

Abstract
We have detected directly the interactions of sarcolipin (SLN) and the sarcoplasmic reticulum Ca-ATPase (SERCA) by measuring fluorescence resonance energy transfer (FRET) between fusion proteins labeled with cyan fluorescent protein (donor) and yellow fluorescent protein (acceptor). SLN is a membrane protein that helps control contractility by regulating SERCA activity in fast-twitch and atrial muscle. Here we used FRET microscopy and spectroscopy with baculovirus expression in insect cells to provide direct evidence for: 1) oligomerization of SLN and 2) regulatory complex formation between SLN and the fast-twitch muscle Ca-ATPase (SERCA1a isoform). FRET experiments demonstrated that SLN monomers self-associate into dimers and higher order oligomers in the absence of SERCA, and that SLN monomers also bind to SERCA monomers in a 1:1 binary complex when the two proteins are coexpressed. FRET experiments further demonstrated that the binding affinity of SLN for itself is similar to that for SERCA. Mutating SLN residue isoleucine-17 to alanine (I17A) decreased the binding affinity of SLN self-association and converted higher order oligomers into monomers and dimers. The I17A mutation also decreased SLN binding affinity for SERCA but maintained 1:1 stoichiometry in the regulatory complex. Thus, isoleucine-17 plays dual roles in determining the distribution of SLN homo-oligomers and stabilizing the formation of SERCA-SLN heterodimers. FRET results for SLN self-association were supported by the effects of SLN expression in bacterial cells. We propose that SLN exists as multiple molecular species in muscle, including SERCA-free (monomer, dimer, oligomer) and SERCA-bound (heterodimer), with transmembrane zipper residues of SLN serving to stabilize oligomeric interactions.
Keywords: Calcium ATPase, Calcium Transport, Cardiac Muscle, Fluorescence Resonance Energy Transfer (FRET), Protein-Protein Interactions, Sarcoplasmic Reticulum, Skeletal Muscle
Introduction
Sarcolipin (SLN)4 is a 3-kDa membrane protein that reversibly inhibits the activity of the calcium-transporting ATPase (SERCA) in sarcoplasmic reticulum (SR) of fast-twitch, slow-twitch, and atrial muscle (1, 2). SERCA is a 110-kDa membrane protein that relaxes muscle by pumping calcium out of the cytoplasm using energy derived from ATP hydrolysis (3). Transgenic mouse studies have demonstrated that knock-out of SLN enhances atrial contractility (4), cross-expression of SLN inhibits ventricular contractility (2, 5, 6), and β-adrenergic stimulation relieves SLN inhibition of contractility (2, 6, 7). Phosphorylation/dephosphorylation of SLN is responsible for SERCA regulation, controlling the rate and amount of calcium loading in SR, which in turn determines the rate of muscle relaxation and the force of subsequent contraction (2, 4–7). SLN gene expression is up-regulated 2–15-fold in patients with skeletal muscle dysferlinopathy and Takotsubo cardiomyopathy, but down-regulated 2–3-fold in patients with heart failure, atrial fibrillation, and congenital heart defects, indicating that SLN acts as a causative or compensatory factor in human diseases of skeletal and cardiac muscle (8–12).
SLN comprises a single transmembrane (TM) helix, plus a small cytoplasmic phosphorylation domain and a short lumenal tail (2, 7, 13–15). Dephosphorylated SLN inhibits the apparent calcium affinity of SERCA (1, 2, 16–19) and decreases the calcium/ATP coupling efficiency (20, 21). Inhibition of SERCA is relieved by phosphorylation of SLN at serine-4 by serine/threonine kinase 16 (STK16) or threonine-5 by calcium/calmodulin-dependent protein kinase II (CaMKII) (2, 6, 7). SLN is monomeric on SDS-PAGE but aggregates when purified in non-ionic detergent, suggesting that SLN oligomerizes in SR membranes (17). When incorporated in thiolipid bilayers, SLN acts as an ATP-activated anion channel selective for inorganic phosphate, with partial functional similarity to the unidentified phosphate channel in SR (22–24). There is ample evidence demonstrating that SLN regulation of SERCA is a major determinant of muscle contractility, but the physiological role and oligomeric structure of the SLN channel are unknown.
SERCA comprises ten TM helices that form two calcium transport sites, plus five short lumenal loops and three large cytoplasmic domains responsible for ATP utilization (nucleotide-binding, phosphorylation, actuator) (3). The SERCA1a isoform is expressed in fast-twitch skeletal muscle, while SERCA2a is expressed in cardiac and slow-twitch skeletal muscle (25). SLN inhibits SERCA1a and SERCA2a similarly (2, 16). The SERCA-SLN regulatory complex is stabilized by interactions between cytoplasmic, lumenal, and TM regions of the two proteins (18, 19, 26–28). Functional mutagenesis indicates that isoleucine-17 (I17) in the TM domain of SLN is involved in SERCA inhibition, either by direct interaction or allosteric effect (26, 28). Molecular modeling predicts that monomeric SLN binds a TM groove of SERCA to form a binary complex (N = 1:1) where I17 of SLN faces lipid hydrocarbon chains (26). Electron microscopy identifies higher order oligomers of SLN (N∼5) interdigitated between strands of SERCA dimers (N = 2) in two-dimensional co-crystals (29). There is ample evidence demonstrating that SLN and SERCA form a regulatory complex, but the subunit stoichiometry and binding interface remain unknown.
It is clear that major questions remain concerning the oligomeric interactions of SLN and SERCA. Definitive answers to these questions require direct and quantitative measurements of SLN-SLN and SERCA-SLN interactions in living cells. Fluorescence resonance energy transfer (FRET) microscopy using fluorescent fusion proteins has been used to detect interactions of SERCA and its other regulatory partner phospholamban (PLB) (30–33). In the present study, we have used FRET microscopy and spectroscopy with fluorescent fusion proteins to detect molecular interactions of SLN with itself (self-association) and SERCA (regulatory complex formation). Relative binding affinities and subunit stoichiometries were determined by FRET analysis and molecular modeling. Site-directed mutagenesis of SLN was used to perturb self-association and SERCA binding to gain insight into their structural determinants. Results obtained for SLN and SERCA1a are compared with the well-studied oligomeric interactions of PLB and SERCA2a (reviewed in Refs. 34–36).
EXPERIMENTAL PROCEDURES
Materials
RNA WizTM total RNA isolation kit, Poly(A)-PureTM mRNA isolation kit, and ProSTARTM RT-PCR cloning kit were purchased from Ambion. cDNA encoding rabbit SERCA1a (Enzyme Collection Number 03.06.03.08) was provided by Dr. David MacLennan (University of Toronto). cDNAs encoding CFP and YFP were purchased from Clontech (37). DNA mutagenesis QuikChangeTM kit was purchased from Stratagene. Baculovirus transfer plasmid pAcSG2 was purchased from Orbigen. BaculoGoldTM baculovirus DNA was purchased from Pharmingen. Sf21 insect cells and bis-arsenical fluorescein (FLASH) were purchased from Invitrogen. Anti-phosphoserine-PLB/SLN antibody (mouse monoclonal) was purchased from Pierce Antibodies (2). Anti-phosphothreonine antibody (rabbit polyclonal) was purchased from Fitzgerald Industries (7). Bacterial expression plasmid pProEx HTc and anti-tetrahistidine antibody (mouse monoclonal) were purchased from Invitrogen.
Cloning and Mutagenesis
RT-PCR was used to clone SLN cDNA from rabbit fast-twitch muscle and PLB cDNA from dog ventricular muscle. The protein sequence of rabbit SLN was the same as previously described (GenBankTM Accession Number U96091) (13). The protein sequence of dog PLB was the same as previously described (GenBankTM Accession Number M16012) (38). The protein sequences of SLN and PLB were aligned manually (supplemental Fig. S1). cDNA encoding CFP or YFP was fused to the initiator methionine codon of SLN cDNA using a restriction enzyme/ligation strategy (Nco1/Sac1) that inserted three amino acids (glycine-glutamate-leucine) (37). Residue alanine-206 of CFP and YFP was mutated to lysine (A206K) to disrupt dimerization by GFP and its mutant derivatives (39). SLN residue isoleucine-17 was mutated to alanine (I17A) to disrupt oligomeric interactions of SLN. SERCA residue arginine-671 was mutated to cysteine (R671C) to create the tetra-cysteine motif (670CRRACC to 670CCRACC) required for labeling with bis-arsenical fluorescein (FLASH) (40).
Molecular Modeling
The molecular model of CFP-SERCA1a in the calcium-free state was constructed using DS Visualizer (Accelrys, San Diego, CA) and Fusion Protein Modeler (FPMOD) as previously described (37). Molecular models for CFP-SLN and YFP-SLN were constructed similarly using the NMR structure determined for human SLN (PDB accession 1JDM), fused to the x-ray crystal structures of CFP (PDB accession 1OXD) and YFP (PDB accession 1YFP). The molecular model of FLASH-SERCA was constructed using the crystal structure of calcium-free SERCA with ATP-bound (PDB accession 3AR4) (41), modifying the structure to include the R671C mutation for fluorescent labeling. For helical wheel analysis, we assumed 3.5 residues per turn for the TM domain of SLN (Fig. 1, supplemental Fig. S2). The model for SLN pentamer assembly was constructed using standard zipper positions a and d, similar to assembly of the PLB pentamer (Fig. 1, supplemental Fig. S2). Two models of the SLN dimer were constructed using alternate modes (symmetric and asymmetric) derived from the SLN pentamer (supplemental Fig. S2).
FIGURE 1.

Molecular models of SLN, SERCA, and PLB. Construction of molecular models is described under “Experimental Procedures.” CFP-SLN: CFP fused to the N terminus of SLN, with CFP in cyan and SLN in red. YFP-SLN: YFP fused to the N terminus of SLN, with YFP in yellow and SLN in red. CFP-SERCA: CFP fused to N terminus of SERCA, with CFP in cyan and SERCA actuator domain in red, nucleotide-binding domain in green, phosphorylation domain in blue, and transmembrane domain in gray (37). TM-SLN: transmembrane domain of SLN as a 3.5 residue-per-turn helix. The I17A mutation is shown in red. The T18A mutation disrupts SLN channel activity (24). TM-PLB: transmembrane domain of PLB as a 3.5 residue-per-turn helix. Isoleucine and leucine zipper residues are shown in red (34).
Baculovirus Expression
CFP-SLN and YFP-SLN were expressed as previously described for CFP-SERCA1a and YFP-PLB (37, 42, 43). Spodoptera frugiperda (Sf21) insect cells were cultured in shaker flasks at 27 °C in the dark. Cells were seeded at 0.5 × 106 cells/ml and passed at 48 h. Recombinant baculoviruses were generated by homologous recombination following cotransfection of Sf21 cells with baculovirus transfer plasmid DNA and linearized Autographica californica nuclear polyhedrosis baculovirus DNA (BaculoGoldTM). Recombinant baculoviruses were isolated by plaque purification and cultured through three rounds of expansion. Final virus stocks were titered using a plaque assay. Sf21 cells were coinfected with 2 viruses per cell for CFP fusion proteins and 3 viruses per cell for YFP fusion proteins. For microscopy, 6.0 × 105 cells were infected in 60 mm2 glass-bottom dishes. For spectroscopy, 9.0 × 106 cells were infected in 75-cm2 culture flask. After 48 h of infection, FRET was measured using acceptor-photobleach microscopy (live cells) or steady-state spectroscopy (cell homogenates). The protein concentration of Sf21 cell homogenates was measured using the Lowry assay with bovine serum albumin (BSA) as standard (44).
SDS-PAGE and Immunoblotting
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was run using pre-cast gels (4–15%) according to manufacturer instructions (Bio-Rad). In-gel fluorescence of fluorescent fusion proteins was quantified using the STORM 840 densitometer (Molecular Devices). Coomassie-stained protein bands were quantified using an Odyssey densitometer (LI-COR Biosciences). Immunoblotting was performed using phosphoserine and phosphothreonine antibodies for SLN expression in Sf21 cells, and a tetrahistidine antibody for SLN expression in Escherichia coli cells. Primary antibodies were labeled using fluorescent secondary antibodies (goat anti-mouse or goat anti-rabbit) from LI-COR Biosciences. Secondary antibody binding was quantified using an Odyssey densitometer. Prior to electrophoresis, gel samples were typically solubilized in 1.0% SDS for 5 min at 23 °C. Gel samples were also solubilized in low SDS (0.1%) or high temperature (42 °C, 65 °C, 100 °C) to perturb oligomer formation of fluorescent fusion proteins (supplemental Fig. S3).
FRET Spectroscopy
Steady-state fluorescence spectroscopy measurements were recorded on a K2 fluorometer (ISS, Inc.) equipped with a Xenon arc lamp. The standard buffer was 100 mm KCl, 3 mm MgCl2, 50 mm MOPS (pH 7.0) with a 2.0 ml volume in a 1 cm2 cuvette. Samples were stirred continuously at 23 °C with a protein concentration of 50 μg/ml. Sf21 homogenates expressing CFP-SLN were excited at 420 nm with an emission spectrum from 450–650 nm, while Sf21 homogenates expressing YFP-SLN were excited at 480 nm with an emission spectrum from 510–650 nm. Homogenates coexpressing CFP-SLN+YFP-SLN or CFP-SLN+R671C-SERCA were excited at 420 nm with emission spectrum of 450–650 nm. SERCA was labeled by adding 0.1 μm FLASH to the cuvette (supplemental Fig. S4) (40).
Confocal Microscopy
Localization of YFP-SLN in Sf21 cells was determined using a Bio-Rad 1024 Multiphoton Confocal Microscope system equipped with a 60× oil-immersion objective (1.40 numerical aperture) (Fig. 2A, supplemental movie S1). A Spectra Physics Tsunami Ti-Sapphire laser was used for two-photon excitation (780 nm), as detected with a 575 ± 150 nm bandpass filter and photomultiplier tube detector. ImageJ software (NIH) was used for three-dimensional image reconstruction of cells expressing YFP-SLN (supplemental movie S1).
FIGURE 2.

Baculovirus expression of fluorescent fusion proteins in Sf21 cells. A, confocal microscopy of YFP-SLN expressed in a live Sf21 cell. B, SDS-PAGE of Sf21 cell homogenates with detection by in-gel fluorescence (left) and Coomassie staining (right). Sf21 cells were infected with recombinant baculoviruses encoding CFP-SERCA1a (lane 1), CFP-SLN (lane 2), YFP-SLN (lane 3), and YFP-I17A (lane 4).
FRET Microscopy
Fluorescence microscopy measurements were recorded on an Eclipse TE200 microscope (Nikon Instruments, Inc.) equipped with a 20× dry objective (0.45 numerical aperture), an X-Cite metal-halide lamp (EXFO Corp.), and a Cascade II CCD camera (Photometrics Corp.) (supplemental Fig. S5). Metamorph software (Molecular Devices Corp.) was used for fluorescence quantitation on a cell-by-cell basis.
FRET was used to quantify the physical interaction between fluorescent fusion proteins with cyan and yellow probes. Energy transfer efficiency (FRET) was calculated from the Förster equation,
where FDA and FD are the fluorescence intensities of the donor in the presence and absence of the acceptor, respectively (45). Here, CFP was used as the donor and YFP was used as the acceptor (39). FDA and FD are the fluorescence of the donor (CFP) before and after selective photobleaching of the acceptor (YFP) (39). YFP was selectively photobleached using full lamp intensity in 12 intervals of 3 min for a total of 36 min. Fluorescence intensities of CFP fusion proteins and YFP fusion proteins, coexpressed in Sf21 insect cells, were recorded after each interval of acceptor-selective photobleaching of YFP (supplemental Fig. S5). FRET is expressed as mean ± standard error of the mean (S.E.), with n indicating the number of cells assayed (Table 1).
TABLE 1.
Summary of FRET microscopy
Mean FRET was calculated using all cells assayed for each interaction (n). Kd values were calculated from FRET binding curves. Oligomer number (N) was determined using a stoichiometry plot of donor recovery versus acceptor photobleaching. Error is reported as S.E. Pairwise comparison of all four protein interactions show significance of p < 0.01 (denoted by **).
| Protein interaction | SLN-SLN | SLN-I17A | SERCA-SLN | SERCA-I17A |
|---|---|---|---|---|
| Mean FRET efficiency | 0.344 ± 0.005** | 0.245 ± 0.005** | 0.273 ± 0.008** | 0.177 ± 0.008** |
| Kd (AU) | 1.00 ± 0.03** | 1.35 ± 0.08** | 2.08 ± 0.18** | 3.01 ± 0.28** |
| Oligomer number (N) | >2 | = 2 | = 2 | = 2 |
| Number of cells (n) | 417 | 355 | 198 | 114 |
FRET Interactions Characterized by a Hyperbolic Binding Model
Fluorescence microscopy was also used to analyze the relative binding affinities of protein complexes. FRET between two fluorescent proteins typically exhibits a hyperbolic dependence on acceptor concentration, permitting the calculation of an apparent dissociation constant (Kd) (39, 45, 46). Kd values were determined by nonlinear curve fitting of binned data with the Origin software (Origin Lab Corp.) using Equation 2,
where Kd is the apparent dissociation constant for the protein-protein interaction, and FRETMAX is the asymptotic value that depends on the donor-acceptor distance in the bound complex. This approach has been used previously to measure relative binding affinities for SERCA-PLB and PLB-PLB interactions (31). Initial analysis demonstrated that FRETMAX values were statistically equivalent between wild-type and mutant paired interactions, indicating that there is no donor-acceptor distance change within the complexes due to I17A mutation. Therefore, we used the FRETMAX values of SLN-SLN and SERCA-SLN to calculate the binding affinities for mutant interactions. Values reported in this study are expressed as mean ± S.E. (n), with n indicating the number of cells assayed. To determine p values for pairwise comparisons, a z score was calculated using Equation 3,
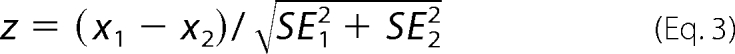 |
where p < 0.05 was considered significant (47). Based on quantitative densitometry of gels, the SLN and SERCA levels in Sf21 cells were typically 4% SLN and 1.5% SERCA by weight (molar ratio of 12 SLN/SERCA), corresponding to a fluorescence intensity of 3.1 AU for YFP-SLN. Expression level of YFP-SLN was normalized to fluorescence units, and the conversion factor (2.48 AU/nmol/mg) was applied to SLN expression in muscle, as indicated on FRET binding curves (Fig. 5).
FIGURE 5.

Binding affinity determined by FRET microscopy. A plot of FRET efficiency versus acceptor expression was fit by a hyperbolic binding curve (Eq.2) for each protein-protein interaction. The dissociation constant (Kd) is the YFP-SLN concentration giving half-maximal FRET. A, SLN-SLN (Kd1) and SLN-I17A (Kd1′). B, SERCA-SLN (Kd2) and SERCA-I17A (Kd2′). Symbols indicate mean ± S.E. (n = 6 for SLN-SLN and n = 4 for SERCA-SLN). All four Kd values show pairwise significance (p < 0.01). Blue arrows indicate the relative level of SLN expression in muscle tissue, as reported for mouse heart (Atria) and rabbit fast-twitch muscle (EDL) (57). Based on gel densitometry, the mean SERCA content of Sf21, atria, and EDL homogenates is 0.107 ± 0.025 nmol/mg, 0.090 ± 0.009 nmol/mg (57), and 0.880 ± 0.065 nmol/mg (57), respectively.
Antibacterial Assay
SLN and PLB were expressed in E. coli strain BL21(DE3) as N-terminal fusion proteins with a six-histidine residue tag (His tag). A plating assay of colony formation was used to determine the effect of SLN and PLB expression on E. coli viability. Expression of SLN and PLB was induced by addition of 0.6 mm IPTG. Two hours after induction, E. coli cells were plated, then incubated for 16 h at 37 °C for colony formation. E. coli cells were also induced with His-tagged chloramphenicol transferase (CAT) as standard control (non-lethal). Antibacterial assay results are reported as mean ± S.E. and were repeated at least three times (n≥3).
RESULTS
Baculovirus Expression of Fluorescent Fusion Proteins in Sf21 Cells
CFP and YFP were fused to the N terminus of SLN in the cytoplasmic phosphorylation domain (Fig. 1, upper). Fluorescent fusion proteins of SLN were expressed in S. frugiperda insect cells (Sf21) via baculovirus infection (Fig. 2). Fluorescence microscopy demonstrated that CFP-SLN and YFP-SLN are localized to endoplasmic reticulum (ER) when expressed in Sf21 cells (Fig. 2A, supplemental Fig. S5, supplemental movie S1). Similarly, SLN labeled with green fluorescent protein at the N terminus (GFP-SLN) is targeted to ER of HEK cells (48). To analyze protein expression, Sf21 cell homogenates were subjected to SDS-PAGE, where Coomassie staining and in-gel fluorescence imaging identified CFP-SLN and YFP-SLN as 30-kDa monomers (Fig. 2B). In-gel fluorescence imaging also identified a low but significant amount of 60-kDa dimers (supplemental Fig. S3, left), an oligomeric form of SLN previously unobserved on SDS-PAGE (17, 49). The amount of dimeric SLN on SDS-PAGE was variable, with a range of 2–16% dimer of total SLN (compare Fig. 2B, left, to supplemental Fig. S3, left). Immunoblotting with phosphoserine and phosphothreonine antibodies (2, 7) demonstrated that SLN is dephosphorylated when expressed in Sf21 cells. Densitometry of Coomassie-stained gels determined that CFP-SLN and YFP-SLN are expressed as ∼5% of the total protein in Sf21 cells (Fig. 2B).
We previously used baculovirus expression and FRET spectroscopy to investigate intramolecular domain movements of SERCA1a with CFP fused to the N terminus in the actuator domain (37) (Fig. 1, upper). Both CFP-SERCA1a and wild-type SERCA are targeted to ER of Sf21 cells following baculovirus infection (37, 50). On SDS-PAGE, CFP-SERCA1a migrated at 115–125 kDa, giving a doublet of Coomassie staining bands, one fluorescent (lower band) and one non-fluorescent (upper band) (Fig. 2B). The fluorescence of CFP-SERCA1a was sensitive to SDS and heat, with the lower fluorescent band converting to the upper non-fluorescent band upon denaturation of CFP (supplemental Fig. S3, right) (see also Ref. 51). Densitometry of Coomassie-stained gels demonstrated that CFP-SERCA1a is expressed at ∼5% of total protein in Sf21 cell homogenates (Fig. 2B, supplemental Fig. S3, right). Protease accessibility experiments indicate that fluorescent fusion proteins of SLN and SERCA exhibit native topology when heterologously expressed in ER membranes (37, 48). We conclude that fluorescent fusion proteins of SLN and SERCA are efficiently expressed, correctly localized, and properly folded in Sf21 cells.
FRET Spectroscopy of SLN-SLN Interactions in Sf21 Cell Homogenates
Self-association of SLN was examined in Sf21 cell homogenates using FRET spectroscopy with CFP-SLN as donor and YFP-SLN as acceptor. When excited at 420 nm, CFP-SLN expressed by itself exhibited 10-fold higher fluorescence than control homogenates, producing an emission maximum at 480 nm with a secondary shoulder at 512 nm (Fig. 3A). When excited at 480 nm, YFP-SLN expressed by itself exhibited 50-fold higher fluorescence than control homogenates, producing an emission maximum at 520 nm (Fig. 3B). Thus, the baculovirus system gives characteristic emission spectra for CFP and YFP fusion proteins and provides an optimal signal-to-noise ratio for fluorescence spectroscopy (Fig. 3).
FIGURE 3.

FRET spectroscopy of SLN-SLN interactions in Sf21 cell homogenates. A, fluorescence of CFP-SLN and control homogenates, with CFP-selective excitation at 420 nm. B, fluorescence of YFP-SLN and control homogenates, with YFP-selective excitation at 480 nm. C, fluorescence of homogenates coexpressing CFP-SLN and YFP-SLN, with CFP-selective excitation at 420 nm. Detergent was added to dissociate (C12E8) or denature (SDS) SLN. D, fluorescence of homogenates coexpressing CFP-SLN and YFP-SLN, with CFP-selective excitation at 420 nm. YFP-SLN and control homogenates were also scanned.
When Sf21 homogenates coexpressing CFP-SLN and YFP-SLN were excited using CFP-selective excitation at 420 nm, CFP-SLN (donor) displayed low fluorescence at 480 nm while YFP-SLN (acceptor) showed high fluorescence at 520 nm, indicating FRET between SLN molecules (Fig. 3, C and D). Addition of the non-ionic detergent C12E8 increased CFP-SLN fluorescence by 10% and decreased YFP-SLN fluorescence by 25%, indicating that C12E8 disrupts self-association and FRET between SLN molecules. Control experiments determined that fluorescence of CFP-SLN homogenates decreased slightly (5–10%) in the presence of C12E8, demonstrating that addition of detergent did not produce the observed increase in donor fluorescence (Fig. 3C). YFP-SLN expressed by itself showed low fluorescence using CFP-selective excitation at 420 nm, but coexpression of CFP-SLN increased YFP-SLN emission by 2.5-fold, another indicator of FRET between SLN molecules (Fig. 3D). Thus, self-association of SLN is demonstrated by multiple parameters of FRET spectroscopy (donor quenching, detergent dissociation, acceptor sensitization).
FRET Microscopy of SLN-SLN Interactions in Live Sf21 Cells
Self-association of SLN was further examined using FRET microscopy with CFP/YFP-tagged proteins coexpressed in live Sf21 cells (Fig. 1, supplemental Fig. S5). Acceptor-selective photobleaching experiments were used to determine mean FRET and subunit stoichiometry of SLN self-association (Equation 1). Progressive acceptor photobleaching decreased fluorescence of YFP-SLN exponentially while increasing the fluorescence of CFP-SLN concurrently, an indicator of FRET between SLN molecules (Fig. 4A). Oligomerization of SLN was examined using a plot of CFP fluorescence recovery as a function of YFP fluorescence bleaching (Fig. 4C, black) (see also Refs. 31, 46). This plot is nonlinear, indicating that SLN forms higher order oligomers (N>2), in addition to monomers and dimers observed on SDS-PAGE (Fig. 2B; supplemental Fig. S3, left). Mean FRET for the SLN-SLN interaction is 0.344 ± 0.005 (n = 417 cells), indicating a high degree of association between CFP-SLN and YFP-SLN (Fig. 4C, black).
FIGURE 4.

FRET microscopy of SLN-SLN and SERCA-SLN interactions in live Sf21 cells. A, B, D, E, acceptor photobleaching and donor recovery in cells coexpressing the indicated proteins. C and F, plots of donor recovery versus acceptor photobleaching to determine mean FRET efficiency and oligomer number, N. Error is reported as mean ± S.E., and all four mean FRET values show pairwise significance (p < 0.01). SLN-SLN experiments were performed using six photobleaches (n = 6) (C) while SERCA-SLN experiments were performed using four photobleaches (n = 4) (F).
To disrupt self-association, SLN residue isoleucine-17 was mutated to alanine (I17A), analogous to the I40A mutation that disrupts pentamer formation of PLB (Fig. 1, bottom; supplemental Fig. S1). YFP-I17A was mostly monomeric on SDS-PAGE (Fig. 2B), but also formed a low amount of dimer, similar to CFP-SLN and YFP-SLN (supplemental Fig. S3, left). CFP-SLN and YFP-I17A were coexpressed in Sf21 cells, and acceptor photobleaching determined a mean FRET of 0.245 ± 0.005 for the SLN-I17A interaction (n = 355 cells) (Fig. 4C, red; Equation 1), a decrease of 0.099 ± 0.007 (p < 0.01) compared with the SLN-SLN interaction. The relation of donor recovery to acceptor bleach for CFP-SLN+YFP-I17A was linear on the stoichiometry plot (Fig. 4C, red), indicating that SLN-I17A associate as dimers (N = 2) but not higher order oligomers. We conclude that SLN monomers show a strong propensity to form dimers and higher order oligomers in ER, and that SLN residue I17 is essential for the formation of higher order oligomers.
FRET Microscopy of SERCA-SLN Interactions in Live Sf21 Cells
To investigate regulatory complex formation, CFP-SERCA1a and YFP-SLN were coexpressed in Sf21 cells. In-gel fluorescence imaging showed that the SERCA-SLN complex is completely dissociated on SDS-PAGE. Progressive acceptor photobleaching produced an exponential decrease of YFP-SLN fluorescence and a concurrent increase in CFP-SERCA fluorescence, indicating FRET between SERCA and SLN molecules (Fig. 4F, black). The mean FRET for the SERCA-SLN interaction is 0.273 ± 0.008 (n = 198 cells) (Fig. 4F, black; Equation 1), less than that observed for SLN self-association (Fig. 4C, black). The relation of donor recovery to acceptor photobleach was linear on the stoichiometry plot, indicating that SERCA-SLN form a heterodimer (N = 2) (Fig. 4F), in contrast to SLN-SLN homodimers and higher order oligomers (Fig. 4C). FRET spectroscopy also demonstrated substantial FRET (E∼0.22) between CFP-SLN and FLASH-labeled SERCA in Sf21 cell homogenates, similar to FRET between CFP-SERCA and YFP-SLN, indicating that SERCA-SLN association is not an artifact of dimerization by CFP and YFP (supplemental Fig. S4).
To determine whether SLN residue I17 is involved in SERCA binding, YFP-I17A was coexpressed with CFP-SERCA1a in Sf21 cells. FRET microscopy determined that mean FRET for the SERCA-I17A interaction is 0.177 ± 0.008 (n = 114 cells) (Fig. 4F, red; Equation 1), a decrease of 0.096 ± 0.011 compared with the SERCA-SLN interaction (p < 0.01) (Fig. 4F, black). Stoichiometry plot analysis demonstrated that SERCA-I17A form a heterodimer similar to SERCA-SLN (Fig. 4F). We conclude that SLN monomers bind SERCA monomers to form heterodimers in ER, and that SLN residue I17 is involved in stabilizing the SERCA-SLN interaction.
Binding Affinity Determined by FRET Microscopy
Binding curves for the SLN-SLN and SERCA-SLN interactions were constructed using FRET versus acceptor expression (Fig. 5) (31, 32, 39). Both interactions showed increasing FRET with increasing acceptor levels, revealing a hyperbolic dependence. SLN-SLN exhibits higher maximum FRET (0.575 ± 0.020) than SERCA-SLN (0.459 ± 0.059) (Fig. 5, Equation 2), indicating that SLN dimers and oligomers are more compact on average than SERCA-SLN dimers (p < 0.05) (Fig. 4).
The dissociation constant (Kd) for each protein-protein interaction was defined as the concentration of acceptor at which half-maximal FRET efficiency was calculated (Eq. 2). SLN-SLN exhibited a dissociation constant of 1.00 ± 0.03 AU (Kd1) (Fig. 5A, black). SLN-I17A showed a dissociation constant of 1.35 ± 0.08 AU (Kd1′) (Fig. 5A, red), demonstrating that the I17A mutation decreases the binding affinity of SLN self-association (p < 0.01) in addition to destabilizing higher order oligomers formed by wild-type SLN (Fig. 4C). SERCA-SLN exhibited a dissociation constant of 2.08 ± 0.18 AU (Kd2) (Fig. 5B, black), a 2-fold lower apparent binding affinity than SLN-SLN (p < 0.01). SERCA-I17A showed a Kd of 3.01 ± 0.28 AU (Kd2′) (Fig. 5B, red), demonstrating that the I17A mutation decreases the binding affinity of SERCA-SLN dimers (p < 0.01). Kd1 and Kd2 are linked equilibria, so Kd1 is an inherent component of Kd2 for our observed SERCA-SLN binding (Fig. 5).
Antibacterial Assay of SLN and PLB
To obtain independent information about the oligomeric state of SLN, we expressed SLN and I17A in E. coli and measured cell viability, reasoning that higher order oligomers are more likely to increase membrane permeability and cell death than monomers and dimers. Both SLN and PLB show channel activity when reconstituted in lipid bilayers (22–24, 52–54). Expression of PLB in E. coli results in cell lysis, presumably due to formation of pentameric ion channels (55). Here, we tested whether SLN is lethal to E. coli using a plating assay to measure cell viability (Fig. 6). Expression of chloramphenicol transferase (CAT) slightly increased colony formation (120%), while expression of PLB decreased colony formation to 40% (p < 0.01) (Fig. 6), consistent with previous results demonstrating that PLB exhibits antibacterial activity (55). Expression of SLN decreased colony formation to 5% (Fig. 6), indicating that SLN shows greater antibacterial activity than PLB (p < 0.01). In contrast, the I17A mutation in SLN completely restored E. coli viability (p < 0.01) (Fig. 6), probably due to disruption of higher order oligomers, which decrease membrane permeability (56). Thus, the antibacterial assay provides additional evidence, independent of FRET data on fluorescent fusion proteins in insect cells (Fig. 4C), that SLN forms higher order oligomers stabilized by residue I17.
FIGURE 6.

Antibacterial assay of SLN and PLB. His-tagged proteins were expressed in E. coli. Cell viability was determined by a plating assay of colony formation. Proteins expressed were chloramphenicol transferase (CAT), PLB, SLN, and I17A. Cell viability was determined after protein expression, as normalized to the number of colonies before induction (CON). Immunoblotting using His tag antibody demonstrated similar expression levels for PLB, SLN, and I17A. Error is reported as mean ± S.E. (n ≥ 3). While CON and CAT show statistical equivalence, all other pairwise comparisons are statistically different (p < 0.01).
DISCUSSION
Oligomerization of SLN
SLN has been found to be primarily monomeric in SDS and DPC micelles (N = 1) (14, 18, 49, 57). Self-association of SLN, including higher order oligomers, has been observed after cross-linking in detergent solution (17). Our FRET microscopy data in living cells, which is the first measurement of SLN-SLN interactions in membranes, clearly show substantial oligomeric interactions of SLN in the nonphosphorylated state (Fig. 4). Indeed, the curvature of Fig. 4C (black) provides direct evidence for higher order oligomers (N>2). Gel assays typically preclude measurement of membrane protein interactions, so direct measurement of such interactions by FRET yields the highest resolution for the detection of protein-protein binding.
There is strong evidence that our detected oligomeric interactions of SLN are not artifacts of aggregation by fluorescent fusions proteins. First, the addition of detergent decreases these interactions, as shown by SDS-PAGE (Fig. 2) and by the loss of FRET in the presence of the non-ionic detergent C12E8 (Fig. 3C). Second, all FRET microscopy assays in this study were performed with CFP and YFP derivatives that contain mutations to prevent dimerization (39, 58). Third, mutation of SLN decreases FRET, indicating that SLN residues are responsible for oligomerization of fluorescent fusion proteins. It is conceivable that the fluorescent fusion proteins could cause steric hindrance, resulting in underestimates of native SLN oligomer formation. However, this seems unlikely, in light of the substantial oligomeric interactions detected for both SLN-SLN and SERCA-SLN (Figs. 4 and 5). Because of limitations in molecular modeling due to size contributions of CFP and YFP (46), the specific number of SLN subunits in higher order oligomers could not be resolved here. Thus, our FRET assays exhibit a composite dissociation constant defined as Kd1 for assembly of SLN dimers and higher order oligomers (Fig. 7).
FIGURE 7.

Proposed model for oligomeric interactions of SLN and SERCA. SLN monomers self-assemble into dimers and higher order oligomers (Kd1). SLN monomers also bind to SERCA monomers to form dimeric inhibitory complexes (Kd2). The I17A mutant of SLN eliminates higher order oligomers of SLN (Kd1′) and decreases inhibitory complex formation (Kd2′). Higher order oligomers of SLN are responsible for antibacterial activity.
The presence of one I17A mutation decreases FRET of SLN self-association by one-third and converts higher order oligomers of SLN into dimers and monomers (lack of curvature in Fig. 4C, red). Helical wheel analysis and sequence comparison indicate that I17-SLN is analogous to I40-PLB at zipper position d (Fig. 1, lower; supplemental Fig. S1), and homology modeling suggests that SLN forms pentamers like PLB using similar leucine-isoleucine zippers (Fig. 1, supplemental Fig. S2). Structural modeling further suggests that the SLN dimer could occupy two possible conformations: symmetric or asymmetric (supplemental Fig. S2). FRET results for the CFP-SLN+YFP-I17A interaction are consistent with either symmetric or asymmetric dimer assembly. The same ambiguity exists for the I17A-I17A dimer. Additional spectroscopic studies with I17A and other SLN mutants are needed to better define these interacting surfaces.
Further evidence that the I17A mutation disrupts a leucine-isoleucine zipper comes from our antibacterial assay (Fig. 6). SLN and PLB significantly reduce E. coli viability, while the oligomer-destabilizing mutation I17A restores colony formation for SLN (Fig. 6). Both SLN and PLB form oligomeric ion channels (24, 55), suggesting that decreased E. coli viability is caused by increased membrane permeability due to channel formation. For comparison, a leucine zipper enhances cytotoxicity for an antimicrobial peptide (59).
Binding of SLN and SERCA
SERCA and SLN copurify from SR vesicles and HEK cell microsomes when solubilized by non-ionic detergent (16, 49), while SERCA and “proteolipid” (probably SLN) exhibit low FRET (0.05–0.10) by spectroscopy in reconstituted membranes (60). However, none of these studies measured binding stoichiometry. In the present study, we used fluorescence microscopy in live cells to observe robust FRET (0.273 ± 0.008) between CFP-SERCA and YFP-SLN, and to demonstrate that SLN binds SERCA in a 1:1 ratio (linear plot in Fig. 4F, black). The location of SLN in the binary regulatory complex remains unclear, but the substantial decrease in SERCA-SLN FRET due to the I17A mutation indicates that I17 is important not only for stabilization of SLN-SLN interaction, but also for stabilization of SERCA-SLN interaction. Molecular modeling predicts that I17 is lipid-facing in the SERCA-SLN complex (26), but co-immunoprecipitation and functional studies indicate that I17 is important for interaction with SERCA (1, 26). The simplest interpretation is that the I17A mutation induces allosteric effects on SLN structure that disrupt functional interaction with SERCA, but it is also possible that I17 interacts directly with SERCA.
Proposed Model of Oligomeric Interactions of SLN and SERCA in SR
FRET microscopy has enabled the measurement of binding affinities and stoichiometries for SLN-SLN and SERCA-SLN in ER of live Sf21 cells. We propose that SLN in SR exists as multiple molecular species: monomers, dimers, oligomers, and heterodimers with SERCA (Fig. 7). Previous molecular modeling indicates that SERCA has two adjacent TM grooves for possible binding of SLN (26). However, our results indicate that only one SLN is bound per SERCA, even when SLN is expressed in many-fold excess of SERCA (Figs. 4F and 5B). We propose that the SLN monomer is the molecular species that binds to and inhibits SERCA in SR.
Binding affinities of SLN-SLN and SERCA-SLN were determined in insect cell ER in the absence of other muscle proteins that directly interact with SERCA, SLN, or both (16, 27, 61). The dissociation constants described in this study apply to SLN-SLN and SERCA-SLN interactions in all muscle tissues because Kd values were determined using a distribution of expression levels, as produced by baculovirus infection of Sf21 cells. Fig. 5B illustrates that native expression levels of SLN in skeletal and cardiac muscle are lower than the Kd values determined by FRET microscopy, suggesting that less than half of the SERCA molecules in muscle are bound to SLN. It is likely that both dissociation constants (Kd1, Kd2) are regulated by SLN phosphorylation and SERCA ligands. We propose that SLN self-association directly competes with SERCA binding, with SLN-SLN binding affinity approximately equal to that of SERCA-SLN (Figs. 5 and 7).
Comparison of SLN and PLB Oligomeric Interactions
SERCA2a in ventricle is inhibited by PLB, a 6-kDa membrane protein that shows sequence homology to SLN (25, 31, 43, 45, 62–64) (Fig. 1, supplemental Fig. S1). Both SLN and PLB self-associate into higher order oligomers, and both SLN and PLB interact with SERCA in a binary inhibitory complex (Fig. 4 in this study, (62, 65)). I40 of PLB, located at the d position of the leucine-isoleucine zipper, is conserved in SLN as I17 (Fig. 1, supplemental Fig. S2). In PLB, the I40A mutation decreases pentamer formation and increases SERCA-PLB interactions due to formation of additional PLB monomers (31). Here we demonstrated that the homologous mutation I17A decreases both SLN-SLN and SERCA-SLN interactions (Figs. 4 and 5). Thus, SLN′s self-association is similar to that of PLB, but SLN′s interaction with SERCA has unique features.
CONCLUSIONS
We used the baculovirus system and fluorescent fusion proteins to directly detect the oligomeric interactions of SLN and SERCA. We identified the formation of SLN dimers and oligomers, in addition to SERCA-SLN dimers. We demonstrated that both interactions exhibit similar binding affinity and determined that residue Ile-17 of SLN acts as a critical stabilizing residue for both interactions. Important questions remain regarding SLN oligomerization and SERCA binding, including effects of SLN phosphorylation and SERCA ligands.
Supplementary Material
Acknowledgments
We thank Dr. David H. MacLennan (University of Toronto) for providing cDNA encoding rabbit SERCA1a, Bengt Svensson for molecular modeling of fluorescent fusion proteins (using facilities of the Minnesota Supercomputing Institute), Sarah Blakely and Octavian Cornea for administrative assistance, and Suzanne Haydon, Ji Li, and John Oja for helpful discussions on fluorescence microscopy. Microscopy and spectroscopy were performed in the Biophysical Spectroscopy Facility at the University of Minnesota.
This work was supported in part by Grant GM27906 from the National Institutes of Health (to D. D. T.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S5 and movie S1.
- SLN
- sarcolipin
- SERCA
- calcium-transporting ATPase
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein
- I17A-SLN
- sarcolipin mutant with isoleucine-17 to alanine substitution
- I40A-PLB
- phospholamban mutant with isoleucine-40 to alanine substitution
- SR
- sarcoplasmic reticulum
- ER
- endoplasmic reticulum
- TM
- transmembrane
- FRET
- fluorescence resonance energy transfer.
REFERENCES
- 1. Odermatt A., Becker S., Khanna V. K., Kurzydlowski K., Leisner E., Pette D., MacLennan D. H. (1998) J. Biol. Chem. 273, 12360–12369 [DOI] [PubMed] [Google Scholar]
- 2. Gramolini A. O., Trivieri M. G., Oudit G. Y., Kislinger T., Li W., Patel M. M., Emili A., Kranias E. G., Backx P. H., Maclennan D. H. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 2446–2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Toyoshima C., Nakasako M., Nomura H., Ogawa H. (2000) Nature 405, 647–655 [DOI] [PubMed] [Google Scholar]
- 4. Babu G. J., Bhupathy P., Timofeyev V., Petrashevskaya N. N., Reiser P. J., Chiamvimonvat N., Periasamy M. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 17867–17872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Asahi M., Otsu K., Nakayama H., Hikoso S., Takeda T., Gramolini A. O., Trivieri M. G., Oudit G. Y., Morita T., Kusakari Y., Hirano S., Hongo K., Hirotani S., Yamaguchi O., Peterson A., Backx P. H., Kurihara S., Hori M., MacLennan D. H. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 9199–9204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Babu G. J., Bhupathy P., Petrashevskaya N. N., Wang H., Raman S., Wheeler D., Jagatheesan G., Wieczorek D., Schwartz A., Janssen P. M., Ziolo M. T., Periasamy M. (2006) J. Biol. Chem. 281, 3972–3979 [DOI] [PubMed] [Google Scholar]
- 7. Bhupathy P., Babu G. J., Ito M., Periasamy M. (2009) J. Mol. Cell Cardiol. 47, 723–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nef H. M., Möllmann H., Troidl C., Kostin S., Voss S., Hilpert P., Behrens C. B., Rolf A., Rixe J., Weber M., Hamm C. W., Elsässer A. (2009) Eur. Heart J. 30, 2155–2164 [DOI] [PubMed] [Google Scholar]
- 9. Campanaro S., Romualdi C., Fanin M., Celegato B., Pacchioni B., Trevisan S., Laveder P., De Pittà C., Pegoraro E., Hayashi Y. K., Valle G., Angelini C., Lanfranchi G. (2002) Hum Mol. Genet. 11, 3283–3298 [DOI] [PubMed] [Google Scholar]
- 10. Shanmugam M., Molina C. E., Gao S., Severac-Bastide R., Fischmeister R., Babu G. J. (2011) Biochem. Biophys. Res. Commun. 410, 97–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Uemura N., Ohkusa T., Hamano K., Nakagome M., Hori H., Shimizu M., Matsuzaki M., Mochizuki S., Minamisawa S., Ishikawa Y. (2004) Eur. J. Clin. Invest. 34, 723–730 [DOI] [PubMed] [Google Scholar]
- 12. Vittorini S., Storti S., Parri M. S., Cerillo A. G., Clerico A. (2007) Mol. Med. 13, 105–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Odermatt A., Taschner P. E., Scherer S. W., Beatty B., Khanna V. K., Cornblath D. R., Chaudhry V., Yee W. C., Schrank B., Karpati G., Breuning M. H., Knoers N., MacLennan D. H. (1997) Genomics 45, 541–553 [DOI] [PubMed] [Google Scholar]
- 14. Mascioni A., Karim C., Barany G., Thomas D. D., Veglia G. (2002) Biochemistry 41, 475–482 [DOI] [PubMed] [Google Scholar]
- 15. Buffy J. J., Traaseth N. J., Mascioni A., Gor'kov P. L., Chekmenev E. Y., Brey W. W., Veglia G. (2006) Biochemistry 45, 10939–10946 [DOI] [PubMed] [Google Scholar]
- 16. Asahi M., Kurzydlowski K., Tada M., MacLennan D. H. (2002) J. Biol. Chem. 277, 26725–26728 [DOI] [PubMed] [Google Scholar]
- 17. Hellstern S., Pegoraro S., Karim C. B., Lustig A., Thomas D. D., Moroder L., Engel J. (2001) J. Biol. Chem. 276, 30845–30852 [DOI] [PubMed] [Google Scholar]
- 18. Buffy J. J., Buck-Koehntop B. A., Porcelli F., Traaseth N. J., Thomas D. D., Veglia G. (2006) J. Mol. Biol. 358, 420–429 [DOI] [PubMed] [Google Scholar]
- 19. Hughes E., Clayton J. C., Kitmitto A., Esmann M., Middleton D. A. (2007) J. Biol. Chem. 282, 26603–26613 [DOI] [PubMed] [Google Scholar]
- 20. Smith W. S., Broadbridge R., East J. M., Lee A. G. (2002) Biochem. J. 361, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mall S., Broadbridge R., Harrison S. L., Gore M. G., Lee A. G., East J. M. (2006) J. Biol. Chem. 281, 36597–36602 [DOI] [PubMed] [Google Scholar]
- 22. Becucci L., Guidelli R., Karim C. B., Thomas D. D., Veglia G. (2007) Biophys. J. 93, 2678–2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Becucci L., Cembran A., Karim C. B., Thomas D. D., Guidelli R., Gao J., Veglia G. (2009) Biophys. J. 96, L60–L62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Becucci L., Guidelli R., Karim C. B., Thomas D. D., Veglia G. (2009) Biophys. J. 97, 2693–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Periasamy M., Kalyanasundaram A. (2007) Muscle Nerve 35, 430–442 [DOI] [PubMed] [Google Scholar]
- 26. Asahi M., Sugita Y., Kurzydlowski K., De Leon S., Tada M., Toyoshima C., MacLennan D. H. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 5040–5045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gramolini A. O., Kislinger T., Asahi M., Li W., Emili A., MacLennan D. H. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 16807–16812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morita T., Hussain D., Asahi M., Tsuda T., Kurzydlowski K., Toyoshima C., Maclennan D. H. (2008) Biochem. Biophys. Res. Commun. 369, 188–194 [DOI] [PubMed] [Google Scholar]
- 29. Glaves J. P., Gorski P., Young H. S. (2009) Biophys. J. 96, 211a [Google Scholar]
- 30. Robia S. L., Campbell K. S., Kelly E. M., Hou Z., Winters D. L., Thomas D. D. (2007) Circ. Res. 101, 1123–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kelly E. M., Hou Z., Bossuyt J., Bers D. M., Robia S. L. (2008) J. Biol. Chem. 283, 12202–12211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hou Z., Kelly E. M., Robia S. L. (2008) J. Biol. Chem. 283, 28996–29003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hou Z., Robia S. L. (2010) J. Mol. Biol. 402, 210–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Simmerman H. K., Jones L. R. (1998) Physiol. Rev. 78, 921–947 [DOI] [PubMed] [Google Scholar]
- 35. Thomas D. D., Reddy L. G., Karim C. B., Li M., Cornea R., Autry J. M., Jones L. R., Stamm J. (1998) Ann. N.Y. Acad. Sci. 853, 186–194 [DOI] [PubMed] [Google Scholar]
- 36. MacLennan D. H., Kranias E. G. (2003) Nat. Rev. Mol. Cell Biol. 4, 566–577 [DOI] [PubMed] [Google Scholar]
- 37. Winters D. L., Autry J. M., Svensson B., Thomas D. D. (2008) Biochemistry 47, 4246–4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fujii J., Ueno A., Kitano K., Tanaka S., Kadoma M., Tada M. (1987) J. Clin. Invest. 79, 301–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zacharias D. A., Violin J. D., Newton A. C., Tsien R. Y. (2002) Science 296, 913–916 [DOI] [PubMed] [Google Scholar]
- 40. Griffin B. A., Adams S. R., Tsien R. Y. (1998) Science 281, 269–272 [DOI] [PubMed] [Google Scholar]
- 41. Toyoshima C., Yonekura S., Tsueda J., Iwasawa S. (2011) Proc. Natl. Acad. Sci. U.S.A. 108, 1833–1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Skerjanc I. S., Toyofuku T., Richardson C., MacLennan D. H. (1993) J. Biol. Chem. 268, 15944–15950 [PubMed] [Google Scholar]
- 43. Autry J. M., Jones L. R. (1997) J. Biol. Chem. 272, 15872–15880 [DOI] [PubMed] [Google Scholar]
- 44. Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]
- 45. Mueller B., Karim C. B., Negrashov I. V., Kutchai H., Thomas D. D. (2004) Biochemistry 43, 8754–8765 [DOI] [PubMed] [Google Scholar]
- 46. Li M., Reddy L. G., Bennett R., Silva N. D., Jr., Jones L. R., Thomas D. D. (1999) Biophys. J. 76, 2587–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Song Q., Pallikkuth S., Bossuyt J., Bers D. M., Robia S. L. (2011) J. Biol. Chem. 286, 9120–9126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Butler J., Lee A. G., Wilson D. I., Spalluto C., Hanley N. A., East J. M. (2007) Cardiovasc Res. 74, 114–123 [DOI] [PubMed] [Google Scholar]
- 49. MacLennan D. H. (1974) Methods Enzymol. 32, 291–302 [DOI] [PubMed] [Google Scholar]
- 50. Waggoner J. R., Huffman J., Griffith B. N., Jones L. R., Mahaney J. E. (2004) Protein Expr Purif 34, 56–67 [DOI] [PubMed] [Google Scholar]
- 51. Alkaabi K. M., Yafea A., Ashraf S. S. (2005) Appl Biochem. Biotechnol 126, 149–156 [DOI] [PubMed] [Google Scholar]
- 52. Kovacs R. J., Nelson M. T., Simmerman H. K., Jones L. R. (1988) J. Biol. Chem. 263, 18364–18368 [PubMed] [Google Scholar]
- 53. Smeazzetto S., Henkel M., Ferri T., Thiel G., Moncelli M. R. (2010) Biophys. J. 98, 328a [Google Scholar]
- 54. Aly A., Hunter M., Knowles P. F., Colyer J. (2001) Biophys. J. 80, 26A [Google Scholar]
- 55. Cook E. A., Huggins J. P., Sathe G., England P. J., Piggott J. R. (1989) Biochem. J. 264, 533–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wimley W. C. (2010) ACS Chem. Biol. 5, 905–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vangheluwe P., Schuermans M., Zádor E., Waelkens E., Raeymaekers L., Wuytack F. (2005) Biochem. J. 389, 151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Espagne A., Erard M., Madiona K., Derrien V., Jonasson G., Lévy B., Pasquier H., Melki R., Mérola F. (2011) Biochemistry 50, 437–439 [DOI] [PubMed] [Google Scholar]
- 59. Pandey B. K., Srivastava S., Singh M., Ghosh J. K. (2011) Biochem. J. 436, 609–620 [DOI] [PubMed] [Google Scholar]
- 60. Jóna I., Martonosi A. (1989) Acta Physiol. Hung 74, 215–228 [PubMed] [Google Scholar]
- 61. Remppis A., Most P., Löffler E., Ehlermann P., Bernotat J., Pleger S., Börries M., Reppel M., Fischer J., Koch W. J., Smith G., Katus H. A. (2002) Basic Res. Cardiol. 97, Suppl. 1, I56–I62 [DOI] [PubMed] [Google Scholar]
- 62. Kimura Y., Kurzydlowski K., Tada M., MacLennan D. H. (1997) J. Biol. Chem. 272, 15061–15064 [DOI] [PubMed] [Google Scholar]
- 63. Reddy L. G., Jones L. R., Thomas D. D. (1999) Biochemistry 38, 3954–3962 [DOI] [PubMed] [Google Scholar]
- 64. Chen Z., Akin B. L., Stokes D. L., Jones L. R. (2006) J. Biol. Chem. 281, 14163–14172 [DOI] [PubMed] [Google Scholar]
- 65. Cornea R. L., Autry J. M., Chen Z., Jones L. R. (2000) J. Biol. Chem. 275, 41487–41494 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.