standard diet |
Ketogenic diet |
Standard diet |
Ketogenic diet |
||||
| Sham | CCI | Sham | CCI | Sham | CCI | Sham | CCI | |
| pH | 7.36±0.03 | 7.38±0.04 | 7.34±0.04 | 7.36±0.02 | 7.33±0.05 | 7.35±0.04 | 7.38±0.02 | 7.36±0.05 |
| PCO2 (mmHg) | 41.33±1.15 | 41.43±3.84 | 37.67±1.15 | 41.90±2.67 | 42.03±3.44 | 42.77±3.11 | 38.73±2.05 | 41.49±3.10 |
| Glucose (mM) | 9.44±0.58 | 9.08±0.74 | 7.95±0.42* | 8.94±0.58 | 9.52±0.37 | 8.65±1.22 | 6.83±1.18* | 6.20±0.92* |
| Lactate (mM) | 2.32±0.34 | 2.25±0.57 | 1.46±0.20* | 1.86±0.37 | 2.28±0.52 | 2.20±0.55 | 1.13±0.16* | 1.19±0.20* |
| β-OHB (mM) | 0.07±0.02 | 0.12±0.06 | 0.32±0.19* | 0.21±0.11* | 0.12±0.03 | 0.11±0.08 | 1.45±0.51* | 1.39±0.37* |
Data are mean±STD values. *p<0.05 significantly different compared to sham animal with standard diet at respective time point (highlighted in grey).
FIG. 4.

Representative 1H-NMRS spectra of brain β-hydroxybutyrate levels in both standard (A, B) and ketogenic fed (C) PND35 rats. (A) Full spectra width 1H-NMRS spectrum from a standard-fed, PND35 injured rat. (B) Magnified 1H-NMRS spectrum from (A) between 1.19 ppm and 1.51 ppm. (C) Similarly magnified 1H-NMRS spectrum to (B) from a ketogenic fed, PND35injured rat. (Table) Brain levels of β-hydroxybutyrate are expressed as mean±SD (μmol per gram of sample dry protein pellet).
Energy metabolism
There was no significant effect of KG diet on any energy metabolite measured at 6 h after injury in PND35 rats (Fig. 5A). However, by 24 h after injury and the initiation of the KG diet, ATP levels were significantly increased toward sham values by 32% compared to standard-fed, injured rats (Fig. 5A; p<0.05). Similarly, both Cr and PCr levels were enhanced by KG diet to ∼23% above those of standard-fed injured rats, and the PCr/Cr ratio remained in concordance with this. NAD levels remained unaffected by diet.
FIG. 5.

The effect of ketogenic diet on injured brain energy metabolite concentrations. Ketogenic diet metabolite values ([Metabolites]KG DIET) are expressed as a percent change from standard diet values ([Metabolite]SD) at 6 hr (gray bars) and 24 h (black bars) after CCI injury in both (A) PND35 and (B) PND70 rats. #p<0.05, compared to standard-fed, injured rats.
There was no significant effect of KG diet on any brain energy metabolite measured in PND70 rats at 6 h after injury (Fig. 5B). Similarly at 24 h, ATP and Cr levels remained unaffected by diet and whereas PCr was variably increased by 24% because of the KG diet, this was not significant. However, the PCr/Cr ratio was significantly increased by 35% because of the KG diet in PND70 injured rats (p<0.05). In addition, NAD levels rose significantly by 36% after 24 h on the KG diet.
Amino acids
In PND35 rats fed the KG diet, NAA remained at sham levels at 24 h and did not decrease to levels observed in standard-fed PND35 injured brains (Table 3). The elevated Lac levels associated with injured rats on SD were also significantly attenuated by KG diet at 24h. KG diet also significantly ameliorated the deficits in GABA and Asp that were associated with injured rats on SD at 24 h. Concentrations of Glu, Gly, and Tau were also improved toward sham values in injured rats after 24 h on the KG diet, although this was not significant. In contrast, in the PND70 brain there was no significant effect of KG diet on the measured amino acids at any time point assessed after injury (data not shown).
Table 3.
Effect of Ketogenic Diet on Cerebral Amino Acids and Membrane Metabolites at 24hrs after CCI Injury in PND35 Rats
|
Amino acids |
|
Membrane metabolites |
|||
|---|---|---|---|---|---|
| |
CCI-SD diet |
CCI-KG diet |
|
CCI-SD diet |
CCI-KG diet |
| (vs. sham with standard diet) | (vs. sham with standard diet) | ||||
| NAA | −26%* | −14% | Choline | −23%* | −8% |
| Lactate | +108%* | +20% | Myo-Inositol | −23%* | −19% |
| Glutamine | — | +12% | PChol | −17% | −12% |
| Glutamate | −19% | — | PEtn | −26%* | −10% |
| GABA | −23%* | — | GroPChol | −15% | −15% |
| Glycine | −16% | — | GroPEtn | — | −15% |
| Taurine | −21% | −11% | |||
| Aspartate | −22%* | −9% | |||
| Alanine | +9% | — | |||
Data are expressed as % change versus sham-injured standard fed values. “—” indicates less than 5% difference. *p<0.05 significantly different compared to sham animal with standard diet. SD- standard diet, KG- ketogenic diet.
Membrane metabolites
KG diet significantly reduced the deficit in brain PEtn levels at 24 h in PND35 injured rats (p<0.05). Although there was no such improvement in other membrane metabolites, apart from the glycerol-containing compounds, all other choline-containing metabolites were normalized toward sham-injured levels at 24 h compared to SD-fed rats. In the PND70 brain, the levels of membrane metabolites remained unchanged after the diet treatment at the time points examined (data not shown).
Discussion
The current data show that contusion injury in PND35 rats on SD results in deficits in metabolites associated with energy metabolism, amino acids, and cell membranes. However, in comparison to similarly acquired data from injured PND70 adult rats, and to previous work in adult injured rats (Bartnik et al. 2005; Lee et al., 1999; Schuhmann et al., 2003), many of these metabolic deficits in the younger brain occur only at the more chronic, 24 h time point rather than at the earlier time points examined in older rats. Access to a KG diet acutely after injury significantly ameliorated and/or prevented many of the metabolic deficits at 24 h in injured PND35 rats. Although diet significantly increased the PCr/Cr ratio and NAD levels in PND70 rats, this did not coincide with ATP recovery. There were no other significant effects associated with the KG diet in older rats.
The time course of TBI-induced metabolic disruption is age dependent
The results of the current study suggest that the time course of metabolic “crisis” after TBI is different in PND35 than in PND70 rats on SD. Deficits in energy metabolites (ATP, Cr, PCr), amino acids (NAA, Lac, Glu), and membrane metabolites (Chol) present at 6 h in adult injured rats (Lee et al., 1999; Prins et al., 2004; Thomale et al., 2007) were not recorded in PND35 brains until 24 h post-injury. Non-significant perturbations in both PCr and Cr at 6 h in PND35 brains may reflect the early stages of metabolic disturbances as the brain attempts to increase high energy reserves of PCr in the face of increased demand for ATP. However there would need to be further study to determine this. It is notable that PCr and Cr are not resolvable with standard clinical 1H-NMRS, so that early changes in PCr or Cr could well be missed. In addition, the widespread use of the proton-observed total PCr+Cr peak to create in vivo metabolic ratios with other MR-visible, proton-containing metabolites could potentially result in misleading data, especially in the most metabolically vulnerable tissues in the injured brain. The greater deficits in Cr compared to PCr resulted in a significantly increased PCr/Cr ratio at both 6 and 24 h after injury in PND35 rats. This increased ratio is generally considered a marker of heightened energy stores, which may appear to be in conflict with the lowered ATP levels at 24 h. However, in addition to a role as cellular energy buffer, PCr and Cr metabolites may have additional roles under pathologic conditions. Cr has been reported to function as an antioxidant by scavenging superoxide anions and peroxynitrite (Lawler et al., 2002; Sestili et al., 2006) and Cr has been shown to act as a cell osmolyte to prevent hyperosmotic shock in cultured cells (Alfieri et al., 2006). These data are in agreement with previous findings after mild TBI in adult rats in which a significant decrease of total creatine occurred without direct concomitant reductions in PCr/Cr ratio (Signoretti et al., 2010).
Delayed decreases in amino acids were also observed in injured PND35 rats on the SD. NAA has been established as a marker for neuronal integrity (Briellmann et al., 2005; Caramanos et al., 2005; Parsons et al., 2000; Vagnozzi et al., 2010) and correlated with mitochondrial energy metabolism and functional outcome (Bates et al., 1996; Clark, 1998; De Stefano et al., 1995;). Changes in NAA following adult diffuse brain injury were found to be temporally related to ATP levels (Gasparovic et al., 2001; Signoretti et al., 2001, 2010). The early and sustained reduction of NAA in the adult model herein is likely to reflect these ATP reductions as well as compromised neuronal integrity that occurs after CCI injury. In the current study, NAA levels were already reduced by 6 h after trauma in PND70 rats, and these deficits were sustained at the 24-h time point, which is in agreement with previous findings in adult models of TBI (Schuhmann et al., 2003). The delayed deficit in NAA levels seen in the PND35 rats suggests that younger brains may be less susceptible to metabolic crisis and that the window for potential neuroprotective treatments in adults is shorter.
In addition to changes in NAA, Lac and Gln also demonstrate age-related differences in time course after injury. Whereas adult brain lactate levels recovered by 24 h, sustained increase in brain lactate among PND35 rats at both 6 and 24 h suggests ongoing impaired mitochondrial oxidation (Opii et al., 2007; Xiong et al. 1997) or disruptions in cytosolic redox states (Satchell et al., 2003; Sheline et al., 2000). Glu levels remained relatively stable at both 6 and 24 h after injury in the PND35 rats, whereas Gln was transiently and significantly increased at 6 h. Similar findings have been reported at 3.5 h after injury in adult rats (Bartnik et al., 2005) and at 5.5–6 h after injury in juvenile rats (Scafidi et al., 2009) using 13C NMRS following an intravenous (i.v.) infusion of 13C-labelled glucose. Despite the well-documented, excessive efflux of Glu into the extracellular space immediately after TBI (Katayama et al., 1990; Palmer et al., 1993), the technique used in the current study does not distinguish between metabolites contained within the extracellular space and those within cells. It simply indicates that total brain Glu levels remain unchanged in PND35 rats. The raised Gln levels may in fact indicate excesses in extracellular Glu, as the increase in astrocytic production of Gln has been shown to occur when extracellular Glu level is increased (McKenna et al., 1996). One potential mechanism for this has been demonstrated in cultured cortical astrocytes, where high extracellular Glu levels can induce an increase in Gln synthetase (GS), which converts Glu to Gln (Lehmann et al., 2009). Increased GS expression has been known to support neuronal survival under pathological conditions, such as trauma and ischemia (Benton et al., 2000; Gorovits et al., 1997; Petito et al., 1992; Ramonet et al., 2004). In light of this evidence, it appears therefore that the increased Gln level within the first few hours after brain trauma may indicate a protective response against excitotoxicity through the increased astrocytic control of extracellular Glu levels.
The current study also found decreases in Chol, PEtn, and Inos at 24 h in PND35 rats, which is likely reflective of gliosis, inflammation, demyelination, and membrane breakdown (Ashwal et al., 2006; Brooks et al., 1999, 2000, 2001; Cecil et al., 1998;).
It should be noted that the same injury paradigm was used on both younger and older brains in this study. In a previous study conducted on PND17, 35, 45, and 65 rats using the same injury, no difference in the development of cortical contusion volume was observed among different age groups (Prins et al. 2005). This suggests that primary tissue loss induced by the mechanical impact is probably not a factor that contributes to the age-dependent metabolic changes after injury. However, the responses to KG diet in PND35 and 45 rats were different from those in PND17 or 65 rats after the same injury (Prins et al., 2005). The current study has provided crucial evidence that indicates that the susceptibility to metabolic crisis is age dependent. The metabolic profile data demonstrated a slower developing metabolic crisis in the younger brains, which may provide a greater window for therapeutic intervention among PND35 rats.
Early KG diet decreases metabolic defects in young rats but not in adults
After 24 h of access to KG diet, there was a marked normalization of many cerebral metabolites in PND35 rats, but not in PND70 rats. These age-related differences are consistent with previous work that showed a preferential use of ketones rather than glucose as an energy source after brain injury in younger rats but not adults (Prins and Hovda, 2009). Decreased brain lactate levels in PND35 injured rats on the KG diet, is further evidence that cerebral metabolism of glucose has shifted toward the alternative fuel. Because the acute period of hyperglycolysis after brain injury is critical to maintain cellular homeostasis (Hovda et al., 1990; Kawamata et al.,1992; Yoshino et al. 1991), the administration of alternative fuels early following brain trauma could be critical in preventing the post-acute changes in energy metabolism, enabling the longer term structural and functional benefits that result from ketogenic diet in this model (Appelberg et al. 2009; Prins et al. 2005).
There are several possible reasons for the failure of the PND70 rats to show normalization of cerebral metabolites upon administration of the KG diet. First, there are age differences in the magnitude of ketosis achieved on the KG diet within the first 24 h after TBI. PND35 rats have been previously shown to develop plasma levels of ßOHB that are 20–25% greater than those in PND70 rats within 24 h (Prins and Giza, 2006; Prins and Hovda, 2009). Second, previous work has shown that expression of monocarboxylate transporters within the endothelium is greater in younger than in older brains, indicating a greater potential for uptake of ketones (Prins and Giza, 2006). In accordance with this, in the current study, brain concentrations of ßOHB achieved in PND70 rats on the KG diet were 30% less than those achieved in PND35 rats. The slower ability of adult rats to increase brain concentrations of ketones in the face of a hyperacute metabolic crisis likely contributes significantly to the poor outcome. Nonetheless, there was a significant increase of NAD levels in PND70 rats after KG diet. Studies have shown that post-injury free radical production and therefore ADP-ribose polymerase (PARP)-mediated DNA repair process consumes and can deplete NAD pool (Berger, 1985). Repletion of NAD has been indicated as protective in reducing PARP-mediated cell death through preventing glycolytic inhibition (Ying et al., 2003) and apoptosis-inducing factor (AIF) translocation (Alano et al., 2004). The increase of NAD levels in the PND70 brains on KG diet may in part contribute to the normal rates of glycolysis (Prins and Hovda, 2009). However, this did not correlate with recovery of ATP levels, suggesting that energy metabolism was not improved by diet in older rats. It appears that the neuroprotective mechanism of KG diet is directly related to improved cellular bioenergetics. Direct infusion of high-dose ketones through i.v. infusion in injured adult rats resulted in 8.5-fold increase in brain uptake of the βOHB followed by an acute improvement in cerebral ATP levels (Prins et al., 2004). It therefore appears that the mode of administration of ketones is important in obtaining neuroprotective effects when considering different age groups. However, it remains to be seen whether i.v. administration in adult injured rats translates to improved NAA and better long- term outcome. Regardless, these data suggest that intervention with alternative substrates after TBI in older animals will require delivery mechanisms within a shorter time window.
Cerebral metabolism of ketones has been previously shown to affect numerous cellular mechanisms that contribute to improved recovery after brain injury. Although it remains unclear which mechanisms contribute to enhanced recovery in the PND35 rats after injury, the current study reveals that enhanced recovery is, in part, related to improved cerebral bioenergetics. The partial recovery of the membrane metabolites Inos, Chol, and PEtn levels observed after 24-h KG diet in the CCI-PND35 brains would appear consistent with the idea that cell viability is improved with normalized metabolism. It is unknown whether the therapeutic effects of ketones after injury occur solely by enhancing energy metabolism or through a multifaceted mechanism. Future studies are required to determine the relative contribution of other mechanisms including decreases in inflammation (Ruskin et al., 2009), cell death (Hu et al., 2009), and oxidative damage (Haces et al., 2008). Enriched ketone diet is a rich source for acetyl-CoA production, for generation of important biosynthesis materials for membrane or myelin (Morell and Jurevics, 1996). It remains unclear if KG diet enables catabolism of plasma membranes following TBI. Doing so would imply a restorative mechanism of ketones after brain injury, and this alone might widen the therapeutic window to obtain functional benefits.
Conclusion
In summary, brain injury-induced deficits in brain energy, amino acid, and membrane metabolism was delayed until 24 h in young (PND35) rats compared to adults (PND70), in which deficits occurred much earlier. Administration of KG diet significantly ameliorated these deficits, but only in the younger animals.
Acknowledgments
We thank Dr. Brenda Bartnik-Olson for helpful comments on the manuscript and Dr. David McArthur for help with statistics. This work was supported by the UCLA Brain Injury Research Center and National Institutes of Health Award Numbers NS055910 and NS058489 from the National Institute of Neurological Disorders and Stroke (NINDS). The content is the sole responsibility of the authors and does not necessarily represent official views of the NINDS or the National Institutes of Health.
Author Disclosure Statement
No competing financial interests exist. This material has been reviewed by the Walter Reed Army Institute of Research. There is no objection to its presentation and/or publication. The opinions or assertions contained herein are the private views of the author, and are not to be construed as official, or as reflecting true views of the Department of the Army or the Department of Defense.
References
- Alano C.C. Ying W. Swanson R.A. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J. Biol. Chem. 2004;279:18,895–18,902. doi: 10.1074/jbc.M313329200. [DOI] [PubMed] [Google Scholar]
- Alessandri B. Reinert M. Young H.F. Bullock R. Low extracellular (ECF) glucose affects the neurochemical profile in severe head-injured patients. Acta Neurochir. Suppl. 2000;76:425–430. doi: 10.1007/978-3-7091-6346-7_88. [DOI] [PubMed] [Google Scholar]
- Alfieri R.R. Bonelli M.A. Cavazzoni A. Brigotti M. Fumarola C. Sestili P. Mozzoni P. De Palma G. Mutti A. Carnicelli D. Vacondio F. Silva C. Borghetti A.F. Wheeler K.P. Petronini P.G. Creatine as a compatible osmolyte in muscle cells exposed to hypertonic stress. J. Physiol. 2006;576:391–401. doi: 10.1113/jphysiol.2006.115006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen B.J. Marmarou A. Functional compartmentalization of energy production in neural tissue. Brain Res. 1992;585:190–195. doi: 10.1016/0006-8993(92)91206-t. [DOI] [PubMed] [Google Scholar]
- Appelberg K.S. Hovda D.A. Prins M.L. The effects of a ketogenic diet on behavioral outcome after controlled cortical impact injury in the juvenile and adult rat. J. Neurotrauma. 2009;26:497–506. doi: 10.1089/neu.2008.0664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashwal S. Babikian T. Gardner–Nichols J. Freier M.C. Tong K.A. Holshouser B.A. Susceptibility-weighted imaging and proton magnetic resonance spectroscopy in assessment of outcome after pediatric traumatic brain injury. Arch. Phys. Med. Rehabil. 2006;87:S50–58. doi: 10.1016/j.apmr.2006.07.275. [DOI] [PubMed] [Google Scholar]
- Bartnik B.L. Sutton R.L. Fukushima M. Harris N.G. Hovda D.A. Lee S.M. Upregulation of pentose phosphate pathway and preservation of tricarboxylic acid cycle flux after experimental brain injury. J. Neurotrauma. 2005;22:1052–1065. doi: 10.1089/neu.2005.22.1052. [DOI] [PubMed] [Google Scholar]
- Bates T.E. Strangward M. Keelan J. Davey G.P. Munro P.M. Clark J.B. Inhibition of N-acetylaspartate production: implications for 1H MRS studies in vivo. Neuroreport. 1996;7:1397–1400. [PubMed] [Google Scholar]
- Benton R.L. Ross C.D. Miller K.E. Glutamine synthetase activities in spinal white and gray matter 7 days following spinal cord injury in rats. Neurosci. Lett. 2000;291:1–4. doi: 10.1016/s0304-3940(00)01362-8. [DOI] [PubMed] [Google Scholar]
- Berger N.A. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat. Res. 1985;101:4–15. [PubMed] [Google Scholar]
- Bergsneider M. Hovda D.A. Shalmon E. Kelly D.F. Vespa P.M. Martin N.A. Phelps M.E. McArthur D.L. Caron M.J. Kraus J.F. Becker D.P. Cerebral hyperglycolysis following severe traumatic brain injury in humans: a positron emission tomography study. J. Neurosurg. 1997;86:241–251. doi: 10.3171/jns.1997.86.2.0241. [DOI] [PubMed] [Google Scholar]
- Briellmann R.S. Wellard R.M. Jackson G.D. Seizure-associated abnormalities in epilepsy: evidence from MR imaging. Epilepsia. 2005;46:760–766. doi: 10.1111/j.1528-1167.2005.47604.x. [DOI] [PubMed] [Google Scholar]
- Brooks W.M. Friedman S.D. Gasparovic C. Magnetic resonance spectroscopy in traumatic brain injury. J. Head Trauma Rehabil. 2001;16:149–164. doi: 10.1097/00001199-200104000-00005. [DOI] [PubMed] [Google Scholar]
- Brooks W.M. Jung R.E. Ford C.C. Greinel E.J. Sibbitt W.L., Jr Relationship between neurometabolite derangement and neurocognitive dysfunction in systemic lupus erythematosus. J. Rheumatol. 1999;26:81–85. [PubMed] [Google Scholar]
- Brooks W.M. Stidley C.A. Petropoulos H. Jung R.E. Weers D.C. Friedman S.D. Barlow M.A. Sibbitt W.L., Jr. Yeo R.A. Metabolic and cognitive response to human traumatic brain injury: a quantitative proton magnetic resonance study. J. Neurotrauma. 2000;17:629–640. doi: 10.1089/089771500415382. [DOI] [PubMed] [Google Scholar]
- Caramanos Z. Narayanan S. Arnold D.L. 1H-MRS quantification of tNA and tCr in patients with multiple sclerosis: a meta-analytic review. Brain. 2005;128:2483–2506. doi: 10.1093/brain/awh640. [DOI] [PubMed] [Google Scholar]
- Cecil K.M. Lenkinski R.E. Meaney D.F. McIntosh T.K. Smith D.H. High-field proton magnetic resonance spectroscopy of a swine model for axonal injury. J. Neurochem. 1998;70:2038–2044. doi: 10.1046/j.1471-4159.1998.70052038.x. [DOI] [PubMed] [Google Scholar]
- Clark J. B. N-acetyl aspartate: a marker for neuronal loss or mitochondrial dysfunction. Dev. Neurosci. 1998;20:271–276. doi: 10.1159/000017321. [DOI] [PubMed] [Google Scholar]
- De Stefano N. Matthews P.M. Arnold D.L. Reversible decreases in N-acetylaspartate after acute brain injury. Magn. Reson. Med. 1995;34:721–727. doi: 10.1002/mrm.1910340511. [DOI] [PubMed] [Google Scholar]
- Dixon C.E. Clifton G.L. Lighthall J.W. Yaghmai A.A. Hayes R.L. A controlled cortical impact model of traumatic brain injury in the rat. J. Neurosci. Methods. 1991;39:253–262. doi: 10.1016/0165-0270(91)90104-8. [DOI] [PubMed] [Google Scholar]
- Faden A.I. Demediuk P. Panter S.S. Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- Gasparovic C. Arfai N. Smid N. Feeney D.M. Decrease and recovery of N-acetylaspartate/creatine in rat brain remote from focal injury. J. Neurotrauma. 2001;18:241–246. doi: 10.1089/08977150151070856. [DOI] [PubMed] [Google Scholar]
- Glonek T. Kopp S.J. Kot E. Pettegrew J.W. Harrison W.H. Cohen M.M. P-31 nuclear magnetic resonance analysis of brain: the perchloric acid extract spectrum. J. Neurochem. 1982;39:1210–1219. doi: 10.1111/j.1471-4159.1982.tb12557.x. [DOI] [PubMed] [Google Scholar]
- Goodman J.C. Valadka A.B. Gopinath S.P. Uzura M. Robertson C.S. Extracellular lactate and glucose alterations in the brain after head injury measured by microdialysis. Crit. Care Med. 1999;27:1965–1973. doi: 10.1097/00003246-199909000-00041. [DOI] [PubMed] [Google Scholar]
- Gorovits R. Avidan N. Avisar N. Shaked I. Vardimon L. Glutamine synthetase protects against neuronal degeneration in injured retinal tissue. Proc. Natl. Acad. Sci. U. S. A. 1997;94:7024–7029. doi: 10.1073/pnas.94.13.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti A. Cheney D.L. Trabucchi M. Doteuchi M. Wang C. Focused microwave radiation: a technique to minimize post mortem changes of cyclic nucleotides, dopa and choline and to preserve brain morphology. Neuropharmacology. 1974;13:1115–1122. doi: 10.1016/0028-3908(74)90061-6. [DOI] [PubMed] [Google Scholar]
- Haces M.L. Hernandez–Fonseca K. Medina–Campos O.N. Montiel T. Pedraza–Chaverri J. Massieu L. Antioxidant capacity contributes to protection of ketone bodies against oxidative damage induced during hypoglycemic conditions. Exp. Neurol. 2008;211:85–96. doi: 10.1016/j.expneurol.2007.12.029. [DOI] [PubMed] [Google Scholar]
- Harris N.G. Plant H.D. Briggs R.W. Jones H.C. Metabolite changes in the cerebral cortex of treated and untreated infant hydrocephalic rats studied using in vitro 31P-NMR spectroscopy. J. Neurochem. 1996;67:2030–2038. doi: 10.1046/j.1471-4159.1996.67052030.x. [DOI] [PubMed] [Google Scholar]
- Harris N.G. Plant H.D. Inglis B.A. Briggs R.W. Jones H.C. Neurochemical changes in the cerebral cortex of treated and untreated hydrocephalic rat pups quantified with in vitro 1H-NMR spectroscopy. J. Neurochem. 1997;68:305–312. doi: 10.1046/j.1471-4159.1997.68010305.x. [DOI] [PubMed] [Google Scholar]
- Hovda D.A. Yoshino A. Kawamata T. Katayama Y. Fineman I. Becker D.P. The increase in local cerebral glucose utilization following fluid percussion brain injury is prevented with kynurenic acid and is associated with an increase in calcium. Acta Neurochir. Suppl. (Wien) 1990;51:331–333. doi: 10.1007/978-3-7091-9115-6_112. [DOI] [PubMed] [Google Scholar]
- Hu Z.G. Wang H.D. Jin W. Yin H.X. Ketogenic diet reduces cytochrome c release and cellular apoptosis following traumatic brain injury in juvenile rats. Ann. Clin. Lab. Sci. 2009;39:76–83. [PubMed] [Google Scholar]
- Katayama Y. Becker D.P. Tamura T. Ikezaki K. Early cellular swelling in experimental traumatic brain injury: a phenomenon mediated by excitatory amino acids. Acta Neurochir. Suppl. (Wien) 1990;51:271–273. doi: 10.1007/978-3-7091-9115-6_92. [DOI] [PubMed] [Google Scholar]
- Katayama Y. Maeda T. Koshinaga M. Kawamata T. Tsubokawa T. Role of excitatory amino acid-mediated ionic fluxes in traumatic brain injury. Brain Pathol. 1995;5:427–435. doi: 10.1111/j.1750-3639.1995.tb00621.x. [DOI] [PubMed] [Google Scholar]
- Kawamata T. Katayama Y. Hovda D.A. Yoshino A. Becker D.P. Administration of excitatory amino acid antagonists via microdialysis attenuates the increase in glucose utilization seen following concussive brain injury. J. Cereb. Blood Flow Metab. 1992;12:12–24. doi: 10.1038/jcbfm.1992.3. [DOI] [PubMed] [Google Scholar]
- Kocher M. Metabolic and hemodynamic activation of postischemic rat brain by cortical spreading depression. J. Cereb. Blood Flow Metab. 1990;10:564–571. doi: 10.1038/jcbfm.1990.99. [DOI] [PubMed] [Google Scholar]
- Lawler J.M. Barnes W.S. Wu G. Song W. Demaree S. Direct antioxidant properties of creatine. Biochem. Biophys. Res. Commun. 2002;290:47–52. doi: 10.1006/bbrc.2001.6164. [DOI] [PubMed] [Google Scholar]
- Lee S.M. Wong M.D. Samii A. Hovda D.A. Evidence for energy failure following irreversible traumatic brain injury. Ann. N. Y. Acad. Sci. 1999;893:337–340. doi: 10.1111/j.1749-6632.1999.tb07849.x. [DOI] [PubMed] [Google Scholar]
- Lehmann C. Bette S. Engele J. High extracellular glutamate modulates expression of glutamate transporters and glutamine synthetase in cultured astrocytes. Brain Res. 2009;1297:1–8. doi: 10.1016/j.brainres.2009.08.070. [DOI] [PubMed] [Google Scholar]
- Maalouf M. Sullivan P.G. Davis L. Kim D.Y. Rho J.M. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience. 2007;145:256–264. doi: 10.1016/j.neuroscience.2006.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklund N. Salci K. Ronquist G. Hillered L. Energy metabolic changes in the early post-injury period following traumatic brain injury in rats. Neurochem. Res. 2006;31:1085–1093. doi: 10.1007/s11064-006-9120-0. [DOI] [PubMed] [Google Scholar]
- Mautes A.E. Thome D. Steudel W.I. Nacimiento A.C. Yang Y. Shohami E. Changes in regional energy metabolism after closed head injury in the rat. J. Mol. Neurosci. 2001;16:33–39. doi: 10.1385/JMN:16:1:33. [DOI] [PubMed] [Google Scholar]
- McKenna M.C. Sonnewald U. Huang X. Stevenson J. Zielke H.R. Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J. Neurochem. 1996;66:386–393. doi: 10.1046/j.1471-4159.1996.66010386.x. [DOI] [PubMed] [Google Scholar]
- Medina M.A. Jones D.J. Stavinoha W.B. Ross D.H. The levels of labile intermediary metabolites in mouse brain following rapid tissue fixation with microwave irradiation. J. Neurochem. 1975;24:223–227. doi: 10.1111/j.1471-4159.1975.tb11868.x. [DOI] [PubMed] [Google Scholar]
- Miccheli A. Aureli T. Delfini M. Di Cocco M.E. Viola P. Gobetto R. Conti F. Study on influence of inactivation enzyme techniques and extraction procedures on cerebral phosphorylated metabolite levels by 31P NMR spectroscopy. Cell Mol. Biol. 1988;34:591–603. [PubMed] [Google Scholar]
- Morell P. Jurevics H. Origin of cholesterol in myelin. Neurochem. Res. 1996;21:463–470. doi: 10.1007/BF02527711. [DOI] [PubMed] [Google Scholar]
- Nehlig A. de Vasconcelos A.P. Boyet S. Quantitative autoradiographic measurement of local cerebral glucose utilization in freely moving rats during postnatal development. J. Neurosci. 1988;8:2321–2333. doi: 10.1523/JNEUROSCI.08-07-02321.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell M.T. Seal A. Nortje J. Al-Rawi P.G. Coles J.P. Fryer T.D. Menon D.K. Pickard J.D. Hutchinson P.J. Glucose metabolism in traumatic brain injury: a combined microdialysis and [18F]-2-fluoro-2-deoxy-D-glucose-positron emission tomography (FDG-PET) study. Acta Neurochir. Suppl. 2005;95:165–168. doi: 10.1007/3-211-32318-x_35. [DOI] [PubMed] [Google Scholar]
- Opii W.O. Nukala V.N. Sultana R. Pandya J.D. Day K.M. Merchant M.L. Klein J.B. Sullivan P.G. Butterfield D.A. Proteomic identification of oxidized mitochondrial proteins following experimental traumatic brain injury. J. Neurotrauma. 2007;24:772–789. doi: 10.1089/neu.2006.0229. [DOI] [PubMed] [Google Scholar]
- Palmer A.M. Marion D.W. Botscheller M.L. Swedlow P.E. Styren S.D. DeKosky S.T. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J. Neurochem. 1993;61:2015–2024. doi: 10.1111/j.1471-4159.1993.tb07437.x. [DOI] [PubMed] [Google Scholar]
- Parsons M.W. Li T. Barber P.A. Yang Q. Darby D.G. Desmond P.M. Gerraty R.P. Tress B.M. Davis S.M. Combined (1)H MR spectroscopy and diffusion-weighted MRI improves the prediction of stroke outcome. Neurology. 2000;55:498–505. doi: 10.1212/wnl.55.4.498. [DOI] [PubMed] [Google Scholar]
- Petito C.K. Chung M.C. Verkhovsky L.M. Cooper A.J. Brain glutamine synthetase increases following cerebral ischemia in the rat. Brain Res. 1992;569:275–280. doi: 10.1016/0006-8993(92)90639-q. [DOI] [PubMed] [Google Scholar]
- Prins M. Hovda D. The effects of age and ketogenic diet on local cerebral metabolic rates of glucose after controlled cortical impact injury in rats. J. Neurotrauma. 2009;26:1083–1093. doi: 10.1089/neu.2008.0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins M.L. Cerebral metabolic adaptation and ketone metabolism after brain injury. J. Cereb. Blood Flow Metab. 2008;28:1–16. doi: 10.1038/sj.jcbfm.9600543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins M. L. Giza C.C. Induction of monocarboxylate transporter 2 expression and ketone transport following traumatic brain injury in juvenile and adult rats. Dev. Neurosci. 2006;28:447–456. doi: 10.1159/000094170. [DOI] [PubMed] [Google Scholar]
- Prins M.L. Fujima L.S. Hovda D.A. Age-dependent reduction of cortical contusion volume by ketones after traumatic brain injury. J. Neurosci. Res. 2005;82:413–420. doi: 10.1002/jnr.20633. [DOI] [PubMed] [Google Scholar]
- Prins M.L. Lee S.M. Fujima L.S. Hovda D.A. Increased cerebral uptake and oxidation of exogenous betaHB improves ATP following traumatic brain injury in adult rats. J. Neurochem. 2004;90:666–672. doi: 10.1111/j.1471-4159.2004.02542.x. [DOI] [PubMed] [Google Scholar]
- Ramonet D. Rodriguez M.J. Fredriksson K. Bernal F. Mahy N. In vivo neuroprotective adaptation of the glutamate/glutamine cycle to neuronal death. Hippocampus. 2004;14:586–594. doi: 10.1002/hipo.10188. [DOI] [PubMed] [Google Scholar]
- Rho J.M. Kim D.W. Robbins C.A. Anderson G.D. Schwartzkroin P.A. Age-dependent differences in flurothyl seizure sensitivity in mice treated with a ketogenic diet. Epilepsy Res. 1999;37:233–240. doi: 10.1016/s0920-1211(99)00068-6. [DOI] [PubMed] [Google Scholar]
- Rose M.E. Huerbin M.B. Melick J. Marion D.W. Palmer A.M. Schiding J.K. Kochanek P.M. Graham S.H. Regulation of interstitial excitatory amino acid concentrations after cortical contusion injury. Brain Res. 2002;935:40–46. doi: 10.1016/s0006-8993(02)02445-9. [DOI] [PubMed] [Google Scholar]
- Ruskin D.N. Kawamura M. Masino S.A. Reduced pain and inflammation in juvenile and adult rats fed a ketogenic diet. PLoS One. 2009;4:e8349. doi: 10.1371/journal.pone.0008349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satchell M.A. Zhang X. Kochanek P.M. Dixon C.E. Jenkins L.W. Melick J. Szabo C. Clark R.S. A dual role for poly-ADP-ribosylation in spatial memory acquisition after traumatic brain injury in mice involving NAD+ depletion and ribosylation of 14-3-3gamma. J. Neurochem. 2003;85:697–708. doi: 10.1046/j.1471-4159.2003.01707.x. [DOI] [PubMed] [Google Scholar]
- Scafidi S. O'Brien J. Hopkins I. Robertson C. Fiskum G. McKenna M. Delayed cerebral oxidative glucose metabolism after traumatic brain injury in young rats. J. Neurochem. 2009;109(Suppl. 1):189–197. doi: 10.1111/j.1471-4159.2009.05896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmann M.U. Stiller D. Skardelly M. Bernarding J. Klinge P.M. Samii A. Samii M. Brinker T. Metabolic changes in the vicinity of brain contusions: a proton magnetic resonance spectroscopy and histology study. J. Neurotrauma. 2003;20:725–743. doi: 10.1089/089771503767869962. [DOI] [PubMed] [Google Scholar]
- Sestili P. Martinelli C. Bravi G. Piccoli G. Curci R. Battistelli M. Falcieri E. Agostini D. Gioacchini A.M. Stocchi V. Creatine supplementation affords cytoprotection in oxidatively injured cultured mammalian cells via direct antioxidant activity. Free Radic. Biol. Med. 2006;40:837–849. doi: 10.1016/j.freeradbiomed.2005.10.035. [DOI] [PubMed] [Google Scholar]
- Sharpless N.S. Brown L.L. Use of microwave irradiation to prevent postmortem catecholamine metabolism: evidence for tissue disruption artifact in a discrete region of rat brain. Brain Res. 1978;140:171–176. doi: 10.1016/0006-8993(78)90248-2. [DOI] [PubMed] [Google Scholar]
- Sheline C.T. Behrens M.M. Choi D.W. Zinc-induced cortical neuronal death: contribution of energy failure attributable to loss of NAD(+) and inhibition of glycolysis. J. Neurosci. 2000;20:3139–3146. doi: 10.1523/JNEUROSCI.20-09-03139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signoretti S. Di Pietro V. Vagnozzi R. Lazzarino G. Amorini A.M. Belli A. D'Urso S. Tavazzi B. Transient alterations of creatine, creatine phosphate, N-acetylaspartate and high–energy phosphates after mild traumatic brain injury in the rat. Mol. Cell Biochem. 2010;333:269–277. doi: 10.1007/s11010-009-0228-9. [DOI] [PubMed] [Google Scholar]
- Signoretti S. Marmarou A. Tavazzi B. Lazzarino G. Beaumont A. Vagnozzi R. N-Acetylaspartate reduction as a measure of injury severity and mitochondrial dysfunction following diffuse traumatic brain injury. J. Neurotrauma. 2001;18:977–991. doi: 10.1089/08977150152693683. [DOI] [PubMed] [Google Scholar]
- Suh S.W. Chen J.W. Motamedi M. Bell B. Listiak K. Pons N.F. Danscher G. Frederickson C.J. Evidence that synaptically-released zinc contributes to neuronal injury after traumatic brain injury. Brain Res. 2000;852:268–273. doi: 10.1016/s0006-8993(99)02095-8. [DOI] [PubMed] [Google Scholar]
- Sunami K. Nakamura T. Ozawa Y. Kubota M. Namba H. Yamaura A. Hypermetabolic state following experimental head injury. Neurosurg. Rev. 1989;12(Suppl 1):400–411. doi: 10.1007/BF01790682. [DOI] [PubMed] [Google Scholar]
- Sutton R.L. Hovda D.A. Adelson P.D. Benzel E.C. Becker D.P. Metabolic changes following cortical contusion: relationships to edema and morphological changes. Acta Neurochir. Suppl. (Wien) 1994;60:446–448. doi: 10.1007/978-3-7091-9334-1_122. [DOI] [PubMed] [Google Scholar]
- Sutton R.L. Lescaudron L. Stein D.G. Unilateral cortical contusion injury in the rat: vascular disruption and temporal development of cortical necrosis. J. Neurotrauma. 1993;10:135–149. doi: 10.1089/neu.1993.10.135. [DOI] [PubMed] [Google Scholar]
- Swaab D.F. Pitfalls in the use of rapid freezing for stopping brain and spinal cord metabolism in rat and mouse. J. Neurochem. 1971;18:2085–2092. doi: 10.1111/j.1471-4159.1971.tb05067.x. [DOI] [PubMed] [Google Scholar]
- Thomale U.W. Griebenow M. Mautes A. Beyer T.F. Dohse N.K. Stroop R. Sakowitz O.W. Unterberg A.W. Stover J.F. Heterogeneous regional and temporal energetic impairment following controlled cortical impact injury in rats. Neurol. Res. 2007;29:594–603. doi: 10.1179/016164107X166272. [DOI] [PubMed] [Google Scholar]
- Thomas S. Prins M.L. Samii M. Hovda D.A. Cerebral metabolic response to traumatic brain injury sustained early in development: a 2-deoxy-D-glucose autoradiographic study. J. Neurotrauma. 2000;17:649–665. doi: 10.1089/089771500415409. [DOI] [PubMed] [Google Scholar]
- Trehout D. Desille M. Doan B.T. Mahler S. Fremond B. Malledant Y. Campion J.P. Desbois J. Beloeil J.C. de Certaines J. Clement B. Follow-up by one- and two-dimensional NMR of plasma from pigs with ischemia-induced acute liver failure treated with a bioartificial liver. NMR Biomed. 2002;15:393–403. doi: 10.1002/nbm.794. [DOI] [PubMed] [Google Scholar]
- Vagnozzi R. Signoretti S. Cristofori L. Alessandrini F. Floris R. Isgro E. Ria A. Marziale S. Zoccatelli G. Tavazzi B. Del Bolgia F. Sorge R. Broglio S.P. McIntosh T.K. Lazzarino G. Assessment of metabolic brain damage and recovery following mild traumatic brain injury: a multicentre, proton magnetic resonance spectroscopic study in concussed patients. Brain. 2010;133:3232–3242. doi: 10.1093/brain/awq200. [DOI] [PubMed] [Google Scholar]
- Vink R. Golding E.M. Headrick J.P. Bioenergetic analysis of oxidative metabolism following traumatic brain injury in rats. J. Neurotrauma. 1994;11:265–274. doi: 10.1089/neu.1994.11.265. [DOI] [PubMed] [Google Scholar]
- Xiong Y. Gu Q. Peterson P.L. Muizelaar J.P. Lee C.P. Mitochondrial dysfunction and calcium perturbation induced by traumatic brain injury. J. Neurotrauma. 1997;14:23–34. doi: 10.1089/neu.1997.14.23. [DOI] [PubMed] [Google Scholar]
- Ying W. Garnier P. Swanson R.A. NAD+ repletion prevents PARP-1-induced glycolytic blockade and cell death in cultured mouse astrocytes. Biochem. Biophys. Res. Commun. 2003;308:809–813. doi: 10.1016/s0006-291x(03)01483-9. [DOI] [PubMed] [Google Scholar]
- Yoshino A. Hovda D.A. Kawamata T. Katayama Y. Becker D.P. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: evidence of a hyper- and subsequent hypometabolic state. Brain Res. 1991;561:106–119. doi: 10.1016/0006-8993(91)90755-k. [DOI] [PubMed] [Google Scholar]