

Abstract
Background
The transition metal molybdenum is essential for life. Escherichia coli imports this metal into the cell in the form of molybdate ions, which are taken up via an ABC transport system. In E. coli and other Proteobacteria molybdenum metabolism and homeostasis are regulated by the molybdate-responsive transcription factor ModE.
Results
Orthologues of ModE are widespread amongst diverse prokaryotes, but not ubiquitous. We identified probable ModE-binding sites upstream of genes implicated in molybdenum metabolism in green sulphur bacteria and methanogenic Archaea as well as in Proteobacteria. We also present evidence of horizontal transfer of nitrogen fixation genes between green sulphur bacteria and methanogenic Archaea.
Conclusions
Whereas most of the archaeal helix-turn-helix-containing transcription factors belong to families that are Archaea-specific, ModE is unusual in that it is found in both Archaea and Bacteria. Moreover, its cognate upstream DNA recognition sequence is also conserved between Archaea and Bacteria, despite the fundamental differences in their core transcription machinery. ModE is the third example of a transcriptional regulator with a binding signal that is conserved in Bacteria and Archaea.
Background
The transition metal molybdenum is essential for life on earth. It is at the catalytic centre of over 30 enzymes, which are involved in the nitrogen, carbon, and sulphur cycles [1]. Molybdenum is found in the nitrogenase complex, which fixes dinitrogen gas, and in nitrate reductase, which reduces nitrate to nitrite. Other molybdo-proteins include xanthine oxidase, aldehyde oxidase, formate dehydrogenase, sulphite oxidase, nitrite reductase, DMSO reductase, pyridoxal dehydrogenase, xanthine dehydrogenase, and pyrogallol transhydrolase.
Molybdenum is available to organisms in the form of the tetraoxyanion molybdate, which is transported into Escherichia coli cells by an ABC-type transport system encoded by the modABCD operon [2]. A ModE-molybdate complex binds specific DNA target sequences and thus represses or activates transcription of several operons in response to molybdate concentration. The ModE protein from E. coli consists of two domains [3]. At the N terminus is the DNA-binding domain (Pfam accession PF02573, HTH_9) containing a winged helix-turn-helix (HTH) motif. At the C-terminal end is the molybdate-binding domain (Pfam accession PF03459, TOBE), which consists of two sub-domains of OB-folds with exchanging C-terminal strands. In E. coli, ModE bound to molybdate has been shown to contribute to the regulation of modABCD [4], napFDAGHBC [5], moaABCDE [6], and dmsABC operons [7]. These operons encode proteins involved in molybdenum homeostasis and metabolism.
Homologues of ModE are known in γ-Proteobacteria (e.g. E. coli, Haemophilus influenzae) and α-Proteobacteria (e.g. Rhodobacter capsulatus). Now that over 100 complete genome sequences are available, we wished to discover whether this regulatory system is found more widely in organisms other than Proteobacteria and whether we could gain any insights into the evolution of the molybdenum regulon.
Results and Discussion
Taxonomic distribution of ModE homologues and protein domain organisation of their sequences
Probable orthologues of the molybdenum-responsive regulator ModE were found in several Proteobacteria as well as in the green sulphur bacterium Chlorobium tepidum. Sequence analysis using Pfam [9] revealed that these proteins contained the HTH_9 domain at the N terminus and two TOBE sub-domains at the C terminus (Figure 2A). In most cases, only a single ModE homologue was found in each complete genome sequence. However, in the α-proteobacterium Rhodobacter capsulatus, there are two ModE homologues. One is associated with molybdate transport genes in the operon modEABC, whilst the second homologue is encoded by modE2, located upstream of vnfA2, the gene encoding one of two homologues of the nitrogenase regulatory protein, VnfA. The methanogenic Archaea Methanosarcina mazei and M. acetivorans each encode a protein that resembles ModE, but has only one rather than two C-terminal TOBE sub-domains (Figure 2B). No orthologues of ModE were found in the complete genome sequences from other lineages, such as the Gram-positive Bacteria and Cyanobacteria.
Figure 2.
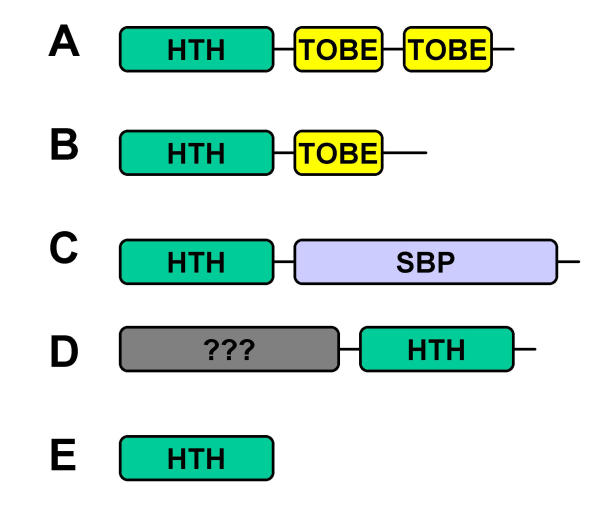
Domain architectures of some example members of the HTH_9 family as defined by Pfam (Bateman et al., 2002). The domains are described in the text. A. Q9I631 (Pseudomonas aeruginosa), Q88AA0 (Ps. syringae), Q88QX4 (Ps. putida), Q9CMR6 (Pasteurella multocida), MODE_HAEIN (Haemophilus influenzae), Q8Z8A6 (Salmonella Typhi), Q8ZQR8 (Salmonella Typhimurium), MODE_ECOLI (E. coli), MODE_YERPE (Yersinia pestis), Q8EAN6 (Shewanella oneidensis), Q8XQR8 (Ralstonia solanacearum), Q8KC82 (Chlorobium tepidum), MODE_AZOVI (Azotobacter vinelandii), Q9ABA4 (Caulobater crescentus), MOPA_RHOCA (Rhodobacter capsulatus), MOPA_RHOCA (Rhodobacter capsulatus), Q9F4K4 (Herbaspirillum seropedicae). B. Q8PWN4 (Methanosarcina mazei), and Q8TTZ2 (M. acetivorans). C. Q9RBF7 (Alcaligenes eutrophus), Q8XXM1 (Ralstonia solanacearum), and Q8ZZY3 (Pyrobaculum aerophilum). D. Q9PMF6 (Campylobacter jejuni). E. Q97Z66 (Sulfolobus solfataricus), Q97ET9 (Sulfolobus tokodaii), Q8ZYE6 (P. aerophilum), O29240 (Archaeglobus fulgidus), Q8TVF9 (Methanopyrus kandleri), and Q98KI4 (Rhizobium loti).
Several other Bacteria and Archaea encode proteins that contain the HTH_9 domain characteristic of ModE, but completely lack a molybdate-binding TOBE domain. Examples include Q9RBF7, Q8XXM1 (RSC2092), and Q8ZZY3 (PAE0019) from Alcaligenes eutropha, Ralstonia solanacearum, and Pyrobaculum aerophilum respectively. In these proteins, rather than a TOBE domain, there is a domain at the C terminus that is homologous to the SBP_bac_1 family (Pfam accession PF01547). Members of this family [8] bind diverse solutes such as sugars, peptides, and inorganic ions. In Alcaligenes eutropha, Q9RBF7 (FdsR) has been shown to regulate the fdsGBACD operon, which encodes the soluble NAD+ linked formate dehydrogenase molybdo-enzyme [10]. Based on similarity between the C-terminal domain of FdsR and a domain of formate dehydrogenase, Oh and Bowien [10] proposed that this C-terminal domain binds formate, thus modulating DNA-binding activity.
In Bacteria and Archaea, transcriptional regulators containing a HTH exhibit a position-function correlation such that repressors and dual function repressors/activators tend to have the HTH-containing domain at the N terminus whilst activators tend to have the HTH at the C terminus [11,12]. ModE can act as both a repressor and an activator. Consistent with this, ModE and the other proteins described above, the HTH_9 domain occurs at the N terminus. In contrast to this arrangement, Q9PMF6 (Cj1507c) from Campylobacter jejuni has an HTH_9 domain at the N terminus (Figure 2D). This suggests that it might be a transcriptional activator rather than a repressor. Intriguingly the Cj1507c ORF overlaps fdhD encoding the molybdo-enzyme formate dehydrogenase (Cj1508c), strongly suggesting a functional link between this regulatory protein and molybdenum metabolism.
Interestingly several Bacteria and Archaea encode proteins that consist of just the HTH_9 domain, and lack any recognisable molybdate- or other solute-binding domains (Figure 2E). Examples are found in several Archaea and also in Salmonella typhimurium and Agrobacterium tumefaciens. These proteins would certainly not be functional for binding molybdate, but the HTH domain is probably capable of binding DNA and perhaps forming multimeric protein complexes.
Conservation of DNA-recognition elements in ModE homologues
The HTH_9 domain of ModE is responsible for its sequence-specific DNA-binding activity. According to the model proposed by Hall et al. [3] based on the three-dimensional crystal structure of ModE, nine amino acid residues in the HTH directly interact with the target DNA and have a role in sequence-recognition: Ser35, Gln36, Lys39, Ser44, Tyr45, Lys46, Ser47, Trp49, and Asp50 (numbering as in [3]). Interestingly, most of these residues were moderately conserved, even between phylogenetically distant organisms (see alignment in Figure 1). This suggested that the cognate target sequences recognised by these proteins might also be conserved. Therefore we decided to investigate the occurrence of potential ModE-binding sites in a range of prokaryotic genome sequences.
Figure 1.

Alignment of the helix-turn-helix (HTH) domains homologous to the molybdenum-responsive transcriptional regulator ModE. Amino acid residues predicted to directly interact with the target DNA are marked with asterisks (*). The alignment was taken from the Pfam database http://www.sanger.ac.uk/Software/Pfam/.
Identification of ModE-binding sites in genomic DNA sequence
The consensus ModE-recognition sequence has dyad symmetry and can be approximately represented as atCGcTATATAN6TATATAaCGat [5]. As a first step to fully characterising the set of target sequences for this family of transcription factors, we constructed an alignment (Figure 3) of several known and probable ModE-binding sites from γ-Proteobacteria, also illustrated as a sequence logo in Figure 4. Using a position-dependent weight matrix derived from this alignment (Figure 5), we scanned the protein-coding and non-coding regions of a range of complete genomes to find matches to the canonical ModE-binding DNA sequence motif. The results of these searches are summarised in Tables 1 [see Additional File 3], 2, and 3. It is important to remember that the mere presence of DNA sequence similarity to a consensus is not sufficient to demonstrate a biologically significant protein-binding site; in other words this approach of scanning DNA sequence against a weight matrix model will inevitably yield some false-positives. For example, several high-scoring matches were found within protein-coding regions in most of the genomes that we examined; these intragenic sites are unlikely to be functional regulatory sites. Furthermore, many of the intergenic matches fell upstream of genes not obviously involved in molybdenum metabolism and therefore probably not subject to regulation by ModE.
Figure 3.
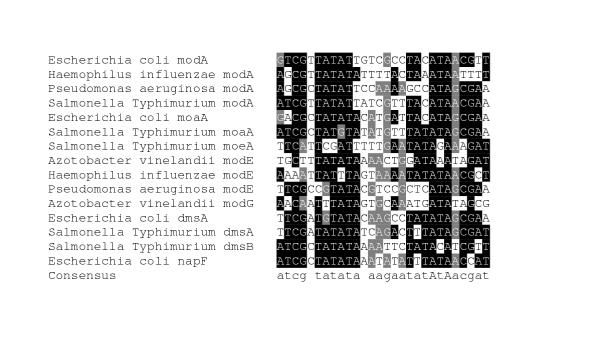
Alignment of known and strongly suspected ModE-binding sites used to generate the weight matrix.
Figure 4.

Sequence logo representation of the alignment of known and strongly suspected ModE-binding in Figure 3. A graphic representation of an aligned set of binding sites. The relative heights of the letters are proportional to the frequencies of bases at each position. The degree of sequence conservation is measured in bits of information and is indicated by the total height of a stack of letters. The vertical scale is in bits, with a maximum of 2 bits possible at each position. The logo was generated using WebLogo [27].
Figure 5.
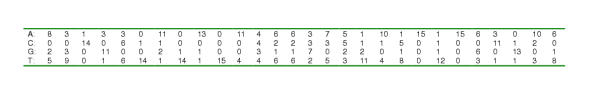
Position-specific weight matrix derived from the alignment of known and strongly suspected ModE-binding sites used to generate the weight matrix in Figure 1.
Table 2.
Potential ModE-binding sites identified by scanning archaeal genome sequences against the weight matrix derived from alignment in Figure 3. Sites that fall immediately upstream of genes implicated in molybdenum metabolism are indicated in bold. Scores are Kullback-Leibler distances that have been normalised such that the maximum possible score is 100. Essentially, the higher the score is, the greater the magnitude of the theoretical binding energy [26]. All sites scoring more than 75 are listed. Distances are given (number of bases) between the downstream end of the putative ModE-binding site and the predicted translational start codon.
| Organism | Sequence | Score | Distance | Annotation of potential target gene |
| Methanosarcina mazei | TGGCGTTATGTTTATTTAAACATAACGAT | 80 | -5 | MM1564 molybdenum containing formylmethanofuran dehydrogenase isoenzyme I subunit E |
| Methanosarcina mazei | TAATGTTATATATCTTAATAAATAACTTT | 78 | 524 | MM1328 Two component response regulator |
| Methanosarcina mazei | TAACGATATATTAATAATTAGATTTAGAT | 78 | 299 | MM1783 hypothetical protein |
| Methanosarcina mazei | TATCCATATACTAATGATTATATATCCAT | 78 | 69 | MM1790 conserved protein |
| Methanosarcina mazei | AATATTTATATAGTACACAATATATCAAT | 78 | 39 | MM2248 conserved protein |
| Methanosarcina mazei | CACCCATATATTAGTTTATAAATAACGCT | 78 | 948 | MM2931 hydrolase |
| Methanosarcina mazei | GAAGCGTATATATAAGAGTATATAAAGAG | 78 | 53 | MM3131 Fructokinase |
| Methanosarcina mazei | TTTCTCTCTATACTAGTCTACAGATAGAG | 78 | 191 | MM3331 conserved protein |
| Methanosarcina mazei | GAACAATATATCGTTAACAATATATCCAT | 77 | 48 | MM0378 sugar phosphate nucleotydyl transferase |
| Methanosarcina mazei | AATCTTTATATCATGGAATTTATCGTGAA | 77 | 290 | MM0957 Ammonium transporter |
| Methanosarcina mazei | CATCTTTTTATACTTCCCTATATACTTAA | 77 | 31 | tlpC MM1658 methyl accepting chemotaxis protein |
| Methanosarcina mazei | AAAACATATATAGAAACATACATATCGCA | 77 | 153 | MM2159 hypothetical protein |
| Methanosarcina mazei | TCTCGTTATATTTCTATTTATATATATTT | 77 | 344 | MM2777 Acylphosphatase |
| Methanosarcina mazei | TTTCGCTGTATATCTTATTTCATAAATAG | 76 | 1436 | MM0016 translation initiation factor 1A |
| Methanosarcina mazei | TGACAGTATATAAATTAATAGTTAGAGAT | 76 | 76 | MM0211 Cysteine proteinase |
| Methanosarcina mazei | AGTCATTATCTATATCAATATAAATAGTT | 76 | 527 | MM0338 putative phosphomethylpyrimidine kinase |
| Methanosarcina mazei | AAACATTAAATATTTTAATACATAGTTAT | 76 | 39 | MM0659 GDP mannose 4,6 dehydratase |
| Methanosarcina mazei | ATTAGGTTTATAAGTCAATAAATAATGAA | 76 | 43 | MM0996 cobalamin biosynthesis protein G |
| Methanosarcina mazei | TTTAGATTTATATAAGAATTTAAAACTAT | 76 | 276 | MM1157 conserved protein |
| Methanosarcina mazei | TATCGGGATATTCTATGGATTATATCGAA | 76 | 13 | MM1281 conserved protein |
| Methanosarcina mazei | AAACGTTATATACAAGCGAATATGAGTAT | 76 | 156 | MM1355 conserved protein |
| Methanosarcina mazei | CATCAATAAATTAATTTCTATATAAGGTA | 76 | 30 | MM1847 hypothetical protein |
| Methanosarcina mazei | GATTGATATATAATTATTTTCAAACTGAT | 76 | 25 | iorA MM2093 Indolepyruvate oxidoreductase, subunit |
| Methanosarcina mazei | AATTAATATATGATGGATTTTATATAGAT | 76 | 290 | MM2747 hypothetical protein |
| Methanosarcina mazei | TAAATATATATAAATAGAAATATAACGAG | 76 | 101 | MM2776 hypothetical protein |
| Methanosarcina mazei | AAGCGTTATTTATAAACTAATATATGGGT | 76 | 35 | MM2932 conserved protein |
| Methanosarcina mazei | AATCGCTCTATAGAAGTAAACTGAGCGGA | 76 | 377 | MM2945 Mannosyltransferase |
| Methanosarcina mazei | AAACTTTATATATTAAAATATAAATAAAA | 76 | 499 | MM3203 hypothetical protein |
| Methanosarcina acetivorans | TATGGTTATGTAATTCTAAACATAACGAA | 81 | 1504 | vnfH MA1213 nitrogenase (iron protein) |
| Methanosarcina acetivorans | AATCGTTATGTTTAGGTATACATAACTAC | 81 | 164 | modA MA2280 molybdenum ABC transporter, solute binding protein |
| Methanosarcina acetivorans | TAAAGTTATTTATTAGTATACATAACTAT | 80 | 764 | cheY2 MA0016 chemotaxis response regulator |
| Methanosarcina acetivorans | AAATGAAATATATATATATAGATAACGAT | 80 | 185 | MA0724 predicted protein |
| Methanosarcina acetivorans | TTTCAATATATATTTCAATATATAAAATA | 80 | 378 | vhtG MA1146 F420 nonreducing hydrogenase |
| Methanosarcina acetivorans | ATTCGTTATGTTTAGAATTACATAACCAT | 80 | 339 | nifI1 MA1212 P II family nitrogen regulatory protein |
| Methanosarcina acetivorans | AATCTGTATATAATTAACCAGATAGAGTT | 80 | 9 | MA2899 conserved hypothetical protein |
| Methanosarcina acetivorans | TGTCGTTATGTTTATTTAAACATAACGGT | 79 | -6 | fmdE MA0304 formylmethanofuran dehydrogenase, subunit E |
| Methanosarcina acetivorans | CACCGTTATGTTTAAATAAACATAACGAC | 79 | 1483 | MA0303 conserved hypothetical protein |
| Methanosarcina acetivorans | ATTAATTATATAAATGTGTATATAAATAT | 79 | 1375 | hypC MA1140 hydrogenase expression/formation protein |
| Methanosarcina acetivorans | GAACCTTATATATTTTTCTACAGAGAGCT | 79 | 114 | MA3892 hypothetical protein (multi domain) |
| Methanosarcina acetivorans | AGTGGCTATATTTAGCTATATATAACAAA | 79 | 710 | MA3957 ABC transporter, ATP binding protein |
| Methanosarcina acetivorans | ACTCGATATATTATTCAACGAATAGTGAT | 78 | 118 | MA1301 predicted protein |
| Methanosarcina acetivorans | ATCCGTTATGTATGAATGAACATAACGTT | 78 | 27 | MA1663 predicted protein |
| Methanosarcina acetivorans | TATCTTTATGTTTATCCGAACATATCGAT | 78 | 6 | MA4536 ABC transporter, solute binding protein |
| Methanosarcina acetivorans | ATTTACTTTATATCTGTATATATATTGAA | 77 | 529 | MA0519 conserved hypothetical protein |
| Methanosarcina acetivorans | TATCGTTATCTATATATATATATTTCATT | 77 | 114 | MA0725 conserved hypothetical protein |
| Methanosarcina acetivorans | CTATTTTATATATTGAAATATATATTGAA | 77 | 122 | MA1145 hypothetical protein (multi domain) |
| Methanosarcina acetivorans | GTTCCTTTTATATTGCAAATCATAACGTT | 77 | 43 | MA2861 response regulator receiver |
| Methanosarcina acetivorans | AAACGTTATATACAAGCGGATATGAGTAT | 76 | 147 | MA0056 conserved hypothetical protein |
| Methanosarcina acetivorans | AATTCTTTTATATAAATCCATATAACGGT | 76 | 130 | MA0459 conserved hypothetical protein |
| Methanosarcina acetivorans | TACCGTTATATGGATTTATATAAAAGAAT | 76 | 229 | MA0458 predicted protein |
| Methanosarcina acetivorans | CTAGGTTATATAACAGAAATCATAAAGAG | 76 | 17 | MA1630 sensory transduction histidine kinase |
| Methanosarcina acetivorans | TTACGATATATATAAATTTATCTAAAAAA | 76 | 87 | MA1757 conserved hypothetical protein |
| Methanosarcina acetivorans | ATTTTTTAGATAAATTTATATATATCGTA | 76 | 441 | MA1756 cell surface protein |
| Methanosarcina acetivorans | ATCCATTAGATACAAATATTTATATAGAA | 76 | 1467 | MA3192 conserved hypothetical protein |
| Methanosarcina acetivorans | TATATTTATATAAAAATCAACATATCTAT | 76 | 555 | cpa MA3604 carboxypeptidase A |
| Methanosarcina acetivorans | GTACAGTATATATTTTAAAATATAGTTAT | 76 | 507 | atpH MA4152 H(+) transporting ATP synthase, subunit H |
Table 3.
Frequencies of matches to the ModE-binding consensus in several complete genomes. The number of sites scoring greater than 75 for similarity to the ModE-binding consensus was counted for each of the complete genome sequences. Note that the consensus sequence is a nearly palindromic, so only sites on on one strand were counted. Matches were designated intragenic if the center of the 28 base site was located within a protein-coding region and intergenic otherwise.
| Organism | Number of intergenic matches (number implicated in molybdenum metabolism) | Number of intragenic matches | Genome Size (Mb) |
| Agrobacterium tumefaciens | 2 (1) | 0 | 2.84 |
| Bradyrhizobium japonicum | 1 (1) | 1 | 9.11 |
| Campylobacter jejuni | 1 (0) | 13 | 1.64 |
| Chlorobium tepidum | 2 (1) | 4 | 2.15 |
| Escherichia coli K12 | 17 (5) | 9 | 4.64 |
| Haemophilus ducreyi | 4 (0) | 10 | 1.70 |
| Haemophilus influenzae | 9 (3) | 13 | 1.83 |
| Helicobacter hepaticus | 6 (2) | 14 | 1.80 |
| Helicobacter pylori 26695 | 0 (0) | 10 | 1.67 |
| Helicobacter pylori J99 | 0 (0) | 6 | 1.64 |
| Mesorhizobium loti | 4 (2) | 2 | 7.04 |
| Methanosarcina acetivorans | 28 (4) | 3 | 5.75 |
| Methanosarcina mazei | 28 (2) | 6 | 4.10 |
| Pasteurella multocida | 13 (3) | 11 | 2.26 |
| Pseudomonas aeruginosa | 3 (1) | 0 | 6.26 |
| Pseudomonas putida | 3 (1) | 1 | 6.18 |
| Pseudomonas syringae pv tomato | 3 (1) | 0 | 6.4 |
| Ralstonia solanacearum | 1 (1) | 0 | 5.80 |
| Salmonella enterica Typhi | 16 (6) | 6 | 4.81 |
| Salmonella enterica Typhimurium | 15 (4) | 7 | 4.86 |
| Shewanella oneidensis | 10 (2) | 12 | 4.97 |
| Shigella flexneri | 20 (5) | 5 | 4.61 |
| Sinorhizobium meliloti | 2 (0) | 1 | 3.65 |
| Yersinia pestis strain CO92 | 19 (5) | 4 | 4.65 |
However, notwithstanding the occurrence of some false-positives, we are confident that our method had some discriminatory value in identifying bona fide ModE-binding sites because of the high proportion of the matches that did fall upstream of molybdenum-linked genes. Despite the importance of molybdenum-dependent processes, relatively few bacterial genes are directly involved in molybdenum metabolism: certainly no more than a few percent of the genome. Nevertheless, a disproportionately high number of our identified matches were located immediately upstream of genes implicated in molybdenum metabolism. For example in E. coli K12, we found a total of 26 high-scoring matches to the ModE-binding consensus (Table 3). Of these 26 sites, 17 occurred in non-coding DNA. Five of these 17 sites were located upstream of genes or operons associated with molybdenum metabolism or molybdo-enzymes (Table 1 [see Additional File 3]). In some cases, these ratios were more impressive: for example in Agrobacterium tumefaciens, Bradyrhizobium japonicum, Chlorobium tepidum, Mesorhizobium loti and Ralstonia solanacearum, at least 50% of the intergenic matches were located immediately upstream of molybdenum-associated genes (Table 3). The frequent occurrence of these ModE operator-like sequences upstream of molybdenum-associated genes is consistent with their being biologically significant rather than mere coincidence.
As expected, we found good matches to the proposed consensus ModE binding sequence immediately upstream of genes implicated in molybdenum metabolism in the γ-proteobacterial genomes. For example, high-scoring sites were detected upstream of homologues of modA, moeA, dmsA, and napF in E. coli and Salmonella species, which correspond to ModE-binding sites. Additional good candidate sites were found upstream of yecK in both E. coli and Haemophilus influenzae. The yecKbisZ operon in E. coli encodes a molybdo-enzyme that forms part of a respiratory system [13]. Therefore it is not surprising that the yecKbisZ operon might be regulated by ModE.
We observed some differences in the repertoires of predicted ModE-binding sites between the closely related strains of enteric bacteria (E. coli, Yersinia pestis, Shigella flexneri, and Salmonella enterica). There appears to be a core ModE-dependent regulon consisting of the dmsABC, moaABCDE, modABCD and napFDAGHBC operons, the only exception being that there is no apparent ModE binding site associated with dmsABC in Y. pestis. Additionally, yecK also has a putative upstream ModE binding site in E. coli and S. flexneri; yecK appears to be absent from S. enterica and Y. pestis. Furthermore, in S. enterica a second dms operon also has sites, whilst Y. pestis has additional sites upstream of pflA and narP. These differences in the predicted ModE-dependent regulon between closely related species suggest some evolutionary plasticity in the regulon. We also observed clear differences between the close relatives Haemophilus influenzae and H. ducreyi, the latter lacking a ModE homologue and lacking conserved ModE operator-like sites upstream of its mod operon.
Our data show that probable ModE-binding sites were found upstream of modA homologues not only in enteric bacteria and H. influenzae as has been previously established, but also in A. tumefaciens, Helicobacter hepaticus, M. loti, Pasteurella multocida, several Pseudomonas strains, R. solanacearum and Shewanellla oneidensis. This indicates that ModE-dependent regulation of the molybdate ABC transporter is widespread amongst proteobacteria.
Interestingly, we found strong potential ModE-binding sites upstream of modA (involved in molybdate transport) and napA (encoding a molybdenum-containing nitrate reductase) in the ε-proteobacterium Helicobacter hepaticus (but not in its close relatives H. pylori and Campylobacter jejuni). The complete genome sequence of H. hepaticus does not encode a full-length ModE homologue; it does encode a 135 amino acid protein (HH0653), consisting of a single HTH_9 domain, which is therefore probably able to specifically bind to the identified sites. However, since it lacks a molybdate-binding TOBE domain, it cannot directly use molybdenum availability to modulate DNA-binding activity. Interestingly, a potential ModE-binding site is also observed upstream of HH0158, predicted to encode a molydenum-containing periplasmic nitrate reductase in H. hepaticus.
A similar scenario is observed in Agrobacterium tumefaciens. A ModE-binding site is found upstream of modA but A. tumefaciens has no full-length ModE homologue, only a 131 amino acid protein (ATU2654, Q8UCD4) consisting of a HTH_9 domain. It is possible that the ModE-dependent regulatory system is in a process of degeneration in these organisms. However, it is equally possible that these proteins are functional and that DNA-binding is modulated by protein-protein interaction with some unknown factor or factors.
Strikingly, we also found a good candidate ModE-binding sites associated with molybdenum metabolism in some more distantly related organisms. One striking example was a site upstream of an open reading frame (CT1544) annotated as a probable periplasmic molybdenum-binding protein component of a molybdenum ABC transporter complex in the green sulphur bacterium C. tepidum [14]. This suggested that the full-length orthologue of ModE (CT1543, Q8KC82) functions in regulation of molybdenum transport in C. tepidum in a similar manner to ModE in E. coli.
The genome of C. tepidum [14] has at least three loci that contain genes implicated in molybdenum homeostasis and metabolism. The first of these loci includes a mod transport system and genes involved in synthesis of the molybdopterin genes cofactor, moaCBmoeEmobBAmoaA. A second includes CT1765, encoding a homologue of the ModG molybindin of Azotobacter vinelandii that binds molybdate. CT1765 has been misleadingly annotated as a molybdopterin-binding protein, as have most other molybdate-binding proteins in the public databases. It is probably involved in molybdate homeostasis. The third locus includes moaCB, moeE, mobBA, moaA and the nif genes that encode nitrogenase. Structural genes for the nitrogenase system in C. tepidum most closely resemble those of the Archaea rather than those of other nitrogen-fixing bacteria [14]. This raises the question of horizontal transfer between Bacteria and Archaea. However, a strong candidate σ54-dependent promoter is located upstream of C. tepidum nifH [15], which encodes the nitrogenase. σ54-dependent transcription of nitrogenase genes is common among Proteobacteria, but σ54 is not found in Archaea. Taken together, the σ54-dependent promoter and the ModE binding site centred just upstream of CT1544 suggest that the regulation of nitrogenase in C. tepidum shares much in common with that in Proteobacteria such as A. vinelandii. Does this tell us anything about the direction of horizontal transfer of nitrogen-fixation genes between Archaea and green sulphur bacteria? If the green sulphur bacteria acquired nitrogen-fixing genes from an archaeon, then this would imply that the recruitment of σ54 to the regulation of nitrogen fixation has occurred at least twice independently. The alternative, and more likely, scenario is that the Archaea acquired nitrogen-fixation genes from a relative of the green sulphur bacteria.
Even more striking was our finding that good candidate ModE-binding sites occur immediately upstream of several genes implicated in molybdenum metabolism in the methanogenic Archaea (Table 2). For example, we found high-scoring matches to ModE-binding sites in Methanosarcina acetivorans upstream of modA and fmdE, which encodes a molybdenum-containing hydrogenase. We also found sites upstream of vnfH and nifI1, implicated in nitrogen-fixation. MA1213 (vnfH) is annotated as an iron-containing nitrogenase, VnfH [16]. However, examination of this sequence suggests that it is more likely to be a molybdenum-containing nitrogenase [17]. The predicted ModE-binding site upstream of fmdE is also conserved in Methanosarcina mazei. Therefore it is possible that the ModE-like proteins Q8PWN4 and Q8TTZ2 from Methanosarcina mazei and Methanosarcina acetivorans respectively are functional for molybdate-responsive regulation of these molybdo-enzymes.
Conclusions
Transcriptional regulation of molybdo-enzymes, and genes involved in molybdenum metabolism and homeostasis, is performed by the molybdate-responsive transcription factor ModE in E. coli and related Proteobacteria. We found that homologues of ModE are also found in the green sulphur bacterium C. tepidum, and in methanogenic Archaea. Moreover, we found that its cognate DNA recognition element is also highly conserved even between Bacteria and Archaea. As far as we are aware this is only the third report of a regulatory DNA element whose sequence appears to be conserved in bacteria and Archaea. The other two examples are the regulator of biotin metabolism, BirA [18], and the metal dependent regulator MDR1/DtxR [24].
Although the basal transcription apparatus in Archaea is similar to eukaryotic RNAPII rather than to bacterial RNAP, the emerging view is that Archaea use a varied repertoire of regulatory mechanisms that includes both eukaryal and bacterial paradigms (e.g. [19-22]). Archaea contain large numbers of HTH-containing domains, which are more similar to bacterial HTH domains rather than to eukaryotic proteins [23]. However, most of the Archaeal HTH-containing proteins form Archaea-specific families [23] and so the archaeal and bacterial HTH domains may share very ancient common origins [11]. The high-affinity molybdate transport system, which has a small size and simple organisation, may have a very ancient origin. The fact that the ModE regulon is conserved across large phylogenetic distances also suggests an ancient common origin followed by loss in multiple lineages rather than multiple transfer events. This may be further supported by the observation of partially degenerated ModE regulatory systems in bacteria such as A. tumefaciens and H. hepaticus.
Methods
Domain architectures were inferred from protein sequences using Pfam [9] release 10.0.
Matches to ModE-binding sites were identified in non-coding regions of complete genome sequences using a standard position-specific weight matrix scoring approach. A frequency matrix was generated from the alignment of known binding site sequences using the make_matrix.pl script [see Additional File 2]. This matrix contained the relative frequencies of each base being found at each position in the alignment (see Figure 5). The matrix can be considered to be a statistical model of the DNA sequence motif. Another way of visualising the DNA sequence motif is as a sequence logo [25] as in Figure 4.
Matches to this DNA motif were detected in a genome sequence by the following method. The entire genome sequence was scanned such that every window of 28 bases was assigned a score using the matrix and the promscan.pl script [see Additional File 1]. This score, known as the Kullback-Leibler distance, reflects the theoretical binding energy of the DNA protein interaction [26] and is calculated using the formula in Figure 6. The maximum possible score for a window, i.e. that given by a perfect match to the consensus, differs slightly from genome to genome according to percentage G+C content. Therefore, all scores were normalised such that 100 is the highest possible score for a 28 base window, and the scores were rounded to the nearest integer. Incidentally, we would expect a site scoring 100 only once in about 7.2 × 1016 bases of random DNA sequence (assuming 50 G+C content), so it is perhaps not surprising that no sites were found to have a score of 100.
Figure 6.
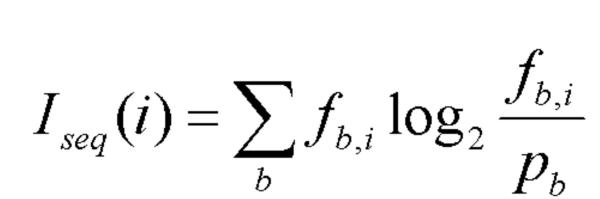
Formula used to calculate Iseq, the Kullback-Leibler distance [26], where i is the position within the site, pb is the frequency of that base in the genome, and fb,i is the observed frequency of each base at that position (from the weight matrix). Values for pb were calculated from the percentage G+C content of the genome sequence.
We chose 75 as a cut-off threshold. That is, we only considered sequences that scored greater than 75. This threshold value was chosen as all of the known (and strongly suspected) ModE binding sites scored greater than 75.
The Perl scripts used for the analysis are available as Additional Files 1 and 2.
Authors' contributions
DJS carried out the computational analyses. Both authors contributed equally to the conception and design of the study and writing the manuscript.
Supplementary Material
In Adobe Acrobat Reader format (Portable Document Format). Potential ModE-binding sites identified by scanning bacterial genome sequences against the weight matrix derived from alignment in Figure 2. Sites that fall immediately upstream of genes implicated in molybdenum metabolism are indicated in bold. Scores are Kullback-Leibler distances that have been normalised such that the maximum possible score is 100. Essentially, the higher the score is, the greater the magnitude of the theoretical binding energy [26]. All sites scoring more than 75 are listed. Distances are given (number of bases) between the downstream end of the putative ModE-binding site and the predicted translational start codon.
This is the Perl script, in plain text format, used to generate a scoring matrix from a Clustal alignment.
This is the Perl script, in plain text format, used to scan genome sequences against the ModE binding site matrix.
Acknowledgments
Acknowledgements
We are grateful to John A. Leigh for his helpful comments. Work in RNP's laboratory is partially supported by Australian Research Council Discovery grant DP0344143. DJS is supported by the MRC.
Contributor Information
David J Studholme, Email: ds2@sanger.ac.uk.
Richard N Pau, Email: r.pau@unimelb.edu.au.
References
- Stiefel EI. Molybdenum enzymes, cofactors and chemistry. In: Stiefel EI, Coucouvanis D, Newton WE, editor. In Molybdenum enzymes, cofactors and model systems. Washington DC:American Chemical Society; pp. 1–18. [Google Scholar]
- Self WT, Grunden AM, Hasona A, Shanmugam KT. Molybdate transport. Res Microbiol. 2001;152:311–321. doi: 10.1016/S0923-2508(01)01202-5. [DOI] [PubMed] [Google Scholar]
- Hall DR, Gourley DG, Leonard GA, Duke EM, Anderson LA, Boxer DH, Hunter WN. The high-resolution crystal structure of the molybdate-dependent transcriptional regulator (ModE) from Escherichia coli: a novel combination of domain folds. EMBO J. 1999;18:1435–1346. doi: 10.1093/emboj/18.6.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunden AM, Ray RM, Rosentel JK, Healy FG, Shanmugam KT. Repression of the Escherichia coli modABCD (molybdate transport) operon by ModE. J Bacteriol. 1996;178:735–744. doi: 10.1128/jb.178.3.735-744.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicholas PM, Gunsalus RP. The molybdate-responsive Escherichia coli ModE transcriptional regulator coordinates periplasmic nitrate reductase (napFDAGHBC) operon expression with nitrate and molybdate availability. J Bacteriol. 2002;184:3253–3259. doi: 10.1128/JB.184.12.3253-3259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicholas PM, Rech SA, Gunsalus RP. Characterization of the ModE DNA-binding sites in the control regions of modABCD and moaABCDE of Escherichia coli. Mol Microbiol. 1997;23:515–524. doi: 10.1046/j.1365-2958.1997.d01-1864.x. [DOI] [PubMed] [Google Scholar]
- McNicholas PM, Chiang RC, Gunsalus RP. Anaerobic regulation of the Escherichia coli dmsABC operon requires the molybdate-responsive regulator ModE. Mol Microbiol. 1998;27:197–208. doi: 10.1046/j.1365-2958.1998.00675.x. [DOI] [PubMed] [Google Scholar]
- Tam R, Saier MH., Jr Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol Rev. 1993;57:320–346. doi: 10.1128/mr.57.2.320-346.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JI, Bowien B. Dual control by regulatory gene fdsR of the fds operon encoding the NAD+-linked formate dehydrogenase of Ralstonia eutropha. Mol Microbiol. 1999;34:365–376. doi: 10.1046/j.1365-2958.1999.01613.x. [DOI] [PubMed] [Google Scholar]
- Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Rueda E, Collado-Vides J. Common history at the origin of the position-function correlation in transcriptional regulators in archaea and bacteria. J Mol Evol. 2001;53:172–179. doi: 10.1007/s002390010207. [DOI] [PubMed] [Google Scholar]
- Perez-Rueda E, Gralla JD, Collado-Vides J. Genomic position analyses and the transcription machinery. J Mol Biol. 1998;275:165–170. doi: 10.1006/jmbi.1997.1465. [DOI] [PubMed] [Google Scholar]
- S Gon, JC Patte, V Mejean, C Iobbi-Nivol. The torYZ (yecK bisZ) operon encodes a third respiratory trimethylamine N-oxide reductase in Escherichia coli. J Bacteriol. 2000;182:5779–5786. doi: 10.1128/JB.182.20.5779-5786.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JA, Nelson KE, Paulsen IT, Heidelberg JF, Wu M, Dodson RJ, Deboy R, Gwinn ML, Nelson WC, Haft DH, Hickey EK, Peterson JD, Durkin AS, Kolonay JL, Yang F, Holt I, Umayam LA, Mason T, Brenner M, Shea TP, Parksey D, Nierman WC, Feldblyum TV, Hansen CL, Craven MB, Radune D, Vamathevan J, Khouri H, White O, Gruber TM, Ketchum KA, Venter JC, Tettelin H, Bryant DA, Fraser CM. The complete genome sequence of Chlorobium tepidum TLS, a photosynthetic, anaerobic, green-sulfur bacterium. Proc Natl Acad Sci USA. 2002;99:9509–9514. doi: 10.1073/pnas.132181499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studholme DJ, Dixon R. Domain architectures of σ54-dependent transcriptional activators. J Bacteriol. 2003;185:1757–1767. doi: 10.1128/JB.185.6.1757-1767.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan JE, Nusbaum C, Roy A, Endrizzi MG, Macdonald P, FitzHugh W, Calvo S, Engels R, Smirnov S, Atnoor D, Brown A, Allen N, Naylor J, Stange-Thomann N, DeArellano K, Johnson R, Linton L, McEwan P, McKernan K, Talamas J, Tirrell A, Ye W, Zimmer A, Barber RD, Cann I, Graham DE, Grahame DA, Guss AM, Hedderich R, Ingram-Smith C, Kuettner HC, Krzycki JA, Leigh JA, Li W, Liu J, Mukhopadhyay B, Reeve JN, Smith K, Springer TA, Umayam LA, White O, White RH, Conway de Macario E, Ferry JG, Jarrell KF, Jing H, Macario AJ, Paulsen I, Pritchett M, Sowers KR, Swanson RV, Zinder SH, Lander E, Metcalf WW, Birren B. The genome of M. acetivorans reveals extensive metabolic and physiological diversity. Genome Res. 2002;12:532–542. doi: 10.1101/gr.223902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler PS, McLarnan J, Leigh JA. Nitrogenase phylogeny and the molybdenum dependence of nitrogen fixation in Methanococcus maripaludis. J Bacteriol. 1997;179:541–543. doi: 10.1128/jb.179.2.541-543.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov DA, Mironov AA, Gelfand MS. Conservation of the biotin regulon and the BirA regulatory signal in Eubacteria and Archaea. Genome Res. 2002;12:1507–1516. doi: 10.1101/gr.314502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrpides NC, Ouzounis CA. Transcription in archaea. Proc Natl Acad Sci USA. 1999;96:8545–8550. doi: 10.1073/pnas.96.15.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SD, Jackson SP. Mechanism and regulation of transcription in Archaea. Curr Opin Microbiol. 2001;4:208–213. doi: 10.1016/S1369-5274(00)00190-9. [DOI] [PubMed] [Google Scholar]
- Soppa J. Transcription initiation in Archaea: facts, factors and future aspects. Mol Microbiol. 1999;31:1295–1305. doi: 10.1046/j.1365-2958.1999.01273.x. [DOI] [PubMed] [Google Scholar]
- Leigh JA. Transcriptional regulation in Archaea. Curr Opin Microbiol. 1999;2:131–134. doi: 10.1016/S1369-5274(99)80023-X. [DOI] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. DNA-binding proteins and evolution of transcription regulation in the archaea. Nucleic Acids Res. 1999;27:4658–4670. doi: 10.1093/nar/27.23.4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SD, Cairns SS, Robson RL, Jackson SP. Transcriptional regulation of an archaeal operon in vivo and in vitro. Mol Cell. 1999;4:971–982. doi: 10.1016/s1097-2765(00)80226-9. [DOI] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stormo GD. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- WebLogo http://weblogo.berkeley.edu/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In Adobe Acrobat Reader format (Portable Document Format). Potential ModE-binding sites identified by scanning bacterial genome sequences against the weight matrix derived from alignment in Figure 2. Sites that fall immediately upstream of genes implicated in molybdenum metabolism are indicated in bold. Scores are Kullback-Leibler distances that have been normalised such that the maximum possible score is 100. Essentially, the higher the score is, the greater the magnitude of the theoretical binding energy [26]. All sites scoring more than 75 are listed. Distances are given (number of bases) between the downstream end of the putative ModE-binding site and the predicted translational start codon.
This is the Perl script, in plain text format, used to generate a scoring matrix from a Clustal alignment.
This is the Perl script, in plain text format, used to scan genome sequences against the ModE binding site matrix.