

Abstract
Under the conditions of ruthenium catalyzed transfer hydrogenation employing isopropanol as terminal reductant, π-unsaturated compounds (1,3-dienes, allenes, 1,3-enynes and alkynes) reductively couple to aldehydes to furnish products of carbonyl addition. In the absence of isopropanol, π-unsaturated compounds couple directly from the alcohol oxidation level to form identical products of carbonyl addition. Such “alcohol-unsaturate C-C couplings” enable carbonyl allylation, propargylation and vinylation from the alcohol oxidation level in the absence of stoichiometric organometallic reagents or metallic reductants. Thus, direct catalytic C-H functionalization of alcohols at the carbinol carbon is achieved.
Introduction
We have found that hydrogenation of unsaturates in the presence of carbonyl compounds and imines enables reductive C-C bond formation.1–5,6a More recently, under the conditions of iridium4b,6b,7 or ruthenium catalyzed8 transfer hydrogenation, C-C coupling between unsaturates and carbonyl partners was observed. In such “transfer hydrogenative C-C couplings,” hydrogen embedded within a donor alcohol, typically isopropanol, mediates unsaturate-carbonyl reductive coupling. Of greater significance, an alcoholic reactant may serve a dual role: as hydrogen donor and precursor to the carbonyl electrophile. Thus, by exploiting alcohols and unsaturates as redox partners, carbonyl addition may be conducted from the alcohol oxidation level.
Transfer hydrogenative carbonyl addition adds significantly to the evolution of carbonyl addition chemistry. First, C-C bond formation from either the alcohol or aldehyde oxidation level enhances step economy by avoiding the redox manipulations typically required to convert alcohols to aldehydes. Secondly, the use of unsaturates as nonstabilized carbanion equivalents circumvents the use of preformed organometallic reagents and, hence, the generation of stoichiometric metallic byproducts. In this account, the formation of C-C bonds under the conditions of ruthenium catalyzed transfer hydrogenation is reviewed (Scheme 1).
Scheme 1.

Catalytic C-C bond formation under the conditions of hydrogenation and transfer hydrogenation.
Diene-Carbonyl Coupling from the Alcohol or Aldehyde Oxidation Level
Under the conditions of ruthenium catalyzed transfer hydrogenation employing RuHCl(CO)(PPh3)3 as the precatalyst, direct coupling of acyclic dienes to carbonyl partners is achieved with complete regiocontrol from the alcohol or aldehyde oxidation.8a Butadiene, isoprene and 2,3-dimethylbutadiene couple to benzylic alcohols to provide products of carbonyl crotylation, isoprenylation, and reverse 2-methyl-prenylation, respectively. The presence of an acid cocatalyst (m-NO2BzOH) is essential as only trace quantities of product are observed otherwise. Additionally, added acetone (2.5 mol%) and phosphine ligand have a beneficial effect upon the reaction efficiency. Using this first generation catalytic system, simple unactivated aliphatic alcohols engage in C-C coupling, as demonstrated by the union of isoprene and 1-nonanol (Scheme 2). For diene-aldehyde coupling, m-NO2BzOH and acetone are not needed, though larger loadings of diene are required. Isopropanol or formic acid may serve as terminal reductant. The observed branched regioselectivity complements the linear regioselectivity observed in related Ni-catalyzed diene-aldehyde reductive couplings. 9,10 A limitation of this first-generation catalytic system involves control of relative and absolute stereochemistry (Table 1).
Scheme 2.

Ruthenium catalyzed transfer hydrogenative coupling of isoprene to an unactivated aliphatic alcohol.
Table 1.
Ruthenium catalyzed transfer hydrogenative coupling of conjguated dienes to alcohols and aldehydes to furnish homo-allylic alcohols.
 | ||
|---|---|---|
| Diene | Product | Isolated Yield, Diastereoselectivity |
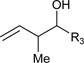 |
R3 = p-NO2Ph, 84% Yield, 2:1 dr (B) R3 = Ph, 61% Yield, 2:1 dr (B) R3 = p-MeOPh, 76% Yield, 2:1 dr (C) |
|
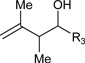 |
R3 = p-NO2Ph, 84% Yield, 2:1 dr (A) R3 = Ph, 93% Yield, 2:1 dr (C) R3 = p-MeOPh, 84% Yield, 2:1 dr (C) |
|
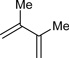 |
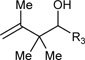 |
R3 = p-NO2Ph, 89% Yield, 2:1 dr (B) R3 = Ph, 91% Yield, 2:1 dr (B) R3 = p-MeOPh, 67% Yield, 2:1 dr (B) |
 | ||
| Terminal Reductant: HCO2H (200 mol%) or i-PrOH (400 mol%) R3 = p-NO2Ph, 89% Yield, 2:1 dr (A, IPA); 84% Yield, 2:1 dr (A, HCO2H) R3 = Ph, 64% Yield, 2:1 dr (C, IPA); 82% Yield, 2:1 dr (C, HCO2H) R3 = p-MeOPh, 68% Yield, 2:1 dr (C); 68% Yield, 2:1 dr (C, HCO2H) | ||
Conditions A employ no added ligand: Conditions B employ (p-MeOPh)3P (15 mol%) as ligand; Conditions C employ rac-BINAP (5 mol%) as ligand.
The coupling of isoprene to d2-benzyl alcohol results in transfer of a benzylic deuteride to the allylic methyl (19% 2H) and allylic methine (32% 2H). These data are consistent with reversible hydrometallation of the less substituted olefin to form a secondary σ-allyl. Conversion to the more stable primary σ-allyl haptomer occurs in advance of carbonyl addition, which proceeds through a closed six-centered transition state with allylic inversion to deliver the branched product of carbonyl allylation. In related aldehyde couplings employing d8-isopropanol as the terminal reductant, incorporation of deuterium is observed at the allylic methyl (19%2 H) and allylic methine (10% 2H) (Scheme 3).
Scheme 3.

Isotopic labeling experiments in ruthenium catalyzed transfer hydrogenative couplings of isoprene to benzyl alcohol and benzaldehyde.
Based on the hypothesis that coordinative unsaturation should promote oxidation of the initially formed homoallylic alcohol, a ruthenium complex possessing a counter ion less strongly coordinating than chloride was sought. Exposure of RuH2(CO)(PPh3)3 to acids HX is known to generate RuHX(CO)(PPh3)3 or RuX2(CO)(PPh3)2.11 Through an assay of acidic additives, it was found that diene-alcohol couplings performed using a catalyst generated in situ from RuH2(CO)(PPh3)3 (5 mol%) and trifluoroacetic acid (TFA) (5 mol%) produce the β,γ-unsaturated ketones in good to excellent isolated yields.8d Butadiene, isoprene, 2,3-dimethylbutadiene and myrcene may serve as nucleophilic partners. In each case, complete branch regioselectivity is observed. Under identical conditions, isoprene couples to aldehydes to provide β,γ-unsaturated ketones in good to excellent isolated yields (Table 2).8d,12
Table 2.
Ruthenium catalyzed transfer hydrogenative coupling of conjugated dienes to alcohols and aldehydes to furnish β,γ-unsaturated ketones.a
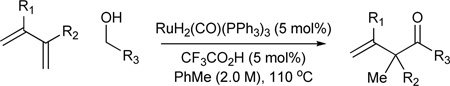 | ||
|---|---|---|
| Diene | Product | Isolated Yield |
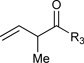 |
R3 = p-MeOPh, 96% Yield R3 = m-MeOPh, 75% Yield R3 = p-BrPh, 62% Yieldb |
|
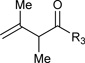 |
R3 = p-MeOPh, 81% Yield R3 = m-MeOPh, 70% Yield R3 = p-BrPh, 80% Yield |
|
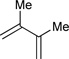 |
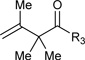 |
R3 = p-MeOPh, 80% Yield R3 = m-MeOPh, 93% Yield R3 = p-BrPh, 91% Yield |
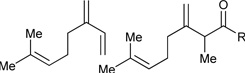 |
R3 = p-MeOPh, 96% Yield R3 = m-MeOPh, 91% Yield R3 = p-BrPh, 85% Yield |
|
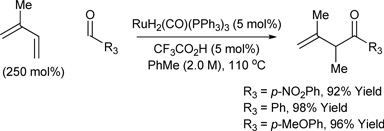 | ||
Butadiene (800 mol%), isoprene (250 mol%), 2,3-dimethylbutadiene (300 mol%), myrcene (300 mol%).
The reaction product was contaminated with approximately 10% of the α,β-unsaturated ketone.
Coupling of isoprene to d2-benzyl alcohol under standard conditions employing the catalyst generated in situ from RuH2(CO)(PPh3)3 (5 mol%) and trifluoroacetic acid (TFA) (5 mol%) provides the indicated β,γ-unsaturated ketone, which incorporates deuterium primarily at the allylic methyl (34%), and the allylic methine (12%). This pattern of deuterium incorporation closely matches that obtained in the analogous coupling reaction of isoprene to d2-benzyl alcohol to provide the homoallylic alcohol. Hence, a similar mechanism appears operative: hydrometallation of the less substituted olefin of isoprene to deliver the secondary σ-allyl metal haptomer, followed by carbonyl addition from the more stable primary σ-allyl haptomer through a six-centered transition structure with β-hydride elimination of the resulting alkoxide to deliver the ketone (Scheme 4).
Scheme 4.

Ruthenium catalyzed transfer hydrogenative coupling of isoprene to d2-benzyl alcohol under oxidative conditions.
Our collective studies of ruthenium catalyzed diene-alcohol and diene-aldehyde transfer hydrogenative C-C coupling demonstrate that carbonyl addition may be achieved from the alcohol or aldehyde oxidation level to deliver either the homoallylic alcohol or the β,γ-unsaturated ketone. Thus, all oxidations levels of substrate (alcohol or aldehyde) and product (homoallyl alcohol or β,γ-unsaturated ketone) are accessible (Scheme 5).
Scheme 5.

Transcending oxidation level via ruthenium catalyzed diene-alcohol and diene-aldehyde transfer hydrogenative C-C coupling
Just as diene hydrometallation enables generation of transient allyl metal species, so should allene hydrometallation. Consequently, allenes were explored as nucleophilic partners in ruthenium catalyzed transfer hydrogenative carbonyl addition. To our delight, upon exposure of 1,1-disubstituted allenes to paraformaldehyde and higher aldehydes in the presence of [RuBr(CO)3(η3-C3H5)] (5 mol%), t-BuPPh2 (15 mol%) and isopropanol (400 mol%), branched products of carbonyl allylation bearing all-carbon quaternary centers are formed as single regioisomers.8c Related ruthenium catalyzed allene-alcohol transfer hydrogenative couplings are currently under investigation. Again, a limitation of this first-generation catalytic system involves control of relative and absolute stereochemistry (Table 3).
Table 3.
Ruthenium catalyzed transfer hydrogenative coupling of allenes to paraformaldehyde and higher aldehydes to furnish homoallylic alcohols.a
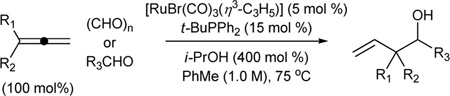 | ||
|---|---|---|
| Allene | Product | Isolated Yield |
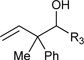 |
R3 = H, 86% yield R3 = p-NO2Ph, 87% yield, 2:1 dr R3 = CH2OBn, 71% yield, 1:1 dr |
|
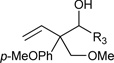 |
R3 = H, 74% yield R3 = p-NO2Ph, 76% yield, 1:1 dr R3 = CH2OBn, 72% yield, 2:1 dr |
|
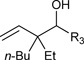 |
R3 = H, 77% yield R3 = p-NO2Ph, 82% yield, 1:1 dr R3 = CH2OBn, 70% yield, 1:1 dr |
|
For couplings to paraformaldehyde: allene (100 mol%), paraformaldehyde (400 mol%). For couplings to higher aldehydes: allene (200 mol%), paraformaldehyde (200 mol%)
Enyne-Carbonyl Coupling from the Alcohol or Aldehyde Oxidation Level
Carbonyl propargylation based on C-C bond forming transfer hydrogenation is potentially achieved using conjugated enynes as surrogates to preformed allenyl metal reagents. The outcome of such couplings was uncertain, as enynes engage in C-C bond formation at the acetylenic terminus under the 4 conditions of rhodium2a, b, e, g and nickel13 catalysis. Nevertheless, it was found that exposure of 1,3-enynes to alcohols in the presence of catalyst prepared in situ from RuHCl(CO)(PPh3)3 and DPPF provides the desired products of carbonyl propargylation as single regioisomers.8b Iridium complexes also catalyze enyne-mediated propargylation, but ruthenium catalysts were found to be superior.
This carbonyl propargylation protocol exhibits remarkably broad substrate scope. Benzylic alcohols, allylic alcohols and unactivated aliphatic alcohols participate in enyne mediated carbonyl propargylation in good to excellent yield and with exceptional regiocontrol. Additionally, a variety of 1,3-enynes are tolerated (Table 4, top). Under related transfer hydrogenation conditions employing isopropanol as the terminal reductant, carbonyl propargylation is achieved from the aldehyde oxidation level, although in certain cases the coupling product is contaminated by small quantities (≤ 10%) of alkyne reduction to form the cis-alkene (Table 4, bottom). Thus, through ruthenium catalyzed transfer hydrogenative coupling, carbonyl propargylation can be achieved in the absence of preformed allenyl metal reagents from the alcohol or aldehyde oxidation level. Stereocontrolled enyne-mediated propargylation represents an important goal of ongoing research.
Table 4.
Carbonyl propargylation via ruthenium catalyzed transfer hydrogenative coupling of 1,3-enynes to alcohols and aldehydes.
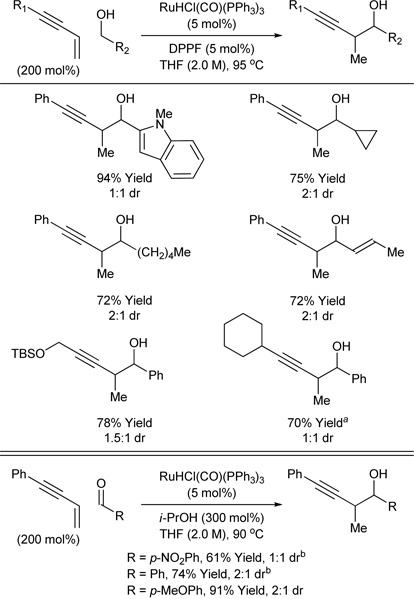 |
m-NO2BzOH (5 mol%) employed as additive.
Product was accompanied by ca. 10% alkyne reduction.
Enyne coupling to d2-benzyl alcohol results in transfer of a benzylic deuteride to the allylic methyl (56% 2H) and allylic methine (24% 2H). Deuterium is completely retained at the benzylic methine of the coupling product (Scheme 6). These results are consistent with a catalytic cycle involving aldehyde generation via alcohol dehydrogenation followed by reversible alkene hydrometallation from the resulting ruthenium hydride to furnish an allenylmetal species. Carbonyl addition with propargylic transposition delivers the propargylic alcohol.
Scheme 6.

Isotopic labeling experiments in ruthenium catalyzed transfer hydrogenative couplings of 1,3-enynes.
Alkyne-Carbonyl Coupling from the Alcohol Oxidation Level
Allylic alcohols are important building blocks in organic synthesis. One important strategy for their synthesis involves the catalytic asymmetric addition of vinyl-metal reagents to aldehydes.14 More recently, direct carbonyl vinylation has been achieved via metal catalyzed alkyne-aldehyde reductive coupling.2b–h,15 Transfer hydrogenative alkyne-alcohol coupling would enable direct carbonyl vinylation in the absence of any stoichiometric reductant. In a preliminary set of experiments, it was found that exposure of alcohols to butyne in the presence of Ru(TFA)(CO)(PPh3)2 results in formation of the desired allylic alcohols.8e Thus, carbonyl vinylation is achieved from the alcohol oxidation level in absence of any stoichiometrically preformed vinyl metal reagents (Scheme 7).
Scheme 7.

Carbonyl vinylation via ruthenium catalyzed transfer hydrogenative coupling of butyne to alcohols.
Conclusion and Outlook
Under the conditions of ruthenium catalyzed transfer hydrogenation, one may perform carbonyl allylation, propargylation and vinylation from the aldehyde or alcohol oxidation level. These initial findings evoke numerous possibilities, especially in the case of alcohol-unsaturate coupling. For example, transfer hydrogenative coupling of ethylene and alcohols would enable carbonyl ethylation from the alcohol oxidation level, eliminating the need for diethylzinc–a pyrophoric liquid. Ethylene-ethanol coupling would represent a by-product-free method for the preparation of sec-butanol, an attractive biofuel (Scheme 8).
Scheme 8.

By-product-free conversion of ethanol and ethylene to sec-butanol via ruthenium catalyzed transfer hydrogenative coupling.
Many challenges remain. Activation of isolated olefins in transfer hydrogenative C-C coupling is an especially important objective. Additionally, the development of stereoselective couplings and imine addition from the amine oxidation level will be required for applications in the area of fine chemical synthesis. It is the author’s hope that the work described in this account will itself catalyze further progress toward these goals.
Acknowledgments
Acknowledgment is made to Merck, Umicore, the Robert A. Welch Foundation, the ACS-GCI Pharmaceutical Roundtable, the Donald D. Harrington Fellows Program and the NIH-NIGMS (RO1-GM069445) for partial support of this research.
References and Notes
- 1.For selected reviews of hydrogenative C-C coupling, see: Ngai M-Y, Kong J-R, Krische MJ. J. Org. Chem. 2007;72:1063. doi: 10.1021/jo061895m. Iida H, Krische MJ. Top. Curr. Chem. 2007;279:77. Skucas E, Ngai M-Y, Komanduri V, Krische MJ. Acc. Chem. Res. 2007;40:1394. doi: 10.1021/ar7001123.
- 2.For hydrogenative C=X vinylation, see: Kong J-R, Cho C-W, Krische MJ. J. Am. Chem. Soc. 2005;127:11269. doi: 10.1021/ja051104i. Kong J-R, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2006;128:718. doi: 10.1021/ja056474l. Rhee J-U, Krische MJ. J. Am. Chem. Soc. 2006;128:10674. doi: 10.1021/ja0637954. Kong J-R, Krische MJ. J. Am. Chem. Soc. 2006;128:16040. doi: 10.1021/ja0664786. Komanduri V, Krische MJ. J. Am. Chem. Soc. 2006;128:16448. doi: 10.1021/ja0673027. Cho C-W, Krische MJ. Org. Lett. 2006;8:3873. doi: 10.1021/ol061485k. Hong Y-T, Cho C-W, Skucas E, Krische MJ. Org. Lett. 2007;9:3745. doi: 10.1021/ol7015548. Ngai M-Y, Barchuk A, Krische MJ. J. Am. Chem. Soc. 2007;129:280. doi: 10.1021/ja0670815. Skucas E, Kong J-R, Krische MJ. J. Am. Chem. Soc. 2007;129:7242. doi: 10.1021/ja0715896. Barchuk A, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2007;129:8432. doi: 10.1021/ja073018j. Ngai M-Y, Barchuk A, Krische MJ. J. Am. Chem. Soc. 2007;129:12644. doi: 10.1021/ja075438e.
- 3.For hydrogenative aldol and Mannich addition, see: Jung C-K, Garner SA, Krische MJ. Org. Lett. 2006;8:519. doi: 10.1021/ol052859x. Jung C-K, Krische MJ. J. Am. Chem. Soc. 2006;128:17051. doi: 10.1021/ja066198q. Garner SA, Krische MJ. J. Org. Chem. 2007;72:5843. doi: 10.1021/jo070779w. Bee C, Han SB, Hassan A, Iida H, Krische MJ. J. Am. Chem. Soc. 2008;130:2746. doi: 10.1021/ja710862u.
- 4.For hydrogenative acyl substitution via reductive hydroacylation, see: Hong Y-T, Barchuk A, Krische MJ. Angew. Chem. Int. Ed. 2006;128:6885. doi: 10.1002/anie.200602377.
- 5.For iridium catalyzed hydrogenative and transfer hydrogenative carbonyl allylations employing allenes as allyl donors, see: Skucas E, Bower JF, Krische MJ. J. Am. Chem. Soc. 2007;129:12678. doi: 10.1021/ja075971u. Bower JF, Skucas E, Patman RL, Krische MJ. J. Am. Chem. Soc. 2007;129:15134. doi: 10.1021/ja077389b.
- 6.For rhodium and iridium catalyzed couplings of dienes to carbonyl compounds under hydrogenative and transfer hydrogenative conditions, respectively, see: Jang H-Y, Huddleston RR, Krische MJ. Angew. Chem. Int. Ed. 2003;42:4074. doi: 10.1002/anie.200351986. Bower JF, Patman RL, Krische MJ. Org. Lett. 2008;10:1033. doi: 10.1021/ol800159w.
- 7.For iridium catalyzed transfer hydrogenative carbonyl allylations employing allyl acetate as an allyl donor, see: Kim IS, Ngai M-Y, Krische MJ. J. Am. Chem. Soc. 2008;130:6340. doi: 10.1021/ja802001b. Kim IS, Ngai M-Y, Krische MJ. 2008 Submitted for Publication.
- 8.For ruthenium catalyzed transfer hydrogenative coupling, see: Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:6338. doi: 10.1021/ja801213x. Patman RL, Williams VM, Bower JF, Krische MJ. Angew. Chem. Int. Ed. 2008;47:5220. doi: 10.1002/anie.200801359. Ngai M-Y, Skucas E, Krische MJ. Org. Lett. 2008;10:2705. doi: 10.1021/ol800836v. Shibahara F, Bower JF, Krische MJ. 2008 Submitted for Publication. Chauligain M, Patman RL, Williams VM, Krische MJ. Manuscript in Preparation
- 9.Nickel catalytic intermolecular diene-aldehyde reductive coupling: Kimura M, Ezoe A, Shibata K, Tamaru Y. J. Am. Chem. Soc. 1998;120:4033. Takimoto M, Hiraga Y, Sato Y, Mori M. Tetrahedron Lett. 1998;39:4543. Kimura M, Fujimatsu H, Ezoe A, Shibata K, Shimizu M, Matsumoto S, Tamaru Y. Angew. Chem. Int. Ed. 1999;38:397. doi: 10.1002/(SICI)1521-3773(19990201)38:3<397::AID-ANIE397>3.0.CO;2-Y. Kimura M, Shibata K, Koudahashi Y, Tamaru Y. Tetrahedron Lett. 2000;41:6789. Kimura M, Ezoe A, Tanaka S, Tamaru Y. Angew. Chem. Int. Ed. 2001;40:3600. doi: 10.1002/1521-3773(20011001)40:19<3600::aid-anie3600>3.0.co;2-n. Loh T-P, Song H-Y, Zhou Y. Org. Lett. 2002;4:2715. doi: 10.1021/ol026216i. Sato Y, Sawaki R, Saito N, Mori M. J. Org. Chem. 2002;67:656. doi: 10.1021/jo0106086. Bareille L, Le Gendre P, Moïse C. Chem. Commun. 2005;775 doi: 10.1039/b414322a. Kimura M, Ezoe A, Mori M, Iwata K, Tamaru Y. J. Am. Chem. Soc. 2006;128:8559. doi: 10.1021/ja0608904. Yang Y, Zhu S-F, Duan H-F, Zhou C-Y, Wang L-X, Zhou Q-L. J. Am. Chem. Soc. 2007;129:2248.. doi: 10.1021/ja0693183. Sato Y, Hinata Y, Seki R, Oonishi Y, Saito N. Org. Lett. 2007;9:5597. doi: 10.1021/ol702543m.
- 10.For reviews encompassing nickel-catalyzed diene-aldehyde reductive coupling, see: Tamaru Y. J. Organomet. Chem. 1999;576:215. Ikeda S-i. Angew. Chem. Int. Ed. 2003;42:5120. doi: 10.1002/anie.200301673. Montgomery J. Angew. Chem. Int. Ed. 2004;43:3890. doi: 10.1002/anie.200300634. Tamaru Y, editor. Modern Organonickel Chemistry. Weinheim, Germany: Wiley-VCH; 2005. Kimura M, Tamaru Y. Top. Curr. Chem. 2007;279:173.
- 11.Dobson A, Robinson SD, Uttley MF. J. Chem. Soc. Dalton Trans. 1975:370. [Google Scholar]
- 12.For intermolecular hydroacylation of dienes catalyzed by ruthenium, see: Kondo T, Hiraishi N, Morisaki Y, Wada K, Watanabe Y, Mitsudo T-a. Organometallics. 1998;17:2131.
- 13.For nickel catalyzed couplings of conjugated enynes, see: For nickel catalyzed reductive coupling of 1,3-enynes to carbonyl compounds, see: Miller KM, Luanphaisarnnont T, Molinaro C, Jamison TF. J. Am. Chem. Soc. 2004;126:4130. doi: 10.1021/ja0491735. Miller KM, Jamison TF. Org. Lett. 2005;7:3077. doi: 10.1021/ol051075g. Miller KM, Colby EA, Woodin KS, Jamison TF. Adv. Synth. Catal. 2005;347:1533.
- 14.For enantioselective catalytic addition of vinyl-metal reagents to aldehydes, see: Oppolzer W, Radinov R. Helv. Chim. Acta. 1992;75:170. Oppolzer W, Radinov R. J. Am. Chem. Soc. 1993;115:1593. Soai K, Takahashi KJ. Chem. Soc., Perkin Trans. 1994;1:1257. Wipf P, Xu W. Tetrahedron Lett. 1994;35:5197. Oppolzer W, Radinov RN, De Brabander J. Tetrahedron Lett. 1995;36:2607. Wipf P, Ribe S. J. Org. Chem. 1998;63:6454. Oppolzer W, Radinov RN, El-Sayed E. J. Org. Chem. 2001;66:4766. doi: 10.1021/jo000463n. Dahmen S, Bräse S. Org. Lett. 2001;3:4119. doi: 10.1021/ol016954r. Chen YK, Lurain AE, Walsh PJ. J. Am. Chem. Soc. 2002;124:12225. doi: 10.1021/ja027271p. Ji J-X, Qiu L-Q, Yip CW, Chan ASC. J. Org. Chem. 2003;68:1589. doi: 10.1021/jo026551k. Lurain AE, Walsh PJ. J. Am. Chem. Soc. 2003;125:10677. doi: 10.1021/ja035213d. Ko D-H, Kang S-W, Kim KH, Chung Y, Ha D-C. Bull. Korean. Chem. Soc. 2004;25:35. Sprout CM, Richmond ML, Seto CT. J. Org. Chem. 2004;69:6666. doi: 10.1021/jo049160+. Jeon S-J, Chen YK, Walsh PJ. Org. Lett. 2005;7:1729. doi: 10.1021/ol050255n. Lauterwasser F, Gall J, Hoefener S, Bräse S. Adv. Synth. Catal. 2006;348:2068. Jeon S-J, Fisher EL, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2006;128:9618. doi: 10.1021/ja061973n. Salvi L, Jeon S-J, Fisher EL, Carroll PJ, Walsh PJ. J. Am. Chem. Soc. 2007;129:16119. doi: 10.1021/ja0762285.
- 15.For selected reviews encompassing intra- and intermolecular direct reductive coupling of alkynes to carbonyl partners, see: Ojima I, Tzamarioudaki M, Li Z, Donovan RJ. Chem. Rev. 1996;96:635. doi: 10.1021/cr950065y. Montgomery J. Acc. Chem. Res. 2000;33:467. doi: 10.1021/ar990095d. Montgomery J, Amarashinghe KKD, Chowdhury SK, Oblinger E, Seo J, Savchenko AV. Pure Appl. Chem. 2002;74(6):129. Ikeda S-I. Angew. Chem. Int. Ed. 2003;42:5120. doi: 10.1002/anie.200301673. Miller KM, Molinaro C, Jamison TF. Tetrahedron Asymm. 2003:3619. Montgomery J. Angew. Chem. Int. Ed. 2004;43:3890. doi: 10.1002/anie.200300634. Moslin RM, Miller-Moslin K, Jamison TF. Chem. Comm. 2007:4441. doi: 10.1039/b707737h. Montgomery J, Sormunen GJ. Top. Curr. Chem. 2007;279:1.