

Abstract
The integral membrane protein BlaR1 of methicillin-resistant Staphylococcus aureus senses the presence of β-lactam antibiotics in the milieu and transduces the information to the cytoplasm, where the biochemical events that unleash induction of antibiotic resistance mechanisms take place. We report herein by two-dimensional and three-dimensional NMR experiments of the sensor domain of BlaR1 in solution and by determination of an x-ray structure for the apo protein that Lys-392 of the antibiotic-binding site is posttranslationally modified by Nζ-carboxylation. Additional crystallographic and NMR data reveal that on acylation of Ser-389 by antibiotics, Lys-392 experiences Nζ-decarboxylation. This unique process, termed the lysine Nζ-decarboxylation switch, arrests the sensor domain in the activated (“on”) state, necessary for signal transduction and all the subsequent biochemical processes. We present structural information on how this receptor activation process takes place, imparting longevity to the antibiotic-receptor complex that is needed for the induction of the antibiotic-resistant phenotype in methicillin-resistant S. aureus.
Keywords: Antibiotics, Bacteria, Computation, Crystal Structure, Signal Transduction, Signal Transduction, Antibiotic Resistance, Bacteria, Structure
Introduction
An antibiotic-resistant bacterium, which emerged first in the United Kingdom in 1961, disseminated rapidly globally and came to be known as methicillin-resistant Staphylococcus aureus (MRSA)3 (1, 2). Modern forms of MRSA are broadly resistant to antibiotics of various classes, but resistance to β-lactam antibiotics (penicillins, cephalosporins, and carbapenems, among others) is most insidious as it encompasses virtually all commercially available members of the class (3–5). The mechanistic basis of resistance to β-lactam antibiotics in MRSA is complex for which the antibiotic sensor/signal transducer protein BlaR1 is a key player (4). The surface domain of BlaR1 detects the presence of the β-lactam antibiotic in the milieu. Once the antibiotic complex with the sensor domain forms, it initiates transduction of the information on antibiotic recognition to the cytoplasmic domain, which is a zinc-dependent protease (Fig. 1A) (6). The protease domain degrades the gene repressor BlaI to derepress expression of the antibiotic resistance genes for β-lactams, including that for BlaR1 itself, for manifestation of the MRSA antibiotic resistance phenotype (6, 7).
FIGURE 1.

A, a schematic of the events in manifestation of β-lactam resistance by BlaR1 in MRSA. The sensor domain binds to the antibiotic covalently, whereby the message is transduced to the cytoplasm to activate the zinc protease domain by a yet unknown mechanism. The activated protease degrades BlaI to derepress the genes involved in antibiotic resistance. B, N-carboxylation and N-decarboxylation of Lys-392 of BlaRS.
The recognition at the surface domain is not merely a complex formation between the antibiotic and the sensor domain, but rather involves covalent chemistry. Antibiotic acylates Ser-389, resulting in an acyl-protein complex. Furthermore, we had detected by spectroscopy that the sensor domain experiences the uncommon posttranslational modification of N-carboxylation at the side chain of Lys-392, a residue within the antibiotic-binding site (8, 9). This is the product of reaction of carbon dioxide with the side-chain amine of the lysine (Fig. 1B). Conspicuously, the N-carboxylated lysine has not been seen in the x-ray structures for the sensor domain (10–12).
We document herein for the first time by x-ray crystallography that the apo form of the sensor domain of BlaR1 from S. aureus is indeed N-carboxylated at the side chain of Lys-392. We also document by a series of two- and three-dimensional solution NMR experiments that contiguity exists in the covalent connectivities of the side-chain atoms of N-carboxylated Lys-392, an arrangement that is lost on binding to the β-lactam antibiotics, leading to unmodified Lys-392. NMR experiments revealed that the side chain of N-carboxylated Lys-392 in the native protein is highly mobile in solution in the picosecond time regime, the mechanistic significance of which was investigated by molecular dynamics simulations. We demonstrate that the complex of the antibiotic with the sensor domain does not experience N-recarboxylation at the side chain of Lys-392 even in the presence of excess carbon dioxide, a critical event that is at the root of BlaR1 serving as a sensor domain of β-lactam antibiotics. This unique property, for which we provide herein a structural basis, is at the root of the antibiotic sensing event, central in induction of resistance in MRSA.
EXPERIMENTAL PROCEDURES
Detailed experimental techniques used for site-directed mutagenesis, expression, and purification of BlaRS are provided with the supplemental data. Computational methods used are also described there together with the associated references and additional figures.
Crystallization, Data Collection, Structure Determination, and Refinement
Crystals of BlaRS were grown at 24 °C from either 30% PEG 8000, 0.2 m ammonium sulfate at pH 6.5 buffered with sodium cacodylate (native protein and protein with imipenem and meropenem) or 30% PEG 4000, 0.2 m ammonium sulfate at pH 8.5 buffered with Tris (protein with CBAP) using protein concentrations of 3 mg/ml. Prior to data collection, crystals were briefly transferred to mother liquor that had been supplemented with 20% glycerol for cryo-protection, and then they were flash-frozen in the liquid nitrogen.
Antibiotics were dissolved in the cryo-protective solution to a concentration of 3–5 mg/ml. For soaking, BlaRS crystals were soaked in these solutions for ∼2 h. Although this strategy was successful with imipenem and meropenem, soaking was unsuccessful with CBAP, leading instead to crystal cracking and dissolution within 2 min. Crystals of BlaRS complexed with CBAP were thus obtained by co-crystallization, with 75 μl of CBAP in water at 5 mg/ml added to 900 μl of crystallization buffer.
Diffraction data were collected at beamlines 19BM and 21ID at the Advanced Photon Source. Oscillation frames were integrated and scaled with HKL2000 (13). The native structure was solved by molecular replacement with MOLREP (14), using the previously determined native protein structure (Protein Data Bank (PDB) entry 1XKZ; molecule A) as a search model. The structures with antibiotics were subsequently solved using the fully refined new native structure as a starting model. Rigid body refinement and TLS refinement followed by multiple steps of restrained refinement were performed with Refmac5 (15) as incorporated in CCP4-6.0.2. Anisotropic and bulk solvent corrections were performed throughout the refinement. Waters were added using ARP/wARP (16). Graphical evaluation of the model and fitting to maps were performed using Coot (17) and XtalView (18). A composite omit map, a composite of density maps in which 5% of the structure was iteratively omitted and maps calculated using simulated annealing and energy minimization to reduce bias, were calculated using the CNS module as implemented in Discovery Studio 3.0 (Accelrys, Inc.) and used to validate the electron density for the N-carboxylated lysine in the apo BlaRS protein.
The quality of the structures during and after refinement was monitored with the Coot validation engine, Procheck (19), and the WHATIF template structure check (20). The side-chain orientations of Gln, Asn, and His were validated using MolProbity (21). Data collection and refinement statistics are presented in supplemental Table S1.
NMR Spectroscopy
U-15N/80% 2H-labeled BlaRS was overexpressed at 25 °C in Escherichia coli BL21(DE3) cells (Novagen), transformed with the expression vector pET24a(+)_BlaRS. We used the protocol of Marley et al. (22), with the initial growth in rich LB medium followed by overexpression in M9 minimal media containing [15N]-NH4Cl as the sole nitrogen source and 100% D2O for perdeuteration. BlaRS was then purified chromatographically from the bacterial lysate per reported procedures (9). For lysine N-carboxylation, we included 20 mm NaH13CO3 as described previously (9), which gave 550 μm 13CO2. This 13CO2 concentration is well above the Kd for carbon dioxide binding to BlaRS Kd = 0.6 ± 0.2 μm (9) and the final BlaRS concentration (150 μm). The final NMR buffer conditions (after NaH13CO3 addition) were 30 mm sodium phosphate, 20 mm NaCl, 0.2% NaN3, 10% D2O, maintaining pH at 7.5.
NMR spectra were recorded at 16.4 and 18.8 T, using Bruker Avance spectrometers equipped with cryogenically cooled TCI probes. Three-dimensional HNCO and two-dimensional heteronuclear single quantum correlation spectra for the carbamate assignment and antibiotic additions (see Fig. 3) were recorded at 16.4 T, 295 K and used 1H-detected transverse relaxation optimized spectroscopy schemes (23, 24). Carrier positions optimized for carbamate spectra were 172 ppm (13CO), 80 ppm (15N), and 4.7 ppm (1H). We carried out the 15N spin relaxation measurements at 18.8 and 16.4 T, 295 K, using sensitivity-enhanced, two-dimensional 1H-detected pulse schemes (25–27). The R2 experiments used the Carr-Purcell-Meiboom-Gill spin lock, corrected for cross-correlated relaxation artifacts (28, 29). The Carr-Purcell-Meiboom-Gill carrier was applied on-resonance with the carbamate 15N and used 160-μs 15N refocusing pulses with interpulse delays of 900 μs. The relaxation delays were 0.86, 0.17, 0.34, 0.52 (2×), 0.86, 1.03, 1.46, 1.98, and 2.58 s for the R1 series and 8.5, 17.0 (2×), 25.4, 33.9, 42.4, 50.9, 59.4, 76.3, 84.8, and 93.3 ms for the R2 series. The experiments produced files of cross-peak intensity versus relaxation delay, which were then fit to single exponential decays using the Levenberg-Marquardt algorithm. Analysis of the 15N R2/R1 ratios for the backbone NHs (26) gave an estimated overall rotational correlation time τm = 15.0 ± 0.2 ns/radian. The fits of the decay curves, the subsequent extraction of the Lipari-Szabo internal motion parameters (S2 and τe), and the attendant Monte Carlo error analyses based on duplicate spectra were performed using previously described methods and in-house software (30).
FIGURE 3.

NMR resonances from carbamate moiety of N-carboxylated Lys-392 and their decrease upon the addition of two different penicillins. Spectra were recorded at 295 K, 16.4 T. A, cross-peak from the NζH bond of the carbamate moiety. U-15N/80%2H-labeled BlaRS in buffer containing 13C bicarbonate was used as the CO2 source. The two-dimensional spectrum is a 1H-13CO projection of the three-dimensional HNCO, and the single cross-peak (blue) has the carbamate 13CO and 1H chemical shifts. The faint contours on the right are residual water signals. B and C, cross-sections of 1H-15N heteronuclear single quantum correlation spectra showing the decrease of the carbamate 1H resonance after the addition of antibiotic. B, penicillin G addition at 0 min (black, 100%), + 2 h (blue, 60%), and + 36 h (green, 20%). C, CBAP addition at 0 min (black, 100%), + 2 h (blue, 72%), and + 36 h (green, 30%). The decarboxylation is slower than that observed in previous kinetic measurements (9), reflecting the use of the higher protein concentration (150 μm) for NMR versus kinetic (1 μm) experiments.
RESULTS
Structure of the Native BlaRS Reveals Lys-392 N-Carboxylated
Screening of the conditions for growth of crystals of the sensor domain (hereafter referred to as BlaRS), including those previously shown to result in diffracting crystals (11), generated cubic crystals that diffracted to no better than 4-Å resolution at synchrotron sources, similar to what had been reported earlier (10). As the wild-type sequence failed to crystallize to high resolution, we pursued a different strategy in which select surface lysines were replaced with alanine to minimize entropic factors inhibiting optimal crystal growth. This type of strategy has been successful in other systems (31). The gene for a triple mutant variant of the sensor domain (K369A/K370A/K372A) was cloned. The protein was expressed and purified to homogeneity. This mutant variant gave crystals that diffracted to 2.2-Å resolution and permitted determination of the structure by molecular replacement (supplemental Table S1).
The overall structure was the same as those reported previously for BlaRS from S. aureus and Bacillus licheniformis (10–12), consisting of an α/β fold that closely resembles structures of class D β-lactamases. There were two molecules in the asymmetric unit, related by a non-crystallographic two-fold axis of symmetry. The structures of the two molecules were nearly identical, superimposing with backbone and all-atom root mean square deviation values of 0.3 and 0.7 Å, respectively. Despite these similarities, a key difference was seen for Lys-392 within the antibiotic-binding sites of the two. Although Lys-392 in the first molecule was unmodified, as seen in the earlier structure for BlaRS from S. aureus (11), the second protein molecule exhibited additional density contiguous to the Lys-392 Nζ atom. The presence of this additional density was confirmed through the calculation of an unbiased composite omit map (supplemental Fig. S1). This additional density was larger and more unusually shaped than typically seen for a water molecule but too small to accommodate additional ligands such as sulfate or glycerol. Further, the peak density was situated too close to the lysine side-chain nitrogen to permit a non-covalent interaction. Overall, this density was most compatible with the presence of an N-carboxylated lysine, the existence of which was alluded to in the earlier spectroscopic studies (Fig. 2A). Its observation in just one of two molecules in the asymmetric unit, together with the previous structure showing only an unmodified lysine (11), is consistent with a reversible N-carboxylation reaction; as will be elaborated, this reversibility has mechanistic implications for the activation of the sensor domain.
FIGURE 2.

The structure of BlaRS protein. A, view of the active site for the N-carboxylated molecule of the apo protein, with 2Fo − Fc electron density contoured at 1 σ shown for select residues. Contiguous density for the N-carboxylated Lys-392 (KCX392) is clearly visible. B, superimposition of the carboxylated and non-carboxylated molecules in the structure, with select active-site residues shown. The different positioning of the carbamate of the N-carboxylated lysine and the primary amine of the unmodified lysine is evident. Carbons of the N-carboxylated protein are green, and carbons of the non-carboxylated protein are magenta. C, superimposition of CBAP-acylated BlaRS onto the N-carboxylated apo protein. Carbons of CBAP are dark gray, and the covalent connection to Ser-389 is clearly visible. Carbons of active site side chains in the CBAP-acylated protein are light blue. Carbons of the N-carboxylated apo protein are green. The sulfate anion present in the apo protein is absent in the CBAP-acylated protein. See Fig. S3 and S4 for structures of imipenem and meropenem bound to BlaRS and chemical structures of these antibiotics.
In the protein molecule with the unmodified lysine, the Nζ of Lys-392 points toward solvent and forms a number of hydrogen bonds with polar side chains within the active site, including those of Asn-439 and Ser-389. The protein with the unmodified Lys-392 is not active, as disclosed in an earlier study (8). Lys-392 has rotated nearly 180° about its δ carbon in the protein molecule with N-carboxylated lysine. This conformation does not allow an interaction with the side chain of Ser-389, but one with Asn-439 is still retained (Fig. 2B).
It is known that N-carboxylated Lys-392 promotes/activates Ser-389 for acylation by the antibiotic (8); hence, this crystallographic conformation for N-carboxylated Lys-392 was initially a puzzle. As we will see by the forthcoming discussion on NMR analysis of the protein, N-carboxylated Lys-392 is highly mobile within the antibiotic-binding site, a feature critical for activation of Ser-389 for the acylation event. The observed NMR mobility of the residue is supported by the crystallographic B-factors for the atoms of the side chain of the N-carboxylated Lys-392, which are as much as three standard deviations above the average for the protein (this calculation excludes the atoms of the first seven amino acid residues, for which only weak electron density was observed) (supplemental Fig. S2).
High Mobility of the N-Carboxylated Lys-392 in BlaRS
The presence of N-carboxylated Lys-392 in BlaRS was also documented by two-dimensional- and three-dimensional-NMR experiments in solution (see supplemental Results for details). The resonance line widths of the carbamate and amide 15N-1H bond vectors are governed by the same spin relaxation mechanisms. However, the carbamate 15N and 1H line-widths were much narrower than most of the BlaRS backbone NH groups, suggesting enhanced local mobility. To quantify this, we measured 15N spin relaxation parameters at 295 K (spin lattice relaxation rate, R1 = 1/T1 and spin-spin relaxation rate, R2 = 1/T2 at 18.8 T and R1 at 16.4 T) for the individual NH bond vectors of BlaRS. For the carbamate NζH bond, we extracted the dynamics information from these rates using the formalism of Lipari and Szabo (32, 33), which assigns each NH bond vector a site-specific order parameter S2 and internal motion correlation time, τe. These parameters describe the amplitude (S2) and time scale (τe) for the re-orientational motions of the NH bond vector due to conformational dynamics. S2 lies between 1 and 0, becoming smaller for larger amplitude motions. We found for the carbamate that S2 = 0.16 ± 0.02, and τe = 210 ± 50 ps. These values indicated that the carbamate moiety underwent large amplitude internal motions on the picosecond time scale. Further evidence of high local mobility was the observation that 1H-1H NOE cross-relaxation between the carbamate NζH proton and other protons (within ∼5 Å) occurred only at lower temperature (278 K). At 295 K, these 1H-1H NOE cross-peaks disappeared. This reveals that at 295 K, the 1H-1H NOE cross-relaxation is averaged out by large amplitude motion of the N-carboxylated lysine. To our knowledge, the NMR study reported herein is the first experimental investigation of the dynamics of N-carboxylated lysine in proteins. Collectively, these experimental data reveal that the NζH, and thus, the side chain of N-carboxylated Lys-392, undergoes large amplitude subnanosecond motion.
N-Decarboxylation of Lys-392 on Binding of β-Lactam Antibiotics
NMR spectra of BlaRS showed that the addition of β-lactam antibiotic causes N-decarboxylation of Lys-392. Specifically, we added either penicillin G or CBAP to BlaRS (17 mm penicillin G or 16 mm CBAP and 150 μm BlaRS) and then monitored the effects on the carbamate 15N-1H cross-peak in heteronuclear single quantum correlation spectra. For both reactions, the cross-peak intensity decreased steadily over time. Fig. 3, B and C, show 1H cross-sections of this cross-peak for time points subsequent to the addition of the antibiotic. At the end of monitoring, the cross-peak had diminished by 80% for penicillin G and 70% for CBAP. The progressive decrease in the resonance is a direct consequence of the disappearance of the carbamate species, caused by N-decarboxylation of Lys-392. It is important to note that these experiments were performed in the presence of excess of 13CO2, which indicates that on acylation of the protein by the antibiotic and the attendant N-decarboxylation of Lys-392, the carbamate cannot readily reform. The implication of this observation is that N-decarboxylation of Lys-392 keeps the sensor domain in the acylated (“on”) state.
Simulations of the Motion of N-Carboxylated Lys-392
The mobility of the N-carboxylated Lys-392 side chain, as suggested by crystallographic temperature factors and directly observed through NMR, was simulated by molecular dynamics (Figs. 4 and 5). The present x-ray crystal structure places the side chain of N-carboxylated Lys-392 in an extended conformation in the active site. This extended conformation positions the side chain away from the Ser-389 side-chain hydroxyl, resulting in no interactions between the two. We additionally note that in class D OXA10 β-lactamase, among other class D enzymes, the active-site lysine corresponding to Lys-392 of BlaR1 is N-carboxylated and that the N-carboxylated side chain is in hydrogen-bonding contact with the active-site serine, an arrangement not seen in the x-ray structure of BlaR1 reported herein (Figs. 2A and 4A).
FIGURE 4.

Stereo representation of the antibiotic-binding site of BlaR1 (A) and the class D OXA10 β-lactamase (B) (PDB code: 1K57). The active-site residues are shown as capped sticks. The protein backbone is shown as green ribbon. Carbon atoms are colored in purple (BlaR1 x-ray) and gray (BlaR1 model from molecular dynamics simulations) in panel A and orange (OXA10 β-lactamase x-ray) in panel B. Oxygen atoms are in red, nitrogen is in blue, hydrogen is in cyan, and sulfur is in yellow.
FIGURE 5.
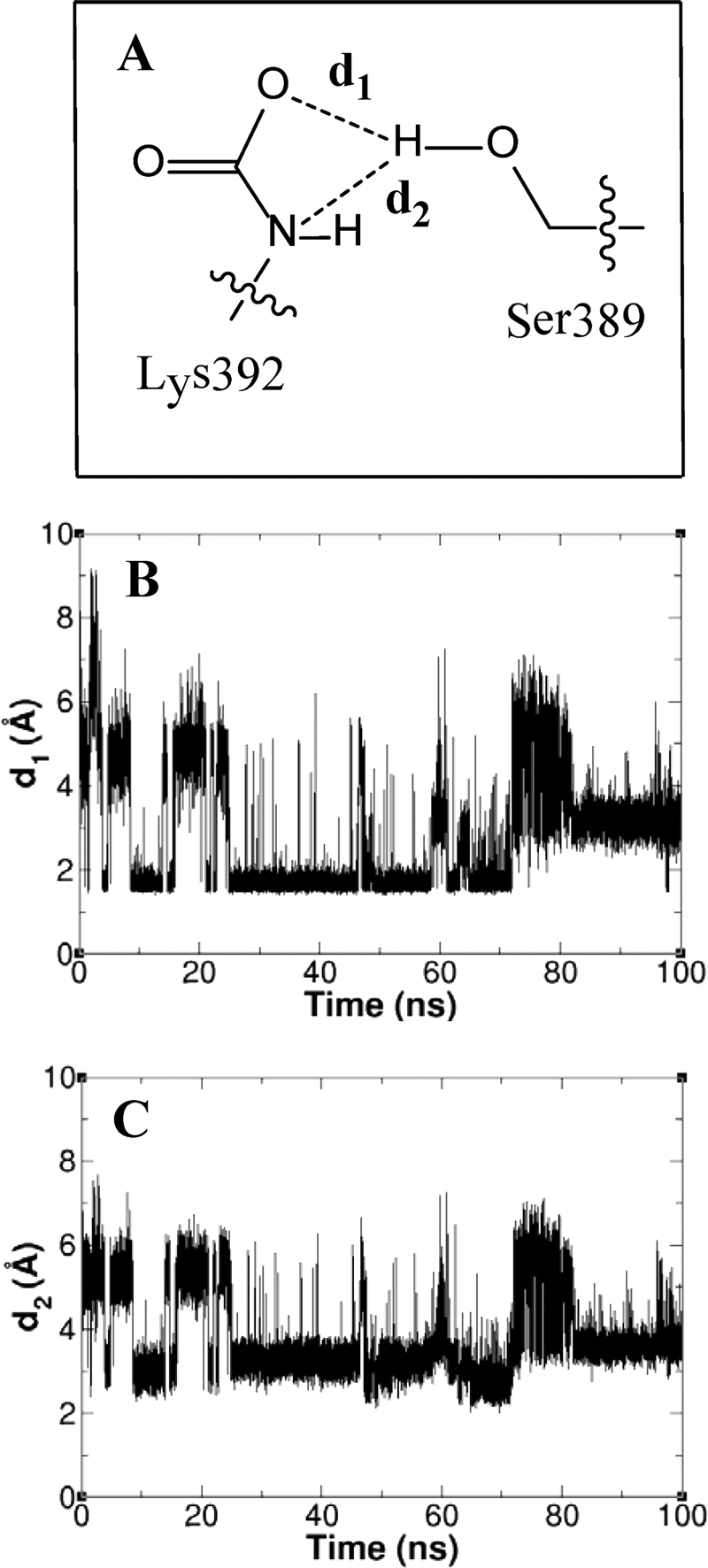
A, the interactions between the Lys-392 group and Ser-389, with two distances for bifurcated hydrogen bonding defined. B and C, the hydrogen-bonding distances d1 (B) and d2 (C) fluctuate as a function of time in the course of the 100 ns of simulations.
Consistent with the NMR findings, our molecular dynamics simulations show that N-carboxylated Lys-392 is highly mobile. The side chain moves from the extended conformation seen in the x-ray structure in the direction of Ser-389 to achieve a conformation that facilitates hydrogen bonding with serine hydroxyl. Approximately 50% of the 50,000 conformations sampled over a 100-ns period demonstrate hydrogen-bonding contact between the hydroxyl group of Ser-389 and one oxygen of the N-carboxylated Lys-392 or the other (Fig. 5A).
Consistent with the experimental picosecond time regime measured by NMR for mobility of the N-carboxylated Lys-392, the 100 ns of simulations revealed that hydrogen bonds between Ser-389 and Lys-392 form and break repeatedly (Fig. 5, B and C). The hydrogen bond between the N-carboxylate oxygen of Lys-392 and the side-chain amide of Asn-439, as seen in the x-ray crystal structure, was conserved in 55% of the snapshots from the simulations.
DISCUSSION
The collective information from the x-ray structures, the NMR experiments, and the simulations are revealing as to the mechanism of action of the sensor domain in detection of β-lactam antibiotics by BlaR1. Notwithstanding the high similarity of the structures of class D β-lactamases and the sensor domain of BlaR1, it is clear that the former is an enzyme with catalytic ability (many turnovers), whereas the latter is a receptor and devoid of turnover chemistry. For the sensor domain to function as a receptor, it should experience acylation by the antibiotic, but the complex should have longevity such that mobilization of induction of resistance in the bacterium could take place. We see this exactly in the complex of antibiotics and BlaRS. What is the structural basis that arrests the events at the acyl-protein complex stage for BlaR1, in contrast to the case of class D β-lactamases, which experience deacylation in completing the catalytic turnover process?
The structures of the class D β-lactamases are highly similar to that of BlaRS, both in the overall fold and within the antibiotic-binding sites. We now know that the corresponding active-site lysines in class D β-lactamases (34–41) and BlaR1 (this study) are N-carboxylated. However, the conformations of the N-carboxylated lysines, as seen from the respective x-ray structures, are different in the two cases. In both class D β-lactamases (38, 39) and BlaRS (8), the role of N-carboxylated lysine is the critical promotion of the active-site serine for acylation by the antibiotic. As class D β-lactamases enjoy symmetry in catalysis (42), N-carboxylated lysine promotes/activates a water molecule for the deacylation event as well, a function that BlaR1 is incapable of performing as it experiences N-decarboxylation on acylation by the antibiotic. It is known that the N-decarboxylated Lys-392 of BlaR1 is incapable of promoting the deacylation step (8). However, our simulations of the dynamics of the BlaR1 sensor domain showed that N-carboxylated Lys-392 is well poised to promote acylation of Ser-389 only after it assumes a conformation that closely approximates that of the class D β-lactamase.
The NMR experiment in this study showed that acylation of the sensor domain by antibiotics entails loss of N-carboxylation at Lys-392, which incidentally cannot readily be reversed, even in the presence of excess carbon dioxide. This lack of reversal is at the root of the BlaR1 function as a receptor. We have termed this process the “lysine N-decarboxylation switch.” This event locks the sensor domain in its activated (on) state. One simple and plausible explanation for this lack of reversal could be that on N-decarboxylation, the side chain of Lys-392 becomes protonated and charged. Our molecular dynamics simulations, however, shed light on an additional possibility. Of the many interactions that we monitored within the antibiotic-binding site of the sensor domain, the hydrogen-bonding interaction between the side chains of Lys-392 and Asn-439 and a water molecule stood out. We noted that Asn-439 is conserved in all BlaR1 (and in the related MecR1) sequences available, except for BlaR1 from B. licheniformis and Brevibacillus brevis, where it is substituted by threonine; hence, the hydrogen-bonding ability by the side chain at position 439 is retained in all cases. We observed that these hydrogen bonds exist regardless of whether Lys-392 is N-carboxylated or not. If Lys-392 is N-carboxylated, the hydrogen-bonding contact is between one of the carbamate oxygen atoms and hydrogen atoms of Nδ of Asn-439. If Lys-392 is N-decarboxylated, the hydrogen bond is between hydrogen atoms on Nζ of Lys-392 and Oδ of Asn-439. These hydrogen bonds solvate the side chain of N-decarboxylated Lys-392 in the antibiotic complex and attenuate its ability to experience N-recarboxylation. Hence, the activated surface domain enjoys sufficient longevity that lasts the duration of the doubling time by MRSA. This mechanistic basis is at the root of evolution of function of BlaR1 as an antibiotic sensor.
We would like to close with one final observation of general importance. It has been predicted that nature might have a mere 2700 unique folds for proteins (not all discovered yet) (43). Considering that most genomes (microbial or more complex organisms) have more than 2700 open reading frames, this implies that nature uses the same structural template multiples of times for evolution of disparate functions in proteins. This work is a clear example of this thesis in that BlaR1 shares a nearly identical structure with those of class D β-lactamases, yet the two classes of proteins have diverged in their functions entirely; one is an antibiotic receptor protein, and the other is an antibiotic resistance enzyme.
Supplementary Material
Acknowledgments
Results shown in this study are derived from work performed at the Structural Biology Center and LS-CAT at the Advanced Photon Source of Argonne National Laboratory. Use of the Advanced Photon Source was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract Number DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corp. and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817).
This work was supported, in whole or in part, by National Institutes of Health Grants AI33170 (to S. M.) and GM085109 (to J. P.).
The atomic coordinates and structure factors (codes 3Q7V, 3Q7Z, 3Q81, and 3Q82) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Results and Methods, Table S1, and Figs. S1–S4.
- MRSA
- methicillin-resistant S. aureus
- CBAP
- 2-(2′-carboxyphenyl)-benzoyl-6-aminopenicillanic acid
- T
- teslas.
REFERENCES
- 1. Jevons M. P. (1961) Br. Med. J. 1, 124–125 [Google Scholar]
- 2. Rolinson G. N. (1961) Br. Med. J. 1, 125–126 [Google Scholar]
- 3. Fisher J. F., Meroueh S. O., Mobashery S. (2005) Chem. Rev. 105, 395–424 [DOI] [PubMed] [Google Scholar]
- 4. Llarrull L. I., Fisher J. F., Mobashery S. (2009) Antimicrob. Agents Chemother. 53, 4051–4063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Llarrull L. I., Testero S. A., Fisher J. F., Mobashery S. (2010) Curr. Opin. Microbiol. 13, 551–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang H. Z., Hackbarth C. J., Chansky K. M., Chambers H. F. (2001) Science 291, 1962–1965 [DOI] [PubMed] [Google Scholar]
- 7. Llarrull L. I., Prorok M., Mobashery S. (2010) Biochemistry 49, 7975–7977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thumanu K., Cha J., Fisher J. F., Perrins R., Mobashery S., Wharton C. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 10630–10635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Golemi-Kotra D., Cha J. Y., Meroueh S. O., Vakulenko S. B., Mobashery S. (2003) J. Biol. Chem. 278, 18419–18425 [DOI] [PubMed] [Google Scholar]
- 10. Birck C., Cha J. Y., Cross J., Schulze-Briese C., Meroueh S. O., Schlegel H. B., Mobashery S., Samama J. P. (2004) J. Am. Chem. Soc. 126, 13945–13947 [DOI] [PubMed] [Google Scholar]
- 11. Wilke M. S., Hills T. L., Zhang H. Z., Chambers H. F., Strynadka N. C. (2004) J. Biol. Chem. 279, 47278–47287 [DOI] [PubMed] [Google Scholar]
- 12. Kerff F., Charlier P., Colombo M. L., Sauvage E., Brans A., Frere J. M., Joris B., Fonzé E. (2003) Biochemistry 42, 12835–12843 [DOI] [PubMed] [Google Scholar]
- 13. Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 14. Vagin A., Teplyakov A. (1997) J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 15. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 16. Perrakis A., Sixma T. K., Wilson K. S., Lamzin V. S. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 448–455 [DOI] [PubMed] [Google Scholar]
- 17. Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 18. McRee D. E. (1999) J. Struct. Biol. 125, 156–165 [DOI] [PubMed] [Google Scholar]
- 19. Laskowski R. A., Macarthur M. W., Moss D. S., Thornton J. M. (1993) J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 20. Vriend G. (1990) J. Mol. Graph. 8, 52–56 [DOI] [PubMed] [Google Scholar]
- 21. Word J. M., Lovell S. C., Richardson J. S., Richardson D. C. (1999) J. Mol. Biol. 285, 1735–1747 [DOI] [PubMed] [Google Scholar]
- 22. Marley J., Lu M., Bracken C. (2001) J. Biomol. NMR 20, 71–75 [DOI] [PubMed] [Google Scholar]
- 23. Salzmann M., Pervushin K., Wider G., Senn H., Wüthrich K. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 13585–13590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pervushin K., Riek R., Wider G., Wüthrich K. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 12366–12371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dayie K. T., Wagner G. (1994) J. Magn. Reson. Series A. 111, 121–126 [Google Scholar]
- 26. Kay L. E., Torchia D. A., Bax A. (1989) Biochemistry 28, 8972–8979 [DOI] [PubMed] [Google Scholar]
- 27. Palmer A. G., Rance M., Wright P. E. (1991) J. Am. Chem. Soc. 113, 4371–4380 [Google Scholar]
- 28. Palmer A. G., Skelton N. J., Chazin W. J., Wright P. E., Rance M. (1992) Mol. Phys. 75, 699–711 [Google Scholar]
- 29. Kay L. E., Nicholson L. K., Delaglio F., Bax A., Torchia D. A. (1992) J. Magn. Reson. 97, 359–375 [Google Scholar]
- 30. Peng J. W., Wagner G. (1992) Biochemistry 31, 8571–8586 [DOI] [PubMed] [Google Scholar]
- 31. Cooper D. R., Boczek T., Grelewska K., Pinkowska M., Sikorska M., Zawadzki M., Derewenda Z. (2007) Acta Crystallogr. D Biol. Crystallogr. 63, 636–645 [DOI] [PubMed] [Google Scholar]
- 32. Lipari G., Szabo A. (1982) J. Am. Chem. Soc. 104, 4546–4559 [Google Scholar]
- 33. Lipari G., Szabo A. (1982) J. Am. Chem. Soc. 104, 4559–4570 [Google Scholar]
- 34. Johnson J. W., Gretes M., Goodfellow V. J., Marrone L., Heynen M. L., Strynadka N. C., Dmitrienko G. I. (2010) J. Am. Chem. Soc. 132, 2558–2560 [DOI] [PubMed] [Google Scholar]
- 35. Sun T., Nukaga M., Mayama K., Braswell E. H., Knox J. R. (2003) Protein Sci. 12, 82–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schneider K. D., Karpen M. E., Bonomo R. A., Leonard D. A., Powers R. A. (2009) Biochemistry 48, 11840–11847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Docquier J. D., Benvenuti M., Calderone V., Giuliani F., Kapetis D., De Luca F., Rossolini G. M., Mangani S. (2010) Antimicrob. Agents Chemother. 54, 2167–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bou G., Santillana E., Sheri A., Beceiro A., Sampson J. M., Kalp M., Bethel C. R., Distler A. M., Drawz S. M., Pagadala S. R., van den Akker F., Bonomo R. A., Romero A., Buynak J. D. (2010) J. Am. Chem. Soc. 132, 13320–13331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Golemi D., Maveyraud L., Vakulenko S., Samama J. P., Mobashery S. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 14280–14285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maveyraud L., Golemi D., Kotra L. P., Tranier S., Vakulenko S., Mobashery S., Samama J. P. (2000) Structure 8, 1289–1298 [DOI] [PubMed] [Google Scholar]
- 41. Docquier J. D., Calderone V., De Luca F., Benvenuti M., Giuliani F., Bellucci L., Tafi A., Nordmann P., Botta M., Rossolini G. M., Mangani S. (2009) Chem. Biol. 16, 540–547 [DOI] [PubMed] [Google Scholar]
- 42. Golemi D., Maveyraud L., Vakulenko S., Tranier S., Ishiwata A., Kotra L. P., Samama J.-P., Mobashery S. (2000) J. Am. Chem. Soc. 122, 6132–6133 [Google Scholar]
- 43. Liu X., Fan K., Wang W. (2004) Proteins 54, 491–499 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.