

Abstract
Lentinan, a cell wall β-glucan from the fruiting bodies of Lentinus edodes, is well known to be a biological defense modifier, but the signal transduction pathway(s) induced by Lentinan have not been elucidated. In this study, we extracted Lentinan (LNT-S) by ultrasonication from Lentinus edodes and report that, in murine RAW 264.7 macrophages, LNT-S glucan activated NF-κB p65 and triggered its nuclear translocation as determined by Western blotting. Moreover, LNT-S enhanced NF-κB-luciferase activity in the Dual-Luciferase gene system assay. Its upstream signaling molecules, MAPKs such as ERK1/2 and JNK1/2, were shown to be activated by assessing the level of phosphorylation in a time- and concentration-dependent manner, but its downstream proinflammatory enzyme, inducible NOS, was not observed. The data evaluated using a TNF-α ELISA kit and Griess reagent further demonstrated that no proinflammatory mediators such as TNF-α and NO were produced by LNT-S stimulation in RAW 264.7 cells. In contrast, LPS significantly induced inducible NOS expression and increased NO and TNF-α production, which are associated with activation of the NF-κB p65/p50 heterodimer complex. It is possible that LNT-S did not activate NF-κB p65/p50, and the activation of NF-κB p65 was not sufficient to stimulate cytokine production. These data demonstrate that LNT-S glucan carries out its immunomodulating activity by activating MAPK signaling pathways without secretion of TNF-α and NO.
Keywords: ERK, Jun N-terminal Kinase (JNK), Macrophages, MAP Kinases (MAPKs), NF-κB, Signal Transduction, Lentinan, β-Glucan
Introduction
β-Glucans are known to possess significant biological and physiological activities, including antitumor activity (1, 2), antiviral activity (3, 4), antibacterial activity (5–7), antifungal activity (8), and immunomodulating activity (9–12). In addition, β-glucans are effective in wound healing (3) and in lowering blood cholesterol (13) and glucose (14) concentrations. Therefore, β-glucan is a promising product in biochemical and medical applications.
Among the numerous β-glucans derived from different sources such as plants, animals, and microbials, Lentinan from Lentinus edodes is especially remarkable for its anticancer and immunomodulating activities (15–17) and is a β-(1,3)-glucan with one β-(1,6)-glucose branch every five glucose residues. Lentinan is currently used clinically as an antitumor agent (18, 19). It has been shown to stimulate natural killer cell activity (20–22), macrophage/monocyte functions (secreting IL-1 and superoxide anion), phagocytosis, and cytotoxicity (23–28). Lentinan was found to elevate the cytotoxic activity and TNF secretion of macrophages in vitro and in vivo (16, 29). Pretreatment of bone marrow macrophages with Lentinan results in increased production of NO in vitro (16). However, Masihi et al. (30) demonstrated that pretreatment of Lentinan before LPS administration induces a striking inhibition of up to 89% of circulating TNF-α in bacillus Calmette-Guérin-primed mice. It is possible that the different immunomodulatory reaction was highly dependent on the genotype of the host. To our knowledge, most of the research has focused on secretion of proinflammatory cytokines or mediators such as TNF-α, IL-12, IFN-γ, and NO and on inflammatory mRNA expression from Lentinan-stimulated macrophages or monocytes (16, 29–31), and there have been few studies involved in the signal transduction for macrophage activation by Lentinan. In contrast, the signal transduction of β-glucans, in particular water-insoluble zymosan or yeast glucan from Saccharomyces cerevisiae as a model of immunomodulatory glucans, has been partly elucidated (32, 33). It has been reported that, after activation of receptor dectin-1 by zymosan, spleen tyrosine kinase (Syk) is recruited and induces the assembly of a scaffold consisting of the Card9 (caspase recruitment domain 9) protein and the adaptor proteins Bcl10 and Malt1; the Card9-Bcl10-Malt1 scaffold then couples dectin-1 to the canonical NF-κB pathway by activation of the IκB kinase complex, leading to nuclear translocation of NF-κB subunit p65 (32). Yan and co-workers have demonstrated that yeast glucan particles amplify phagocyte killing of iC3b-opsonized tumor cells via the complement receptor 3-Syk-PI3K pathway (33) and activate murine resident macrophages to secrete proinflammatory cytokines, including TNF-α, MCP-1 (monocyte chemotactic protein-1), and IL-6, through MyD88- and Syk-dependent pathways (34). Therefore, in this study, we explored activation of RAW 264.7 macrophages induced by Lentinan to clarify the signal transduction pathways used by Lentinan.
EXPERIMENTAL PROCEDURES
Sample Preparation
The dried fruiting bodies of L. edodes, a commercial product cultivated in the Fujian Province of China, were cut into small pieces and refluxed in ethyl acetate and then acetone for 8 h. After refluxing, the dried residues were dipped into 0.9% NaCl aqueous solution with continuous stirring at 25 °C for 24 h and centrifuged. This process was repeated three times, and the residues were extracted in hot water (120 °C) three times again. After the hot water extraction, the final residues were ultrasonicated (500 watts, 18 min, 25 °C) in pure water. The collected supernatant was treated with 30% H2O2 to decolorize and subjected to the method of Sevag (35) to remove free proteins, followed by dialysis against distilled water (Mr cutoff = 8000) for 10 days. The solution was then filtered, concentrated by a rotary evaporator at reduced pressure below 45 °C, and precipitated into acetone. The precipitates were redissolved in pure water and lyophilized to obtain white pure flakes coded as Lentinan (LNT-S)2 with a yield of 0.4%. LNT-S was identified as β-(1,3)-d-glucan with β-(1,6)-branches by GC-MS and NMR. LNT-S dissolved in PBS was sterilized at 121 °C for 20 min and used in the following experiments.
Reagents and Antibodies
DMEM (glutamine, high glucose), penicillin, streptomycin, and LPS from Escherichia coli 0111:B4 were purchased from Sigma. Antibodies were obtained from the following sources: mouse monoclonal anti-inducible NOS (iNOS), BD Transduction Laboratories; rabbit polyclonal anti-NF-κB p65, rabbit polyclonal anti-phospho-ERK1/2, rabbit polyclonal anti-phospho-JNK1/2, and rabbit polyclonal anti-phospho-p38, Cell Signaling Technology (Beverly, MA); and mouse monoclonal anti-β-actin, Santa Cruz Biotechnology (Santa Cruz, CA).
Cell Culture
RAW 264.7 cells (a murine macrophage/monocyte-like cell line; American Type Culture Collection) were maintained in DMEM (glutamine, high glucose) supplemented with penicillin (100 units/ml), streptomycin (100 μg/ml), and 10% heat-inactivated FBS (Sekisui Medical Co., Ltd., Tokyo, Japan) at 56 °C for 30 min. Subculturing was done by dislodging the cells with trypsin (0.25%) and EDTA·2Na·2H2O (0.02%), followed by centrifugation and seeding into the culture flask or dish, which was incubated at 37 °C under a humidified atmosphere of 95% air and 5% CO2.
Western Blot Analysis
The cytoplasmic and nuclear proteins were extracted following the procedure described previously (36) with slight modifications. Briefly, RAW 264.7 cells were plated in a 60-mm culture dish at a density of 5 × 106 cells/dish in DMEM with 10% FBS for 24 h. The cells were then rinsed with sterilized PBS and exposed to LPS (100 ng/ml) and LNT-S (0∼200 μg/ml) in DMEM without FBS. At the end of treatment, the cells were harvested by scrapping and treated with 200 μl of lysis buffer (50 mm KCl, 0.5% Nonidet P-40, 25 mm HEPES (pH 7.8), 1 mm PMSF, 10 μg/ml leupeptin, 20 μg/ml aprotinin, and 100 mm dithiothreitol) on ice for 10 min. After a 5-min centrifugation at 14,000 rpm, the supernatant was saved as the cytoplasmic extract. The remaining nuclear pellet was washed once with the same volume of buffer without Nonidet P-40. The nuclear pellet was then treated with 40 μl of extraction buffer (500 mm KCl and 10% glycerol with the same concentrations of HEPES, PMSF, leupeptin, aprotinin, and dithiothreitol as in lysis buffer) on ice for 40 min with pipetting every 10 min. After centrifugation at 14,000 rpm for 10 min, the supernatant was harvested as the nuclear protein extract. Both the cytoplasmic and nuclear protein extracts were preserved at −70 °C for Western blot assay. The protein concentration was determined using Bradford protein assay reagent (Bio-Rad) and BSA (Nacalai Tesque, Kyoto, Japan) as the reference.
Both cytoplasmic and nuclear lysates were mixed with 4× SDS sample buffer and denatured in boiling water for 5 min. Aliquots of 10∼15 μg of denatured cytoplasmic proteins and 15∼25 μg of denatured nuclear proteins were separated by SDS-PAGE on a 10% polyacrylamide gel and then electrically transferred to a PVDF membrane (BioTrace, Pall Corp., Port Washington, NY). After blocking with 5% (w/v) BSA in TBS (10 mm Tris-HCl (pH 8.0) and 150 mm NaCl) containing 0.1% Tween 20 at room temperature for 1 h, the membranes were then incubated with an appropriate specific primary antibody (anti-iNOS, 1:8000; anti-ERK1/2, 1:1000; anti-JNK1/2, 1:1000; anti-p38, 1:1000; anti-NF-κB p65, 1:1000; or anti-β-actin, 1:20,000) overnight at 4 °C. The reactive bands were visualized with an HRP-conjugated secondary antibody (1:20;000; Santa Cruz Biotechnology) via ECL Western blot detection reagent on a LightCapture II system (ATTO, Tokyo) according to the manufacturer's instructions.
NF-κB-Luciferase Assay
RAW 264.7 cells in 24-well plates (3 × 105 cells/well) were preincubated for 12 h and then transfected with 0.8 μl of NF-κB-driven luciferase reporter vectors using 1.56 μl of Lipofectamine (Invitrogen) in 26 μl of Opti-MEM (Invitrogen) per well. After a 6-h transfection, the cells were rinsed with PBS and incubated for an additional 6 h in DMEM with 10% FBS. The cells were then stimulated with LPS (100 ng/ml) and LNT-S (200 μg/ml) in DMEM without FBS. Following 12-h stimulations, cells were lysed with Passive Lysis Buffer, and firefly and Renilla luciferase activities were assessed using a Dual-Luciferase reporter assay (Promega, Madison, WI) and a Wallac microplate luminometer (1420 ARVO Sx, PerkinElmer Life Sciences) according to the manufacturers' instructions. NF-κB activity was expressed as -fold induction relative to the control cells without LPS treatment.
Nitrite Determination
NO production was determined based on the amount of nitrite present in the conditioned medium, a stable end product of NO, by the Griess reaction. Briefly, RAW 264.7 cells were seeded (5 × 105 cells/well) in 24-well plates and incubated for 24 h. The cells were rinsed with PBS and exposed to LPS (100 ng/ml) and LNT-S in DMEM without FBS. After a 24-h stimulation, each supernatant (100 μl) was mixed with an equal volume of Griess reagent (50 μl of 1% sulfanilamide in 5% phosphoric acid and 50 μl of 0.1% N-(1-naphthyl)ethylenediamine dihydrochloride in distilled water) at room temperature. The absorbance was measured at 570 nm using the Wallac microplate luminometer (1420 ARVO Sx), and the concentration of NO was quantified with a standard curve generated with sodium nitrite in the range of 0–100 μm.
TNF-α Assays by ELISA
RAW 264.7 cells (5 × 105 cells/well) were seeded in 24-well plates and incubated for 24 h before stimulation. At the end of the preincubation period, cells were rinsed with PBS, and the medium was exchanged to DMEM without FBS. The cells were exposed to LPS (100 ng/ml) and LNT-S (200 μg/ml), respectively. After a 24-h stimulation, the conditioned medium was collected and centrifuged, and TNF-α levels in the supernatant were assessed using an ELISA kit (Thermo Scientific) according to the manufacturer's instructions.
Statistical Analysis
All data are presented as the mean ± S.E. from at least three independent experiments unless specified otherwise. Student's t test was performed, and the differences were considered statistically significant at p < 0.05.
RESULTS
LNT-S Activates ERK1/2 and JNK1/2 in Murine RAW 264.7 Cells
Fig. 1 shows the concentration and time courses of iNOS, ERK1/2, and JNK1/2 activation by LNT-S in murine RAW 264.7 macrophage cells as measured by Western blotting. LPS was also used in the stimulation of RAW 264.7 cells as a positive control. Similar to the positive control, ERK1/2 was phosphorylated by LNT-S, and activation became stronger with an increase in LNT-S concentration (Fig. 1A) in 24-h stimulated RAW 264.7 macrophages. In the positive control, LPS induced not only ERK1/2 activation (evaluated by the level of phosphorylation, labeled by phospho-ERK1/2) but also iNOS production. However, LNT-S itself could not induce notable iNOS production at the different concentrations used in this experiment (Fig. 1A). The phosphorylation of two other MAPKs, phospho-JNK1/2 and phospho-p38, was not observed (data not shown).
FIGURE 1.
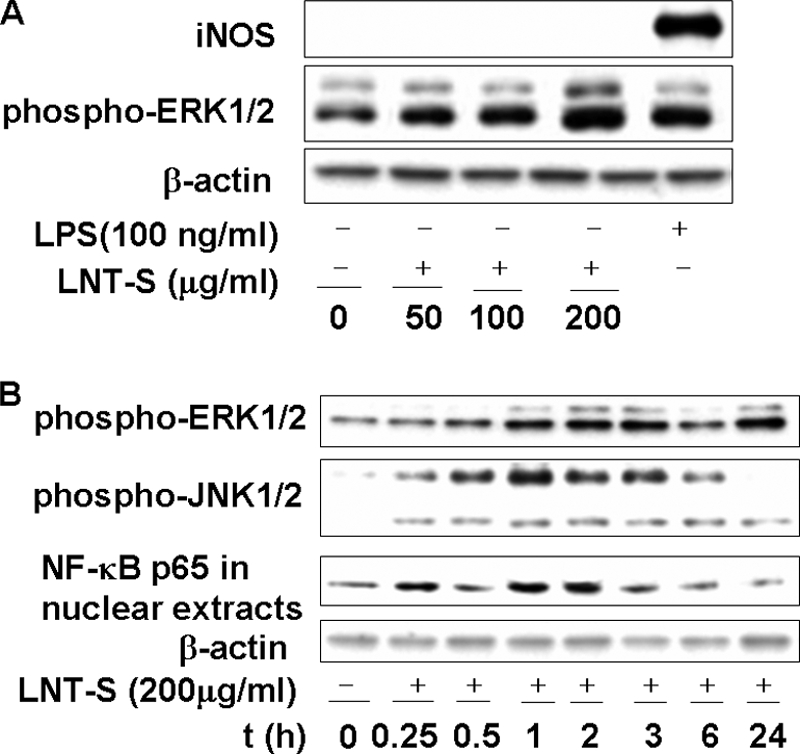
LNT-S activates the phosphorylation of MAPKs and enhances the nuclear translocation of NF-κB. RAW 264.7 macrophages were treated with LNT-S for 24 h (A) and at different time point (B). Phospho-ERK1/2, phospho-JNK1/2, NF-κB p65, iNOS, and β-actin were detected by Western blot analysis using their specific antibodies. The concentrations of LNT-S and LPS used are indicated. Data are representative of three independent experiments.
In the case of the time course, both phospho-JNK1/2 and phospho-ERK1/2 protein bands were observed after a 15-min stimulation by LNT-S (Fig. 1B). The density of the phospho-JNK1/2 protein band peaked at 1 h and then gradually declined thereafter (Fig. 1B), which can be explained as degradation of the protein. The phosphorylation level of phospho-ERK1/2 also reached the maximal value at 1 h of stimulation and almost leveled off within 24 h. The activation of p38 MAPK was not detectable in the time course experiments (data not shown). These data demonstrate that, in murine RAW 264.7 macrophage cells, LNT-S directly triggers a rapid and strong activation of JNK1/2 and ERK1/2, but not iNOS.
LNT-S Enhances the Nuclear Translocation of NF-κB p65 and NF-κB-Luciferase Activity
The transcription factor NF-κB is largely involved in immune responses and expression of proinflammation elicited by a variety of mediators, including LPS. Fig. 1B shows that the amount of NF-κB p65 observed after a 15-min activation increased with stimulation time and almost peaked at 1∼2 h and then decreased due to protein degradation. It has been reported that zymosan significantly activates NF-κB at 2 h after stimulation (36).
To verify that NF-κB in the nucleus was transactivated, the NF-κB activity was assessed by transfecting an NF-κB-luciferase reporter into RAW 264.7 cells using the Dual-Luciferase reporter assay. Reporter activity in LNT-S-stimulated RAW 264.7 cells was elevated relative to that in unstimulated cells, similar to the positive control (LPS-stimulated cells) (Fig. 2). LNT-S directly triggers NF-κB activation in RAW 264.7 macrophage cells.
FIGURE 2.
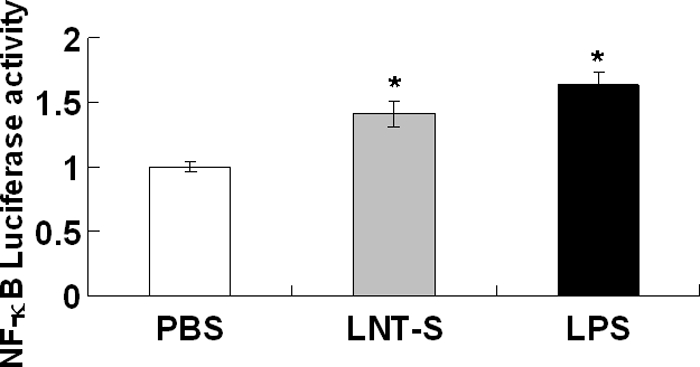
NF-κB-luciferase activity in LNT-S- and LPS-stimulated RAW 264.7 cells. RAW 264.7 macrophages were transfected with an NF-κB-luciferase reporter vector using Lipofectamine. Transiently transfected cells were stimulated LPS (100 ng/ml) and LNT-S (200 μg/ml) for 12 h. NF-κB-luciferase activity was measured as described under “Experimental Procedures” and is expressed relative to the respective controls. Each value represents the mean ± S.E. of three independent experiments. *, p < 0.05 versus the control (PBS).
NO and TNF-α Production
Murine macrophages activated by LPS in vitro express iNOS to synthesize NO (37), which is a key molecule in the immunopharmacology of β-glucans. ERK and JNK are thought to regulate production of inflammatory cytokines and mediators such as TNF-α and NO (38–41). Moreover, activation of the classic NF-κB transcription factor is often associated with the production of inflammatory cytokines (42–44). Because LNT-S triggers NF-κB activation and MAPKs ERK1/2 and JNK1/2, proinflammatory mediators such as NO and TNF-α were evaluated (Fig. 3). Surprisingly, LNT-S itself could not induce NO (Fig. 3A) or TNF-α (Fig. 3B) production, in contrast to LPS, which stimulated RAW 264.7 cells to produce a large amount of NO and TNF-α (Fig. 3). We can thus conclude that LPS induces production of NO and TNF-α, whereas LNT-S does not, indicating that LNT-S is not contaminated with significant levels of endotoxin.
FIGURE 3.
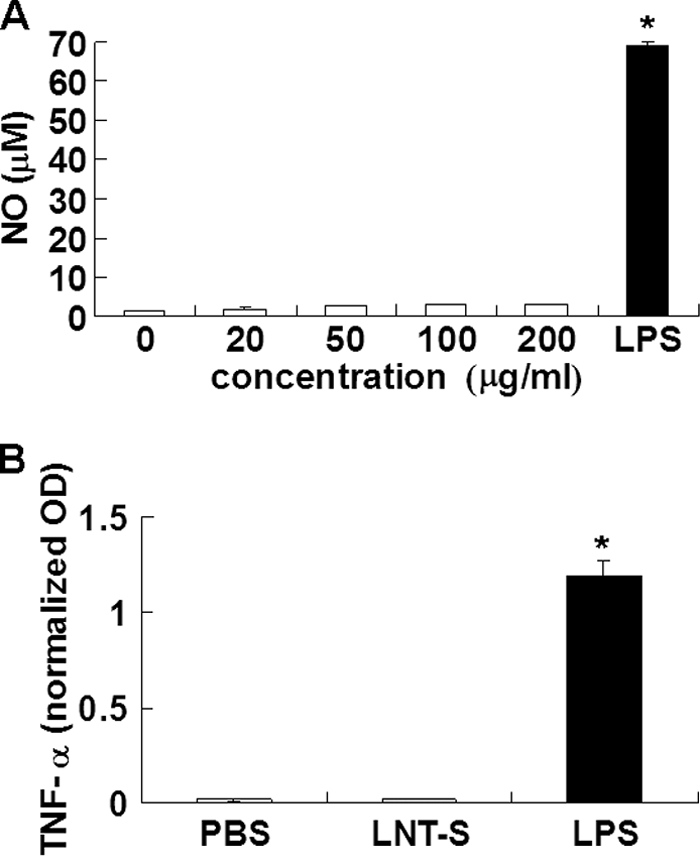
Concentration dependence of NO production in 24-h stimulated RAW 264. 7 macrophages by LNT-S (A) and TNF-α release in 24-h stimulated RAW 264.7 cells by 200 μg/ml LNT-S (B). Each value represents the mean ± S.E. of three independent experiments. *, p < 0.01 versus the control (PBS).
DISCUSSION
In this work, β-glucan from L. edodes (LNT-S) was shown to stimulate murine RAW 264.7 macrophage cells by activation of MAPKs ERK1/2 and JNK1/2 and transcription factor NF-κB (Fig. 1). Moreover, activation occurred in a time- and concentration-dependent manner. Western blotting indicated that the nuclear titer of NF-κB p65 increased in response to LPS and LNT-S treatment of the RAW 264.7 cells (Fig. 1B), which was probably due to its translocation from the cytoplasm. The increase in NF-κB-luciferase activity further demonstrated that the nuclear transcription factor was really activated by LNT-S. LPS is known to induce MAPK-dependent phosphorylation, thereby activating multiple transcription factors to translocate to the nucleus, facilitating DNA-binding activity, and leading to up-regulation of iNOS expression (41, 45). In particular, NF-κB is a major activator for TNF-α production in macrophages and a central target for activators or inhibitors of iNOS expression (46, 47). For example, zymosan-induced TNF-α production in RAW 264.7 macrophage cells is associated essentially with activation of NF-κB, similar to LPS (36). However, as shown in Figs. 1A and 3, LNT-S showed a major difference compared with LPS and zymosan; LNT-S could not induce iNOS expression or production of NO and TNF-α regardless of its activation of MAPKs (ERK1/2 and JNK1/2) and NF-κB. This phenomenon is very similar to that of water-soluble PGG-glucan, a β-(1,3)-glucan with β-(1,6)-branches that activates NF-κB p65 without inducing inflammatory cytokines such as TNF-α and IL-6 (48). This was attributed to the fact that PGG-glucan increased the titer of only p65, but not other NF-κB subunits such as p50, p52, p68, and p75, and that p65 formed a heterodimer complex (called NF-κB-like transcription factor) with another unknown unit.
It is well known that the activated form of NF-κB is a heterodimer, which usually consists of two proteins, a p65 (also called RelA) subunit and a p50 subunit. Other subunits, including C-Rel, RelB, v-Rel, and p52, may also be part of activated NF-κB (48, 49). Of these, the classic p65/p50 heterodimer complex has been demonstrated to be involved in the transcriptional regulation of the proinflammatory cytokines IL-1β and TNF-α (42–44) and the proinflammatory mediator NO. On the basis of the fact discussed above and the study of PGG-glucan, we can attribute this to the following: LNT-S increased the titer of p65, which could not form a heterodimer with p52 but maybe a complex called NF-κB-like transcription factor with another unknown unit and was not sufficient to stimulate inflammatory cytokine mRNA transcription and cytokine production. Whether this is true or not will be determined in future work.
As we know, the well studied zymosan particle activates β-glucan receptor dectin-1, followed by recruiting Syk, leading to activation of NF-κB and proinflammatory cytokine production, whereas water-soluble PGG-glucan activates NF-κB without production of proinflammatory cytokines. LNT-S is more similar to PGG-glucan. Water solubility likely plays an important role in the regulation of immunomodulating activity. This work is one of the first reports demonstrating that Lentinan activates ERK1/2, JNK1/2, and NF-κB p65 without production of proinflammatory cytokines. Study of the upstream signaling pathway will be continued in our future work.
In summary, our results suggest that the β-glucan extracted from L. edodes by ultrasonication directly stimulated macrophages by inducing phosphorylation of MAPKs ERK1/2 and JNK1/2, but not p38. Subunit p65 of nuclear transcription factor NF-κB was shown to translocate from the cytoplasm to the nucleus, and NF-κB-luciferase activity was also enhanced compared with the blank (PBS) and the positive control (LPS stimulation). However, TNF-α and NO were not produced in the supernatant of RAW 264.7 cells, which may be explained by the lack of formation of the active NF-κB p65/p50 heterodimer complex.
This work was supported by Japan Society for the Promotion of Science Postdoctoral Fellowship 20.08431 (to X. X.), National Natural Science Foundation Grant 20874078, and Youth Technology Chenguang Project of Wuhan 200950431193.
- LNT-S
- Lentinan
- iNOS
- inducible NOS.
REFERENCES
- 1. Ross G. D., Vetvicka V., Yan J., Xia Y., Vetvicková J. (1999) Immunopharmacology 42, 61–74 [DOI] [PubMed] [Google Scholar]
- 2. Vetvicka V., Yvin J. C. (2004) Int. Immunopharmacol. 4, 721–730 [DOI] [PubMed] [Google Scholar]
- 3. Mayell M. (2001) Altern. Med. Rev. 6, 48–60 [PubMed] [Google Scholar]
- 4. Chen J., Seviour R. (2007) Mycol. Res. 111, 635–652 [DOI] [PubMed] [Google Scholar]
- 5. Markova N., Kussovski V., Drandarska I., Nikolaeva S., Georgieva N., Radoucheva T. (2003) Int. Immunopharmacol. 3, 1557–1562 [DOI] [PubMed] [Google Scholar]
- 6. Markova N., Michailova L., Kussovski V., Jourdanova M., Radoucheva T. (2005) Pharmazie 60, 42–48 [PubMed] [Google Scholar]
- 7. Kournikakis B., Mandeville R., Brousseau P., Ostroff G. (2003) MedGenMed 5, 1. [PubMed] [Google Scholar]
- 8. Herre J., Willment J. A., Gordon S., Brown G. D. (2004) Crit. Rev. Immunol. 24, 193–203 [DOI] [PubMed] [Google Scholar]
- 9. Wasser S. P. (2002) Appl. Microbiol. Biotechnol. 60, 258–274 [DOI] [PubMed] [Google Scholar]
- 10. Cleary J. A., Kelly G. E., Husband A. J. (1999) Immunol. Cell Biol. 77, 395–403 [DOI] [PubMed] [Google Scholar]
- 11. Vetvicka V., Vetvickova J., Frank J., Yvin J. C. (2008) Biomed. Pharmacother. 62, 283–288 [DOI] [PubMed] [Google Scholar]
- 12. Shimojoh M., Kojima T., Nakajima K., Hatta K., Katoh A., Kurita K. (2010) Biomacromolecules 11, 1212–1216 [DOI] [PubMed] [Google Scholar]
- 13. Nicolosi R., Bell S. J., Bistrian B. R., Greenberg I., Forse R. A., Blackburn G. L. (1999) Am. J. Clin. Nutr. 70, 208–212 [DOI] [PubMed] [Google Scholar]
- 14. Lo H. C., Tsai F. A., Wasser S. P., Yang J. G., Huang B. M. (2006) Life Sci. 78, 1957–1966 [DOI] [PubMed] [Google Scholar]
- 15. Fang N., Li Q., Yu S., Zhang J., He L., Ronis M. J., Badger T. M. (2006) J. Altern. Complement. Med. 12, 125–132 [DOI] [PubMed] [Google Scholar]
- 16. Kupfahl C., Geginat G., Hof H. (2006) Int. Immunopharmacol. 6, 686–696 [DOI] [PubMed] [Google Scholar]
- 17. Zheng R., Jie S., Hanchuan D., Moucheng W. (2005) Int. Immunopharmacol. 5, 811–820 [DOI] [PubMed] [Google Scholar]
- 18. Suga T., Shiio T., Maeda Y. Y., Chihara G. (1984) Cancer Res. 44, 5132–5137 [PubMed] [Google Scholar]
- 19. Okamura K., Suzuki M., Chihara T., Fujiwara A., Fukuda T., Goto S., Ichinohe K., Jimi S., Kasamatsu T., Kawai N. (1986) Cancer 58, 865–872 [DOI] [PubMed] [Google Scholar]
- 20. Taguchi T., Kaneko Y. (1986) in Host Defense Mechanisms against Cancer (Urushizaki I., Aoki T., Tsubura E. eds) p. 221, Excerpta Medica, Amsterdam [Google Scholar]
- 21. Nanba H., Kuroda H. (1987) Chem. Pharm. Bull. 35, 2459–2464 [DOI] [PubMed] [Google Scholar]
- 22. Péter G., Károly V., Imre B., János F., Kaneko Y. (1988) Immunopharmacol. Immunotoxicol. 10, 157–163 [DOI] [PubMed] [Google Scholar]
- 23. Fruehauf J. P., Bonnard G. D., Herberman R. B. (1982) Immunopharmacology 5, 65–74 [DOI] [PubMed] [Google Scholar]
- 24. Akiyama T., Kashima S., Hayami T., Izawa M., Mitsugi K., Hamuro J. (1987) in Manipulation of Host Defense Mechanism (Acki T., Uneshizaki I., Tsubura E. eds) pp. 227–238, Excerpta Medica, Amsterdam [Google Scholar]
- 25. Abel G., Szöllösi J., Chihara G., Fachet J. (1989) Int. J. Immunopharmacol. 11, 615–621 [DOI] [PubMed] [Google Scholar]
- 26. Herlyn D., Kaneko Y., Powe J., Aoki T., Koprowski H. (1985) Gann 76, 37–42 [PubMed] [Google Scholar]
- 27. Nanba H., Mori K., Toyomasu T., Kuroda H. (1987) Chem. Pharm. Bull. 35, 2453–2458 [DOI] [PubMed] [Google Scholar]
- 28. Ladányi A., Tímár J., Lapis K. (1993) Cancer Immunol. Immunother. 36, 123–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kerékgyártó C., Virág L., Tankó L., Chihara G., Fachet J. (1996) Int. J. Immunopharmacol. 18, 347–353 [DOI] [PubMed] [Google Scholar]
- 30. Masihi K. N., Madaj K., Hintelmann H., Gast G., Kaneko Y. (1997) Int. J. Immunopharmacol. 19, 463–468 [DOI] [PubMed] [Google Scholar]
- 31. Liu F., Ooi V. E., Fung M. C. (1999) Life Sci. 64, 1005–1011 [DOI] [PubMed] [Google Scholar]
- 32. Gross O., Gewies A., Finger K., Schäfer M., Sparwasser T., Peschel C., Förster I., Ruland J. (2006) Nature 442, 651–656 [DOI] [PubMed] [Google Scholar]
- 33. Li B., Allendorf D. J., Hansen R., Marroquin J., Ding C., Cramer D. E., Yan J. (2006) J. Immunol. 177, 1661–1669 [DOI] [PubMed] [Google Scholar]
- 34. Li B., Cramer D., Wagner S., Hansen R., King C., Kakar S., Ding C., Yan J. (2007) Clin. Immunol. 124, 170–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sevag M. G. (1934) Biochem. Z. 273, 419–429 [Google Scholar]
- 36. Young S. H., Ye J., Frazer D. G., Shi X., Castranova V. (2001) J. Biol. Chem. 276, 20781–20787 [DOI] [PubMed] [Google Scholar]
- 37. Lorsbach R. B., Murphy W. J., Lowenstein C. J., Snyder S. H., Russell S. W. (1993) J. Biol. Chem. 268, 1908–1913 [PubMed] [Google Scholar]
- 38. Hambleton J., Weinstein S. L., Lem L., DeFranco A. L. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 2774–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Han J., Lee J. D., Bibbs L., Ulevitch R. J. (1994) Science 25, 808–811 [DOI] [PubMed] [Google Scholar]
- 40. Lee J. C., Laydon J. T., McDonnell P. C., Gallagher T. F., Kumar S., Green D., McNulty D., Blumenthal M. J., Heys J. R., Landvatter S. W., Strickler J. E., McLaughlin M. M., Siemens I. R., Fisher S. M., Livi G. P., White J. R., Adams J. L., Young P. R. (1994) Nature 372, 739–746 [DOI] [PubMed] [Google Scholar]
- 41. Kim J. W., Kim C. (2005) Biochem. Pharmacol. 70, 1352–1360 [DOI] [PubMed] [Google Scholar]
- 42. Baldwin A. S., Jr. (1996) Annu. Rev. Immunol. 14, 649–683 [DOI] [PubMed] [Google Scholar]
- 43. Sweet M. J., Hume D. A. (1996) J. Leukocyte Biol. 60, 8–26 [DOI] [PubMed] [Google Scholar]
- 44. Barnes P. J., Karin M. (1997) N. Engl. J. Med. 336, 1066–1071 [DOI] [PubMed] [Google Scholar]
- 45. Tebo J. M., Chaoqun W., Ohmori Y., Hamilton T. A. (1994) J. Immunol. 153, 4713–4720 [PubMed] [Google Scholar]
- 46. Ghosh S., May M. J., Kopp E. B. (1998) Annu. Rev. Immunol. 16, 225–260 [DOI] [PubMed] [Google Scholar]
- 47. Xie Q. W., Kashiwabara Y., Nathan C. (1994) J. Biol. Chem. 269, 4705–4708 [PubMed] [Google Scholar]
- 48. Adams D. S., Pero S. C., Petro J. B., Nathans R., Mackin W. M., Wakshull E. (1997) J. Leukocyte Biol. 62, 865–873 [DOI] [PubMed] [Google Scholar]
- 49. Gringhuis S. I., den Dunnen J., Litjens M., van der Vlist M., Wevers B., Bruijns S. C., Geijtenbeek T. B. (2009) Nat. Immunol. 10, 203–213 [DOI] [PubMed] [Google Scholar]