

Abstract
Background
Viral replication as well as an immunopathological component are assumed to be involved in the development of coxsackie B virus (CBV)-induced myocarditis. We observed that mycophenolic acid (MPA), the active metabolite of the immunosuppressive agent mycophenolate mofetil (MMF), inhibits coxsackie B3 virus (CBV3) replication in primary Human myocardial fibroblasts. We therefore studied whether MMF, which is thus endowed with a direct antiviral as well as immunosuppressive effect, may prevent CBV-induced myocarditis in a murine model.
Results
Four week old C3H-mice were infected with CBV3 and received twice daily, for 7 consecutive days (from one day before to 5 days post-virus inoculation) treatment with MMF via oral gavage. Treatment with MMF resulted in a significant reduction in the development of CBV-induced myocarditis as assessed by morphometric analysis, i.e. 78% reduction when MMF was administered at 300 mg/kg/day (p < 0.001), 65% reduction at 200 mg/kg/day (p < 0.001), and 52% reduction at 100 mg/kg/day (p = 0.001). The beneficial effect could not be ascribed to inhibition of viral replication since titers of infectious virus and viral RNA in heart tissue were increased in MMF-treated animals as compared to untreated animals.
Conclusion
The immunosuppressive agent MMF results in an important reduction of CBV3-induced myocarditis in a murine model.
Keywords: enterovirus, myocarditis, antiviral, coxsackie
Background
Viral myocarditis is a common pathological condition detected in approximately 1% of unselected asymptomatic individuals [1]. Many viruses have been shown to be involved as causative agents, but the principal agents belong to enteroviruses in general and coxsackieviruses in particular [2,3]. The proportion of cases of myocarditis with coxsackieviral ethiology varies but can in 25–50% of the cases be attributed to coxsackie B group viruses (CBV) [4,5]. Although most enterovirus-related cardiac illnesses are subclinical, severe arrythmias and sudden cardiac death may appear. About 10 to 20% of symptomatic patients will eventually develop chronic cardiac disease, which may progress to dilated cardiomyopathy, a severe pathological condition, often requiring heart transplantation [6-8]. Overall, the enteroviral genome is present in the hearts of 15–20% of patients with dilated cardiomyopathy [4,5,9,10]. Both direct viral injury and the immune response of the host are believed to play a role in the pathogenesis of viral heart disease [11]. Recent studies in mouse models show that direct viral-induced damage to the heart is necessary to cause CBV3-induced myocarditis [12,13] and that a scenario of 'molecular mimicry' is believed to be unlikely [14]. There is a prepondorance of evidence that cytotoxic T lymphocytes are involved in CBV3-induced myocarditis [15,16]. A therapeutic strategy against coxsackievirus myocarditis may therefore ideally be a combination of antiviral and immunosuppressive, ideally lymphocyte-selective, therapy. Mycophenolate mofetil (MMF), [the morpholinoethyl ester of mycophenolic acid (MPA)] is an immunosupressive agent that is used with success in the prevention of acute renal allograft rejection and in the treatment of refractory rejection in renal, heart and liver transplant patients [17,18]. MMF is hydrolysed to MPA, the active immunosuppressive molecule, which is a potent inhibitor of inosine monophosphate dehydrogenase (IMP-DH). The latter enzyme is key in de novo purine synthesis and is responsible for the conversion of IMP through XMP to GMP. Since IMP represents an important intermediate in the generation of guanine-based nucleotides, MPA causes depletion of intracellular guanine nucleotide pools, which are responsible for the immunosuppressive effect of the drug [19]. T and B lymphocytes are singularly dependent on the de novo pathway for purine synthesis, what explains the lymphocyte-selective antiproliferative effects of MPA [19-23]. We earlier reported on the broad-spectrum antiviral activity of MPA and demonstrated that the molecule inhibits CBV replication in vitro [24]. Since MMF is thus endowed with both a direct inhibitory effect on CBV replication (in vitro), as well as with an immunosuppressive effect, we decided to study the effect of treatment with MMF in a murine model for CBV-induced myocarditis.
Results
In vitro anti-CBV3 activity of MPA
Both MPA and ribavirin inhibit the in vitro replication of CBV3 in Vero and HMF cells. The EC50-values for inhibition of CBV3 replication in Vero cells by ribavirin was 117 μg/ml and 55 μg/ml by MPA. In HMF cells EC50-values of ribavirin and MPA were 250 μg/ml and 80 μg/ml, respectively. In virus yield assays, MPA and ribavirin (both at 100 μg/ml) reduced viral yield by 89% and 87%, respectively (Table 1).
Table 1.
Effect of ribavirin and MPA on CBV3 replication in Vero cells and human myocardial fibroblasts (HMF)
| EC50 (μg/ml)a | Yield reductionb (%) | MTC (μg/ml)c | ||
| Vero | HMF | |||
| MPA | 55 | 80 | 89 | 200 |
| Ribavirin | 117 | 250 | 87 | >500 |
aConcentration of compound that reduces viral induced cytopathic effect by 50% bReduction of virus yield in Vero cells in the presence of 100 μg/ml of either compound cAs determined in Vero cells. Minimal toxic concentration, or correlation required to other normal cell morphology
Effect of MMF on CBV3-induced myocarditis
C3H/HeNHsd mice were infected intraperitoneally with CBV3 and were treated by oral gavage twice daily for a period of 6 consecutive days (starting one day before virus inoculation) with either 100, 200 or 300 mg/kg/day of MMF. The interferon inducer Poly IC [15 mg/kg/day once a day, given intraperitoneally for 3 consecutive days, i.e. on day one before virus inoculation, on the day of virus inoculation and one day after virus inoculation] was used as a positive control. No signs of toxicity of the compounds were noted in treated animals. In a parallel toxicity experiment, the body weight of drug-treated uninfected young mice was monitored. Animals that received treatment with MMF at 200 or 300 mg/kg/day showed some growth retardation, but appeared otherwise healthy. The bodyweight of mice that received either 200 mg/kg/day or 300 mg/kg/day of MMF for 5 consecutive days was respectively 117% and 108% of their body weight on day 0. The bodyweight of mice that had received vehicle only for 5 consecutive days was 132% of their body weight on day 0. All infected mice were sacrificed on day 7 post-virus inoculation and the severity of the myocarditis was assessed by morphometrical analysis (Fig. 1). A dose of 300 mg/kg/day MMF resulted in 78% reduction of the myocarditis score (p < 0.001), a 200 mg/kg/day dose of MMF resulted in 65% reduction (p < 0.001), and a dose of 100 mg/kg/day of MMF resulted in a reduction of 52% of the myocarditis foci (p = 0.001). The interferon inducer poly IC reduced the severity of myocarditis by 97% (p < 0.001). Prednisone, another immunosuppressive agent was included as a control. Mice received the compound at a dose of 15 mg/kg/day by intraperitoneal injections starting one day before the virus inoculation, until day 5 post-virus inoculation. In contrast to what was observed for MMF, treatment with prednisone resulted in an aggravation of the myocarditis, with a 75% increase in the number of myocardial foci (p = 0.004).
Figure 1.
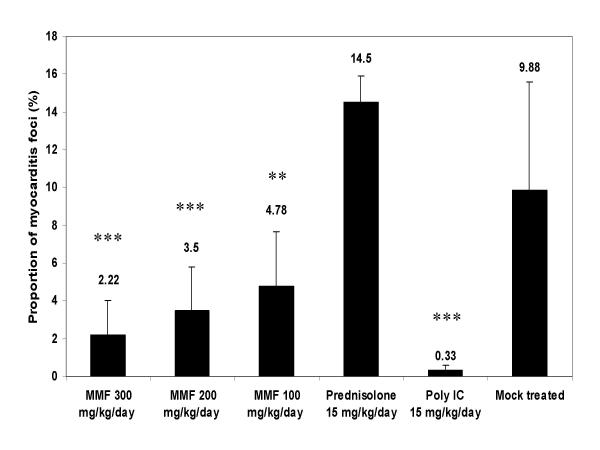
Effect of treatment with MMF or the interferon inducer poly IC on coxsackie virus B3 induced myocarditis in C3H mice. Infected mice (two independent experiments) were treated via oral gavage for 7 consecutive days (-1, 0, 1, 2, 3, 4, 5 relative to virus inoculation) with MMF at the indicated doses [(100 (n = 9), 200 (n = 10) or 300 (n = 6) mg/kg/day)]. Poly IC was given intraperitoneally at 15 mg/kg/day on days -1, 0, 1 relative to virus inoculation. Animals were sacrificed on day 7 post-virus inoculation and the severity of the myocarditis lesions was assessed morphometrically. The virus control group consisted of 8 animals. Statistical differences were calculated between each experimental condition and the mock treated controls (*: p < 0.05; **: p < 0.01: ***: p < 0.001).
Immunosuppressive effect of MMF
To verify whether MMF at the highest and most effective dose used (i.e. 300 mg/kg/day), indeed suppressed the immune system in C3H mice, a flow cytometric analysis was carried out on cells isolated from spleen and blood of (i) CBV3 infected mice (at day 7 post-virus inoculation) that had been treated with 300 mg/kg/day of MMF for 7 consecutive days (n = 2) (ii) uninfected mice that had received treatment with 300 mg/kg/day of MMF for 7 consecutive days (n = 2) (iii) CBV3 infected but untreated mice (n = 2) and (iv) uninfected untreated mice (n = 2). A significant drop (50%) in the number of CD19+ cells was observed in spleen of MMF treated infected mice as compared to untreated infected mice (Fig. 2), indicating severe B-cell depletion. No significant effect of MMF administration on the numbers of CD4+ and CD8+ cells in spleen in the different groups of mice was observed (data not shown).
Figure 2.
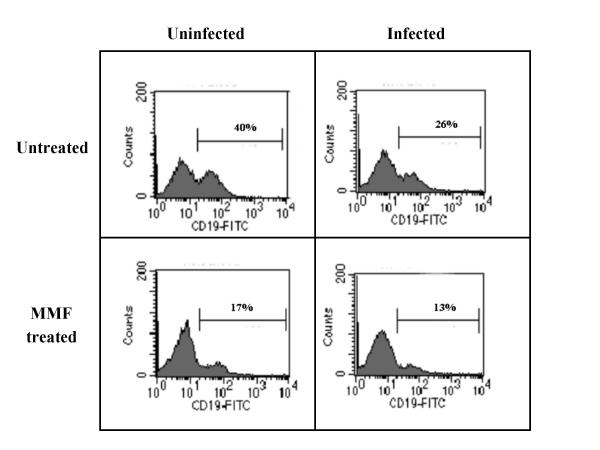
Effect of MMF on the number of CD19+ cells in the spleen of C3H mice. Flow cytometric analysis on cells isolated from the spleen of (i) uninfected untreated mice (ii) CBV infected mice [at day 7 post infection] that had been treated with 300 mg/kg/day of MMF for 7 consecutive days (iii) uninfected mice that had received treatment with 300 mg/kg/day of MMF for 7 consecutive days and (iv) CBV3 infected but untreated mice. Samples from two mice were pooled for analysis.
Effect of MMF on viral titers in heart
C3H/HeNHsd mice were infected intraperitoneally with CBV3 and were treated two times daily for a period of 7 consecutive days (starting one day before virus inoculation) with either 100, 200 or 300 mg/kg/day of MMF. Hearts were dissected on day 6 post-virus inoculation and viral load was determined by monitoring (i) the titer of infectious virus, as well as (ii) the levels of viral RNA by quantitative real-time RT-PCR. In contrast to expectations, a significant increase was noted in viral RNA levels in heart homogenates of animals that had been treated with either 100 mg/kg/day of MMF (28%, p < 0.001), 200 mg/kg/day of MMF (17%, p = 0.04) or 300 mg/kg/day of MMF (16%, p = 0.003) as compared to infected untreated controls (Fig. 3).
Figure 3.
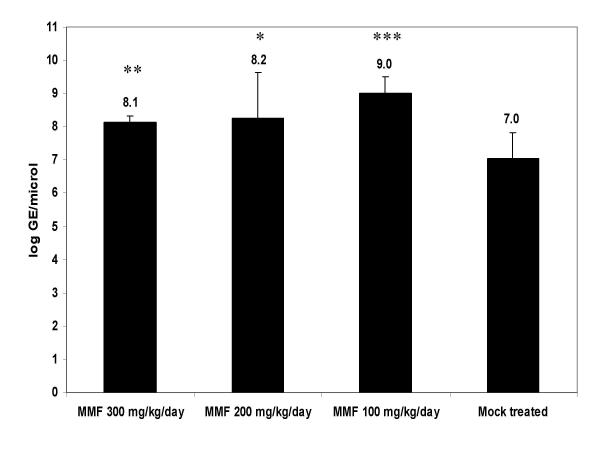
Effect of MMF on viral RNA load monitored by quantitative RT-PCR in the heart of CBV3-infected C3H mice. The different groups consisted of infected mice that had either been left untreated or that had been treated with MMF (at 100, 200 or 300 mg/kg/day). Each group consisted of 4 animals (*: p < 0.05; **: p < 0.01: ***: p < 0.001).
Discussion
Recent studies have shown that both direct viral induced damage, as well as immunopathological factors, are involved in coxsackievirus induced myocarditis [11,12]. We therefore hypothesised that a molecule that would have an inhibitory effect on both the immune system and on viral replication, may be efficient in preventing the development of CBV-induced myocarditis. Earlier we observed that the replication of various RNA viruses can be inhibited by mycophenolic acid (MPA), the active component of mycophenolate mofetil [24]. MPA, which is a potent inhibitor of IMP-dehydrogenase depletes intracellular GTP pools thereby inhibiting viral RNA synthesis and as a consequence viral replication. MMF is a well known immunosuppressive agent that is being used in a variety of organ transplantations [25]. In the murine model for acute myocarditis treatment with MMF resulted in a marked and dose dependent reduction in the severity of the virus induced myocarditis. In marked contrast to MMF, prednisone, which was included as an example of another immunosuppressive agent, resulted in a significant increase in the number of myocarditis lesions. This is consistent with earlier studies where it was shown that corticosteroids increase the severity of the disease during the acute phase of viral myocarditis in murine models [26,27]. In contrast to expectations, and despite the protective effect of MMF on the progression of the myocarditis, RNA levels in heart tissue were markedly increased in MMF treated animals. This can only be explained if the antiviral effect of MPA is not potent enough or if the pharmacokinetic profile of the molecule is not favourable enough to allow MPA to elicit an antiviral effect in vivo. In addition, the immunosuppressive effect of MMF may "neutralize" its own antiviral effect since viral replication will not, or not more efficiently, be controlled by the immune system of the host. It remains puzzling, however, why MMF (despite the fact that it results in an increase in viral RNA levels in the heart), is able to reduce the severity of viral induced myocarditis, whereas prednisone, another immunosuppressive agent, aggravates the disease in the same model. Other studies with murine models for CBV3 induced myocarditis show that therapy with immunosuppressive drugs, such as FK-506 and cyclophosphamide early in the course of acute viral myocarditis tend to be unfavorable for the host, resulting in higher virus titers in the heart, higher mortality rate but sometimes with a apparent reduction of myocardial infiltration [28-30]. The role of virus-induced damage in initiating CBV3-induced myocarditis and the benefit of reducing the level of viremia early during infection has been recently stressed [12,14]. We here observed an important B-cell, but not T-cell depletion in MMF treated infected mice. B lymphocytes appear central in the interaction between Coxsackieviruses (CVB) and their mammalian hosts. Lack of antibody production by agammaglobulinemic humans is responsible for their enhanced susceptibility to chronic infections by CVB's [31,32] and related enteroviruses [33]. Several studies have shown that B lymphocytes can be productively infected by CVB, suggesting the role of these cells in virus dissemination and/or modulation of the host immune response [34-36]. Antibodies directed against CBV resulte, when administered to CBV infected to mice in either (i) a protective effect [37], (ii) induction of cardiopathologic alterations [38], (iii) initial protection via virus clearance with subsequent stimulation of proinflammatry reactions and sustained myocarditis [39], and (iv) antibody-mediated immune enhancement [40,41]. Our observations support this complex picture of the role of B lymphocytes in CVB3-induced myocarditis. The effects of WIN 54954, a specific inhibitor of enteroviral replication, in CBV-induced myocarditis in a mouse model, were attributed to its inhibitory effects on viral replication without interference with cellular or humoral immunity [42]. Recently, we demonstrated that also 2-(3,4-dichlorophenoxy)-5-nitrobenzonitrile (MDL-860), a broad spectrum anti-picornavirus compound is able to markedly reduce the development of CBV3-induced myocarditis [43]. Also the interferon inducers Ampligen and poly IC were shown to efficiently inhibit CBV3 replication in the heart and to have, as a result, an important inhibitory effect on progression of viral myocarditis [44]. From these studies and from the data presented, it may be assumed that the combined use of a potent and selective inhibitor of CBV replication, together with MMF may result in a protective synergistic effect on the progression of the viral myocarditis. Although we initially hypothesized that MMF may have a dual effect (i.e. reduction of the immunopathological component as well as reduction of viral titers) on CBV-induced myocarditis, the protective effect of MMF as noted here, is apparently solely attributable to a reduction of the immunopathological component.
Conclusions
We conclude that the immunosuppressive agent MMF results in a marked and dose dependent reduction in the severity of CBV3-induced myocarditis in a murine model. Howerer, we do not advise the use of this drug as a single therapy for this pathological condition because of the increase in the viral titers in the heart under MMF treatment. However, combination therapy with selective antivirals, if these would become available, may be an interesting option. Despite the fact that MPA, the metabolically active component of MMF, elicits in vitro activity against CBV3, the protective effect of MMF on CBV3-induced myocarditis in mice is apparently solely due to its immunosuppressive effects.
Methods
Cells and viruses
Coxsackievirus B3 (CBV3) (Nancy strain) was obtained from the American Type Culture Collection (ATCC: VR-30). CBV3 was propagated in Vero cells. Human myocardial fibroblasts of pediatric origin (HMF) were prepared from parts of myocardial tissue [kindly provided by Dr. B. Meyns (Cardiovascular Surgery Unit, University Hospitals, Leuven, Belgium), and obtained from the surgery for Fallot-tetralogy] that was digested with trypsine (Gibco, Life Technologies, Rockville, MD). HMF and Vero cells were propagated in minimal essential medium (MEM; Gibco, Life Technologies, Rockville, MD) supplemented with 10% fetal calf serum (FCS; Integro, Zaandam, The Netherlands), 1% L-glutamine (Gibco, Life Technologies, Rockville, MD), and 0.3% sodium bicarbonate (Gibco, Life Technologies, Rockville, MD).
Compounds
Mycophenolic acid (MPA) was purchased from Sigma (St. Louis, Mo., USA). MMF was kindly provided by Roche (Palo Alto, California, USA).
Antiviral assays
Human myocardial fibroblasts (HMF) or Vero cells were grown to confluency in microtiter trays (final cell density: ≈ 105 cell/well) and were infected with an input of CBV3 that caused ≈ 100% CPE at 5 days postinfection (500 CCID50). Virus was removed following a 2-h virus adsorption period after which serial dilutions of the compounds were added. Virus-induced cytopathic effect (CPE) in HMF was recorded microscopically at 5 days post infection following fixation with 70% ethanol and staining with a 2% Giemsa solution (Merck, Darmstadt, Germany). The CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (MTS) was used to determine antiviral activity in Vero cells and proliferation of uninfected Vero cells. The MTS assay was performed according to the instructions of the manufacturer (Promega Corporation, Florida, USA). Briefly, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) is bioreduced by cells into a formazan that is soluble in tissue culture medium after which absorbance of the formazan is read at 490 nm. To measure the effect on cell growth, cells were seeded (in MEM containing 10% FCS) at a density of 4000 cell/well in the presence of serial dilutions of the compounds. Cells were allowed to proliferate for 3 days after which the assay was read by means of the MTS-method.
Virus yield assay
Confluent cultures of Vero cells were infected with a titer of CBV3 that caused 100% CPE at 5 days post infection and were treated (or left untreated) with serial dilutions of the drugs. Cultures were harvested and frozen (-80°C) at 5 days postinfection. Following another freezing-thawing cycle, cell debris was removed by centrifugation and serial dilutions were inoculated on confluent Vero cell cultures. Virus titers were determined 5 days later.
Virus titration from heart homogenate
Hearts were dissected on day 6 post-virus inoculation, a time at which the highest viral titers were detected in untreated controls in preliminary experiments (data not shown). Serial dilutions of 10% (w/v) heart homogenates were inoculated on confluent Vero cell cultures and virus titers were determined 5 days later.
In vivo toxicity experiments
NMRI male mice [Centre d'Elevage, R. Janvier, France] weighing 15 g were treated for 6 consecutive days, by means of oral gavage, with mycophenolate mofetil (MMF) at 200 mg/kg/day or 300 mg/kg/day in 0.2 ml vehicle. Vehicle consisted of sodium chloride (0.9%), sodium carboxymethylcellulose (0.5%), polysorbate 80 (0.4%), benzyl alcohol (0.9%) and distilled water (97.3%).
Treatment of CBV3 infected mice
Four-week-old male C3H/HeNHsd mice (Harlan Laboratories, The Netherlands) weighing 15 g, were inoculated intraperitoneally (i.p.) with 107 CCID50 of CBV3. MMF was administered by oral gavage, twice daily (starting one day before the virus inoculation until day 5 post-virus inoculation) at doses of 100, 200 or 300 mg/kg/day in the same vehicle as described for the toxicity experiments. Poly IC was dissolved in sterile PBS and was administered intraperitoneally, once a day for 3 consecutive days (one day before virus inoculation, the day of virus inoculation and one day after virus inoculation) at a dose of 15 mg/kg/day. Body weight was monitored daily. To assess the severity of acute CVB3-induced myocarditis, all animals were sacrificed on day 7 post-virus inoculation (following ether anesthesia). Hearts were fixed in 6% buffered formaldehyde. The heart was sliced parallel to the basis of the organ into three pieces. Each of these were embedded in paraffin and sectioned at 5 μm. Sections were subsequently stained with H & E and examined by light microscopy.
Morphometry
The extension of myocarditis was determined in H & E-stained sections of the heart of untreated, MMF-treated and polyIC-treated animals by means of a conventional point-counting method using an ocular grid containing 121 equally spaced points. Per grid 5 constant points were counted either hitting or non-hitting an affected area and this was done for 15 stratified random positions of the grid. A myocarditis "score" was defined as the proportion of points hitting the myocarditis area and is expressed in %. The counting was performed for 3 sections per heart. The counting resulted in a total of 75 points per heart. Sections were read at × 200.
Flow cytometric analysis
Spleens were removed and passed through cell strainers (BD Labware, Franclin Lakes, NJ). Blood, taken by heart puncture was collected on heparin. Erythrocytes were removed from blood and splenocyte suspensions by lysis with NH4Cl (0.83% in 0.01 M Tris-HCl, ph 7.2; two consecutive incubations of 5 and 3 minutes at 37°C). Remaining cells were washed, resuspended in cold PBS, counted and stained for flow cytometric analysis as described [45]. Briefly, aliquots of 2 × 105 cells in 0.2 ml were stained with either FITC-conjugated anti-CD8 and PE-conjugated anti-CD4 or stained with FITC-conjugated anti-CD19 and PE-conjugated anti-Mac-1 (CD11b). All antibodies were purchased from R&D Systems Europe, Abingdon, U.K. Cells were analysed with a FACScan flow cytometer (BD Biosciences).
RNA isolation and cDNA synthesis
Heart homogenate of 10% (w/v) was used for the analysis. RNA extraction was performed using the QIAamp RNA Mini Kit (QIAGEN, Hilden, Germany) according to the procedure described by the manufacturer. cDNA was generated at 42°C for 45 minutes using 200 units M-MLV reverse transcriptase (Gibco, Life Technologies, Rockville, MD), 40 units rRNasin Rnase Inhibitor (Promega Corporation, Florida, USA), 5 μM random hexamer primers (Amersham Pharmacia Biotech, Roosendal, The Netherlands), 1 mM dNTPs (Gibco, Life Technologies, Rockville, MD) and buffer containing 250 mM Tris HCl (pH 8.3), 375 mM KCl and 15 mM Mg2+ (Gibco, Life Technologies, Rockville, MD) following denaturation at 70°C for 10 minutes. The reaction was terminated by heating at 99°C for 3 minutes.
Quantitative analysis of CBV3 RNA by real-time RT-Taqman PCR
Real time PCR was performed on the ABI Prism™ 7700 Sequence Detection System (Applied Biosystems, Roche, Branchburg, New Jersey, USA). Primers and probes were developed using Primer Express software (Applied Biosystems, Roche, Branchburg, New Jersey, USA). The primers used are: forward primer 5'-ACGAATCCCAGTGTGTTTTGG-3', reverse primer 5'-TGCTCAAAAACGGTATGGACAT-3' and Taqman probe 5'-CGAGGGAAACGCCCCGCC-3'. The Taqman probe was labelled at the 5' end with the reporter dye molecule FAM (emission wavelength 518 nm) and at the 3' end with the quencher dye TAMRA (emission wavelength 582 nm). The 3' end of the probe was additionally phosphorylated to prevent extension during PCR. Each PCR was performed in 25 μl of PCR reagent mixture containing 0.25 μl of each primer (900 nM), 1 μl specific Taqman probe (200 nM), 12.5 μl 2 x universal Master Mix (Applied Biosystems, Roche, Branchburg, New Jersey, USA), 6 μl water and 5 μl of sample. The PCR-reaction consisted of a decontamination step (5 minutes at 50°C), Taq-activation step (10 minutes at 94°C) and 50 cycles of denaturation (10 seconds at 94°C) and annealing (1 minute at 60°C). For each PCR run, negative template and positive template samples were used. The cycle threshold value (Ct-value) is defined as the number of PCR cycles for which the signal exceeds the baseline, which defines a positive value. The sample was considered to be positive if the Ct-value was <50. Results are expressed as genomic equivalents (GE), being the amount of cDNA corresponding to the amount of viral RNA.
Statistics
To assess differences in the number of heart lesions in treated versus untreated animals, data were analysed by means of the Student's t test.
Authors' contributions
EP carried out the in vitro and in vivo experiments as well as molecular biological studies. EV participated in the histological evaluation of the in vivo experiments. PM participated in the design and interpretation of flow cytometric data. JLA participated in the design and analysis of quantification of CBV3 RNA by real-time RT-Taqman PCR. EDC and JN are the principal investigators and conceived of the study, and participated in its design and coordination. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
This work was supported by the Geconcerteerde Onderzoeksactie (GOA 0/12). We thank Mr. W. Zeegers for assistance with the animal experiments, Mrs T. Mitera for assistance with the flow cytometric analysis, critical reading, Mrs. D. Brabants for kind editorial help and Dr. C. Morrell for critical reading.
Contributor Information
Elizaveta Padalko, Email: epadalk1@jhmi.edu.
Erik Verbeken, Email: Erik.Verbeken@med.kuleuven.ac.be.
Patrick Matthys, Email: Patrick.Matthys@rega.kuleuven.ac.be.
Joeri L Aerts, Email: Ajoeriaerts@aol.com.
Erik De Clercq, Email: erik.declercq@rega.kuleuven.ac.Be.
Johan Neyts, Email: johan.neyts@rega.kuleuven.ac.be.
References
- Gravanis MB, Sterby NH. Incidence of myocarditis: a 10-year autopsy study from Malmo, Sweden, Arch Pathol Lab Med. 1991;115:390–392. [PubMed] [Google Scholar]
- Friman G, Wesslen L, Fohlman J, Karjalainen J, Rolf C. The epidemiology of infectious myocarditis, lymphocytic myocarditis and dilated cardiomyopathy. Eur Heart J. 1995;16:36–41. doi: 10.1093/eurheartj/16.suppl_o.36. [DOI] [PubMed] [Google Scholar]
- Leslie K, Blay R, Haidsch C, Lodge A, Weller A, Huber S. Clinical and experimental aspects of viral myocarditis. Clin Microbiol Rev. 1989;2:191–203. doi: 10.1128/cmr.2.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baboonian C, Davies MJ, Booth JC, McKenna WJ. Coxsackie B viruses and human heart disease. Curr Top Microbiol Immunol. 1997;223:31–52. doi: 10.1007/978-3-642-60687-8_3. [DOI] [PubMed] [Google Scholar]
- Baboonian C, Treasure T. Meta-analysis of the association of enteroviruses with human heart disease. Heart. 1997;78:539–543. doi: 10.1136/hrt.78.6.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laks H, Marelli D, Odim J, Fazio D. Heart Transplantation in the young and elderly. Heart Fail Rev. 2001;6:221–226. doi: 10.1023/A:1011406022657. [DOI] [PubMed] [Google Scholar]
- Fujioka S, Kitaura Y. Coxsackie B virus infection in idiopathic dilated cardiomyopathy: clinical and pharmacological implications. BioDrugs. 2001;15:791–799. doi: 10.2165/00063030-200115120-00002. [DOI] [PubMed] [Google Scholar]
- Sole MJ, Liu P. Viral myocarditis: a paradigm for understanding the pathogenesis and treatment of dilated cardiomyopathy. J Am Coll Cardiol. 1993;22:99A–105A. doi: 10.1016/0735-1097(93)90470-l. [DOI] [PubMed] [Google Scholar]
- Grumbach IM, Heim A, Pring-Akerblom P, Vonhof S, Hein WJ, Muller G, Figulla HR. Adenoviruses and enteroviruses a pathogens in myocarditis and cardiomyopathy. Acta Cardiol. 1999;54:83–88. [PubMed] [Google Scholar]
- Satoh M, Tamura G, Segawa I, Hiramori K, Satodate R. Enteroviral RNA in dilated cardiomyopathy. Eur Heart J. 1994;15:934–939. doi: 10.1093/oxfordjournals.eurheartj.a060613. [DOI] [PubMed] [Google Scholar]
- Knowlton KU, Badorff C. The immune system in viral myocarditis: maintaining the balance. Circ Res. 1999;85:559–561. doi: 10.1161/01.res.85.6.559. [DOI] [PubMed] [Google Scholar]
- Horwitz MS, La Cava A, Fine C, Rodrigez E, Ilic A, Sarvetnick N. Pancreatic expression of interferon-gamma protects mice from lethal coxsackievirus B3 infection and subsequent myocarditis. Nat Med. 2000;6:693–697. doi: 10.1038/76277. [DOI] [PubMed] [Google Scholar]
- Kawai C. From myocarditis to cardiomyopathy: mechanisms of inflammation and cell death: learning from the past for the future. Circulation. 1999;99:1091–1100. doi: 10.1161/01.cir.99.8.1091. [DOI] [PubMed] [Google Scholar]
- Rose NR. Viral damage or 'molecular mimicry' – placing the blame in myocarditis. Nat Med. 2000;6:631–632. doi: 10.1038/76199. [DOI] [PubMed] [Google Scholar]
- Henke A, Huber SA, Stelzner A, Whitton JL. The role of CD8+ T lymphocytes in coxsackievirus B3-induced myocarditis. J Virol. 1995;69:6720–6728. doi: 10.1128/jvi.69.11.6720-6728.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto C, Hiraoka Y, Takada H. T cell-mediated immune response enhances the severity of myocarditis in secondary cardiotropic virus infection in mice. Basic Res Cardiol. 2001;96:439–445. doi: 10.1007/s003950170025. [DOI] [PubMed] [Google Scholar]
- Behrend M. Mycophenolate mofetil: suggested guidelines for use in kidney transplantation. BioDrugs. 2001;15:37–53. doi: 10.2165/00063030-200115010-00004. [DOI] [PubMed] [Google Scholar]
- Vasquez EM, Sifontis NM, Pollak R, Benedetti E. Impact of mycophenolate mofetil on recurrent rejection in kidney transplant patients. Clin Transplant. 2001;15:253–257. doi: 10.1034/j.1399-0012.2001.150406.x. [DOI] [PubMed] [Google Scholar]
- Allison AC, Eugui EM. The design and development of an immunosuppressive drug, mycophenolate mofetil. Springer Semin Immunopathol. 1993;14:353–380. doi: 10.1007/BF00192309. [DOI] [PubMed] [Google Scholar]
- Eugui EM, Mirkovich A, Allison AC. Lymphocyte-selective antiproliferative and immunosuppressive effects of mycophenolic acid in mice. Scand J Immunol. 1991;33:175–183. doi: 10.1111/j.1365-3083.1991.tb03747.x. [DOI] [PubMed] [Google Scholar]
- Sintchak MD, Fleming MA, Futer O, Raybuck SA, Chambers SP, Caron PR, Murcko MA, Wilson KP. Structure and mechanism of inosine monophosphate dehydrogenase in complex with the immunosuppressant mycophenolic acid. Cell. 1996;85:921–930. doi: 10.1016/s0092-8674(00)81275-1. [DOI] [PubMed] [Google Scholar]
- Eugui EM, Almquist SJ, Muller CD, Allison AC. Lymphocyte-selective cytostatic and immunosuppressive effects of mycophenolic acid in vitro: role of deoxyguanosine nucleotide depletion. Scand J Immunol. 1991;33:161–173. doi: 10.1111/j.1365-3083.1991.tb03746.x. [DOI] [PubMed] [Google Scholar]
- Kilic M, Kahan BD. New trends in immunosuppression. Drugs of Today. 2000;36:395–410. doi: 10.1358/dot.2000.36.6.584260. [DOI] [PubMed] [Google Scholar]
- Neyts J, Andrei G, De Clercq E. Differential antiviral activity of several IMP dehydrogenase inhibitors. Tenth International Conference on Antiviral Research, Atlanta, Georgia, USA, 6–11 April 1997. Antiviral Res. 1997;34:A87. doi: 10.1016/S0166-3542(97)83304-9. abstract no 163. [DOI] [Google Scholar]
- Mele TS, Halloran PF. The use of mycophenolate mofetil in transplant recipients. Immunopharmacology. 2000;47:215–245. doi: 10.1016/S0162-3109(00)00190-9. [DOI] [PubMed] [Google Scholar]
- Matsumori A, Tomioka N, Kawai C. Viral myocarditis: immunopathogenesis and the effect of immunosuppressive treatment in a murine model. Jpn Circ J. 1989;53:58–60. doi: 10.1253/jcj.53.58. [DOI] [PubMed] [Google Scholar]
- Tomioka N, Kishimoto C, Matsumori A, Kawai C. Effects of prednisolone on acute viral myocarditis in mice. J Am Coll Cardiol. 1986;7:868–872. doi: 10.1016/s0735-1097(86)80349-7. [DOI] [PubMed] [Google Scholar]
- Herzum M, Huber SA, Weller R, Grebe R, Maisch B. Treatment of experimental murine coxsackie B3 myocarditis. Eur Heart J. 1991;12:200–202. doi: 10.1093/eurheartj/12.suppl_d.200. [DOI] [PubMed] [Google Scholar]
- Hiraoka Y, Kishimoto C, Kurokawa M, Ochiai H, Sasayama S. The effects of FK-506, novel and potent immunosuppressant, upon murine coxsackievirus B3 myocarditis. J Pharmacol Exp Ther. 1991;260:1386–1391. [PubMed] [Google Scholar]
- Kishimoto C, Throp KA, Abelmann WH. Immunosuppression with high doses of cyclophosphamide reduces the severity of myocarditis but increases the mortality in murine coxsackievirus B3 myocarditis. Circulation. 1990;82:989–989. doi: 10.1161/01.cir.82.3.982. [DOI] [PubMed] [Google Scholar]
- Geller TJ, Condie D. A case of protracted coxsackie virus meningoencephalitis in a marginally immunodeficient child treated successfully with intravenous immunoglobulin. J Neurol Sci. 1995;129:131–3. doi: 10.1016/0022-510X(94)00261-L. [DOI] [PubMed] [Google Scholar]
- Hertel NT, Pedersen FK, Heilmann C. Coxsackie B3 virus encephalitis in a patient with agammaglobulinaemia. Eur J Pediatr. 1989;148:642–3. doi: 10.1007/BF00441520. [DOI] [PubMed] [Google Scholar]
- Misbah SA, Spickett GP, Ryba PC, Hockaday JM, Kroll JS, Sherwood C, Kurtz JB, Moxon ER, Chapel HM. Chronic enteroviral meningoencephalitis in agammaglobulinemia: case report and literature review. J Clin Immunol. 1992;12:266–70. doi: 10.1007/BF00918150. [DOI] [PubMed] [Google Scholar]
- Anderson DR, Wilson JE, Carthy CM, Yang D, Kandolf R, McManus BM. Direct interactions of coxsackievirus B3 with immune cells in the splenic compartment of mice susceptible or resistant to myocarditis. J Virol. 1996;70:4632–45. doi: 10.1128/jvi.70.7.4632-4645.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingel K, Stephan S, Sauter M, Zell R, McManus BM, Bultmann B, Kandolf R. Pathogenesis of murine enterovirus myocarditis: virus dissemination and immune cell targets. J Virol. 1996;70:8888–95. doi: 10.1128/jvi.70.12.8888-8895.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mena I, Perry CM, Harkins S, Rodriguez F, Gebhard J, Whitton JL. The role of B lymphocytes in coxsackievirus B3 infection. Am J Pathol. 1999;155:1205–15. doi: 10.1016/S0002-9440(10)65223-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho CT, Feng KK, McCarthy VP, Lenahan MF. Role of antiviral antibodies in resistance against coxsackievirus B3 infection: interaction between preexisting antibodies and an interferon inducer. Infect Immun. 1982;37:720–7. doi: 10.1128/iai.37.2.720-727.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauntt CJ, Arizpe HM, Higdon AL, Wood HJ, Bowers DF, Rozek MM, Crawley R. Molecular mimicry, anti-coxsackievirus B3 neutralizing monoclonal antibodies, and myocarditis. J Immunol. 1995;154:2983–95. [PubMed] [Google Scholar]
- Gauntt CJ, Arizpe HM, Higdon AL, Rozek MM, Crawley R, Cunningham MW. Anti-Coxsackievirus B3 neutralizing antibodies with pathological potential. Eur Heart J. 1991;12:124–9. doi: 10.1093/eurheartj/12.suppl_d.124. [DOI] [PubMed] [Google Scholar]
- Girn J, Kavoosi M, Chantler J. Enhancement of coxsackievirus B3 infection by antibody to a different coxsackievirus strain. J Gen Virol. 2002;83:351–8. doi: 10.1099/0022-1317-83-2-351. [DOI] [PubMed] [Google Scholar]
- Kishimoto C, Kurokawa M, Ochiai H. Antibody-mediated immune enhancement in coxsackievirus B3 myocarditis. J Mol Cell Cardiol. 2002;34:1227–38. doi: 10.1016/S0022-2828(02)92087-0. [DOI] [PubMed] [Google Scholar]
- Fohlman J, Pauksen K, Hyypia T, Eggertsen G, Ehrnst A, Ilback NG, Friman G. Antiviral treatment with WIN 54 954 reduces mortality in murine coxsackievirus B3 myocarditis. Circulation. 1996;94:2254–2259. doi: 10.1161/01.cir.94.9.2254. [DOI] [PubMed] [Google Scholar]
- Padalko E, Verbeken E, De Clercq E, Neyts J. Inhibition of Coxsackie B3 virus induced myocarditis in mice by 2-(3,4-dichlorophenoxy)-5-nitrobenzonitrile. J Med Virol. [DOI] [PubMed]
- Padalko E, Nuyens D, Verbeken E, Aerts JL, De Clercq E, Carmeliet P, Neyts J. The inter-feron inducer Ampligen® [poly(I)-poly(C12U)] markedly protects against Coxsackie B3 virus-induced myocarditis in mice. Antimicrobiol Agents Chemother. [DOI] [PMC free article] [PubMed]
- Matthys P, Vermeire K, Mitera T, Heremans H, Huang S, Schols D, De Wolf-Peeters C, Billiau A. Enhanced autoimmune arthritis in IFN-gamma receptor-deficient mice is conditioned by mycobacteria in Freund's adjuvant and by increased expansion of Mac-1+ myeloid cells. J Immunol. 1999;164:3505–3510. [PubMed] [Google Scholar]