

Abstract
The peptidoglycan recognition protein PGRP-S is an innate immunity molecule that specifically interacts with microbial peptidoglycans and other pathogen-associated molecular patterns. We report here two structures of the unique tetrameric camel PGRP-S (CPGRP-S) complexed with (i) muramyl dipeptide (MDP) at 2.5 Å resolution and (ii) GlcNAc and β-maltose at 1.7Å resolution. The binding studies carried out using surface plasmon resonance indicated that CPGRP-S binds to MDP with a dissociation constant of 10−7 m, whereas the binding affinities for GlcNAc and β-maltose separately are in the range of 10−4 m to 10−5 m, whereas the dissociation constant for the mixture of GlcNAc and maltose was estimated to be 10−6 m. The data from bacterial suspension culture experiments showed a significant inhibition of the growth of Staphylococcus aureus cells when CPGRP-S was added to culture medium. The ELISA experiment showed that the amount of MDP-induced production of TNF-α and IL-6 decreased considerably after the introduction of CPGRP-S. The crystal structure determinations of (i) a binary complex with MDP and (ii) a ternary complex with GlcNAc and β-maltose revealed that MDP, GlcNAc, and β-maltose bound to CPGRP-S in the ligand binding cleft, which is situated at the interface of molecules C and D of the homotetramer formed by four protein molecules A, B, C, and D. In the binary complex, the muramyl moiety of MDP is observed at the C-D interface, whereas the peptide chain protrudes into the center of tetramer. In the ternary complex, GlcNAc and β-maltose occupy distinct non-overlapping positions belonging to different subsites.
Keywords: Crystal Structure, Innate Immunity, Pathogen-associated Molecular Pattern (PAMP), Surface Plasmon Resonance (SPR), X-ray Crystallography, MDP, PGRP-S
Introduction
Peptidoglycan recognition proteins (PGRPs)2 of the innate immune system provide the first line of defense to hosts against infecting microbes. These proteins specifically recognize pathogen-associated molecular patterns (PAMPs) such as peptidoglycan (PGN), LPS, lipoteichoic acid (LTA), mycolic acid, and etc. It is well known that PGN is present on the cell walls of both Gram-negative and -positive bacteria. PGN is a polymer of alternating GlcNAc and N-acetylmuramic acid (MurNAc) in a β(1→4) linkage, cross-linked by short peptide stems composed of alternating l- and d-amino acids. The glycan moiety of PGN is conserved among almost all of the bacteria, but the peptide part displays a considerable diversity (1). According to the amino acid residue at number three position of the peptide stem, PGNs are divided into two major categories, (i) lysine type (Lys-type) and (ii) meso-diaminopimelic acid type. The peptidoglycans or their fragments are recognized by the host through innate immunity proteins, particularly PGRPs (2).
PGRPs are highly conserved molecules (3–7), which bind to PGN and other PAMPs as well as to their fragments and derivatives with varying affinities (8). There are four types of mammalian PGRPs designated as short PGRP (PGRP-S), PGRP-L, PGRP-Iα, and PGRP-Iβ representing short (molecular mass, 20–25 kDa), long (molecular mass, up to 90 kDa), and intermediate (molecular mass, 40–45 kDa), respectively. PGRP-S contains ∼200 amino acid residues and represents a single PGRP domain, wheraes PGRP-L and PGRP-I also contain a similar C-terminal PGRP domain. PGRP-S shows direct bacterial recognition. The potency of its recognition determines its strength in controlling bacterial infections. Initially, PGRP-S was detected in bone marrow (6), whereas subsequently, it was also observed as a soluble protein in the granules of polymorphonuclear leukocytes (9). This observation implied that PGRP-S might have a role as an antibacterial agent (6). Recently, its presence has also been shown in milk of certain species of mammals (10) and in the intestinal M cells (11). As far as mammary gland secretions are concerned, so far, PGRP-S has been detected in the mammary secretions of porcine and camel only (10). As far as structural studies are concerned, the crystal structures of truncated human PGRP-S (HPGRP-S) (amino acid residues from 9 to 191) and a full-length (amino acid residues from 1 to 171) camel PGRP-S (CPGRP-S) have been reported (12, 13) showing truncated HPGRP-S as a monomer (although it is expected to be covalently linked dimer), whereas CPGRP-S was found to be a stable homotetramer (13). The ligand-binding site in HPGRP-S is located on the surface of the monomeric protein, whereas that in CPGRP-S is observed deep inside tetramer that is connected through a channel to the surface of the protein. To establish the wide range of specificities of the binding site in tetrameric CPGRP-S for various PAMPs, we have determined two crystal structures of CPGRP-S complexed with (i) a single compound muramyl dipeptide (MDP), which is a moiety of PGN and (ii) two compounds GlcNAc and β-maltose. Previously, it has been shown that the PAMP-binding site in CPGRP-S was capable of recognizing both Gram-negative and -positive bacteria through its binding to LPS and LTA with equally high affinities (14). From the structure analysis of native CPGRP-S and its complexes with LPS and LTA, it was shown that multiple binding subsites were available for the recognition of PAMPs in the ligand binding site in the CPGRP-S tetramer. Here, we provide further structural evidence of a wide range binding capability of the PAMP-binding site in CPGRP-S.
EXPERIMENTAL PROCEDURES
Purification
CPGRP-S was isolated from the fresh samples of camel milk, which were obtained from the National Research Center on Camels (Bikaner, India), and purified it to homogeneity using the procedure described earlier (13). The purity of protein was established using SDS-PAGE and MALDI-TOF.
Antibacterial Activity Assay
The inhibition of bacterial growth was assessed by suspension assay in the absence and presence of PGN. The strains of Staphylococcus aureus were grown to mid-log phase in 1× TSB (3% w/v; 0.5% NaCl) at 37 °C. The 10-μl aliquots of the cells were added to 2 ml TSB. The purified CPGRP-S was added to a final concentration of 25 μg/ml either alone or supplemented with 100 μg/ml PGN or LPS (Sigma Aldrich). The tubes were shaken at 300 rpm for 5 h. The bacterial growth was monitored by measuring the OD at 600 nm at the intervals of one hour. To minimize the effect of bacterial aggregation on OD, the cell suspensions were stirred for 1 min before each measurement.
Binding Studies Using Surface Plasmon Resonance Spectroscopy
All of the surface plasmon resonance measurements were carried out using Biacore-2000 (Pharmacia Biosensor AB, Uppsala, Sweden) at 25 °C in which a biosensor-based system has been used for real-time specific interaction analysis. The sensor chip CM-5 (surface of which was covered with thin layer of gold coated with carboxymethyl dextran residues for covalent protein immobilization), surfactant P20, the amine coupling kit containing N-hydroxysuccinimide, N-ethyl-N′-3-(diethylaminopropyl)carbodiimide, and ethanolamine hydrochloride (Pharmacia Biosensor AB, Uppsala, Sweden) were used in the experiment. The immobilization of CPGRP-S was performed at a flow rate of 10 μl/min using an amine-coupling kit. The dextran on the chip was equilibrated with running buffer and carboxymethylated matrix was activated with an N-ethyl-N′-3-(diethylaminopropyl)carbodiimide/N-hydroxysuccinimide mixture and 210 μl of PGRP (4.2 μg/ml) in 10 mm sodium acetate (pH 3.5), the unreacted groups were blocked by injecting ethanolamine (pH 8.5). The surface plasmon resonance signal for immobilized CPGRP-S reached 4357.8 resonance units. Three concentrations of analytes (4.5 μm, 2.25 μm, and 0.9 μm) were passed over immobilized PGRP at a flow rate of 10 μl/min. The regeneration of the protein surface from bound analytes was done by using 1 mm NaOH. The association (Kon) and dissociation (Koff) rate constants for the analytes bindings to CPGRP-S were calculated and the values of dissociation constants (Kd) were determined by the mass action relation Kd = Koff/Kon using BIA evaluation software (version 3.0) provided by the manufacturer.
Induction of TNF-α and IL-6
The human peripheral blood was taken from a healthy donor and peripheral blood mononuclear cells (PBMCs) were isolated from heparinized blood by Ficoll-Hypaque gradient centrifugation and suspended in complete RPMI 1640 with 10% FCS at optimum culture conditions of 5% CO2, at 37 °C for 6 h. Cells were stimulated with medium alone and with 10 μg/ml PGN without and with 10 μg/ml CPGRP-S. The culture supernatants were collected after 6 h of stimulation at optimum culture conditions and assayed for TNF-α and IL-6 concentrations by ELISA according to the manufacturer's instructions. The data were expressed as mean values ± S.D. The statistical differences in the results were evaluated by Student's t test.
Crystallization
CPGRP-S was crystallized at room temperature using hanging drop vapor diffusion method. The crystals, suitable for diffraction measurements were obtained after 4 weeks. In all of the crystallization setups, 4 μl of protein solution at a concentration of 12 mg/ml was mixed with 4 μl of reservoir solution, which consisted of 10% PEG-3350, 200 mm potassium sodium tartrate in buffer of 50 mm Tris-HCl, pH 7.5. The 8-μl drops of protein solution were equilibrated against 2 ml of reservoir solutions. The crystals of native protein were obtained after 3 weeks. These crystals were soaked in the reservoir solutions that contained (i) a mixture of PGN fragment and muramyl dipeptide and (ii) a mixture of GlcNAc and β-maltose. The concentrations of (i) MDP and (ii) GlcNAc and β-maltose were prepared at 12 mg/ml. The soaking was carried out for 24 h. The soaked crystals were flash cooled in liquid nitrogen for 30 s in the cryoprotection solution consisting of 20% glycerol (v/v) in the reservoir solution.
X-ray Intensity Data Collections
Two independent x-ray intensity data sets were collected on the crystals soaked (i) in the solution containing MDP and (ii) in the solution containing the equimolar mixture of GlcNAc and β-maltose using DBT-sponsored x-ray beamline BM14 at the European Synchrotron Radiation Facility (Grenoble, France). To minimize the radiation damage, the crystals were mounted in nylon loops and kept at 100 K in a liquid nitrogen stream during the measurements. The data were indexed, integrated, scaled, and merged using the HKL-2000 package (15). The crystals belong to orthorhombic space group I222 with unit cell dimensions of (i) a = 87.1, b = 102.0, and c = 161.6 Å and (ii) a = 87.1, b = 100.8, and c = 161.8 Å with four molecules in the asymmetric unit in both structures. The data collection and processing statistics for the two crystals are given in Table 1.
TABLE 1.
Data collection and refinement statistics for the structures of the complexes of peptidoglycan recognition protein (CPGRP-S) with MDP, GlcNAc, and maltose
The values in parentheses correspond to the values in the highest resolution shell.
| Parameters | CPGRP-S+MDP | CPGRP-S+GlcNAc and maltose |
|---|---|---|
| PDB code | 3NW3 | 3NG4 |
| Space group | I222 | I222 |
| Unit cell dimensions | a = 87.1, b = 102.0, and c = 161.6 Å | a = 87.1, b = 100.8, and c = 161.8 Å |
| Number of molecules in the asymmetric unit | 4 | 4 |
| Resolution range (Å) | 50–2.5 | 85.5–1.7 |
| Total number of measured reflections | 717,180 | 1,562,425 |
| Number of unique reflections | 24,772 | 70,076 |
| Rsym (%)a | 6.5 (25.2) | 4.7 (48.4) |
| I/σ(I) | 31.2 (3.4) | 32.7 (2.4) |
| Overall completeness of data (%) | 97.9 (98.1) | 99.1 (99.0) |
| Rcryst (%)b | 23.6 | 20.2 |
| Rfree (%) | 24.9 | 24.4 |
| Protein atoms | 5348 | 5348 |
| Water oxygen atoms | 400 | 735 |
| Atoms of glycerol | 6 | 6 |
| Atoms of tartrate | 10 | 10 |
| Atoms of ligands (MDP and GlcNAc/maltose) | 34 | 23/15 |
| R.m.s.d in bond lengths (Å) | 0.02 | 0.02 |
| R.m.s.d in bond angles | 1.9° | 1.9° |
| R.m.s.d in torsion angles | 15.3° | 15.0° |
| Wilson B-factor | 52.8 | 20.7 |
| Mean B-factor for main chain atoms (Å2) | 48.9 | 21.3 |
| Mean B-factor for side chain atoms (Å2) | 51.3 | 26.8 |
| Mean B-factor for all atoms(Å2) | 50.2 | 24.3 |
| Ramachandran ϕ, ψ map | ||
| Residues in most favored regions (%) | 89.4 | 88.7 |
| Residues in additionally allowed regions (%) | 10.6 | 11.3 |
a Rsym = ΣhklΣi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl-).
b Rcryst = Σhkl|Fo(hkl) − Fc(hkl)|/Σhkl|Fo(hkl)|, where Fo and Fc are observed and calculated structure factors, respectively.
Structure Determination and Refinement
Because structures of both the complexes of CPGRP-S with (i) MDP and (ii) GlcNAc and β-maltose were isomorphous to the structure of native CPGRP-S (PDB 3C2X), the model of the native structure was subjected to several rounds of simulated annealing/positional refinement using reflection data from sets (i) (structure 1) and (ii) (structure 2), respectively, with the program CNS (16). These were followed by B-factor refinements. The model buildings were carried out using the program O (17) on the Silicon Graphics O2 Work station. As the refinements progressed and the Rcryst factors dropped to <26%, whereas Rfree factors dropped to <29%, the electron density maps (2Fo − Fc) and (Fo − Fc) were calculated in both cases. The characteristic non-protein electron densities were observed at the interface of molecules C and D at >2.5σ cut-off. MDP was modeled into the electron density in structure 1 (see Fig. 1A), whereas molecules GlcNAc and β-maltose were fitted nicely in structure 2 (Fig. 1B). The coordinates of MDP were included in the subsequent cycles of refinement in structure 1, whereas those of GlcNAc and β-maltose were included in structure 2. After further cycles of refinements, electron density maps were calculated intermittently, and manual model buildings were carried out. These electron density maps were also used for determining the positions of 400 water oxygen atoms in structure 1 and 735 water oxygen atoms in structure 2. After additional cycles of refinement with water oxygen atoms, the correctness of water oxygen atoms were examined, and only those water oxygen atoms were retained whose thermal B-factors were <70 Å2 and made hydrogen bonds with protein atoms. The final cycles of refinements using REFMAC (18) converged with Rcryst and Rfree values of 0.236 and 0.249 for structure 1 and 0.202 and 0.244 for structure 2. As indicated by calculations using program PROCHECK (19), there were no non-glycine residues in the disallowed regions of the Ramachandran map (20). The refinement statistics of both structures are summarized in Table 1.
FIGURE 1.

The initial|Fo − Fc| electron density maps at 2.5σ cut-off for muramyl dipeptide in the binary complex with CPGRP-S (A). The compound is located at C-D interface and N-acetylglucosamine and β-maltose (B) in the ternary complex with CPGRP-S. With reference to the center of the tetrameric complex, β-maltose is located at the distal site of the C-D interface, whereas GlcNAc occupies a proximal position.
RESULTS
Antibacterial Activity of CPGRP-S
The addition of an excess amount of LPS or MDP (100 μg/ml) completely blocked the CPGRP-S (25 μg/ml) mediated growth inhibition of S. aureus cells (Fig. 2). Similar results were also obtained when the mixture of GlcNAc and β-maltose were used (data not shown). In the suspension assay, CPGRP-S produces a dose dependent inhibition of S. aureus. The dose of 25 μg/ml of CPGRP-S was found enough to inhibit the growth of bacteria completely. However, when LPS or MDP was added to the culture at 100 μg/ml, it completely reversed the inhibitory effects of CPGRP-S as the bacterial density curve obtained with the addition of CPGRP-S and LPS/MDP fully matched with the curve, which was obtained when additives were not used. Similar conclusions could be drawn with GlcNAc and β-maltose. These results clearly indicated that CPGRP-S inhibited the bacterial growth by interacting with the molecules associated with bacterial cell walls. The binding studies have shown that all the compounds, LPS, MDP, GlcNAc, and maltose bind to CPGRP-S directly with significant binding affinities.
FIGURE 2.
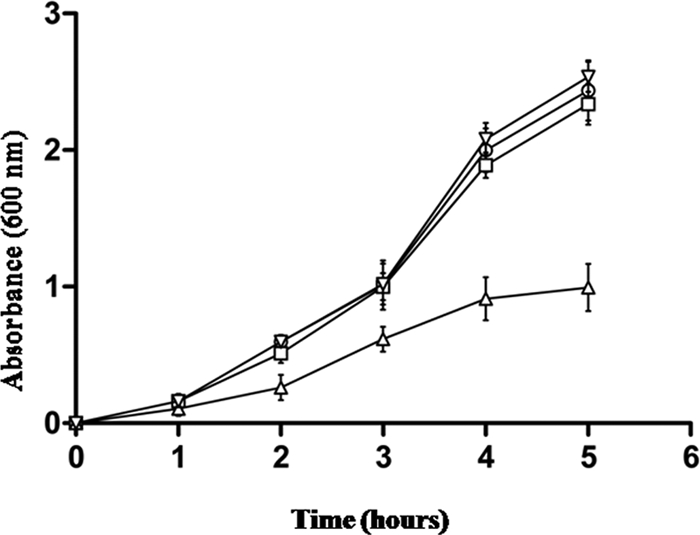
The suspension assay. Bacteria were incubated in 1× TSB (3% w/v) with 25 μg/ml CPGRP-S, either alone (△) or supplemented with 100 μg/ml S. aureus MDP (○), 100 μg/ml LPS (□), or no additives (▿). Tubes were shaken at 300 rpm for 5 h, and bacterial density was monitored by measurement of optical density at 600 nm at 1-h intervals. The data represent the mean ± S.D. values of three independent experiments.
Determination of KD by Surface Plasmon Resonance
The molecular interactions between CPGRP-S and its binding partners MDP, GlcNAc, and β-maltose were studied in real-time using surface plasmon resonance spectroscopy. The sensograms for the interactions of CPGRP-S with (i) MDP and (ii) GlcNAc and β-maltose are shown in supplemental Fig. S1, A and B. The increase of resonance units from the base line represents the binding of ligands to the immobilized protein. The plateau line represents the steady state equilibrium phase of interactions between protein and the ligands, whereas the decrease in resonance units from the plateau represents the dissociation phase. As seen from Fig. 3A, the dissociation phase is slower in case of MDP than other two analytes as calculated using BIA evaluation software. This showed that MDP has the higher affinity than the other two individual ligands. The value of the dissociation constant (KD) for MDP was calculated to be 3.2 × 10−7 m, whereas the KD values for GlcNAc and maltose mixture was estimated to be 3.7 × 10−6 m.
FIGURE 3.

Effect of CPGRP-S on the MDP or MDP+LTA-induced release of cytokines in the human blood as estimated in peripheral blood mononuclear cells. Cells were treated with MDP or mixture of MDP+LTA, either in the presence or absence of CPRP-S. TNF-α (A) and IL-6 (B) in medium were measured after 24 h of the treatment. The data are expressed as mean ± S.D. values (n = 6).
Inhibition of Cytokine Induction in Human PBMCs
Because of the bacterial cell wall components such as MDP are potent inducers of cytokine production in human PBMCs, CPGRP-S was expected to have an effect on the activation of macrophages as it has a high affinity toward MDP. Thus, the effect of CPGRP-S was evaluated on the production of TNF-α and IL-6 by MDP augmented human PBMCs. The 5 μg/ml of CPGRP-S was able to inhibit MDP-induced TNF-α and IL-6 production by 75 and 85%, respectively (Fig. 3). These are very significant results showing reduction in the levels of proinflammatory cytokines on addition of CPGRP-S to the activated PBMCs and indicate the potential of CPGRP-S in the intervention of bacterial infection induced inflammation or the septic shock.
Molecular Structure of CPGRP-S
The crystal structures of two complexes of CPGRP-S (one binary complex with MDP designated as structure 1 and another ternary complex with GlcNAc and β-maltose designated as structure 2) were determined at high resolutions. CPGRP-S forms an asymmetrical homotetramer in which molecules A and B are tightly held through a series of intermolecular hydrogen bonds and hydrophobic interactions. The most critical hydrogen bonds at this interface are formed by involving amino acid residues Ser-A8, Ile-B9, Glu-B14, Arg-A122, Arg-B122, Asn-A126, and Asn-B126. In comparison, molecules C and D are held relatively loosely as the interface has direct intermolecular hydrogen bonds and hydrophobic interactions only in a small region on the outer side, whereas the remaining three-fourths of the region of the unevenly formed interface is filled with water molecules. The prominent amino acid residues that form hydrogen bonds at the C-D interface include His-C37, His-D37, Tyr-C59, Tyr-D59, Trp-C66, glu-C150, Glu-D150, Pro-C151, Thr-D152, and Leu-D153. The dimers A-B and C-D further associate to give rise to a homotetramer in an asymmetrical manner. The molecules A and C interact with each other involving nearly 50% length of their interface covering outer part of it. The residues that are involved in intermolecular contacts from molecule A are Arg-A31, Tyr-A32, Trp-A98, Arg-A138, Ser-A139, Asn-A140, and Arg-A170, whereas those from molecule C are Glu-C21, Arg-C23, Glu-C24, and Gly-C95. On the other hand, internal faces of molecules B and D are rich in positively charged amino acid residues and hence do not form direct intermolecular interactions resulting in the formation of a channel-like structure, which appears to be stereochemically compatible for the diffusion of PAMPs. The residues of molecule B at the interface are Pro-B3, Arg-B23, Arg-B25, Arg-B28, Pro-B29, Gly-B135, and Arg-B138, whereas those on the opposite surface of molecule D are Pro-D29, Arg-D31, Arg-D138, Asn-D140, and Arg-D170, indicating a lower possibility of direct intermolecular contacts between molecules B and D. The wide channel is loosely filled with water molecules that are easily displaced by PAMPs. It may be noted that the B-factors of residues in this region are relatively higher than those in other parts of the protein, indicating a higher degree of mobility.
Structure of Complex of CPGRP-S with MDP
The structure of the complex of CPGRP-S with MDP (CPGRP-S-MDP, designated as structure 1) was determined at 2.5 Å resolution (PDB 3NW3) (supplemental Fig. S2). The structure consists of four crystallographically independent protein molecules designated as A, B, C, and D, one MDP molecule bound with occupancy of 1.0 to the tetrameric complex of CPGRP-S and 400 structured water molecules. The superpositions of four protein molecules A, B, C, and D gave a root mean square deviations (r.m.s.d.) of <0.4 Å for 171 α-carbon atoms, indicating that the four protein molecules in the homotetramer have identical conformations. Furthermore, when the Cα traces of four molecules A, B, C, and D from structure 1 were superimposed on those of the unbound CPGRP-S (PDB 3C2X), the r.m.s.d. shifts were found to be only up to 0.8 Å, showing that the binding of MDP to CPGRP-S did not perturb the conformation of the protein molecule appreciably. However, the orientations of several side chains of amino acid residues in the proximity of binding of MDP were found to be considerably altered although their main chain conformations were not disturbed significantly. The prominent residues that contributed to the binding of MDP include, Arg-C85, His-C93, Gly-C95, Asn-C99, Val-C149, Gln-C150, Leu-C164, Thr-D95, Lys-D144, Asp-D148, and Asp-D149 (supplemental Table S1). Of these residues, the most critical interactions with PAMPs were provided by Trp-C66, Asn-C99, Gln-C150, Val-D149, and Gln-D150 (Fig. 4).
FIGURE 4.

Hydrogen-bonded interactions between CPGRP-S and MDP from molecules C and D.
Structure of Ternary Complex of CPGRP-S with GlcNAc and β-Maltose
The structure of CPGRP-S-GlcNAc-maltose (designated as structure 2) was determined at 1.7 Å resolution (PDB 3NG4) (supplemental Fig. S3). In this structure also, there were four protein molecules A, B, C, and D in the form of a homotetramer in an asymmetrical arrangement. Two compounds, GlcNAc and β-maltose were found bound to the tetrameric protein complex at the interface of molecules C and D. As indicated by refinement of the structure both compounds bound to the protein with full occupancies of 1.0. The positions of 735 water molecules were also determined, the majority of which were found interacting with protein atoms. The wide channel between molecules B and D was sparsely filled with water molecules. When the Cα traces of CPGRP-S from the present ternary complex were superimposed on the Cα traces of unbound CPGRP-S (13), the overall r.m.s.d. was <0.9 Å, indicating that the conformations of protein molecules were not disturbed due to the binding of GlcNAc and β-maltose at the interface of C-D molecules in the tetrameric complex of CPGRP-S. However, the conformations of some of the side chains in the proximity of the binding of these ligands altered considerably. The residues that interacted with GlcNAc included Asn-A140, Lys-C90, Lys-C91, Asn-C99, Ala-D94, Pro-D96, Thr-D97, Asn-D99, and Gln-D150 (supplemental Table S2) (Fig. 5A). The residues that formed hydrogen bonds with β-maltose included Trp-C66, Ala-C94, Gln-C150, Gly-D65, Lys-D90, and Asn-D99 (supplemental Table S3) (Fig. 5B). As seen from supplemental Fig. S3, molecules GlcNAc and β-maltose occupy non-overlapping distinct positions in the PAMP-binding cleft. There is no common residue that interacts with both compounds but several identical residues from protein molecules C and D that contribute to the binding of two ligands. These include residues Lys-C90, Asn-C99, Ala-D94, and Gln-D150, which interact with GlcNAc, and Ala-C94, Gln-C150, Lys-D90, and Asn-D99, which interact with β-maltose. It is also noteworthy that the binding affinities of compounds, GlcNAc and β-maltose were similar even though they occupied different positions in the binding cleft. It clearly shows that the ligand binding site consists of several specific binding regions in the cleft that allows an equally efficient recognition of different PAMPs.
FIGURE 5.

Hydrogen-bonded interactions between CPGRP-S and GlcNAc (A) from molecules A, C, and D and CPGRP-S and β-maltose from molecules C and D (B).
DISCUSSION
Crystal structure determinations have shown that CPGRP-S forms a stable asymmetrical homotetramer involving four identical protein molecules A, B, C, and D, whereas HPGRP-S exists as a monomer. A comparison of amino acids present at the C-D interface shows that CPGRP-S has Ala-94, Pro-96, and Pro-151, which favor dimerization (14, 21). The corresponding residues in HPGRP-S are Ser-94, His-96, and Arg-151, which are not known to favor dimerization (21). Therefore, it is quite evident from the amino acid sequence that molecules C and D in CPGRP-S may prefer to form a dimer, whereas the residues in HPGRP-S may not induce dimerization. Further sequence comparisons of PGRP-S from various other species (supplemental Fig. S4) shows that the corresponding amino acid residues in them do not appear to be as suitable for dimerization as in CPGRP-S. However, the final confirmation of the formation of such a multimeric state can be ascertained only by crystal structure determinations. As indicated by the contact regions between the pairs of molecules, it appears that the tetrameric structure has evolved as a dimer of two dimers formed between molecules A and B and molecules C and D. This arrangement is unique and because a cleft is formed inside the tetramer, it has functional implications. The overall design of the cleft involves contributions from all the four molecules with final binding regions residing at the interface of molecules C and D. The outer side of the C-D interface is closed as several intermolecular interactions between molecules C and D are formed on the distal site. The inner side, which is free from intermolecular contacts, opens into the void at the center of the tetramer. Because the dimers A-B and C-D are slightly shifted with respect to each other in the direction perpendicular to the C-D interface, the inner opening is contiguous to the A-C interface, which is also closed on the outer side with several intermolecular interactions between molecules A and C. The cleft comprising partial C-D and A-C interfaces is directly connected to B-D interface, which forms a long and wide channel between the center of the tetrameric complex and the surface of the protein molecules. The entry to the cleft presumably takes place through the opening at the B-D interface. The flexible N termini (residues, Glu-1 to Ser-8) of molecules B and D form flaps at the two ends of the diffusion channel, one on the surface and another inside at the entrance to the cleft which is connected to the channel via the center of the tetramer. These may help in filtering out non-PAMP molecules lacking stereochemical complimentarity. Furthermore, the shape of the B-D channel is more like a funnel with a wider orifice at the outer side that helps in capturing the PAMPs effectively. Further recognition of PAMPs is carried out by the internal surfaces of molecules B and D. The internal walls of the channel are lined up with a series of positively charged residues, Arg-B13, Arg-B23, Arg-B25, Arg-B28, and Asn-B88 from molecule B and Arg-D31, Tyr-D32, Arg-D138, Ser-D139, Asn-D140, and Arg-D170 from molecule D. The main PAMPs-binding site is comprised of an extended C-D interface along with a portion of the A-C interface. As observed in the case of MDP, the main interactions were provided by molecules C and D. The major interactions involving GlcNAc in the ternary complex came from amino acid residues of molecules A, C, and D with a majority of amino acid residues from molecule C. On the other hand, β-maltose interacted predominantly with residues from molecule D. The analysis of residues that interacted with three ligands shows an interesting pattern where MDP is in contact with two residues from molecule C, Trp-C66 and Asn-C99, and four residues from molecule D, Trp-D98, Asp-D148, Val-D149, and Glu-D150. In the case of GlcNAc, one residue, Asn-A140 from molecule A, three residues from molecule C, Lys-C90, Pro-C96, and Asn-C99 and one residue, Glu-D150, from molecule D. Of six residues that are involved in the binding to ligands, MDP and GlcNAc in the two complexes only two residues, Asn-C99 and Gln-D150 are common, whereas the remaining four residues are different. Similarly, the interactions of GlcNAc and β-maltose that form hydrogen-bonded interactions with six residues, of which only one residue Ala-C94 is common, the rest of the five residues are different. These observations clearly show that the binding subsites are distinct and recognize different chemical entities. It may be mentioned here that the multiligand specific binding sites have also been observed in other multimeric proteins such as a ubiquitously distributed cellular protein A from S. aureus (22), laminin-binding lectin (23), and some MHC tetramers (24).
It is clearly observed that the three compounds MDP, GlcNAc, and β-maltose interact with different residues in the binding site because these are accommodated in different subregions of the PAMP-binding site (Fig. 6). Similarly, a comparison of interactions of CPGRP-S with LPS and LTA also showed notable differences involving a number of common and different amino acid residues (14). These observations show that the PAMP-binding site in CPGRP-S is able to accommodate different types of PAMPs with significant specificities to each PAMP because it contains multiple subsites contributed by three molecules A, C, and D, whereas the supporting diffusion channel is formed by molecules B and D. Thus the mode of binding of CPGRP-S involves direct interactions with bacteria through its cell wall surface molecules by sequestering them effectively and then disallowing their communications for further cellular growth. This mechanism of action is distinct from the effects of membrane permeabilizing antibacterial peptides, peptidoglycan-lytic enzymes, and inhibitors of peptidoglycan biosynthesis. It appears that CPGRP-S essentially targets cell wall molecular patterns rather than cell membranes. This means that CPGRP-S may be useful protein antibiotic and may not suffer from bacterial resistance.
FIGURE 6.

Grasp representation of the binding cleft in the tetrameric CPGRP-S showing the positions of MDP (A) and GlcNAc and β-maltose (B).
Supplementary Material
This work was supported by grants from the Departments of Science and Technology and Department of Biotechnology, Ministry of Science and Technology (government of India, New Delhi) and the Council of Scientific and Industrial Research, New Delhi (to P. S. and M. S.).
The atomic coordinates and structure factors (codes 3NG4 and 3NW3) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1–S3 and Figs. S1–S4.
- PGRP
- peptidoglycan recognition protein
- CPGRP-S
- camel PGRP-S
- PBMC
- peripheral blood mononuclear cell
- PAMP
- pathogen-associated molecular pattern
- PGN
- peptidoglycan
- MDP
- muramyl dipeptide
- LTA
- lipoteichoic acid
- PDB
- Protein Data Bank
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. Schleifer K. H., Kandler O. (1972) Bacteriol. Rev. 36, 407–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dziarski R., Gupta D. (2005) J. Endotoxin. Res. 11, 304–310 [DOI] [PubMed] [Google Scholar]
- 3. Kang D., Liu G., Lundström A., Gelius E., Steiner H. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 10078–10082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ochiai M., Ashida M. (1999) J. Biol. Chem. 274, 11854–11858 [DOI] [PubMed] [Google Scholar]
- 5. Werner T., Liu G., Kang D., Ekengren S., Steiner H., Hultmark D. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 13772–13777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu C., Xu Z., Gupta D., Dziarski R. (2001) J. Biol. Chem. 276, 34686–34694 [DOI] [PubMed] [Google Scholar]
- 7. Christophides G. K., Zdobnov E., Barillas-Mury C., Birney E., Blandin S., Blass C., Brey P. T., Collins F. H., Danielli A., Dimopoulos G., Hetru C., Hoa N. T., Hoffmann J. A., Kanzok S. M., Letunic I., Levashina E. A., Loukeris T. G., Lycett G., Meister S., Michel K., Moita L. F., Muller H. M., Osta M. A., Paskewitz S. M., Reichhart J. M., Rzhetsky A., Troxler L., Vernick K. D., Vlachou D., Volz J., von Mering C., Xu J., Zheng L., Bork P., Kafatos F. C. (2002) Science 298, 159–165 [DOI] [PubMed] [Google Scholar]
- 8. Liu C., Gelius E., Liu G., Steiner H., Dziarski R. (2000) J. Biol. Chem. 275, 24490–24499 [DOI] [PubMed] [Google Scholar]
- 9. Lu X., Wang M., Qi J., Wang H., Li X., Gupta D., Dziarski R. (2006) J. Biol. Chem. 281, 5895–5907 [DOI] [PubMed] [Google Scholar]
- 10. Kappeler S. R., Heuberger C., Farah Z., Puhan Z. (2004) J. Dairy Sci. 87, 2660–2668 [DOI] [PubMed] [Google Scholar]
- 11. Lo D., Tynan W., Dickerson J., Mendy J., Chang H. W., Scharf M., Byrne D., Brayden D., Higgins L., Evans C., O'Mahony D. J. (2003) Cell Immunol. 224, 8–16 [DOI] [PubMed] [Google Scholar]
- 12. Guan R., Wang Q., Sundberg E. J., Mariuzza R. A. (2005) J. Mol. Biol. 347, 683–691 [DOI] [PubMed] [Google Scholar]
- 13. Sharma P., Singh N., Sinha M., Sharma S., Perbandt M., Betzel C., Kaur P., Srinivasan A., Singh T. P. (2008) J. Mol. Biol. 378, 923–932 [DOI] [PubMed] [Google Scholar]
- 14. Sharma P., Dube D., Singh A., Mishra B., Singh N., Sinha M., Dey S., Kaur P., Mitra D. K., Sharma S., Singh T. P. (2011) J. Biol. Chem. 286, 16208–16217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 16. Brünger A. T., Adams P. D., Rice L. M. (1997) Structure 5, 325–336 [DOI] [PubMed] [Google Scholar]
- 17. Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. (1991) Acta Crystallogr. A 47, 110–119 [DOI] [PubMed] [Google Scholar]
- 18. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 19. Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) J. Appl. Cryst. 26, 283–291 [Google Scholar]
- 20. Ramachandran G. N., Sasisekharan V. (1968) Adv. Protein Chem. 23, 283–438 [DOI] [PubMed] [Google Scholar]
- 21. Kini R. M., Evans H. J. (1995) Biochem. Biophys. Res. Commun. 212, 1115–1124 [DOI] [PubMed] [Google Scholar]
- 22. Nguyen T., Ghebrehiwet B., Peerschke E. I. (2000) Infect. Immun. 68, 2061–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bao Z. Z., Muschler J., Horwitz A. F. (1992) J. Biol. Chem. 267, 4974–4980 [PubMed] [Google Scholar]
- 24. Xu X. N., Screaton G. R. (2002) J. Immunol. Methods 268, 21–28 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.