

Abstract
Integration of mouse mammary tumor virus (MMTV) at the common integration site Int6 occurs in the gene encoding eIF3e, the p48 subunit of translation initiation factor eIF3. Integration is at any of several introns of the Eif3e gene and causes the expression of truncated Eif3e mRNAs. Ectopic expression of the truncated eIF3e protein resulting from integration at intron 5 (3e5) induces malignant transformation, but by an unknown mechanism. Because eIF3e makes up at least part of the binding site for eIF4G, we examined the effects of 3e5 expression on protein synthesis. We developed an NIH3T3 cell line that contains a single copy of the 3e5 sequence at a predetermined genomic site. Co-immunoprecipitation indicated diminished binding of eIF3 to eIF4G, signifying a reduction in recruitment of the mRNA-unwinding machinery to the 43 S preinitiation complex. Cell growth and overall protein synthesis were decreased. Translation driven by the eIF4G-independent hepatitis C virus internal ribosome entry sequence (HCV IRES) in a bicistronic mRNA was increased relative to cap-dependent translation. Endogenous mRNAs encoding XIAP, c-Myc, CYR61, and Pim-1, which are translated in a cap-independent manner, were shifted to heavier polysomes whereas mRNAs encoding GAPDH, actin, L32, and L34, which are translated in a cap-dependent manner, were shifted to lighter polysomes. We propose that expression of 3e5 diminishes eIF4G interaction with eIF3 and causes abnormal gene expression at the translational level. The correlation between up-regulation of cap-independent translation and MMTV-induced tumorigenesis contrasts with the well established model for malignant transformation involving up-regulation of highly cap-dependent translation.
Keywords: Mammary Gland, mRNA, Oncogenic Viruses, Protein Synthesis, Translation Initiation Factors
Introduction
Mouse mammary tumor virus (MMTV)3 induces mammary carcinomas in mice, but unlike other acute transforming retroviruses, it does not encode an oncogene (1). Rather, it causes malignant transformation by insertional mutagenesis and clonal expansion of cells with transforming virus-induced mutations. In most of the cases, integration of MMTV activates genes adjacent to the integration site (e.g. Wnt, Fgf, and Rspo) via the effect of strong enhancers present in the viral long terminal repeats on the expression of previously silent genes in the mammary gland (2, 3). However, for the common integration sites Notch-4 and Int6, the viral integration events occur within the gene (4, 5). Viral integration into the gene for Notch-4, a transcriptional regulator (6), corresponds to a gain-of-function mutation. Integration at Int6 (7) occurs in the gene for eIF3e (8) at any of several introns and leads to the synthesis of truncated mRNAs (5). Expression the eIF3e protein resulting from truncation of the mRNA at intron 5 (3e5; amino acid residues 1–157) is sufficient to cause malignant transformation of both mammary and non-mammary cell lines (9, 10) and the formation of mammary tumors in transgenic mice (11), suggesting that expression of 3e5 per se, rather than the loss of one of the Eif3e alleles, is responsible for tumorigenesis.
eIF3e is the p48 subunit of the translational initiation factor eIF3 (8). eIF3 is a heteromultimeric protein composed of 6 subunits in Saccharomyces cerevisiae (12) and 13 subunits in mammals, 6 of which (a, b, c, e, f, and h) make up a functional core (13, 14). It forms a multifactor complex with eIF1, eIF5, and the eIF2·GTP·Met-tRNA ternary complex that binds to the 40 S ribosomal subunit to form the 43 S preinitiation complex (PIC). eIF3 also prevents the premature joining of the 60 S ribosomal subunit until the start codon is recognized (15, 16).
Another function of eIF3 is recruitment of eIF4G-associated mRNAs to the 43 S PIC to form the 48 S PIC and initiate mRNA scanning (17). The portion of eIF4G-1 that binds eIF3 has been mapped to amino acid residues 975–1078 (18, 19). We previously presented evidence that the portion of eIF3 that binds elF4G includes the eIF3e subunit (20). When elF4G(975–1078) is used in a pull-down assay with partially proteolyzed eIF3, fragments of eIF3e are enriched. Free eIF3e binds directly to eIF4G(975–1078) and competes with the binding of intact eIF3. Finally, free eIF3e reduces the rate of protein synthesis in an in vitro translation system and shifts a reporter mRNA from heavy to light polysomes, suggesting an effect on initiation rather than elongation/termination. We proposed that free eIF3e inhibits protein synthesis by interfering with the interaction between eIF3 and eIF4G (20). Ribosomal protein L13a also blocks 43 S PIC recruitment by interacting with eIF4G at the eIF3-binding site in an interferon-γ-dependent mechanism (21). Overexpression of eIF3e, but not eIF3j, blocks the interferon-γ-dependent binding between L13a and eIF4G.
The mechanism by which 3e5 causes malignant transformation is not known. Based on previous in vitro findings with full-length eIF3e (20), we propose that 3e5 interferes with the binding of eIF4G to eIF3 and therefore with the normal recruitment of cap-dependent mRNAs to the 48 S PIC. In the present work, we tested this hypothesis by expressing 3e5 in NIH3T3 cells from a single copy gene. We demonstrate that expression of 3e5 reduces the amount of eIF3 co-immunoprecipitated with eIF4G and vice versa. Overall translation is diminished, but eIF4G-independent translation of a reporter mRNA driven by the internal ribosome entry sequence (IRES) of hepatitis C virus (HCV) is less affected than cap-dependent translation from the same reporter. Moreover, the polysomal distribution of individual endogenous mRNAs differs between control and 3e5-expressing cells: mRNAs known to be cap-dependent (actin, GAPDH, L32, and L34) are on lighter polysomes in 3e5-expressing cells than control cells, but mRNAs known to be cap-independent (XIAP, c-Myc, CYR61, and Pim-1) are on heavier polysomes. The latter mRNAs encode proteins that either induce cellular transformation or promote survival under apoptotic conditions.
EXPERIMENTAL PROCEDURES
Reagents
Perfecta SYBR Green Supermix was purchased from Quanta Biosciences. [35S]Met was purchased from MP Biomedicals. The Flp-InTM system was purchased from Invitrogen. Solutions R and V for nucleoporation were obtained from Lonza. Rabbit anti-eIF3e (22) and anti-eIF4G-1 (peptide 7) (23) antibodies were described previously. Goat anti-eIF3b antibodies were from Santa Cruz Biotechnology and mouse anti-rabbit light chain-specific antibodies, from Jason ImmunoResearch.
Plasmids
Plasmid pKS/FF/HCV/Ren for the synthesis of bicistronic mRNAs was generously donated by Jerry Pelletier, McGill University, Montreal (24). Plasmid pluc-A60 was described previously (25). For the construction of plasmid pEF6/3e5, the sequence of Eif3e mRNA truncated at intron 5 was amplified from Eif3e cDNA, obtained by reverse transcription of total mRNAs isolated from NIH3T3 cells, using primer set 1 (supplemental Table S1), and inserted into pEF6/V5-His6 (Invitrogen). pcDNA5/FRT/3e5 was constructed by amplifying the 3e5 sequence from pEF6/3e5 with primer set 2 and inserting into pcDNA5/FRT.
RNA Synthesis
A bicistronic RNA encoding cap-dependent firefly (FF) luciferase and HCV IRES-dependent Renilla (Ren) luciferase, and a monocistronic RNA encoding FF luciferase, were synthesized in vitro as previously described (25) with BamHI-linearized pKS/FF/HCV/Ren (24) or HpaI-linearized pluc-A60 (25), respectively, as template. RNAs were purified using an E.Z.N.A. total RNA kit (Omega Bio-Tek).
Cell Lines and Culture Conditions
3T3–3e5, 3T3–3e9, and 3T3-Ø cells were constructed by insertion of sequences into Flp-In NIH3T3 cells (Invitrogen), which contain a single site for homologous recombination (FRT) located in a transcriptionally active region. Flp-In NIH3T3 cells expressing 3e5 under control of the CMV promoter (3T3–3e5) were constructed by cotransfection of 2 × 106 cells with pOG44 and pcDNA5/FRT/3e5 at a ratio of 9:1 (4.5 μg of pOG44 plus 0.5 μg of pcDNA5/FRT/3e5). Flp-In NIH3T3 cells containing the identical sequences except those of 3e5 mRNA (3T3-Ø) were constructed in a similar manner except that pcDNA5/FRT was used. A similar cell line was created by the same method but with primer set 17 in which Eif3e mRNA truncated at intron 9 was expressed (3T3–3e9), but since its properties were indistinguishable from those of 3T3–3e5, only data on the latter are presented here, with the exception of Fig. 5B. Transfected cells were selected with 0.3 mg/ml hygromycin in complete DMEM media. Hygromycin, and normocin were omitted from all media 72 h before assays were conducted to measure rate of protein synthesis, luciferase production, or polysomal distribution of endogenous mRNAs. All transfections were performed using nucleoporation technology (Lonza) with solution R and program U-030. Cells were maintained in complete DMEM media containing 10% fetal bovine serum, 100 μg/ml normocin, 300 μg/ml hygromycin, penicillin, and streptomycin.
FIGURE 5.
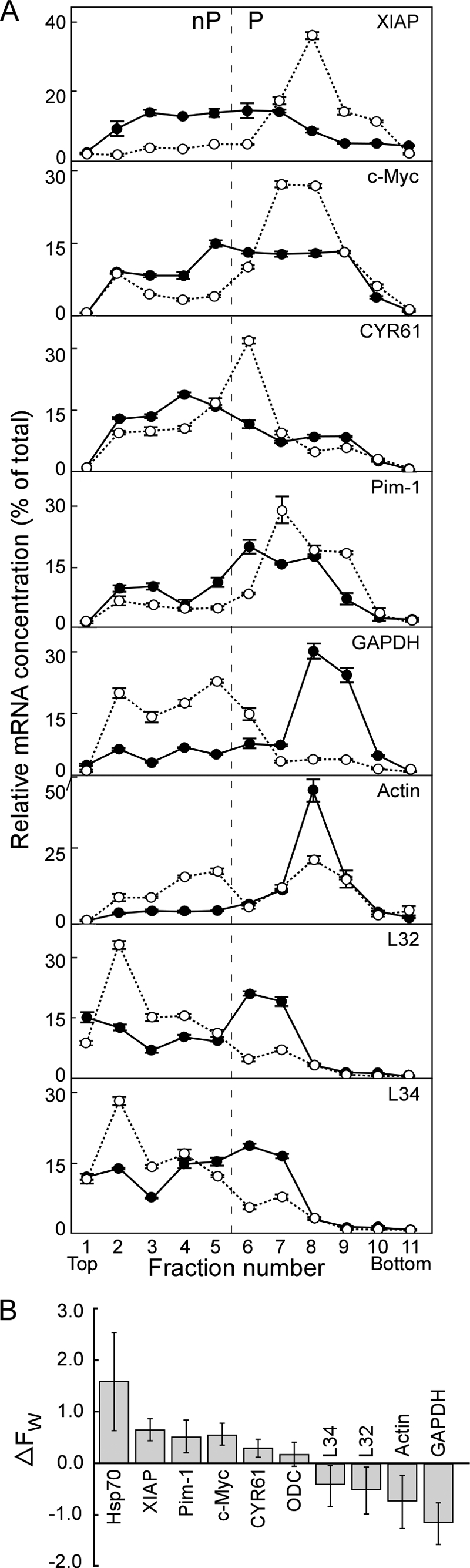
Cap-independent mRNAs are shifted to heavier polysomes, and cap-dependent mRNAs are shifted to lighter polysomes, by 3e5 expression. A, extracts of 3T3-Ø (filled circles) and 3T3–3e5 (open circles) cells were subjected to ultracentrifugation for 2 h and the concentrations of the indicated mRNAs determined by qRT-PCR. Each point represents the amount of a given mRNA expressed as a percentage of the total for that mRNA in all 11 fractions. Fractions 1–5 represent the nP region and fractions 6–11, the P region (see Fig. 3D). B, multiple experiments were performed to compare extracts of 3T3-Ø to those of either 3T3–3e5 or 3T3–3e9 cells in the same ultracentrifugation run. Weighted averages (FW) for the distribution of a given mRNA were calculated with Eq. 1 for each cell line. The FW for 3T3-Ø cells was subtracted from the FW for either 3T3–3e5 or 3T3–3e9 cells (ΔFW) for extracts analyzed in the same ultracentrifugation run. The number of separate ultracentrifugation and qRT-PCR experiments represented in the figure is: actin, 4; c-Myc, 6; CYR61, 3; GAPDH, 6; Hsp70, 2; L32, 3; L34, 3; ODC, 4; Pim-1, 3; and XIAP, 8.
Immunological Assays
3T3-Ø and 3T3–3e5 cells were cultured in 15-cm dishes to 80% confluency, washed in cold PBS twice, and harvested by scraping. Cells were washed again once in cold PBS, centrifuged at 5,000 × g for 2 min, resuspended in 500 μl of lysis buffer (20 mm MOPS, 100 mm KCl, 1 mm EDTA, 2 mm EGTA, 0.5 mm DTT, and protease inhibitors), lysed by maceration in a liquid nitrogen-cooled mortar and pestle, and centrifuged at 13,000 × g for 15 min at 4 °C. Immunoprecipitation (IP) was performed with equal amounts of protein per cell line ranging from 100 to 300 μg (26) by adding 2–4 μg of antibodies overnight at 4 °C followed by 20 μl of either protein A- or protein G-agarose (for rabbit or goat antibodies, respectively) for 2 h at room temperature. The beads were washed four times with 20 volumes of lysis buffer. Protein was eluted with 15 μl of loading buffer at 90 °C for 5 min, separated by SDS-PAGE on a 4–12% gradient gel (Novex, Invitrogen), and transferred to PVDF membranes overnight at 4 °C. The secondary antibody used for eIF3e detection was HRP-conjugated mouse anti-rabbit light chain. Secondary antibodies for elF4G and elF3b were conjugated with alkaline phosphatase. Bands were quantified with ImageQuant 5.0 software (Molecular Dynamics).
Dual-luciferase Assay
Bicistronic mRNA (2 μg in 100 μl of solution R) was introduced into duplicate batches of 2 × 106 cells by nucleoporation (Lonza; program U-030). After washing and resuspending in warm complete DMEM media, cells were incubated with gentle shaking at 37 °C, collected at different times, washed in cold PBS, and frozen in dry ice. The activities of FF and Ren luciferase were determined with a dual-luciferase reporter assay kit (Promega).
Rate of Protein Synthesis
Cells at 70–80% confluency in 35-mm dishes were treated with media containing 100–200 μCi/ml of [35S]Met for 30, 60, or 90 min. The incorporation of radioactivity was stopped by three washes in cold PBS. Cell extracts were prepared by sonicating in lysis buffer [25 mm Tris-HCl, pH 7.8, 300 mm NaCl, 5% glycerol, 0.5% Tween-20, and 1× protease inhibitor mixture (Roche Diagnostics)] and centrifuging at 14,000 × g for 14 min at 4 °C. Radioactivity was measured in 5–10 μg of protein (26) as described previously (20).
Analysis of Polysomes
The polysomal distributions of endogenous mRNAs in 3T3-Ø and 3T3–3e5 cells at 80% confluency was determined as described previously (25) except that 10–45% sucrose gradients were used. To determine the P/nP ratios, OD260 traces were digitally scanned and aligned. The baseline was determined from a control gradient onto which only lysis buffer was loaded. The area above the baseline was divided into non-polysomal (nP, fractions 1–5) and polysomal (P, fractions 6–11) areas. TpsDIG 2.12 software (SUNY Stony Brook) was used to measure the pixel content of the two areas and calculate P/nP ratios.
Quantitation of mRNA
RNA was purified from 100-μl aliquots of polysome gradient fractions with an E-Z 96 Total RNA kit (Omega Bio-Tek). FF luciferase mRNA (100 ng) was added to each aliquot before RNA isolation as an internal standard. Purified RNAs were treated with RQ1 DNase (Promega), after which the DNase was inactivated by heating at 65 °C for 10 min in 2 mm EGTA. cDNA was synthesized with the TaqMan® reverse transcription kit (Applied Biosciences). Quantitative reverse-transcriptase-real-time PCR (qRT-PCR) was performed as previously described (25) with primers listed in supplemental Table S1. The amount of a given mRNA in each fraction was calculated by the ΔCT method with luciferase mRNA as internal standard as described in User Bulletin No. 2 for the ABI Prism 7700 Sequence Detection System. Weighted averages were calculated using equation 1. For determination of 3e5 and Eif3e mNA levels, total mRNAs were extracted from 3T3–3e5 cells, treated with RQ1 DNase (Promega), and 400 ng used for cDNA synthesis. An equal volume of cDNA was used for PCR reactions using primer sets 6 and 7 (supplemental Table S1) for the amplification of 3e5 and endogenous Eif3e mRNA, respectively. Reactions were run in 1% agarose gels and intensities of bands determined digitally with FluorChem Q SA software.
RESULTS
Development of an NIH3T3 Cell Line Containing a Single Copy of the 3e5 Sequence
Previously, several cell culture models were developed to understand the mechanism of tumorigenesis by 3e5. HC11, MCF10A, and NIH3T3 cells stably transfected with a vector expressing 3e5 formed colonies in soft agar, and subcutaneous injection of transformed MCF10A and HC11 cells into nude or BALB/c mice, respectively, formed nodular growths (9). In another study, NIH3T3 cells expressing 3e5 formed foci, grew in soft agar, and were more resistant to serum starvation-induced apoptosis (10). However, these models had limitations for our study of possible 3e5-induced changes in protein synthesis. First, the copy number of 3e5 sequences is unknown in these cell lines, yet there is a single copy of the 3e5 sequence per diploid cell in mouse mammary tumors in which MMTV has inserted into the Eif3e gene. Second, the cells expressing 3e5 in these previous studies are maintained in G418, an inhibitor of protein synthesis, but the parent cell line used for comparison is not. It is possible that components of the protein synthesis machinery exist at different levels in 3e5-expressing cells compared with the parent because of the long-term presence of G418. Third, the 3e5-expressing cells developed to date represent a mixed population selected for G418 resistance after transfection with a plasmid vector that expresses 3e5, but a control cell line was not created that contains the same vector without the 3e5 sequences. Thus, differences we might observe in protein synthesis between 3e5-expressing and control cells could conceivably be due to vector sequences.
We therefore created two new NIH3T3 cell lines by homologous recombination, one containing a vector expressing the 3e5 sequence (3T3–3e5 cells) and the other containing the empty vector inserted at the same site (3T3-Ø cells). A single copy of the 3e5 sequence (Fig. 1A) was inserted into a Flp-In recombination target site located in a transcriptionally active region (Fig. 1B). This nearly mimics the situation with MMTV integration: a single-copy of the truncated Eif3e gene is present for both virus-infected cells and 3T3–3e5 cells. However, there is only one wild-type Eif3e allele in virus-infected cells but two in 3T3–3e5 cells. Both 3T3–3e5 and 3T3-Ø clonal cell lines were maintained under the same selection pressure, the antibiotic hygromycin in this case. (Hygromycin was removed 72 h before any experiments on cell growth or protein synthesis.) Single-copy integration was confirmed by a PCR-based method (27). Briefly, DNA sequences from 3T3-Ø and 3T3–3e5 cells were assayed by quantitative real-time PCR (qPCR) with primer set 8 (supplemental Table S1), which amplifies the same sequence from both the endogenous Eif3e gene and the 3e5 transgene (open arrows in Fig. 1A). The concentration of Eif3e sequences in each cell line was normalized by GAPDH sequences (Table 1). The results indicated that the Eif3e sequence amplified by primer set 8 was 1.64 ± 0.21 more concentrated in 3T3–3e5 DNA than 3T3-Ø DNA, indicating a single insertion per diploid genome (theoretical: 1.50).
FIGURE 1.

Construction of NIH3T3 cells expressing 3e5. A, 3e5 and full-length eIF3e. The mRNA produced by insertion of MMTV at intron 5 of the Eif3e gene encodes eIF3e truncated after 157 amino acid residues (3e5) whereas the full-length protein contains 445 amino acid residues. The black box to the right of 3e5 represents vector sequences. Open arrows show positions of primer set 8 on the corresponding mRNA, which amplifies sequences in both 3e5 and Eif3e. Solid arrows show positions of primer set 6, which amplifies sequences only in 3e5 because the reverse primer is complementary to vector sequences. Cross-hatched arrows show positions of primer set 7, which amplifies sequences only in Eif3e. NES, nuclear export signal. NLS, nuclear localization signal. PCI, the 26 S proteasome-COP9 signalosome-eIF3 domain. B, construction of 3T3-Ø and 3T3–3e5 cells. Insertion of 3e5 and vector sequences by homologous recombination. C, detection of 3e5 and Eif3e mRNAs by RT-PCR. RNA extracted from 3T3-Ø and 3T3–3e5 cells was subjected to PCR with either 3e5-specific primers (primer set 6; lanes 1–4) or Eif3e-specific primers (primer set 7; lanes 6–8). Reverse transcriptase was omitted in lanes 1, 3, 5, and 7. D, relative mRNA levels of 3e5 and endogenous Eif3e mRNA levels were quantitated as described under “Experimental Procedures” for four independent experiments.
TABLE 1.
Determination of gene copy number
DNA concentrations were measured by absorbance at 260 nm. Standard curves of CT versus log DNA concentration were obtained for endogenous Eif3e and GAPDH genes by performing qPCR on DNA from 3T3-Ø and 3T3-3e5 cells with primer sets 8 and 10, respectively (supplemental Table S1). Sequences from Eif3e and GAPDH genes were amplified in quintuplicate from 16 ng of DNA for each cell line. The CT values were used to calculate the concentrations of these sequences from the appropriate standard curve. The concentration of Eif3e sequences was then normalized by the concentration of GAPDH sequences for a given cell line. Finally, the copy number of Eif3e in 3T3-3e5 DNA was calculated relative to the copy number in 3T3-Ø DNA.
| Cell Line | Eif3e | GAPDH | Eif3e normalized to GAPDH | Eif3e copy number normalized to 3T3-Ø |
|---|---|---|---|---|
| 3T3-Ø | 17.45 | 14.33 | ||
| 16.51 | 14.41 | |||
| 18.64 | 13.82 | |||
| 21.51 | 13.97 | |||
| 21.99 | 19.28 | |||
| Average | 19.22 | 15.16 | 1.27 | 1 |
| 3T3-3e5 | 39.01 | 16.41 | ||
| 27.26 | 12.78 | |||
| 30.95 | 15.34 | |||
| 24.55 | 14.79 | |||
| 23.24 | 10.55 | |||
| Average | 29.00 | 13.97 | 2.08 | 1.64 ± 0.21 |
Previous attempts to detect 3e5 by Western blotting have been unsuccessful (9, 10), although a fusion protein of 3e5 and GFP was detected (9). We were similarly unable to detect 3e5 by Western blotting in 3T3–3e5 cells with an antibody raised to a peptide corresponding to amino acids 59–73 (22), which falls within the 3e5 sequence (Fig. 1A). Instead, we demonstrated expression of 3e5 mRNA by reverse transcription and PCR with a forward primer complementary to Eif3e sequences and a reverse primer complementary to vector sequences (Fig. 1C). The level of 3e5 mRNA was approximately half that of Eif3e mRNA (Fig. 1D), perhaps resulting from the single copy of the 3e5 gene compared with two copies of the endogenous Eif3e gene. This result suggests that the truncation does not have unexpected effects on transcription rate or mRNA stability, e.g. through nonsense-mediated decay (28).
Binding of eIF3 to eIF4G Is Diminished in 3T3–3e5 Cells
We carried out IP experiments to test whether 3e5 interferes with the interaction of eIF4G and eIF3 (Fig. 2A). IP of cell lysates with anti-eIF4G antibodies brought down less eIF3 in 3T3–3e5 than 3T3-Ø cells, as detected in Western blots with an antibody against the core eIF3b subunit (Fig. 2, B and D, left panel; p < 0.001). [The multiple bands in the eIF4G Western blot are due to isoforms of eIF4G produced by alternative translation initiation sites (29, 30).] Diminished eIF4G-eIF3 interaction was also observed when the IP was performed with anti-eIF3b antibodies and eIF4G detected by Western blotting (Fig. 2, C and D, middle panel; p < 0.04). The quantitative differences between the eIF4G IP and eIF3b Western blot (Fig. 2D, left panel; ∼42% decrease) versus the reciprocal experiment (Fig. 2D, middle panel; ∼20% decrease) may be due to different pool sizes of free eIF4G and eIF3. Two models that could explain decreased eIF4G-eIF3 interaction in 3T3–3e5 cells are (i) 3e5 binds to eIF4G and prevents its interaction with intact eIF3, and (ii) 3e5 displaces intact eIF3e from eIF3, and the altered eIF3 is unable to bind eIF4G. Evidence against the latter model is provided by the finding that the same amount of eIF3e is brought down by co-IP with eIF3b from lysates of 3T3–3e5 and 3T3-Ø cells (Fig. 2, C and D, right panel; p = 0.2).
FIGURE 2.

Expression of 3e5 antagonizes the interaction between eIF4G and eIF3. A, model for competition between truncated eIF3e (residues 1–157; 3e5) and eIF3 for binding to eIF4G. 3e5 is depicted as a fragment of eIF3e that retains the ability to bind eIF4G (G) and prevents recruitment of mRNAs to the 43 S preinitiation complex (PIC), inhibiting cap-dependent translation. 2, eIF2; 3, eIF3; 3e, eIF3e; A, eIF4A; E, eIF4E; PABP, the poly(A)-binding protein; 40, 40 S ribosomal subunit. B, immunoprecipitation (IP) of extracts from 3T3-Ø (Ø) and 3T3–3e5 (3e5) cells with anti-eIF4G antibodies. Western blotting (WB) was performed with anti-eIF4G (upper panel) or anti-eIF3b (lower panel) antibodies. C, IP as in B except with anti-eIF3b antibodies. WB was performed with anti-eIF3b (upper panel), anti-eIF4G (middle panel), or eIF3e (lower panel) antibodies. D, quantification of initiation factor subunits. Left, the eIF3b bands detected by WB in B were quantified by ImageQuant software and normalized by the eIF4G signal. Average of six experiments. p < 0.001. Middle, the eIF4G bands detected in C were quantified and normalized by the eIF3b signal. Average of four experiments. p < 0.04. Right, the eIF3e bands detected in C were quantified and normalized by the eIF3b signal. Average of four experiments. p = 0.2. Error bars represent S.E.
Cell Growth and Protein Synthesis Are Decreased in 3T3–3e5 Cells
Interfering with the binding of eIF4G to eIF3 would be expected to reduce the overall rate of protein synthesis and, consequently, cell growth. The rate of cell growth is slower for 3T3–3e5 cells (Fig. 3A), with population doubling time of 26.4 ± 3.3 h for 3T3–3e5 cells versus 20.0 ± 2.0 h for 3T3-Ø cells (p < 0.001). The rates of protein synthesis in the two lines were determined by pulse-labeling with [35S]Met (Fig. 3B). The results indicated that 3T3–3e5 cells synthesize proteins 34% slower than 3T3-Ø cells. As observed previously (9, 10), 3T3–3e5 cells formed more colonies in soft agar (data not shown).
FIGURE 3.

Expression of 3e5 alters growth and protein synthesis in NIH3T3 cells. A, logarithmic growth of 3T3-Ø (filled circles) and 3T3–3e5 (open circles) cells. Equal numbers of cells were seeded on 35-mm plates and counted every 24 h. B, rate of protein synthesis. 3T3-Ø and 3T3–3e5 cells were cultured to 70–80% confluency and then labeled for 30 min with [35S]Met. Preliminary experiments showed that incorporation of radioactivity into trichloroacetic acid-insoluble increased linearly over this period. C, differential expression of proteins in 3T3-Ø and 3T3–3e5 cells. The same labeled cell extracts used in B (2 × 105 cpm) were separated by SDS-PAGE on a 4–12% gel, stained with Coomassie Blue (left), and labeled proteins detected by autoradiography for 24 h (middle). Portions of the autoradiogram are enlarged (right). Open arrowheads indicate proteins labeled more strongly in 3T3-Ø cells. Closed arrowheads indicate proteins labeled more strongly in 3T3–3e5 cells. D, analysis of ribosomal subunit distribution between the nonpolysomal fraction (nP) and polysomes (P). Extracts of 3T3-Ø (black trace) and 3T3–3e5 (red trace) cells were subjected to ultracentrifugation for 2 h. Peaks at fractions 2.5, 3.5, and 4.5 were shown by qRT-PCR to be 40 S subunits, 60 S subunits, and 80 S monosomes, respectively.
The spectrum of proteins synthesized in the two cell lines in Fig. 3B was compared by SDS-PAGE (Fig. 3C). Equal radioactivity rather than equal protein was loaded on each lane. Because the rate of [35S]Met incorporation is less for 3T3–3e5 cells (Fig. 3B), the Coomassie Blue staining for this cell line is stronger (Fig. 3C, left panel). The autoradiogram (Fig. 3C, middle panel) shows a similar pattern of proteins synthesized in 30 min, but there are subtle differences. Some proteins are labeled more strongly in 3T3-Ø cells (Fig. 3C, right panels, open arrowheads) whereas others are labeled more strongly in 3T3–3e5 cells (closed arrowheads).
The reduction in protein synthesis in 3T3–3e5 cells (Fig. 3B) could be due to inhibition of either initiation or elongation/termination. Inhibiting initiation causes a shift of mRNAs from larger to smaller polysomes or free mRNPs, whereas inhibition of elongation/termination causes the opposite (31). We investigated how 3e5 expression affects overall protein synthesis by sedimentation analysis of polysomes, monitored by A260 (Fig. 3D). In 3T3–3e5 extracts, there is a decrease in polysomes (P) relative to non-polysomal ribosomal subunits (nP), indicating a decrease in initiation relative to elongation/termination. In three replicate experiments, the P/nP ratio was 1.0 ± 0.2 for 3T3–3e5 compared with 2.2 ± 0.4 for 3T3-Ø (p < 0.03).
Cap-dependent Translation Is Inhibited More than HCV-IRES-driven Translation in 3T3–3e5 Cells
Joining of eIF4G to eIF3 recruits the cap-dependent RNA unwinding machinery to the 43 S PIC (Fig. 2A). However, some RNAs are translated in a cap-independent manner. The HCV IRES contains sequences that bind directly to eIF3 and the 40 S ribosomal subunit (32). Translation driven by the HCV IRES does not require eIF4G but does require eIF2 and eIF3 (33). If the decrease in overall protein synthesis in 3T3–3e5 cells is due to interference with the binding of eIF4G to eIF3, one would expect that translation driven by the HCV IRES would be less affected than cap-dependent translation (Fig. 4A). To test this, we introduced into 3T3-Ø and 3T3–3e5 cells a bicistronic mRNA encoding FF and Ren luciferase with their open reading frames separated by the HCV IRES (Fig. 4A). Synthesis of both types of luciferase was lower in 3T3–3e5 cells, but FF was more severely affected (∼38% of 3T3-Ø) than Ren (∼88% of 3T3-Ø). To compensate for variations between experiments, Ren/FF ratios for a given experiment were calculated and the ratios for three experiments averaged (Fig. 4B). The Ren/FF ratios were higher for 3T3–3e5 cells than 3T3-Ø cells, supporting the idea that 3e5 preferentially inhibits cap-dependent translation.
FIGURE 4.

Translation from a reporter mRNA driven by the HCV IRES is increased relative to cap-dependent translation by 3e5 expression. A, model for the effects of 3e5 on translation of a bicistronic mRNA containing cistrons for FF and Ren luciferase separated by the HCV IRES. The FF cistron is recruited to the 48S PIC through a mechanism that requires the association of eIF3 and eIF4G, which is antagonized by 3e5. The Ren cistron is recruited to the 48S PIC by direct binding of the HCV IRES to eIF3 and the 40 S ribosomal subunit. Abbreviations are as in Fig. 2A. B, dual luciferase assay. The bicistronic mRNA was introduced into 3T3-Ø and 3T3–3e5 cells by nucleoporation and the activities of FF and Ren luciferase measured after 20, 40, and 60 min. For a given cell line, the Ren/FF ratios were similar at each time point, so they were averaged. The mean ± S.D. is shown for three experiments. p < 0.003.
Expression of 3e5 Changes the Polysomal Distribution of Individual Endogenous mRNAs
The foregoing results suggest that 3e5 interferes with cap-dependent translation. Cap-independent translation is well understood in the case of viral IRESes (34), but there are also cellular mRNAs that are translated by cap-independent mechanisms (35). These mRNAs often encode growth factors, growth factor receptors, or proteins that promote survival and antagonize apoptosis.
We examined the polysomal distribution of four such mRNAs in extracts of 3T3–3e5 and 3T3-Ø cells: XIAP (36), c-Myc (37), CYR61 (38), and Pim-1 (39). As controls, we chose four mRNAs that are translated by the canonical cap-dependent pathway: GAPDH, actin, L32, and L34. Analysis of polysomes was performed by sedimentation on sucrose gradients (Fig. 5). The A260 profiles (similar to Fig. 3D) indicated that fractions 1 to 5 correspond to the nP region and fractions 6 to 11, to the P region. RNA prepared from each of the 11 fractions for a given cell line was used to make cDNA, and then the relative concentration of all eight mRNAs was determined by qRT-PCR of the same cDNA preparation. The results indicated that XIAP mRNA is translated more efficiently in 3T3–3e5 cells (Fig. 5A, open circles; ∼85% of XIAP mRNA in the P region) than in 3T3-Ø cells (closed circles; ∼50% in the P region). c-Myc, CYR61, and Pim-1 mRNAs are also translated more efficiently in 3T3–3e5 cells than 3T3-Ø cells. The opposite result is seen for the cap-dependent GAPDH mRNA; it is translated less efficiently in 3T3–3e5 cells (∼25% in the P region) than in 3T3-Ø cells (∼76% in the P region). Similar results are seen with actin, L32, and L34 mRNAs. The percentages of all eight mRNAs in nP and P regions are given in Table 2.
TABLE 2.
Percentages of individual mRNAs in nP and P regions based on the qRT-PCR analysis shown in Fig. 5A
| mRNA | Region | 3T3-Ø | 3T3-3e5 |
|---|---|---|---|
| XIAP | nP | 49.8 | 15.4 |
| P | 50.2 | 84.6 | |
| c-Myc | nP | 41. 8 | 19.9 |
| P | 58. 2 | 80.1 | |
| CYR61 | nP | 61.6 | 45.8 |
| P | 38.4 | 54.2 | |
| Pim-1 | nP | 36.2 | 21.5 |
| P | 63.8 | 78.5 | |
| GAPDH | nP | 23.7 | 74.7 |
| P | 76.3 | 25.3 | |
| Actin | nP | 15.9 | 46.1 |
| P | 84.1 | 53.9 | |
| L32 | nP | 53.7 | 83.4 |
| P | 46.3 | 16.6 | |
| L34 | nP | 60.9 | 82.1 |
| P | 39.1 | 17.2 |
The distribution of some mRNA on polysomes is highly dependent on growth rate, growth factor status, confluency, and other variables that can differ among various batches of cells (40). To compare polysomal distributions over multiple experiments, we calculated the weighted average (FW) of the polysomal distribution for each mRNA as described in Equation 1,
 |
where w is the percentage of mRNA in each fraction and x is the fraction number. In essence, the fraction number is weighted according to the relative amount of a given mRNA in that fraction. mRNAs found predominantly in the P region have higher FW values than mRNAs found predominantly in the nP region. Fig. 5B presents ΔFW, the difference in FW between 3T3–3e5 and 3T3-Ø cell extracts, or between 3T3–3e9 and 3T3-Ø cell extracts, analyzed for each pair in the same ultracentrifugation run. A positive value for ΔFW means the mRNA is on heavier polysomes in cells expressing truncated eIF3e than in control cells. The results show positive ΔFW values for cap-independent mRNAs (Hsp70, XIAP, Pim-1, c-Myc, CYR61, and ODC) and negative ΔFW values for cap-dependent mRNAs (L32, L34, actin, and GAPDH).
DISCUSSION
Dysregulation of protein synthesis is strongly linked to malignant transformation (41). Although mechanisms have been described involving numerous translation factors, the most extensively studied have been those involved in cap-dependent translation, notably eIF4E (42). This factor is overexpressed in human cancers and can transform cells in culture, and its phosphorylation is also required for lymphomagenesis (41). eIF4E is made more available when its major binding protein, 4E-BP1, is phosphorylated through the mTOR pathway (17). The mechanism by which elevated eIF4E levels, phosphorylation, or availability promote transformation is thought to occur through enhanced translation of a subset of mRNAs with highly structured 5′-UTRs that have a greater requirement for the eIF4G-associated RNA unwinding machinery (42). These mRNAs encode proteins involved in cell cycle progression, angiogenesis, cell growth, and antagonism of apoptosis (41). Many proteins of the translational machinery such as ribosomal proteins L32 and L34 are also up-regulated at the translational level by elevated eIF4E levels (40), but this behavior is conferred by a 5′-terminal oligopyrimidine tract rather than 5′-terminal secondary structure (43).
It is against this backdrop that a mechanism for tumorigenesis involving reduced protein synthesis and down-regulation of cap-dependent protein synthesis seems counterintuitive. Our model is that 3e5, like eIF4E, changes the spectrum of mRNAs translated, but by a different mechanism. eIF3e has been shown to interact with the eIF3, COP9 signalosome, and 26 S proteosome complexes through the PCI domain in its C terminus (44). The 3e5 protein lacks the PCI domain (Fig. 1A) and would not be expected to interact with these three complexes. However, eIF3e also binds to eIF4G, likely via its N-terminal portion, based on peptide fragments detected in pull-down experiments (20). Thus, we speculate that 3e5 binds the eIF3-binding site of eIF4G and inhibits recruitment of mRNAs to the 43 S PIC (Fig. 4A). The mRNAs most affected by this inhibition would be those most dependent on the eIF4G-associated unwinding machinery, i.e. highly cap-dependent mRNAs. Previous attempts to detect 3e5 by Western blotting were unsuccessful (9, 10). Although we could detect the 3e5 mRNA (Fig. 1C), we were also unable to detect the protein immunologically. We speculate that 3e5 is unstable compared with full-length eIF3e. The model we propose, however, does not require a stoichiometric complex of eIF4G and 3e5. In fact, production of high steady-state levels of 3e5 would probably be lethal. Rather, we propose that substoichiometric amounts of 3e5 are sufficient to diminish cap-dependent translation relative to cap-independent translation.
When highly cap-dependent translation is inhibited, the protein synthesis machinery is more available for cap-independent translation. For instance, under hypoxic conditions, tumors utilize cap-independent translation for a subset mRNAs encoding pro-growth, pro-angiogenesis, and survival proteins (41). We studied several mRNAs that have been shown previously to be translated in a cap-independent manner. The cysteine-rich protein 61 (CYR61) is a secreted factor that plays diverse roles in development, cell proliferation, tumorigenesis, and angiogenesis (38). It is overexpressed in invasive and metastatic human breast cancer cells. Pim-1, a serine-threonine protein kinase, is a known proto-oncogene commonly activated in murine T cell lymphomas (39). It cooperates with the oncoprotein c-Myc in cellular transformation and its levels are elevated in human leukemias. Both CYR61 and Pim-1 mRNAs remain associated with heavy polysomes when cap-dependent translation is inhibited by eIF4G cleavage after poliovirus infection of HeLa cells (45). The anti-apoptotic protein XIAP, an important factor for tumor cell survival and resistance to apoptosis after treatment with anti-cancer agents (46–48), is regulated by a translational mechanism (36). Initially it was thought that this mRNA contained an IRES (36), but a subsequent study showed that a 3′ splice site was necessary for apparent IRES function (49). The mRNA encoding c-Myc shifts to heavier polysomes under conditions of reduced cap-dependent translation (45), and evidence has been presented that it contains an IRES (37). Significantly, this IRES can function with only the C-terminal domain of eIF4G (50). The translation of the mRNA for ornithine decarboxylase (ODC) is most actively translated at the G2/M transition of the cell cycle, the point when overall cellular protein synthesis is at its lowest (51). All five of these mRNAs contain 5′-UTR sequences that promote translation of the downstream cistron in bicistronic mRNAs (36, 45, 51, 52). mRNAs encoding heat-shock proteins, including Hsp70, also continue to be translated when eIF4E is knocked down by antisense RNA despite a decrease in general protein synthesis (53).
Several studies have correlated either over- or underexpression of the full-length eIF3e protein with human cancers. Early reports described decreased expression of eIF3e in breast (54) and non-small cell lung (54, 55) carcinomas. More recently, elevated eIF3e expression was correlated with high tumor grade in primary breast carcinomas (56). In the current study, changes in the translation of mRNA subsets was observed, some of which encode proteins involved in apoptosis and mitotic regulation. It is unlikely, however, that the same mechanisms would be responsible for translational shifts caused by full-length eIF3e and 3e5 since the latter lacks the PCI domain necessary for interaction with eIF3 (44). The PCI domain is also missing from the protein predicted to result from MMTV insertion in intron 9 (7), so this model would apply to 3T3–3e9 cells as well. Significantly, no tumors resulting from up-regulation of full-length eIF3e by MMTV insertion have been described (5). A particularly striking comparison between full-length and truncated eIF3e can be drawn from the study of Zhang et al. (57), who found that overexpression of full-length eIF3e in NIH3T3 cells leads to inhibition of proteins synthesis and cell growth but does not promote malignant transformation. Truncated eIF3e, on the other hand, similarly inhibits protein synthesis and cell growth (Fig. 3) but does promote malignant transformation of NIH3T3 cells (9). We propose that it is the truncated form of eIF3e, acting in a dominant-negative manner, that antagonizes cap-dependent translation relative to cap-independent translation. The shift in the spectrum of proteins synthesized ultimately results in MMTV-induced malignant transformation.
Supplementary Material
Acknowledgments
We thank Jerry Pelletier, McGill University, for the plasmid pKS/FF/HCV/Ren, Arrigo de Benedetti, Louisiana State University Health Sciences Center in Shreveport (LSUHSC-S) for helpful suggestions during the development of this study, and the LSUHSC-S Research Core Facility for instrumentation.
This work was supported, in whole or in part, by Grant R01GM20818 from the NIGMS, National Institutes of Health.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1.
- MMTV
- mouse mammary tumor virus
- UTR
- untranslated region
- PIC
- preinitiation complex
- HCV
- hepatitis C virus
- IRES
- internal ribosome entry sequence.
REFERENCES
- 1. Callahan R., Smith G. H. (2008) J. Mammary Gland Biol. Neoplasia 13, 269. [DOI] [PubMed] [Google Scholar]
- 2. Callahan R., Benveniste R. E., Sherr C. J., Schidlovsky G., Todaro G. J. (1976) Proc. Natl. Acad. Sci. U.S.A. 73, 3579–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tekmal R. R., Keshava N. (1997) Front. Biosci. 2, 519–526 [DOI] [PubMed] [Google Scholar]
- 4. Theodorou V., Kimm M. A., Boer M., Wessels L., Theelen W., Jonkers J., Hilkens J. (2007) Nat. Genet. 39, 759–769 [DOI] [PubMed] [Google Scholar]
- 5. Callahan R., Smith G. H. (2008) J. Mammary Gland Biol. Neoplasia 13, 309–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raafat A., Lawson S., Bargo S., Klauzinska M., Strizzi L., Goldhar A. S., Buono K., Salomon D., Vonderhaar B. K., Callahan R. (2009) Oncogene 28, 219–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marchetti A., Buttitta F., Miyazaki S., Gallahan D., Smith G. H., Callahan R. (1995) J. Virol. 69, 1932–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Asano K., Merrick W. C., Hershey J. W. B. (1997) J. Biol. Chem. 272, 23477–23480 [DOI] [PubMed] [Google Scholar]
- 9. Rasmussen S. B., Kordon E., Callahan R., Smith G. H. (2001) Oncogene 20, 5291–5301 [DOI] [PubMed] [Google Scholar]
- 10. Mayeur G. L., Hershey J. W. (2002) FEBS Lett. 514, 49–54 [DOI] [PubMed] [Google Scholar]
- 11. Mack D. L., Boulanger C. A., Callahan R., Smith G. H. (2007) Breast Cancer Res. 9, R42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hinnebusch A. G. (2006) Trends Biochem. Sci. 31, 553–562 [DOI] [PubMed] [Google Scholar]
- 13. Sha Z., Brill L. M., Cabrera R., Kleifeld O., Scheliga J. S., Glickman M. H., Chang E. C., Wolf D. A. (2009) Mol. Cell. Biol. 36, 141–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Masutani M., Sonenberg N., Yokoyama S., Imataka H. (2007) EMBO J. 26, 3373–3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dong Z., Zhang J. T. (2006) Crit. Rev. Oncol. Hematol. 59, 169–180 [DOI] [PubMed] [Google Scholar]
- 16. Siridechadilok B., Fraser C. S., Hall R. J., Doudna J. A., Nogales E. (2005) Science 310, 1513–1515 [DOI] [PubMed] [Google Scholar]
- 17. Jackson R. J., Hellen C. U., Pestova T. V. (2010) Nat. Rev. Mol. Cell Biol. 2, 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lamphear B. J., Kirchweger R., Skern T., Rhoads R. E. (1995) J. Biol. Chem. 270, 21975–21983 [DOI] [PubMed] [Google Scholar]
- 19. Korneeva N. L., Lamphear B. J., Hennigan F. L., Rhoads R. E. (2000) J. Biol. Chem. 275, 41369–41376 [DOI] [PubMed] [Google Scholar]
- 20. LeFebvre A. K., Korneeva N. L., Trutschl M., Cvek U., Duzan R. D., Bradley C. A., Hershey J. W., Rhoads R. E. (2006) J. Biol. Chem. 281, 22917–22932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kapasi P., Chaudhuri S., Vyas K., Baus D., Komar A. A., Fox P. L., Merrick W. C., Mazumder B. (2007) Mol. Cell 25, 113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Diella F., Levi G., Callahan R. (1997) DNA Cell Biol. 16, 839–847 [DOI] [PubMed] [Google Scholar]
- 23. Yan R., Rychlik W., Etchison D., Rhoads R. E. (1992) J. Biol. Chem. 267, 23226–23231 [PubMed] [Google Scholar]
- 24. Bordeleau M. E., Matthews J., Wojnar J. M., Lindqvist L., Novac O., Jankowsky E., Sonenberg N., Northcote P., Teesdale-Spittle P., Pelletier J. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 10460–10465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grudzien E., Kalek M., Jemielity J., Darzynkiewicz E., Rhoads R. E. (2006) J. Biol. Chem. 281, 1857–1867 [DOI] [PubMed] [Google Scholar]
- 26. Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 27. Kindich R., Florl A. R., Jung V., Engers R., Müller M., Schulz W. A., Wullich B. (2005) Clin. Chem. 51, 649–652 [DOI] [PubMed] [Google Scholar]
- 28. Maquat L. E., Gong C. (2009) Biochem. Soc. Trans. 37, 1287–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bradley C. A., Padovan J. C., Thompson T. L., Benoit C. A., Chait B. T., Rhoads R. E. (2002) J. Biol. Chem. 277, 12559–12571 [DOI] [PubMed] [Google Scholar]
- 30. Byrd M. P., Zamora M., Lloyd R. E. (2002) Mol. Cell. Biol. 22, 4499–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lodish H. F. (1974) Nature 251, 385–388 [DOI] [PubMed] [Google Scholar]
- 32. Sizova D. V., Kolupaeva V. G., Pestova T. V., Shatsky I. N., Hellen C. U. (1998) J. Virol. 72, 4775–4782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fraser C. S., Hershey J. W., Doudna J. A. (2009) Nat. Struct. Mol. Biol. 16, 397–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jackson R. J. (2005) Biochem. Soc. Trans. 33, 1231–1241 [DOI] [PubMed] [Google Scholar]
- 35. Komar A. A., Hatzoglou M. (2005) J. Biol. Chem. 280, 23425–23428 [DOI] [PubMed] [Google Scholar]
- 36. Holcik M., Lefebvre C., Yeh C., Chow T., Korneluk R. G. (1999) Nat. Cell Biol. 1, 190–192 [DOI] [PubMed] [Google Scholar]
- 37. Jopling C. L., Spriggs K. A., Mitchell S. A., Stoneley M., Willis A. E. (2004) RNA 10, 287–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Menéndez J. A., Mehmi I., Griggs D. W., Lupu R. (2003) Endocr. Relat. Cancer 10, 141–152 [DOI] [PubMed] [Google Scholar]
- 39. Saris C. J., Domen J., Berns A. (1991) EMBO J. 10, 655–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mamane Y., Petroulakis E., Martineau Y., Sato T. A., Larsson O., Rajasekhar V. K., Sonenberg N. (2007) PLoS ONE 2, e242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Silvera D., Formenti S. C., Schneider R. J. (2010) Nat. Rev. Cancer 10, 254–266 [DOI] [PubMed] [Google Scholar]
- 42. De Benedetti A., Graff J. R. (2004) Oncogene 23, 3189–3199 [DOI] [PubMed] [Google Scholar]
- 43. Meyuhas O., Hornstein E. (2000) in Translational Control of Gene Expression (Sonenberg N., Hershey J. W., Mathews M. B. eds), pp. 671–694, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York [Google Scholar]
- 44. Hoareau Alves K., Bochard V., Réty S., Jalinot P. (2002) FEBS Lett. 527, 15–21 [DOI] [PubMed] [Google Scholar]
- 45. Johannes G., Carter M. S., Eisen M. B., Brown P. O., Sarnow P. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 13118–13123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Deveraux Q. L., Reed J. C. (1999) Genes Dev. 13, 239–252 [DOI] [PubMed] [Google Scholar]
- 47. Deveraux Q. L., Takahashi R., Salvesen G. S., Reed J. C. (1997) Nature 388, 300–304 [DOI] [PubMed] [Google Scholar]
- 48. Jin Y., McEwen M. L., Ghandour M. S., Springer J. E. (2004) Cell. Mol. Neurobiol. 24, 853–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Baranick B. T., Lemp N. A., Nagashima J., Hiraoka K., Kasahara N., Logg C. R. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 4733–4738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Spriggs K. A., Cobbold L. C., Jopling C. L., Cooper R. E., Wilson L. A., Stoneley M., Coldwell M. J., Poncet D., Shen Y. C., Morley S. J., Bushell M., Willis A. E. (2009) Mol. Cell. Biol. 29, 1565–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pyronnet S., Pradayrol L., Sonenberg N. (2000) Mol. Cell 5, 607–616 [DOI] [PubMed] [Google Scholar]
- 52. Stoneley M., Subkhankulova T., Le Quesne J. P., Coldwell M. J., Jopling C. L., Belsham G. J., Willis A. E. (2000) Nucleic Acids Res. 28, 687–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Joshi-Barve S., DeBenedetti A., Rhoads R. E. (1992) J. Biol. Chem. 267, 21038–21043 [PubMed] [Google Scholar]
- 54. Marchetti A., Buttitta F., Pellegrini S., Bertacca G., Callahan R. (2001) Int. J. Oncol. 18, 175–179 [DOI] [PubMed] [Google Scholar]
- 55. Buttitta F., Martella C., Barassi F., Felicioni L., Salvatore S., Rosini S., D'Antuono T., Chella A., Mucilli F., Sacco R., Mezzetti A., Cuccurullo F., Callahan R., Marchetti A. (2005) Clin. Cancer Res. 11, 3198–3204 [DOI] [PubMed] [Google Scholar]
- 56. Grzmil M., Rzymski T., Milani M., Harris A. L., Capper R. G., Saunders N. J., Salhan A., Ragoussis J., Norbury C. J. (2010) Oncogene 29, 4080–4089 [DOI] [PubMed] [Google Scholar]
- 57. Zhang L., Pan X., Hershey J. W. B. (2007) J. Biol. Chem. 282, 5790–5800 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.