

Abstract
The snake toxin MT7 is a potent and specific allosteric modulator of the human M1 muscarinic receptor (hM1). We previously characterized by mutagenesis experiments the functional determinants of the MT7-hM1 receptor interaction (Fruchart-Gaillard, C., Mourier, G., Marquer, C., Stura, E., Birdsall, N. J., and Servent, D. (2008) Mol. Pharmacol. 74, 1554–1563) and more recently collected evidence indicating that MT7 may bind to a dimeric form of hM1 (Marquer, C., Fruchart-Gaillard, C., Mourier, G., Grandjean, O., Girard, E., le Maire, M., Brown, S., and Servent, D. (2010) Biol. Cell 102, 409–420). To structurally characterize the MT7-hM1 complex, we adopted a strategy combining double mutant cycle experiments and molecular modeling calculations. First, thirty-three ligand-receptor proximities were identified from the analysis of sixty-one double mutant binding affinities. Several toxin residues that are more than 25 Å apart still contact the same residues on the receptor. As a consequence, attempts to satisfy all the restraints by docking the toxin onto a single receptor failed. The toxin was then positioned onto two receptors during five independent flexible docking simulations. The different possible ligand and receptor extracellular loop conformations were described by performing simulations in explicit solvent. All the docking calculations converged to the same conformation of the MT7-hM1 dimer complex, satisfying the experimental restraints and in which (i) the toxin interacts with the extracellular side of the receptor, (ii) the tips of MT7 loops II and III contact one hM1 protomer, whereas the tip of loop I binds to the other protomer, and (iii) the hM1 dimeric interface involves the transmembrane helices TM6 and TM7. These results structurally support the high affinity and selectivity of the MT7-hM1 interaction and highlight the atypical mode of interaction of this allosteric ligand on its G protein-coupled receptor target.
Keywords: Computer Modeling, G Protein-coupled Receptors (GPCR), Ligand-binding Protein, Protein-Protein Interactions, Toxins, GPCR Dimer, Double Mutant Cycle, Flexible Docking, Muscarinic Acetylcholine Receptor
Introduction
G protein-coupled receptors (GPCRs)5 constitute the largest family of membrane proteins (1) and represent the most important class of targets for current therapeutic agents (2, 3). It is therefore of particular interest to structurally characterize these receptors with the goal of understanding at the atomic level the molecular bases of their ligand recognition and functional versatility. In the last few years, several GPCR x-ray structures were solved, highlighting structural differences between different receptor families and various receptor states (4–13). Despite these encouraging successes, the determination of experimental three-dimensional structures of GPCR-ligand complexes remains a challenge, especially in the case of allosteric modulators. Therefore, we have chosen an alternative way, based on mutagenesis and pharmacological studies combined with molecular modeling and docking procedures, to highlight the molecular bases of the interaction between a GPCR and a highly specific allosteric peptide ligand.
Furthermore, we have addressed the question of the oligomeric state of the GPCR bound to its peptide ligand. Indeed, within the GPCR field, there is still a debate on the receptor oligomeric state in living cells and on the physiological impact of its oligomerization (14). Although a monomeric GPCR can be functional (15–17), some GPCRs can also form dimers or higher order oligomers. Recently, the presence of functional GPCR dimers was demonstrated in vivo (18, 19). We have focused on the interaction of the large allosteric MT7 toxin with its specific muscarinic acetylcholine receptor (mAChR) target. Dimerization of mAChRs was described previously using coimmunoprecipitation experiments (20, 21), resonance energy transfer approaches (22), and fluorescence imaging of single mAChR M1 molecules (23). The effect of various small ligands on the mAChR oligomerization state was explored (20, 22, 24), but the three-dimensional structure of GPCR dimer-ligand complexes remains unsolved. Moreover, the binding mode of a large allosteric ligand has never been described.
The muscarinic toxin MT7 from Dendroaspis angusticeps is the most potent and specific allosteric modulator of the human M1 muscarinic receptor (hM1) (25–29). It is a 65-amino acid protein that folds into a typical “three-finger” β-sheet structure like numerous snake toxins (30). We previously identified MT7 residues involved in hM1 binding on the basis of alanine-scanning studies (31). The MT7 functional residues are mainly located at the tip of its central loop (the positively charged Arg-34 is particularly crucial) and also at the tips of loops I and III. Furthermore, we recently showed by combining both biochemical and biophysical methods that MT7 may bind to a dimeric form of the hM1 receptor (32).
Here we report the use of a data-driven docking procedure to construct a three-dimensional model of the MT7-hM1 complex. We first delineated the binding site of MT7 on hM1 by using chimeras and point mutations. We then performed double mutant cycle experiments on the MT7-hM1 complex and identified several distance restraints between both proteins. These restraints were not consistent with a monomeric hM1 receptor. Therefore, after exploring the conformational flexibility of the free partners, we docked the large MT7 molecule onto two hM1 receptors. Docking simulations from different initial relative positions yielded an ensemble of MT7-hM1 dimer structures consistent with the biochemical data. All these simulations converged toward the same MT7-hM1 dimer structure characterized by a dimer interface formed by TM6 and TM7. On the basis of this model, we discuss the affinity, selectivity, and atypical mode of interaction of MT7 on its GPCR target.
EXPERIMENTAL PROCEDURES
cDNAs Encoding Mutant hM1 and hM3 Receptors
Subcloning, construction, and site-directed mutagenesis of the hM1 and hM3 receptors are described in detail in the supplemental Experimental Procedures.
Stable and Transient mAChR Expression
Heterologous expression of M1 and M3 receptors through transient expression in COS cells as well as membrane preparations from these cell lines is detailed in the supplemental Experimental Procedures.
N-[3H]Methylscopolamine ([3H]NMS) Binding Assays
All binding experiments were done at room temperature in 10 mm sodium phosphate, pH 7.2, 135 mm NaCl, 2.5 mm KCl, pH 7.4 (PBS), 0.1% bovine serum albumin. The effect of various ligands (NMS or toxins) on the equilibrium binding of a fixed concentration of [3H]NMS (0.5 nm) was determined in heterologous inhibition experiments and was analyzed using a classical competitive equation (for NMS) or with the allosteric ternary complex model for the toxins. With [3H]NMS as tracer, membrane proteins at concentrations adjusted so that no more than 10% of added radioligand was specifically bound (around 1500–2000 cpm) were incubated in PBS-BSA at 25 °C for 18–22 h with [3H]NMS and varying concentrations of ligand in a final assay volume of 300 μl. Nonspecific binding was determined in the presence of 50 μm atropine. The reaction was stopped by addition of 3 ml of ice-cold buffer (10 mm Tris, pH 7.8) immediately followed by filtration through Whatman GF/C glass fiber filters presoaked in 0.5% polyethylenimine. The filters were washed once again with 3 ml of ice-cold buffer (PBS) and dried, and the bound radioactivity was counted by liquid scintillation spectrometry. Each experiment was done at least three times. [3H]NMS (78 Ci/mmol) was from PerkinElmer Life Sciences.
Data Analysis
The binding data from individual experiments (n ≥ 3) were analyzed by nonlinear regression analysis using Kaleidagraph 4.0. After subtraction of the nonspecific binding and normalization, data obtained from inhibition binding experiments with the orthosteric radioligand [3H]NMS were analyzed using the Cheng and Prusoff equation for NMS and the allosteric ternary complex model (33) for the toxins.
In this last case, the equation (34) used was
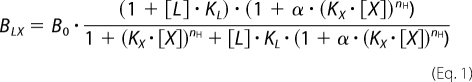 |
where BLX denotes the specific binding of the radioligand L ([3H]NMS) in the presence of the cooperatively interacting agent X. B0 is the binding of L in the absence of X. KL and KX are the affinity constants for the binding of L and X to the unliganded receptors, respectively. α is the cooperative factor for the allosteric interaction between X and L with α > 1, α < 1, and α = 1 indicating positive, negative, and neutral cooperativity, respectively. nH represents the slope of the curve.
Effects of the mutations of hM1 on the Kd values were analyzed by analysis of variance followed by Tukey's post-test with the GraphPad Prism version 5.00 software. For the chimeric receptors, data were analyzed with the Hill equation to estimate the IC50 and the slope factor, nH, of the inhibition curves.
Toxin and hM1 receptor pairs of residues in interaction were identified by cycle mutant experiments, and the energy of this interaction was measured by calculating the variation of free energy of interaction: ΔΔGint(A-B) = ΔΔG(A*;B*) − ΔΔG(A*) − ΔΔG(B*) with ΔΔG(A*) = RT ln(Ki(mut)/Ki(WT)). A ΔΔGint higher than 0.7 kcal/mol was considered the minimum threshold to identify two interacting residues, i.e. two residues separated by less than 7 Å (35).
Molecular Dynamics Simulations of MT7
The x-ray structure of MT7 (Tyr-51 di-iodo MT7, Protein Data Bank code 2VLW) has been solved previously (31). Molecular dynamics simulation of MT7 in water was performed using periodic boundary conditions. The molecule was immersed into a box of 65 × 60 × 50 Å of pre-equilibrated TIP3 water molecules (36) (detailed in the supplemental Experimental Procedures). After heating and equilibration of the system, 2.5 ns of simulation at 300 K were performed. 1000 frames were extracted from the trajectory and clustered into nine groups. One frame of each cluster was selected for docking.
Homology Modeling of hM1
A model of hM1 was generated with MODELLER (37). Available structures of class A GPCR (rhodopsin (Protein Data Bank code 1U19), human β1- (Protein Data Bank code 2VT4) and β2-adrenergic receptors (Protein Data Bank code 2RH1), A2A adenosine receptor (Protein Data Bank code 3EML), CXCR4 chemokine receptor (Protein Data Bank code 2ODO) and dopamine D3 receptor (Protein Data Bank code 3PBL) display a very similar organization of their seven transmembrane helices (Cα r.m.s. deviations, 1.7–2.8 Å). We used the rhodopsin x-ray structure (Protein Data Bank code 1U19 (38)) as a template. The r.m.s. deviations of the hM1 transmembrane helix backbone atoms relative to those of rhodopsin, β1-adrenergic receptor, β2-adrenergic receptor, A2A adenosine receptor, CXCR4 chemokine receptor, and dopamine D3 receptor are 0.5, 1.8, 1.7, 2.2, 2.6, and 2.4 Å, respectively. hM1 loop E2 conformation was predicted with RAPPER (39) (detailed in the supplemental Experimental Procedures).
Sampling of Loop E2 Conformation of hM1
Before simulation, the receptor structure was immersed into a box of 85 × 70 × 55 Å containing equilibrated TIP3 water molecules (36). During the path exploration with distance constraints (PEDC) procedure (40), an r.m.s.d. increment of 0.1 Å was imposed to the loop E2 (residues 169–179) backbone after each 10 ps of simulation up to 10 Å. All the other loops were free. To preserve the structure of the transmembrane region, the backbone atoms (C, N, Cα, and O) of the seven transmembrane helices were maintained by restraints characterized by a force constant of 5 kcal/mol/Å2. During the simulation, 100 frames were collected every 10 ps with an r.m.s. deviation on the loop E2 increasing from 0 to 10 Å compared with the starting structure. The last 36 frames were used in the docking procedure.
Data-driven Docking of MT7 on Two hM1 Receptors
Five docking calculations were performed to explore the relative orientation of the three partners (supplemental Table 1). They mainly followed the same protocol, i.e. a previously described data-driven docking procedure (41–43) based on four-dimensional restraint molecular dynamics (44). To dock one MT7 molecule on two hM1 receptors, we adapted this procedure to three partners. To improve docking accuracy, nine conformations of MT7 and 36 hM1 conformers with different loop E2 conformations were combined for the construction of the initial models. The docking protocol was repeated 30 or 36 times depending on the run. For each docking run, about 10,000 initial configurations were generated. The proximities derived from double mutant cycles were introduced as ambiguous distance restraints via the CHARMM NOE command. To apply these restraints, different low force constants (increasing slowly from 3 to 12, 1 to 4, or 0 to 1 kcal/mol/Å2) were combined with different short upper bonds (2, 4, or 6 Å) depending on the hM1 residues involved in the restraints (detailed in the supplemental Experimental Procedures).
We chose to orient the system with the z axis perpendicular to the membrane. The five runs differed by (i) the sampling method (random or symmetric), (ii) the relative initial positions of both hM1 molecules (i.e. the angle between hM1 protomer principal axes and the z axis), (iii) the rotation of the hM1 protomers around the z axis, and (iv) the ambiguous restraints applied between the two hM1 protomers (supplemental Table 1).
First, two docking runs were performed to sample all the possible dimer interfaces: “symmetric” and “random” runs. In these two docking runs, the starting configuration was composed of two hM1 protomers and one MT7. The two protomers were distant from each other to avoid steric clashes when they rotate to generate various respective orientations: the distance between the center of mass of the two protomers was set to 45 and 35 Å in random and symmetric runs, respectively. To maintain the two protomer principal axes in a parallel orientation, three ambiguous distance restraints between the two promoters were introduced (see supplemental Experimental Procedures). The large grid search of the symmetric run started from a configuration of one MT7 ligand and two distant hM1 receptors related by a cyclic symmetry. Then systematic rotations preserving the C2 symmetry were applied on both hM1 protomers, whereas random rotations were applied on MT7. In the random docking run search, random rotations were applied on the two distant hM1 protomers and MT7 (in that case, initial protomer configuration did not respect C2 symmetry). In the symmetric run, the respective orientations of the two hM1 were systematically explored (rotation from 0 to 360° by 10° steps), whereas in the random run, the relative hM1 orientation was random. In both runs, the MT7 orientation was randomized. Obviously, the random search in which the initial protomer configuration was not symmetric yielded fewer complexes with a C2 symmetry than the grid search in which all the starting configurations respected this symmetry.
Second, three docking runs were performed in which the TM6/TM7 interface was sampled by systematic rotation searches with a sampling interval of 2° and a range of 40°. Focusing on the TM6/TM7 interface, the two protomers can be placed close to each other (supplemental Table 1). In the run “TM6/TM7_sym,” the number of ambiguous distance restraints between the two protomers was set to 16 instead of 3 to improve the protomer contacts (supplemental Table 1). In docking run “TM6/TM7_without,” all ambiguous distance restraints between protomers were removed. In the run “TM6/TM7_cxcr4,” the hM1 protomer orientation was based on the orientation of the two subunits found in the CXCR4 dimer x-ray structure (9), and no ambiguous restraints between hM1 protomers were introduced.
During the refinement, 10,000 steps of minimization were performed under harmonic restraints with a force constant decreasing progressively from 1 to 0 kcal/mol/Å2. During these last steps, the electrostatic energy term was calculated with a distance-dependent dielectric constant.
All docking calculations were performed with CHARMM (45) package version c32b1. CHARMM19 force field was used. The time step was equal to 2 fs, and all bond lengths of hydrogen atoms were constrained using SHAKE (46).
Testing Symmetry of MT7-hM1 Dimer Complex
Docking complexes were subjected to a filter that discards the complexes deviating from the hM1 C2 symmetry. To quantify this symmetry, we calculated the parameter Sdev, which is equal to the average of |dist(A, B′) − dist(A′, B)| (where dist represents distance) over all pairs of α-carbon atoms as proposed by Baker and co-workers (47). Perfect symmetry corresponds to Sdev = 0 Å. The Sdev threshold was set to 2 Å; it was adapted to the magnitude of the uncertainty of the distances derived from double mutant data.
RESULTS
Identification of MT7 Binding Site on hM1 Receptor
Considering the size of the toxin, its high affinity for the hM1 receptor, and the major role of the hM1 receptor extracellular loops in the MT7 interaction (48), we hypothesized that MT7 and hM1 interact via a multipoint binding site involving mainly the extracellular domains of the receptor. Thus, we constructed chimeric receptors by grafting each of the three extracellular loops of the MT7-insensitive M3 receptor onto the M1 receptor and vice versa (Fig. 1), and we studied MT7 binding to these different targets. Exchanging the extracellular loops E1 and E3 did not significantly alter receptor affinity for MT7, whereas exchanging loops E2 drastically affected the binding (Fig. 1C). Grafting the M3 loop E2 onto the M1 receptor (chimera 2) reduced the receptor affinity for MT7 by more than 3 orders of magnitude, whereas the simple introduction of the M1 loop E2 onto the M3 receptor (chimera 5) was sufficient to induce a nanomolar affinity for MT7, corresponding to at least a 2000-fold affinity increase.
FIGURE 1.

Identification of extracellular loops of hM1 receptor involved in MT7 binding. A, schematic representations of the various chimeric receptors constructed by exchanging the extracellular loops between the hM1 and hM3 receptors colored in black and gray, respectively. B, sequence alignments of the extracellular loops E1, E2, and E3 of the hM1 and hM3 receptors. C, IC50(mut)/IC50(WT) values of MT7 toxin on wild-type hM1 and hM3 and chimeric muscarinic receptors. D, ribbon representation of an hM1 structural model indicating the extracellular loops E1, E2, and E3 colored in cyan, magenta, and orange, respectively. The same colors were used to differentiate the three extracellular loops in A, B, and C. N-ter, N terminus; C-ter, C terminus.
Taking these results into account, 17 positions were mutated in the extracellular loops and in the extracellular parts of the transmembrane domains of the hM1 receptor. Residues were mutated either to alanine or to the corresponding residue of the hM3 receptor, and the effects of these mutations on NMS and MT7 interaction were studied by following [3H]NMS binding (Table 1). Mutation of two residues (W91A in E1 and W101A in TM3) impaired NMS binding, whereas mutations at six positions located on the extracellular loops E1 (Trp-91) and E2 (Glu-170, Arg-171, Leu-174, and Tyr-179) or at the top of TM7 (Trp-400) affected toxin binding.
TABLE 1.
Affinity constants of wild-type and mutated hM1 receptors for NMS and MT7 toxin
These values were inferred from competition binding experiments with [3H]NMS and analyzed using the Cheng and Prusoff and ternary complex equations for NMS and MT7, respectively. The effect of the mutations of hM1 on the affinity constant values was analyzed by analysis of variance followed by Tukey's post-test. Mutants in bold are those associated with a significant MT7 affinity decrease (p < 0.05). Data are mean values ± S.E. of at least three experiments.
| hM1 mutant | Region | pKi NMS | pKX MT7 | KX(mut)/KX(WT) |
|---|---|---|---|---|
| WT | 10.22 ± 0.04 | 10.47 ± 0.04 | ||
| H90A | E1 | 10.20 ± 0.09 | 10.66 ± 0.20 | 0.6 |
| W91A | E1 | 9.23 ± 0.33 | 9.30 ± 0.06 | 14.9 |
| W101A | TM3 | 9.29 ± 0.02 | 9.79 ± 0.13 | 4.8 |
| Y166A | E2 | 9.83 ± 0.03 | 10.33 ± 0.32 | 1.4 |
| L167F | E2 | 9.93 ± 0.02 | 10.48 ± 0.11 | 1.0 |
| E170A | E2 | 10.20 ± 0.04 | 10.20 ± 0.07 | 1.9 |
| E170K | E2 | 10.16 ± 0.02 | 9.31 ± 0.11 | 14.6 |
| R171A | E2 | 9.76 ± 0.01 | 9.50 ± 0.29 | 9.4 |
| L174A | E2 | 10.21 ± 0.08 | 10.51 ± 0.01 | 0.9 |
| L174P | E2 | 10.15 ± 0.01 | 9.37 ± 0.14 | 12.5 |
| Q177E | E2 | 10.08 ± 0.04 | 10.58 ± 0.17 | 0.8 |
| Y179F | E2 | 9.85 ± 0.02 | 9.25 ± 0.18 | 16.5 |
| L183A | E2 | 10.00 ± 0.11 | 10.44 ± 0.20 | 1.1 |
| Q185A | E2 | 10.18 ± 0.10 | 10.59 ± 0.37 | 0.8 |
| K392A | E3 | 10.20 ± 0.12 | 10.57 ± 0.18 | 0.8 |
| D393A | E3 | 9.96 ± 0.09 | 10.50 ± 0.23 | 0.9 |
| E397A | E3 | 10.21 ± 0.06 | 10.54 ± 0.20 | 0.9 |
| W400A | TM7 | 10.12 ± 0.04 | 9.43 ± 0.02 | 11.0 |
| E401A | TM7 | 9.76 ± 0.02 | 10.20 ± 0.10 | 1.9 |
Double Mutant Cycle Experiments for Docking MT7 onto hM1
To go further and model the MT7-hM1 complex, we determined proximities between pairs of residues of the toxin and the receptor using the double mutant cycle methodology (35, 49). Indeed, a thermodynamic cycle analysis reveals whether the effects of one mutation of a residue A into A* on the receptor and a second mutation of a residue B into B* on the ligand are coupled or not. A variation of free energy of interaction (ΔΔGint) is measured from ΔΔGint(A-B) = ΔΔG(A*;B*) − ΔΔG(A*) − ΔΔG(B*) and reflects the coupling between the two mutations. Here, the affinity constants of each of the four possible combinations of wild-type and mutant proteins were measured, allowing the calculation of the 61 ΔΔGint values reported in Table 2 and Fig. 2. Using a threshold value of 0.7 kcal/mol (equal to twice the average standard deviation of ΔΔGint), 33 proximities between MT7 and hM1 residues were identified. These proximities mainly involved loop E2 (Glu-170, Arg-171, Leu-174, and Tyr-179; 22 restraints) assisted by Trp-400 in TM7 (six restraints), H90-W91 in loop E1 (four restraints), and Glu-397 in loop E3 (one restraint) of the M1 receptor on the one hand and residues of the MT7 loop II (Tyr-30, Ser-32, Arg-34, Met-35, and Tyr-36; 18 proximities), loop I (Trp-10 and Phe-11; 9 proximities), and loop III (Arg-52; six proximities) on the other hand.
TABLE 2.
Affinity constants and variations in free energy of interaction of wild-type and modified MT7 toxins for wild-type and mutated hM1 receptors
Data were inferred from binding experiments with [3H]NMS and calculated with the ternary complex equation. The free energy of interaction (ΔΔGint) values were calculated from ΔΔGint(A-B) = ΔΔG(A*;B*) − ΔΔG(A*) − ΔΔG(B*). ΔΔGint values exceeding 0.7 kcal/mol are in bold and indicate proximity between the two mutated residues. Data are mean values ± S.E. of at least three experiments (see “Experimental Procedures”).
| hM1 receptor | hM1 region | MT7 toxin | MT7 loop | pKX | ΔΔGint |
|---|---|---|---|---|---|
| kcal/mol | |||||
| WT | WT | 10.47 ± 0.04 | |||
| H90A | E1 | WT | 10.66 ± 0.20 | ||
| W10A | 1 | 11.62 ± 0.28 | −2.08 ± 0.40 | ||
| Y30A | 2 | 9.55 ± 0.28 | +1.14 ± 0.37 | ||
| Y51A | 3 | 10.42 ± 0.13 | +0.20 ± 0.11 | ||
| R52A | 3 | 10.42 ± 0.44 | −0.04 ± 0.59 | ||
| W91A | E1 | WT | 9.30 ± 0.06 | ||
| W10A | 1 | 7.69 ± 0.05 | +1.09 ± 0.07 | ||
| Y30A | 2 | 7.81 ± 0.07 | +1.64 ± 0.09 | ||
| Y51A | 3 | 8.79 ± 0.17 | +0.44 ± 0.24 | ||
| R52A | 3 | 8.54 ± 0.13 | +0.66 ± 0.17 | ||
| W101A | TM3 | WT | 9.79 ± 0.13 | ||
| W10A | 1 | 8.57 ± 0.14 | +0.57 ± 0.19 | ||
| Y30A | 2 | 9.08 ± 0.09 | +0.60 ± 0.11 | ||
| R34A | 2 | 7.74 ± 0.10 | −0.37 ± 0.21 | ||
| R52A | 3 | 9.05 ± 0.19 | +0.62 ± 0.25 | ||
| E170A | E2 | WT | 10.20 ± 0.07 | ||
| W10A | 1 | 8.42 ± 0.20 | +1.33 ± 0.25 | ||
| Y30A | 2 | 8.84 ± 0.28 | +1.47 ± 0.38 | ||
| S32A | 2 | 8.63 ± 0.16 | +1.43 ± 0.22 | ||
| R34A | 2 | 7.50 ± 0.36 | +0.60 ± 0.49 | ||
| M35A | 2 | 8.63 ± 0.13 | +0.87 ± 0.17 | ||
| Y36A | 2 | 7.97 ± 0.13 | +1.24 ± 0.17 | ||
| Y51A | 3 | 9.71 ± 0.31 | +0.41 ± 0.42 | ||
| R52A | 3 | 8.57 ± 0.18 | +1.77 ± 0.27 | ||
| R171A | E2 | WT | 9.50 ± 0.29 | ||
| K5A | 1 | 9.25 ± 0.18 | −0.10 ± 0.24 | ||
| W10A | 1 | 7.76 ± 0.18 | +1.26 ± 0.24 | ||
| Y30A | 2 | 8.22 ± 0.14 | +1.49 ± 0.43 | ||
| S32A | 2 | 8.46 ± 0.08 | +0.72 ± 0.11 | ||
| M35A | 2 | 7.68 ± 0.46 | +1.20 ± 0.62 | ||
| Y36A | 2 | 7.03 ± 0.21 | +1.55 ± 0.28 | ||
| K48A | 2 | 9.48 ± 0.02 | −0.13 ± 0.02 | ||
| Y51A | 3 | 8.67 ± 0.12 | +0.89 ± 0.16 | ||
| R52A | 3 | 8.35 ± 0.17 | +1.17 ± 0.23 | ||
| L174A | E2 | WT | 10.51 ± 0.01 | ||
| W10A | 1 | 9.19 ± 0.20 | +0.71 ± 0.27 | ||
| Y30A | 2 | 9.45 ± 0.17 | +1.08 ± 0.23 | ||
| S32A | 2 | 9.66 ± 0.20 | +0.48 ± 0.27 | ||
| R34A | 2 | 7.52 ± 0.13 | +1.19 ± 0.42 | ||
| M35A | 2 | 8.72 ± 0.10 | +1.16 ± 0.13 | ||
| Y36A | 2 | 8.59 ± 0.23 | +0.83 ± 0.31 | ||
| Y51A | 3 | 10.06 ± 0.23 | +0.37 ± 0.31 | ||
| R52A | 3 | 9.23 ± 0.22 | +1.35 ± 0.29 | ||
| Y179F | E2 | WT | 9.25 ± 0.18 | ||
| K5A | 1 | 9.15 ± 0.15 | −0.28 ± 0.20 | ||
| W10A | 1 | 7.62 ± 0.19 | +1.13 ± 0.25 | ||
| R34A | 2 | <6 | > +1.35 | ||
| K48A | 2 | 9.21 ± 0.01 | −0.09 ± 0.01 | ||
| R52A | 3 | <6 | >+4.00 | ||
| K392A | E3 | WT | 10.57 ± 0.18 | ||
| W10A | 1 | 9.88 ± 0.22 | −0.13 ± 0.29 | ||
| F11A | 1 | 10.19 ± 0.20 | +0.27 ± 0.27 | ||
| D393A | E3 | WT | 10.50 ± 0.23 | ||
| S8A | 1 | 10.05 ± 0.06 | +0.21 ± 0.08 | ||
| W10A | 1 | 10.10 ± 0.02 | −0.53 ± 0.02 | ||
| F11A | 1 | 10.21 ± 0.24 | +0.35 ± 0.09 | ||
| Y51A | 3 | 10.00 ± 0.34 | +0.42 ± 0.46 | ||
| E397A | E3 | WT | 10.54 ± 0.20 | ||
| W10A | 1 | 8.64 ± 0.22 | +1.58 ± 0.31 | ||
| F11A | 1 | 9.89 ± 0.21 | +0.64 ± 0.28 | ||
| W400A | TM7 | WT | 9.43 ± 0.02 | ||
| K5A | 1 | 9.60 ± 0.05 | −0.66 ± 0.07 | ||
| W10A | 1 | 7.61 ± 0.32 | +1.38 ± 0.44 | ||
| F11A | 1 | 8.29 ± 0.25 | +1.29 ± 0.33 | ||
| Y30A | 2 | 8.49 ± 0.08 | +0.90 ± 0.10 | ||
| S32A | 2 | 8.64 ± 0.15 | +0.38 ± 0.20 | ||
| R34A | 2 | <6 | >+1.58 | ||
| M35A | 2 | 8.52 ± 0.13 | −0.11 ± 0.12 | ||
| Y36A | 2 | 7.37 ± 0.29 | +1.00 ± 0.38 | ||
| K48A | 3 | 9.41 ± 0.06 | −0.12 ± 0.07 | ||
| R52A | 3 | 7.73 ± 0.08 | +0.91 ± 0.17 | ||
| E401A | TM7 | WT | 10.20 ± 0.10 | ||
| R34A | 2 | 7.41 ± 0.37 | +0.61 ± 0.45 |
FIGURE 2.

Double mutant cycle analysis of MT7-hM1 complex. Values of ΔΔGint (reported in Table 2) determined from changes in affinity constants are presented in colored bars. The different blue colors correspond to loop I; the different yellow, orange, and red colors correspond to loop II; and the different green colors correspond to loop III of MT7. Inset, ribbon representation of MT7 indicating the loops involved in the binding. Blue, red, and green, MT7 loops I, II, and III, respectively.
Interestingly, some MT7-hM1 receptor proximities highlighted by the double mutant cycle data involved toxin residues such as Trp-10 and Phe-11 on loop I and Arg-52 on loop III that are more than 25 Å apart in the toxin structure (Fig. 2, inset) and yet shared the same partners on the receptor (in this case, Glu-170, Arg-171, Leu-174, Tyr-179, and Trp-400). As a consequence, our attempts to satisfy all the restraints by docking the toxin onto a single receptor failed. They produced models with a positive van der Waals energy and a large distance restraint energy, indicating that these restraints are not compatible with a simple MT7-receptor model. A possible explanation could be that hM1 binding induces large MT7 loop conformational changes, but these are unlikely due to the low flexibility of the three-finger fold (50). A more simple explanation for this observation is suggested by our previous data reporting that MT7 binds to a dimeric form of hM1 receptor (32). To test whether our experimental data can be explained by a model in which the large MT7 ligand binds to a receptor dimer, we decided to calculate such a model based on the proximities issued from the double mutant cycle experiments. The 28 double mutant pairs revealing toxin and receptor residues that are not in contact were used to validate the resulting complexes.
Structure and Dynamics of MT7 Ligand
The x-ray structure of MT7 (Tyr-51 di-iodo MT7, Protein Data Bank code 2VLW) was solved previously (31). Docking accuracy can be significantly enhanced by using an ensemble of conformations picked up in the energy landscape of a molecule instead of a single structure (51–53). To this end, the MT7 conformation was explored by calculating a 2.5-ns molecular dynamics simulation in an explicit water environment (see “Experimental Procedures”). During this simulation, backbone fluctuations remained low in the β-sheet region (r.m.s.d. < 0.7 Å). The amplitudes of the simulated loop backbone motions were larger (r.m.s.d. < 2.2 Å) and reached 4 Å locally in the turn connecting loops I and II. The amplitudes of the side chain motions were higher than 3 Å in three regions: the turn connecting loops I and II and the tips of loops II and III. After clustering the 1000 frames extracted from the trajectory, nine structures of MT7 were selected for the docking calculations (supplemental Fig. S1).
Model of hM1 Receptor and Sampling of Loop E2 Conformations
A model of the muscarinic hM1 receptor was generated by homology modeling and de novo loop building on the basis of the rhodopsin x-ray structure (Protein Data Bank code 1U19). We checked that the helix bundle of our muscarinic hM1 receptor model shows the same organization as those found in various GPCR x-ray structures (Cα r.m.s. deviations = 0.5–2.6 Å) (see “Experimental Procedures”). The extracellular loop E2 of hM1 was initially modeled into a conformation similar to that of the rhodopsin template, covering the receptor cavity and thus blocking access to large ligands (54). It was shown for muscarinic receptors that this loop is a flexible “gatekeeper” adopting either a rhodopsin-like “closed” conformation blocking ligand access or an “open” conformation allowing access of the ligand to the TM-bound crevice (55). To take into account this flexibility, we generated open conformations of loop E2 by molecular dynamics. Time scales accessible by standard molecular dynamics are too short to simulate a large loop motion giving access to the receptor internal cavity. Therefore, an activated molecular dynamics simulation was performed on the hM1 extracellular loop E2 using PEDC (40) (see “Experimental Procedures”). This method allows exploration of low energy conformational pathways. The principle is to drive the molecular dynamics simulation with an additional energy term. The PEDC constraint slowly changes the conformation of the loop E2 from its initial state to more open conformations with a given r.m.s.d. that is increased progressively up to 10 Å during the simulation (supplemental Fig. S2). The last 36 frames of the activated molecular dynamics were used in the docking calculation.
Adapted Docking Protocol
We previously showed that MT7 binds to a dimeric form of the hM1 receptor (32). The double mutant cycle approach provided us with proximities between MT7 and hM1 without any information on which hM1 protomer is involved in the interaction. Therefore, a distance restraint could not be unambiguously assigned to a given toxin residue and a unique hM1 protomer. Ambiguous distance restraints taking into account this uncertainty were used. Docking of MT7 onto hM1 dimer was basically performed following the same protocol that we and other authors used previously (41–43). The protocol was adapted to three partners: MT7 and the two hM1 protomers (see “Experimental Procedures”).
Analysis of structurally characterized homodimers of soluble proteins shows that most of them adopt a cyclic C2 symmetry (56, 57). Moreover, such symmetry was recently observed in the first x-ray structure of a dimeric GPCR (9). Hypothesizing that the hM1 dimer also exhibits a C2 symmetry, the complexes with this symmetry were selected at the end of the five following docking runs (see “Experimental Procedures”).
Dimer Interface Mainly Involves TM6 and TM7 Helices
In the two first runs, different sampling strategies were explored to identify whether our receptor dimerization interface can be defined from the 33 MT7-hM1 proximities. These strategies were based on two different large pools of initial configurations, sampling all the possible dimer interfaces. Orientations of the starting dimer were generated by either random or systematic rotations along their symmetry axes (see “Data-driven Docking of MT7 on Two hM1 Receptors” for a description of symmetric and random runs under “Experimental Procedures” and in supplemental Table 1). In both runs, the MT7 orientation was also randomized. These searches were performed for 36 different hM1 models combined into nine MT7 structures, yielding more than 10,000 initial conformations. Both searches produced MT7-hM1 dimer complexes with restraint energies and total buried accessible areas of the same order of magnitude (data not shown). Fig. 3, A and B, show the variation of the restraint energy versus the global r.m.s.d. for MT7-hM1 dimer complex. Both searches provided distant MT7-hM1 dimer conformations (r.m.s.d. up to 20 Å). Nevertheless, in each run, the lower restraint energy conformations were close to each other: the 22 grid search complexes and the 12 random search complexes with restraint energies lower than 35 and 40 kcal/mol were selected; these complexes had pairwise r.m.s. deviations lower than 6 and 7 Å, respectively. Fig. 3, C–H, show that in both docking runs TM6 and TM7 are located at the dimer interface. Fig. 3, I and J, indicate that all of them present the same interface: the buried residues are located at the N terminus of TM1 and in TM6 and TM7. In conclusion, two different docking simulations based on MT7-hM1 proximities derived from double mutant analysis led to similar hM1 monomer/monomer relative positions characterized by a TM6/TM7 interface.
FIGURE 3.

Identification of dimer interface by two first independent docking calculations. The random search and systematic grid search are on the left and right, respectively. A and B, variation of restraint energy as a function of the global r.m.s. deviation. The global r.m.s.d. was calculated using as reference the structure with the lowest restraint energy and by fitting the Cα atoms of MT7 and hM1 dimer excluding the extracellular loops. Superimpositions of the backbone of the 10 lowest restraint energy structures are shown: C and D, extracellular view; E and F, transverse view; G and H, intracellular view. The TM6 and TM7 domains are colored in orange and yellow, respectively. I and J, basa (in Å2) at the hM1 dimer interface calculated on complexes obtained by random and grid searches, respectively. (Energy thresholds of 40 and 35 kcal/mol were used for random and grid searches, respectively.)
All Docking Runs Converge to Same MT7-hM1 Dimer Conformation
By sampling all the hM1 dimer interfaces, we showed that the hM1-MT7 complex is characterized by a TM6/TM7 interface. Then we focused the conformational sampling on complexes with a TM6/TM7 interface to get more insight at the atomic level on the MT7-hM1 complex. We explored whether different independent docking simulations would reach the same configuration of the MT7-hM1 dimer. By that way, we obtained a reliable estimation of the conformational variability associated to the calculated model. Three docking runs were performed in which the docking conditions varied (dimer restraints, TM6/TM7_sym and TM6/TM7_without runs, respectively; and initial dimer orientation, TM6/TM7_cxcr4 run; see “Experimental Procedures” and supplemental Table 1). All together, these three docking runs yielded about 22,000 structures. These structures were pooled with the structures obtained in the two previous simulations (about 7000 structures). Fig. 4A shows the variation of the restraint energy as a function of the r.m.s. deviation. The observed funnel shape indicates the convergence of the five docking simulations.
FIGURE 4.

Structural model of MT7-dimeric hM1 complex based on experimental double mutant cycle data. A, restraint energy as a function of r.m.s.d. from the MT7-hM1 dimer for about 30,000 calculated complexes obtained by five independent docking simulations (supplemental Table 1). The reference structure was the lowest energy structure. The r.m.s.d. was calculated using the Cα atoms of MT7 and hM1 dimer excluding the extracellular loops. B, histogram of the averaged minimum distances between residue pairs with ΔΔGint >0.7 kcal/mol and involved in ambiguous distance restraints (a) and with ΔΔGint <0.7 kcal/mol and not used in the docking calculations (b). C and D, two perpendicular views of a superimposed ribbon representation of the 10 best structures. E and F, two perpendicular views of a schematic of the lowest restraint energy model. hM1A, hM1B, and MT7 are in red, blue, and green, respectively. hM1 loops are in cyan except E2, which is in magenta.
The 500 structures with lower restraint energy (<40 kcal/mol) were refined (see “Experimental Procedures”), and the 10 structures with the lowest restraint energy were selected and analyzed. Structural statistics (supplemental Table 2) reflect the high quality of these final models. Fig. 4B shows the histogram of the averaged minimum distances between residue pairs. The distances between 29 of 33 pairs of interacting residues (ΔΔGint > 0.7 kcal/mol) were all smaller than 7.5 Å. The average distance between Leu-174 and Met-35 is between 8 and 9 Å, and the distances between Tyr-179 and Arg-52, Trp-400 and Tyr-30, and Trp-400 and Arg-52 are between 11 and 12 Å. As a validation, the distances between the 28 non-interacting pairs of residues (ΔΔGint < 0.7 kcal/mol) not used in the calculation were measured. These distances are all larger than 7.5 Å except that between Glu-170 and Arg-34, which is 4.8 Å.
The 10 structures are displayed in Fig. 4, C and D. They come from the five docking runs: each run provides at least one structure, indicating that all the different docking runs converge to the same structure. Compared with the lowest restraint energy structure, the average r.m.s.d. of the Cα atom of all the residues of the MT7-hM1 dimer complex is 2.5 Å. In these structures, the toxin is located on the extracellular side of a dimeric receptor, interacting with the two protomers (Fig. 4, E and F, and supplemental Fig. S4). The toxin axis (defined as the principal axis of the central β-sheet) makes a constant angle nearly equal to 60° with the axis passing through the centers of gravity of the two receptor protomers (Fig. 4C). The angle between the toxin plane (defined as the β-sheet plane) and the membrane plane varies from 30 to 60° (Fig. 4D). MT7 is flanked by the two extracellular loops E1 and E2. The different loops of MT7 interact with different protomers: loops II and III interact with protomer hM1A, and loop I interacts with protomer hM1B (Fig. 4, E and F). The buried accessible surface between MT7 and the hM1 dimer is equal to 2400 ± 200 Å2.
Model Is in Agreement with Single Point Mutation Data
The following analysis of the buried accessible surface area (basa) was based on the 10 lowest restraint energy structures. First, on the toxin side, the most important residues of MT7 for hM1 binding are Trp-10, Arg-34, Met-35, and Tyr-36 (31). All these MT7 residues have a large buried accessible area (>70Å 2; Fig. 5A), indicating that the present model is in agreement with the MT7 binding site defined from simple mutant binding measurements. Second, on the hM1 receptor side, loop E2 was identified as the main determinant of the MT7-hM1 interaction (Table 1). Four residues of loop E2 (Glu-170, Arg-171, Leu-174, and Tyr-179) appeared to be crucial for MT7 binding. Fig. 5B shows that the two hM1 E2 loops make different contacts with MT7: in protomer hM1A, Glu-170, Arg-171, and Leu-174 are highly buried (basa > 50Å2), whereas they are only slightly buried (basa < 50Å2) in protomer hM1B. Moreover, Tyr-179, which is already largely buried in the free hM1 as compared with Glu-170, Arg-171, and Leu-174, is close to Arg-34 in hM1A but is far from hM1B. Our model is thus in agreement with the major role of loop E2 in the MT7 interaction. Loop E1 of hM1 also contributes to MT7 binding: Trp-91, which was detected as important for the MT7-hM1 interaction by point mutation (Table 1), is one of the two most buried residues in the hM1 E1 loop (His-90 and Trp-91; basa ∼ 40Å2). Lastly, Fig. 5B indicates that the N terminus of the first TM helix of hM1 (Pro-22 and Trp-23) is partially buried. This is only true for a few structures where the N terminus of TM1 is close to the C terminus of MT7 as indicated by the high values of the r.m.s. deviations of the basa values corresponding to the N terminus.
FIGURE 5.

Average buried accessible surface area (in Å2) in 10 structures of MT7-dimeric hM1 complex. A, MT7 residues at the interface with the hM1 receptor; arrows indicate the β-sheets of MT7. B, hM1 residues at the interface with MT7. C, hM1 residues located at the dimer interface. White rectangles symbolize the hM1 TM helices. In each plot, hM1A is at the top, and hM1B at the bottom. The average buried accessible surface area is calculated on the best MT7-dimeric hM1 structures (see text). The error bars indicate the r.m.s. associated with the average values.
Interactions at MT7/hM1 Interface
Fig. 6 shows several specific side chain-side chain proximities observed in our models of the MT7-hM1 complex. First, our model suggests that the functionally most important residues of hM1, Tyr-179 and Trp-400, are involved in several interactions with MT7. Tyr-179 and Trp-400 of protomer hM1A are close to the functionally most important MT7 residues, Arg-34 and Tyr-36, in loop II (Fig. 6A). MT7-R34 interacts with Tyr-179 and Trp-400 of hM1A possibly via a cation-π interaction. This interaction could be protected from the solvent by hydrophobic contacts generated by MT7-Y36 with hM1A-P396 and hM1A-F182. In addition, Tyr-179 and Trp-400 of protomer hM1B are close to the MT7 loop I Trp-10, which suggests major hydrophobic interactions between these residues (Fig. 6B). Second, examination of the proximities between MT7 loop III and loop E2 of hM1A suggests a potential salt bridge between MT7-R52 and hM1A-E170 and an electrostatic interaction between MT7-Y30 and hM1A-E170 (Fig. 6C) in agreement with the double mutant data (Table 2). Finally, proximities between MT7 loop I and hM1B loop E1 were identified. The corresponding interactions mainly involve hydrophobic residues: Trp-10 and Phe-11 of MT7 and His-90 and Trp-91 of hM1B (Fig. 6D).
FIGURE 6.

Specific side chain-side chain interactions in 10 structures of MT7-dimeric hM1 complex. A, tip of the MT7 loop II-hM1A. B, tip of the MT7 loop I-hM1B. C, MT7 loops II and III-hM1A E2. D, MT7 loop I-hM1B E1. hM1A, hM1B, hM1 loop E1, hM1 loop E2, and toxin MT7 are colored in red, blue, cyan, magenta, and green, respectively.
hM1 Dimerization Interface Involves Several Hydrophobic Residues Conserved in Muscarinic Receptors
The TM6/TM7 dimerization interface between the two protomers is exclusively composed of hydrophobic residues with the exception of Lys-362 and Arg-365 on the cytoplasmic side of TM6. The most buried residues are Leu-372, Leu-376, Ile-383, Leu-399, Leu-402, Leu-406, and Leu-420 (basa > 70 Å2) (Fig. 5C). Three interface contacts are supported by the same residues on both protomers: Leu-399, Leu-406, and Leu-376 (Fig. 7). Leu-406 also has contacts with Pro-380 of the facing protomer. Hydrophobic contacts are maintained through a cluster of residues. On the upper part of the dimerization interface, Ile-383 of hM1A forms a hydrophobic cluster with Leu-402 and Trp-405 of hM1B (and symmetrically, Ile-383 of hM1B forms a hydrophobic cluster with Leu-402 and Trp-405 of hM1A). On the lower part of the dimerization interface, another cluster is formed by Leu-372 of hM1A and Ile-413, Cys-417, and Leu-420 of hM1B (and symmetrically, Leu-372 of hM1B forms a cluster with Ile-413, Cys-417, and Leu-420 of hM1A) (Fig. 7).
FIGURE 7.

TM6/TM7 dimerization interface in 10 structures of MT7-hM1 dimer complex. hM1A and hM1B are displayed in red and blue schematics, respectively. The transverse view (A) and extracellular view (B) are on the left and right, respectively. Three contacts at the interface are supported by the same residues on both protomers: residues Leu-376, Leu-399, and Leu-406 are colored in orange, green, and yellow, respectively. The cluster of Ile-383 of hM1A and Leu-402 and Trp-405 of hM1B and the cluster of Ile-383 of hM1B and Leu-402 and Trp-405 of hM1A are colored in pale cyan and cyan, respectively. The cluster of Leu-372 of hM1A and Ile-413, Cys-417, and Leu-420 of hM1B is colored in hot pink, and the cluster of Leu-372 of hM1B and Ile-413, Cys-417, and Leu-420 of hM1A is colored in violet.
Sequence alignment between muscarinic receptors was performed to identify conserved hydrophobic residues presenting a large exposed hydrophobic surface (data not shown). Five of the most conserved residues present accessible surface areas higher than 60 Å2: Phe-121, Ile-161, Ile-383, Leu-406, and Leu-420. Three of them, Ile-383, Leu-406, and Leu-420, are located in TM6-TM7 domains and belong to the most buried residues at the hM1/hM1 interface identified in our MT7-hM1 complexes (Fig. 7). These residues contribute to 20% of the buried dimer interface.
Dimer Interface Modeling Is Driven by Distance between Tips of Loops I and II of MT7
As mentioned previously, the toxin main axis makes a 60° angle with the axis passing through the center of gravity of the two receptor protomers. Because of this orientation, the tips of toxin loops II and III are located on the same vertical axis perpendicular to the membrane plane (Fig. 8A, A axis). The distance between this axis and the vertical axis passing though the tip of loop I (Fig. 8A, B axis) is roughly equal to 20 Å. This distance is similar to the distance between the two MT7 binding sites centered on Tyr-179/Trp-400 of each protomer in our model of MT7-hM1 dimer complex (Fig. 8B). Therefore, there is a nice distance adequacy between the tips of MT7 loops and the two binding sites on the hM1 dimer (Fig. 8, A and B). Two additional simple calculations reinforced this observation. The first one aimed at modeling the MT7-hM1 complex based on the CXCR4 dimer structure. The recent x-ray structure of the CXCR4 chemokine receptor (9) presents a dimer with a different interface (TM3/TM4). Attempts to model the hM1 dimer using the CXCR4 dimer orientation led to structural models with a high level of violations (sum of violations up to 50 Å instead of an average of 1 Å in the present selected complexes). In this case, the distance between the two MT7 binding sites (Tyr-179/Trp-400) is too large (35 Å) to receive the tips of MT7 loops (Fig. 8C). The TM3/TM4 dimer interface found in the CXCR4 chemokine receptor is not consistent with our experimental data on the MT7-hM1 binding. In the second analysis, we calculated the distance between the two MT7 binding sites in hM1 dimer structures generated by exploring all the possible relative positions of the protomers. Fig. 8D shows the variation of the distances between the MT7 binding sites (Tyr-179/Trp-400) of both protomers as a function of the angle of rotation of each protomer around their vertical axis. This figure also indicates which TMs are at the interface when the rotation angle varies. The range of variations of the distance between the toxin binding sites on both protomers is large (from 15 to 52 Å), whereas the range of variations of the distance between the centers of mass of the two protomers is low (37.5 ± 2.5 Å). This is due to the fact that Tyr-179/Trp-400 is not located at the center of mass of the hM1 receptor. Thus, a prerequisite for the interaction of MT7 with a receptor dimer is that the distance between the two binding sites is roughly equal to 20 Å. Fig. 8D indicates that this condition is only observed in one dimer orientation in which the dimer interface is TM6/TM7.
FIGURE 8.

TM interface in MT7-hM1 dimer complex is determined by distance between MT7 loops. A, ribbon representation of MT7 in red with “hot spot” residues Arg-34, Trp-10, and Arg-52 in green. The MT7 principal axis and the orthogonal projection of this axis on the membrane plane are displayed in pink. B, hM1 dimer conformation calculated from our experimental data viewed from the extracellular face (Tyr-179/Trp-400 in green). C, hM1 dimer conformation based on the CXCR4 chemokine structure viewed from the extracellular face (Tyr-179/Trp-400 in yellow). D, measurements of the distances between the two centers of gravity of the protomers of the receptor (○) and the two centers of mass of the MT7 binding sites (Tyr-179/Trp-400) of the protomers (■) during the step by step rotation of each protomer around their vertical axis. Distances are averaged on 10 structures, and the error bars indicate the r.m.s. associated with the average values. Under the graph, the TM involved in the dimer interface is indicated, and in two extreme cases, the localization of the MT7 binding sites in the corresponding dimer configuration is displayed. The hM1 receptor is symbolized by a gray oval, and the MT7 binding site is symbolized by a white circle.
DISCUSSION
Although several GPCR x-ray structures have recently been solved, determination of experimental three-dimensional structures of GPCR-ligand complexes remains a challenge. We have focused on the interaction between the particularly specific allosteric MT7 ligand and the hM1 mAChR. Previous studies from our laboratory identified the MT7 toxin residues critical for its interaction with the hM1 receptor (31) and indicated that MT7 may bind and stabilize a dimeric form of the hM1 receptor, although these results do not exclude an interaction with a monomeric receptor (32). In the present study, we used chimeric and site-directed mutagenesis approaches combined with pharmacological characterization to identify MT7-hM1 receptor proximities. Thus, we revealed that toxin residues such as Trp-10 and Phe-11 on loop I and Arg-52 on loop III, which are more than 25 Å apart in the toxin structure, share the same partners on the receptor. As a consequence, all attempts to satisfy the identified proximities by docking the toxin onto a single receptor failed. However, by taking into account the dimeric state of the hM1 receptor and by using an efficient docking procedure, we were able to show that our experimental data were consistent with a unique family of MT7-hM1 complex structures.
Data-driven Docking Procedure Is Efficient Alternative Approach for Structural Characterization of Ligand-bound GPCR Oligomeric Complexes
During the last decade, data-driven docking procedures have been successfully used to model large protein-protein complex structures (58, 59). In particular, double mutant cycle experiments, which provide information on the proximities between pairs of residues from both proteins of a complex, were combined with restraint molecular dynamics to calculate structures of complexes (35). This procedure was shown to be efficient at driving docking simulations both by our laboratory (41–43) and by several other authors (35, 60–66). We had the opportunity to evaluate the relevance of this approach by comparing our nicotinic acetylcholine receptor-α-neurotoxin complex model (41) with the homologous acetylcholine-binding protein-α-cobratoxin x-ray structure published later (67). The superimposition of the secondary structure elements led to an r.m.s.d. of 2.3 Å between the model and the x-ray structure, corresponding to an appropriate global orientation and location of the toxin on the receptor (68).6
In the present study, we used our previous procedure (41–43) that we adapted to three partners (MT7 and two hM1 receptors). We mainly modified the protocol of the high temperature four-dimensional molecular dynamics under ambiguous restraint steps to introduce an additional dimension in which the third partner does not see the two others at the beginning of the docking calculations (see supplemental Experimental Procedures). A similar data-driven approach designed to build molecular assemblies of more than two partners was recently described by Bonvin and co-workers (69). Moreover, to take into account the flexibility of both partners, a molecular dynamics simulation in explicit solvent was performed on MT7, and a family of hM1 models was generated in which the large movements of the extracellular loop E2 were explored by activated molecular dynamics simulation in explicit solvent.
TM6/TM7 Interface Is Strongly Dependent on Toxin Topology
From the 61 values of ΔΔGint that were experimentally determined, 33 indicate proximities between MT7 and hM1 residues. Different MT7 and hM1 initial conformation sampling procedures were used to calculate independent starting models for the docking simulations. In total, 30,000 complexes were generated by five independent docking simulations under different conditions. The five docking simulations converged to the same configuration of the complex. On this basis, we were able to show that the TM domains present at the dimer interface of the MT7-hM1 dimer complex are TM6 and TM7. This is different from what was observed in the x-ray structure of the dimeric CXCR4 chemokine receptor. Multiple sequence alignment between acetylcholine muscarinic receptor sequences (33 sequences) and CXCR4 chemokine receptor sequences (16 sequences) downloaded from the GPCRDB (70) shows that the TMs of these receptors share 25% sequence identity. It has been shown that the quaternary structure of soluble proteins is well conserved above 50% identity (56, 71, 72). On this basis, no analogy between the quaternary structures of hM1 and CXCR4 dimers can be predicted. Moreover, we have also demonstrated that the dimeric organization of hM1 exists in the context of the binding to MT7 and is strongly dependent on the toxin topology. Therefore it is risky to compare the dimer organization of the MT7-hM1 dimer complex with that observed by x-ray crystallography in the CXCR4 chemokine receptor structure (9).
Multiple Anchor Patches Are Responsible for High Affinity of MT7-hM1 Interaction
Our experimentally based model shows that the tips of MT7 loops II and III interact with one hM1 protomer (protomer hM1A), whereas the tip of loop I binds to the other protomer (protomer hM1B). Therefore, the MT7-hM1 dimer recognition site is composed of three patches on MT7 (the tips of the three loops) and two patches on the hM1 side (the binding sites of the two protomers; Tyr-179/Trp-400), forming a total interface area equal to 2400 Å2. This value is close to the mean value of the protein/protein interface areas reported for two-patch interactions (2500 Å2) (73). The multiplicity of anchor points between the toxin and the receptor dimer generates a high affinity of 29 pm. An analogous approach based on the multiplication of the number of anchor points is adopted in “fragment-based drug design” (74–77) and was successfully used in designing bitopic ligands of muscarinic receptors (78–80).
Molecular Basis of MT7 Binding Specificity
Our model shows that MT7 is flanked by the extracellular loop E2 of each hM1 protomer, a region that plays a crucial role in this interaction (Fig. 4E). Our mutagenesis data underline the important roles of Tyr-179, Glu-170, Leu-174, and Arg-171 in the MT7-hM1 interaction (Table 1). These results complete and enlarge preliminary data identifying the loop E2 of the M1 receptor as critical for the MT7 interaction (48). They confirm the major role of the loop E2 in the binding of allosteric modulators as demonstrated previously for small allosteric ligands interacting with hM2 receptor. In particular, hM2-Y177, which corresponds to hM1-Y179, is critical for the recognition of the allosteric ligands tacrine and Duo3 (81, 82). Furthermore, W4007.35 (Ballesteros and Weinstein numbering in superscript) located at the extracellular top of transmembrane helix 7 is critical for the mAChR binding of all these hM2 allosteric modulators as well as the MT7 toxin (83, 84). Therefore, the MT7 binding site on the hM1 receptor partially corresponds to that previously identified for small allosteric ligands on the hM2 receptor. In particular, we show that hM1 Tyr-179 and Trp-400 residues, which are conserved in all muscarinic receptors, are in close contact with the side chain of Arg-34, which is generally conserved in muscarinic toxins.
Nevertheless, MT7 toxin is highly selective for the M1 subtype of mAChRs, and our model gives some molecular bases to this observation. Swapping the extracellular loops between hM1 and hM3 (Fig. 1, B and C) or introducing point mutations in the hM1 sequence (Table 1) and measuring the binding of these mutants for MT7 indicated that the high affinity of MT7 for the M1 subtype is not due to loop E3 and in particular to the striking mutation E397K (Fig. 1B). The selectivity between hM1 and hM3 is mainly due to loop E2 that includes highly specific M1 residues such as Glu-170, Leu-174, and Ala-175. The charge inversion E170K and the L174P and A175P mutations that may have conformational effects on loop E2 in the hM3 sequence are responsible for the very low affinity of the toxin MT7 for this mAChR subtype. Our model shows that loop E2 interacts with MT7 loop III. The tip of this loop is characterized by the Y51-R52 sequence, which is unique to MT7 when compared with other muscarinic toxins (29). More precisely, our model identified an interaction between hM1A-E170 and MT7-R52. Glu-170 is specific to hM1 when compared with other muscarinic receptors, whereas Arg-52 is specific to MT7. Thus, this electrostatic interaction may be crucial for the specificity of the MT7-hM1 recognition.
Conclusion
In summary, although the organization of GPCRs in multimeric assembly is still a matter of intense debate, several functional properties and biochemical, biophysical, and structural evidence argue for a dimerization/oligomerization of these receptors (for a review, see Ref. 85). Moreover, experimental methods (cysteine cross-linking and bioluminescence resonance energy transfer) used to identify the TM domains present at the dimer interface as well as predictive bioinformatics and molecular modeling approaches applied to GPCR oligomer modeling highlight that GPCR structures are able to oligomerize via multiple interfaces (86). In the present study, we characterized a GPCR dimer conformation observed when the GPCR binds to an allosteric ligand by following an alternative approach that integrates mutagenesis experiments, pharmacological characterizations, and molecular flexible docking. Thus, we obtained a structural model of the MT7-hM1 dimer complex that reveals molecular explanations for the high affinity and selectivity of the MT7-hM1 interaction. In particular, we propose that a major determinant of the MT7-hM1 binding is the cation-π interaction between the conserved hM1A-Y179/W400 and MT7-R34 residues and that a crucial molecular determinant of the MT7-hM1 specificity is the electrostatic interaction between hM1A-E170 and MT7-R52. Furthermore, our model provides an atomic description supporting a TM6/TM7 interface in the hM1 dimer in interaction with MT7. Exploration of all the possible symmetric dimer configurations showed that the topological determinant of the hM1 dimer interface is the distance between the two MT7 functional sites, i.e. the tips of the MT7 loops I and II or III. The proposed model can be used as a starting point to design high affinity ligands that selectively bind specific dimeric forms of the hM1 receptors. Finally, this approach could be generalized to various peptide-GPCR complexes to propose molecular explanations for the pharmacological properties of these interactions.
Supplementary Material
Acknowledgments
We are grateful to Jean-Baptiste Charbonnier, Raphael Guérois, and Marc Le Maire for fruitful discussions.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Experimental Procedures, Tables 1 and 2, and Figs. S1–S4.
B. Gilquin, unpublished results.
- GPCR
- G protein-coupled receptor
- r.m.s.
- root mean square
- r.m.s.d.
- root mean square deviation
- hM1/2/3
- human M1/2/3 muscarinic receptor
- basa
- buried accessible surface area
- NMS
- N-methylscopolamine
- mAChR
- muscarinic acetylcholine receptor
- PEDC
- path exploration with distance constraints
- TM
- transmembrane.
REFERENCES
- 1. Takeda S., Kadowaki S., Haga T., Takaesu H., Mitaku S. (2002) FEBS Lett. 520, 97–101 [DOI] [PubMed] [Google Scholar]
- 2. Wise A., Gearing K., Rees S. (2002) Drug Discov. Today 7, 235–246 [DOI] [PubMed] [Google Scholar]
- 3. Pierce K. L., Premont R. T., Lefkowitz R. J. (2002) Nat. Rev. Mol. Cell Biol. 3, 639–650 [DOI] [PubMed] [Google Scholar]
- 4. Cherezov V., Rosenbaum D. M., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Kuhn P., Weis W. I., Kobilka B. K., Stevens R. C. (2007) Science 318, 1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rosenbaum D. M., Cherezov V., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Yao X. J., Weis W. I., Stevens R. C., Kobilka B. K. (2007) Science 318, 1266–1273 [DOI] [PubMed] [Google Scholar]
- 6. Rasmussen S. G., Choi H. J., Rosenbaum D. M., Kobilka T. S., Thian F. S., Edwards P. C., Burghammer M., Ratnala V. R., Sanishvili R., Fischetti R. F., Schertler G. F., Weis W. I., Kobilka B. K. (2007) Nature 450, 383–387 [DOI] [PubMed] [Google Scholar]
- 7. Warne T., Serrano-Vega M. J., Baker J. G., Moukhametzianov R., Edwards P. C., Henderson R., Leslie A. G., Tate C. G., Schertler G. F. (2008) Nature 454, 486–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jaakola V. P., Griffith M. T., Hanson M. A., Cherezov V., Chien E. Y., Lane J. R., Ijzerman A. P., Stevens R. C. (2008) Science 322, 1211–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu B., Chien E. Y., Mol C. D., Fenalti G., Liu W., Katritch V., Abagyan R., Brooun A., Wells P., Bi F. C., Hamel D. J., Kuhn P., Handel T. M., Cherezov V., Stevens R. C. (2010) Science 330, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chien E. Y., Liu W., Zhao Q., Katritch V., Han G. W., Hanson M. A., Shi L., Newman A. H., Javitch J. A., Cherezov V., Stevens R. C. (2010) Science 330, 1091–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Warne T., Moukhametzianov R., Baker J. G., Nehmé R., Edwards P. C., Leslie A. G., Schertler G. F., Tate C. G. (2011) Nature 469, 241–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rosenbaum D. M., Zhang C., Lyons J. A., Holl R., Aragao D., Arlow D. H., Rasmussen S. G., Choi H. J., Devree B. T., Sunahara R. K., Chae P. S., Gellman S. H., Dror R. O., Shaw D. E., Weis W. I., Caffrey M., Gmeiner P., Kobilka B. K. (2011) Nature 469, 236–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rasmussen S. G., Choi H. J., Fung J. J., Pardon E., Casarosa P., Chae P. S., Devree B. T., Rosenbaum D. M., Thian F. S., Kobilka T. S., Schnapp A., Konetzki I., Sunahara R. K., Gellman S. H., Pautsch A., Steyaert J., Weis W. I., Kobilka B. K. (2011) Nature 469, 175–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hansen J. L., Hansen J. T., Speerschneider T., Lyngsø C., Erikstrup N., Burstein E. S., Weiner D. M., Walther T., Makita N., Iiri T., Merten N., Kostenis E., Sheikh S. P. (2009) J. Biol. Chem. 284, 1831–1839 [DOI] [PubMed] [Google Scholar]
- 15. Chabre M., Cone R., Saibil H. (2003) Nature 426, 30–31 [DOI] [PubMed] [Google Scholar]
- 16. Chabre M., le Maire M. (2005) Biochemistry 44, 9395–9403 [DOI] [PubMed] [Google Scholar]
- 17. Whorton M. R., Bokoch M. P., Rasmussen S. G., Huang B., Zare R. N., Kobilka B., Sunahara R. K. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 7682–7687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rivero-Müller A., Chou Y. Y., Ji I., Lajic S., Hanyaloglu A. C., Jonas K., Rahman N., Ji T. H., Huhtaniemi I. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 2319–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Albizu L., Cottet M., Kralikova M., Stoev S., Seyer R., Brabet I., Roux T., Bazin H., Bourrier E., Lamarque L., Breton C., Rives M. L., Newman A., Javitch J., Trinquet E., Manning M., Pin J. P., Mouillac B., Durroux T. (2010) Nat. Chem. Biol. 6, 587–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Park P. S., Wells J. W. (2003) Biochemistry 42, 12960–12971 [DOI] [PubMed] [Google Scholar]
- 21. Zeng F. Y., Wess J. (1999) J. Biol. Chem. 274, 19487–19497 [DOI] [PubMed] [Google Scholar]
- 22. Goin J. C., Nathanson N. M. (2006) J. Biol. Chem. 281, 5416–5425 [DOI] [PubMed] [Google Scholar]
- 23. Hern J. A., Baig A. H., Mashanov G. I., Birdsall B., Corrie J. E., Lazareno S., Molloy J. E., Birdsall N. J. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 2693–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ilien B., Glasser N., Clamme J. P., Didier P., Piemont E., Chinnappan R., Daval S. B., Galzi J. L., Mely Y. (2009) J. Biol. Chem. 284, 19533–19543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Max S. I., Liang J. S., Potter L. T. (1993) Mol. Pharmacol. 44, 1171–1175 [PubMed] [Google Scholar]
- 26. Olianas M. C., Maullu C., Adem A., Mulugeta E., Karlsson E., Onali P. (2000) Br. J. Pharmacol. 131, 447–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mourier G., Dutertre S., Fruchart-Gaillard C., Ménez A., Servent D. (2003) Mol. Pharmacol. 63, 26–35 [DOI] [PubMed] [Google Scholar]
- 28. Fruchart-Gaillard C., Mourier G., Marquer C., Ménez A., Servent D. (2006) Mol. Pharmacol. 69, 1641–1651 [DOI] [PubMed] [Google Scholar]
- 29. Servent D., Fruchart-Gaillard C. (2009) J. Neurochem. 109, 1193–1202 [DOI] [PubMed] [Google Scholar]
- 30. Galat A., Gross G., Drevet P., Sato A., Ménez A. (2008) FEBS J. 275, 3207–3225 [DOI] [PubMed] [Google Scholar]
- 31. Fruchart-Gaillard C., Mourier G., Marquer C., Stura E., Birdsall N. J., Servent D. (2008) Mol. Pharmacol. 74, 1554–1563 [DOI] [PubMed] [Google Scholar]
- 32. Marquer C., Fruchart-Gaillard C., Mourier G., Grandjean O., Girard E., le Maire M., Brown S., Servent D. (2010) Biol. Cell 102, 409–420 [DOI] [PubMed] [Google Scholar]
- 33. Ehlert F. J. (1988) Mol. Pharmacol. 33, 187–194 [PubMed] [Google Scholar]
- 34. Lazareno S., Birdsall N. J. (1995) Mol. Pharmacol. 48, 362–378 [PubMed] [Google Scholar]
- 35. Schreiber G., Fersht A. R. (1995) J. Mol. Biol. 248, 478–486 [DOI] [PubMed] [Google Scholar]
- 36. Jorgensen W. L. (1981) J. Am. Chem. Soc. 103, 335–340 [Google Scholar]
- 37. Sali A., Blundell T. L. (1993) J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 38. Okada T., Sugihara M., Bondar A. N., Elstner M., Entel P., Buss V. (2004) J. Mol. Biol. 342, 571–583 [DOI] [PubMed] [Google Scholar]
- 39. Depristo M. A., de Bakker P. I., Johnson R. J., Blundell T. L. (2005) Structure 13, 1311–1319 [DOI] [PubMed] [Google Scholar]
- 40. Guilbert C., Perahia D., Mouawad L. (1995) Comput. Phys. Commun. 91, 263–273 [Google Scholar]
- 41. Fruchart-Gaillard C., Gilquin B., Antil-Delbeke S., Le Novère N., Tamiya T., Corringer P. J., Changeux J. P., Ménez A., Servent D. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 3216–3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gilquin B., Racapé J., Wrisch A., Visan V., Lecoq A., Grissmer S., Ménez A., Gasparini S. (2002) J. Biol. Chem. 277, 37406–37413 [DOI] [PubMed] [Google Scholar]
- 43. Gilquin B., Braud S., Eriksson M. A., Roux B., Bailey T. D., Priest B. T., Garcia M. L., Ménez A., Gasparini S. (2005) J. Biol. Chem. 280, 27093–27102 [DOI] [PubMed] [Google Scholar]
- 44. van Schaik R. C., Berendsen H. J., Torda A. E., van Gunsteren W. F. (1993) J. Mol. Biol. 234, 751–762 [DOI] [PubMed] [Google Scholar]
- 45. Brooks B. R., Bruccoleri R. E., Olafson B. D., States D. J., Swaminathan S., Karplus M. (1983) J. Comput. Chem. 4, 187–217 [Google Scholar]
- 46. Ryckaert J. P., Ciccotti G., Berendsen H. (1977) J. Comput. Chem. 23, 327–341 [Google Scholar]
- 47. André I., Strauss C. E., Kaplan D. B., Bradley P., Baker D. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 16148–16152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kukkonen A., Peräkylä M., Akerman K. E., Näsman J. (2004) J. Biol. Chem. 279, 50923–50929 [DOI] [PubMed] [Google Scholar]
- 49. Vaughan C. K., Buckle A. M., Fersht A. R. (1999) J. Mol. Biol. 286, 1487–1506 [DOI] [PubMed] [Google Scholar]
- 50. Gilquin B., Bourgoin M., Ménez R., Le Du M. H., Servent D., Zinn-Justin S., Ménez A. (2003) Protein Sci. 12, 266–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bonvin A. M. (2006) Curr. Opin. Struct. Biol. 16, 194–200 [DOI] [PubMed] [Google Scholar]
- 52. Grünberg R., Leckner J., Nilges M. (2004) Structure 12, 2125–2136 [DOI] [PubMed] [Google Scholar]
- 53. Durrant J. D., de Oliveira C. A., McCammon J. A. (2010) J. Mol. Recognit. 23, 173–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Oliveira L., Hulsen T., Lutje Hulsik D., Paiva A. C., Vriend G. (2004) FEBS Lett. 564, 269–273 [DOI] [PubMed] [Google Scholar]
- 55. Avlani V. A., Gregory K. J., Morton C. J., Parker M. W., Sexton P. M., Christopoulos A. (2007) J. Biol. Chem. 282, 25677–25686 [DOI] [PubMed] [Google Scholar]
- 56. Levy E. D., Boeri Erba E., Robinson C. V., Teichmann S. A. (2008) Nature 453, 1262–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Venkatakrishnan A. J., Levy E. D., Teichmann S. A. (2010) Biochem. Soc. Trans. 38, 879–882 [DOI] [PubMed] [Google Scholar]
- 58. van Dijk A. D., Boelens R., Bonvin A. M. (2005) FEBS J. 272, 293–312 [DOI] [PubMed] [Google Scholar]
- 59. van Dijk A. D., de Vries S. J., Dominguez C., Chen H., Zhou H. X., Bonvin A. M. (2005) Proteins 60, 232–238 [DOI] [PubMed] [Google Scholar]
- 60. Ranganathan R., Lewis J. H., MacKinnon R. (1996) Neuron 16, 131–139 [DOI] [PubMed] [Google Scholar]
- 61. Roisman L. C., Piehler J., Trosset J. Y., Scheraga H. A., Schreiber G. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 13231–13236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Verdier L., Al-Sabi A., Rivier J. E., Olivera B. M., Terlau H., Carlomagno T. (2005) J. Biol. Chem. 280, 21246–21255 [DOI] [PubMed] [Google Scholar]
- 63. Zvi A., Tugarinov V., Faiman G. A., Horovitz A., Anglister J. (2000) Eur. J. Biochem. 267, 767–779 [DOI] [PubMed] [Google Scholar]
- 64. Imredy J. P., MacKinnon R. (2000) J. Mol. Biol. 296, 1283–1294 [DOI] [PubMed] [Google Scholar]
- 65. Harel M., Cohen M., Schreiber G. (2007) J. Mol. Biol. 371, 180–196 [DOI] [PubMed] [Google Scholar]
- 66. Cohavi O., Tobi D., Schreiber G. (2009) J. Mol. Biol. 390, 503–515 [DOI] [PubMed] [Google Scholar]
- 67. Bourne Y., Talley T. T., Hansen S. B., Taylor P., Marchot P. (2005) EMBO J. 24, 1512–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dutertre S., Lewis R. J. (2006) Biochem. Pharmacol. 72, 661–670 [DOI] [PubMed] [Google Scholar]
- 69. Karaca E., Melquiond A. S., de Vries S. J., Kastritis P. L., Bonvin A. M. (2010) Mol. Cell. Proteomics 9, 1784–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Vroling B., Sanders M., Baakman C., Borrmann A., Verhoeven S., Klomp J., Oliveira L., de Vlieg J., Vriend G. (2011) Nucleic Acids Res. 39, D309–D319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bahadur R. P., Chakrabarti P., Rodier F., Janin J. (2004) J. Mol. Biol. 336, 943–955 [DOI] [PubMed] [Google Scholar]
- 72. Dayhoff J. E., Shoemaker B. A., Bryant S. H., Panchenko A. R. (2010) J. Mol. Biol. 395, 860–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chakrabarti P., Janin J. (2002) Proteins 47, 334–343 [DOI] [PubMed] [Google Scholar]
- 74. Shuker S. B., Hajduk P. J., Meadows R. P., Fesik S. W. (1996) Science 274, 1531–1534 [DOI] [PubMed] [Google Scholar]
- 75. Chung F., Tisné C., Lecourt T., Seijo B., Dardel F., Micouin L. (2009) Chemistry 15, 7109–7116 [DOI] [PubMed] [Google Scholar]
- 76. Schulz M. N., Hubbard R. E. (2009) Curr. Opin. Pharmacol. 9, 615–621 [DOI] [PubMed] [Google Scholar]
- 77. Murray C. W., Blundell T. L. (2010) Curr. Opin. Struct. Biol. 20, 497–507 [DOI] [PubMed] [Google Scholar]
- 78. Tränkle C., Weyand O., Voigtländer U., Mynett A., Lazareno S., Birdsall N. J., Mohr K. (2003) Mol. Pharmacol. 64, 180–190 [DOI] [PubMed] [Google Scholar]
- 79. Valant C., Gregory K. J., Hall N. E., Scammells P. J., Lew M. J., Sexton P. M., Christopoulos A. (2008) J. Biol. Chem. 283, 29312–29321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Valant C., Sexton P. M., Christopoulos A. (2009) Mol. Interv. 9, 125–135 [DOI] [PubMed] [Google Scholar]
- 81. Tränkle C., Dittmann A., Schulz U., Weyand O., Buller S., Jöhren K., Heller E., Birdsall N. J., Holzgrabe U., Ellis J., Höltje H. D., Mohr K. (2005) Mol. Pharmacol. 68, 1597–1610 [DOI] [PubMed] [Google Scholar]
- 82. Voigtländer U., Jöhren K., Mohr M., Raasch A., Tränkle C., Buller S., Ellis J., Höltje H. D., Mohr K. (2003) Mol. Pharmacol. 64, 21–31 [DOI] [PubMed] [Google Scholar]
- 83. Prilla S., Schrobang J., Ellis J., Höltje H. D., Mohr K. (2006) Mol. Pharmacol. 70, 181–193 [DOI] [PubMed] [Google Scholar]
- 84. Jäger D., Schmalenbach C., Prilla S., Schrobang J., Kebig A., Sennwitz M., Heller E., Tränkle C., Holzgrabe U., Höltje H. D., Mohr K. (2007) J. Biol. Chem. 282, 34968–34976 [DOI] [PubMed] [Google Scholar]
- 85. Szidonya L., Cserzo M., Hunyady L. (2008) J. Endocrinol. 196, 435–453 [DOI] [PubMed] [Google Scholar]
- 86. Simpson L. M., Taddese B., Wall I. D., Reynolds C. A. (2010) Curr. Opin. Pharmacol. 10, 30–37 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.