

Abstract
The uncoupling proteins UCP2 and UCP3 have been postulated to catalyze Ca2+ entry across the inner membrane of mitochondria, but this proposal is disputed, and other, unrelated proteins have since been identified as the mitochondrial Ca2+ uniporter. To clarify the role of UCPs in mitochondrial Ca2+ handling, we down-regulated the expression of the only uncoupling protein of HeLa cells, UCP3, and measured Ca2+ and ATP levels in the cytosol and in organelles with genetically encoded probes. UCP3 silencing did not alter mitochondrial Ca2+ uptake in permeabilized cells. In intact cells, however, UCP3 depletion increased mitochondrial ATP production and strongly reduced the cytosolic and mitochondrial Ca2+ elevations evoked by histamine. The reduced Ca2+ elevations were due to inhibition of store-operated Ca2+ entry and reduced depletion of endoplasmic reticulum (ER) Ca2+ stores. UCP3 depletion accelerated the ER Ca2+ refilling kinetics, indicating that the activity of sarco/endoplasmic reticulum Ca2+ (SERCA) pumps was increased. Accordingly, SERCA inhibitors reversed the effects of UCP3 depletion on cytosolic, ER, and mitochondrial Ca2+ responses. Our results indicate that UCP3 is not a mitochondrial Ca2+ uniporter and that it instead negatively modulates the activity of SERCA by limiting mitochondrial ATP production. The effects of UCP3 on mitochondrial Ca2+ thus reflect metabolic alterations that impact on cellular Ca2+ homeostasis. The sensitivity of SERCA to mitochondrial ATP production suggests that mitochondria control the local ATP availability at ER Ca2+ uptake and release sites.
Keywords: ATP, Calcium ATPase, Calcium Imaging, Calcium Signaling, Calcium Transport, Mitochondria, Mitochondrial Calcium Uniporter, Uncoupling Proteins
Introduction
Mitochondria are multifunctional organelles that control the life and death of cells. Mitochondria are the site of oxidative phosphorylation and convert reducing equivalents into the ATP that cells use as energy source, release critical factors that induce the programmed cell death of apoptosis, and modulate cell signaling by capturing and subsequently releasing calcium ions. The ability of mitochondria to sequester Ca2+ ions released from the endoplasmic reticulum or entering across plasma membrane channels enables these organelles to shape cytosolic Ca2+ signals and to modulate the activity of membrane channels and transporters (1–3), whereas Ca2+ elevations within the matrix of mitochondria activate three enzymes of the tricarboxylic cycle, thereby boosting oxidative phosphorylation and ATP production. Calcium uptake and release by mitochondria are therefore essential for the patterning of cytosolic Ca2+ signals and for cells to decode Ca2+ signals as either metabolic or death signals. The importance of mitochondrial Ca2+ handling for cell physiology has fostered a long quest to identify the transport molecules that move Ca2+ in and out of mitochondria (4). This quest culminated in recent years with the report of several families of proteins that were proposed to participate in either mitochondrial Ca2+ uptake or mitochondrial Ca2+ extrusion: the uncoupling proteins 2 and 3 (UCP2 2 and UCP3), the leucine zipper EF-hand-containing transmembrane protein 1 (Letm1), the Na+/Ca2+ exchanger (NCLX), the stomatin-like protein 2 (SLP-2), and the mitochondrial calcium uptake protein 1 (MICU1).
Ca2+ enters mitochondria across a mitochondrial Ca2+ uniporter (MCU) that was characterized in 2004 at the electrophysiological level as a highly selective inward rectifying mitochondrial inner membrane Ca2+ channel, inhibited by ruthenium red and Ru360 (5). In 2007, UCP2 and UCP3 were proposed to be fundamental for the MCU (6), based on the altered mitochondrial Ca2+ elevations ([Ca2+]mit) of cells enriched or depleted of UCP2 or UCP3 and on the defective ruthenium red-sensitive Ca2+ uptake of liver mitochondria isolated from UCP2−/− mice. This proposal was subsequently refuted by a study showing that purified liver mitochondria from UCP2−/− and UCP3−/− mice take up calcium normally (7). In 2009, Letm1, a protein previously shown to catalyze mitochondrial K+/H+ exchange (8, 9), was shown to drive mitochondrial Ca2+ uptake by exchanging Ca2+ for H+ with a 1:1 stoichiometry (10). This stoichiometry, however, contradicts earlier studies in isolated mitochondria showing that Ca2+ enters mitochondria together with two positive charges and leaves the matrix in exchange for three protons (reviewed in Ref. 4). Letm1 is associated with Wolf-Hirschhorn syndrome, a severe human neurological disease characterized by mental retardation and seizures (11). In 2010, MICU1, an inner mitochondrial membrane protein with two Ca2+-binding EF-hand domains, was shown to be required for high capacity mitochondrial calcium uptake (12). MICU1 is a single-pass transmembrane protein unlikely to form a channel pore and was proposed to be the calcium sensing regulatory subunit of the MCU because mutations of its EF-hand domains abrogate mitochondrial Ca2+ uptake (13). Two proteins were reported to modulate Ca2+ extrusion from mitochondria. NCLX/NCKX6 was shown to catalyze CGP-37157-sensitive mitochondrial Na+/Ca2+ exchange (14), and SLP-2 was shown to negatively modulate the activity of the mitochondrial sodium-calcium exchanger (15).
The successive reports of proteins essential for mitochondrial Ca2+ uptake have generated some confusion about the identity and mode of operation of the MCU. The first proteins proposed to be fundamental for the MCU, UCP2 and UCP3 (16), are mitochondrial inner membrane protein paralogues of UCP1, the first UCP cloned. UCP1 is almost exclusively expressed in brown adipose tissue, where it catalyzes adaptive thermogenesis by acting as a mitochondrial proton channel, thereby uncoupling oxidative phosphorylation from ATP synthesis (17). Unlike UCP1, UCP2 and UCP3 are expressed in several tissues and are present in ectothermic fishes and plants that do not require thermogenesis. The basal H+ conductance is unchanged in mitochondria isolated from UCP2 or UCP3 null mice (18, 19), and the novel UCP homologues have been proposed to mediate regulated proton leak (20), to modulate insulin secretion (21), and to regulate the export of fatty acids and fatty acid peroxides(22, 23). The ability of UCP2/3 to act as proton leak channels seems strictly related to the presence of specific activators such as alkenyls produced by the peroxidation of membrane phospholipids (24, 25). Thus, there is a broad consensus that UCP2 and UCP3 do not mediate adaptive thermogenesis (25–27). Instead, the novel UCPs might act as mild uncouplers and protect against oxidative damage by attenuating the mitochondrial production of free radicals (28, 29).
The report that the amplitude of [Ca2+]mit elevations in intact cells directly correlates to the expression level of UCP2 and UCP3 (6) might explain the plethoric effects attributed to UCPs because alterations in [Ca2+]mit signals are expected to impact both on mitochondria bioenergetics and on cell signaling. Whether the altered [Ca2+]mit signals are due to defective mitochondrial Ca2+ uptake is unclear due to the conflicting reports obtained in isolated liver mitochondria from UCP2−/− null mice (6, 7). These opposite datasets could be reconciled by postulating a regulatory role for UCP2 and UCP3 in intact cells that would be lost in isolated mitochondria. Importantly, the altered [Ca2+]mit signals reported by Trenker et al. (6) in intact cells depleted or enriched of UCP2/3 have not been confirmed or disproved. To clarify the physiological role of UCPs in Ca2+ homeostasis, we used RNA interference to down-regulate UCP3, the only UCP isoform of HeLa cells (6), and measured the impact of UCP3 depletion on the Ca2+ and ATP levels in different cellular compartments.
EXPERIMENTAL PROCEDURES
Reagents
Minimal essential medium, fetal calf serum, penicillin, streptomycin, and Lipofectamine 2000 transfection reagent were obtained from Invitrogen. Histamine, thapsigargin (TG), antimycin A, and oligomycin were obtained from Sigma. 2,5-Di-t-butyl-1,4-benzohydroquinone (BHQ) were from Aldrich. Fura-2 AM was from Molecular Probes. YC3.6cyto (30), 4mtD3cpv (31), and D1ER (32) constructs were kindly provided by Drs. Amy Palmer and Roger Tsien (University of California, San Diego). Mitochondrial and cytosolic FRET-based ATP indicators (ATeam: adenosine 5′-triphosphate indicator based on the ϵ subunit for Analytical Measurements) were kindly provided by Drs. Hiromi Imamura (Japan Science and Technology Agency, Tokyo) and Hiroyuki Noji (Osaka University) (33). UCP2 and UCP3/mitochondria-targeted DsRed constructs were kindly provided by Dr. Wolfgang Graier (Medical University of Graz).
Cell Culture, Transfection, and RNA Interference
Culturing of HeLa cells has been previously described (34). For all experiments, cells were plated on 25-mm-diameter glass coverslips and co-transfected with the plasmid (1 μg/ml) coding for Ca2+ or pH probe and dsRNA using Lipofectamine 2000. All experiments were performed 2 days after transfection. For silencing UCP3 expression, commercial double-stranded RNAs from Qiagen were used (Hi PerFect-validated siRNA S102780771). For controls, cells were transfected with nonsilencing siRNA (AllStars negative control siRNA, Qiagen 1027281). Knockdown efficiency was verified by Western blots.
Cell Lysis, Mitochondrial Isolation, and Western Blotting
Whole cells were lysed for 30 min on ice in lysis buffer (25 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors (Roche Applied Science). The lysate was centrifuged at 14,000 × g for 20 min, and the protein content of the supernatant was determined using the BCA protein assay (Pierce). The mitochondrial fraction was obtained by differential centrifugation as reported previously (35). 50 μg of total protein (from cell lysate or isolated mitochondria) was loaded per lane of SDS-PAGE. For immunoblotting, proteins were transferred onto nitrocellulose membrane and probed with the following antibodies: anti-UCP3 (Santa Cruz Biotechnology, sc-7756) and anti-Tom20 (Santa Cruz Biotechnology, sc-11415), anti-SERCA2 (Thermo Scientific MA3-919), and anti-actin (Chemicon). Horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences) were used followed by detection by chemiluminescence (Amersham Biosciences).
Mitochondrial, Cytosolic, and Endoplasmic Reticulum Ca2+ Measurements
Experiments were performed in HEPES buffer solution containing (in mm): 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 20 Hepes, 10 glucose, pH 7.4, with NaOH at 37 °C. Ca2+-free solution contained 1 mm EGTA instead of CaCl2. Glass coverslips were inserted in a thermostatic chamber (Harvard Apparatus, Holliston, MA), and solutions were changed by hand. Cells were imaged on an Axiovert s100 TV using a ×40, 1.3 NA oil immersion objective (Carl Zeiss AG, Feldbach, Switzerland) and a cooled, 16-bit CCD back-illuminated frame transfer MicroMax camera (Roper Scientific, Trenton, NJ). Cytosolic Ca2+ was measured with fura-2 (2 μm, 0.2% dimethyl sulfoxide (DMSO), 0.01% Pluronic F127, Invitrogen) followed by 20 min of de-esterification before 10 min of equilibration on the heated stage or with YC3.6cyto. Mitochondrial and endoplasmic reticulum Ca2+ levels were measured with 4mtD3cpv and D1ER, respectively. For dual emission imaging of cameleon constructs (4mtD3cpv, YC3.6cyto, and D1ER), cells were excited at 430 nm through a 455DRLP dichroic and alternately imaged with 480AF30 and 535DF25 emission filters (Omega Optical). Fura-2 was measured simultaneously with 4mtD3cpv and was excited alternately at 340 and 380 nm through a 455DRLP dichroic and 535DF25 emission filter. Images were acquired every 2 s. Fluorescence ratios were calculated in MetaFluor 6.3 (Universal Imaging) and analyzed in Excel (Microsoft) and GraphPad Prism 4 (GraphPad). [Ca]ER was calculated from D1ER ratios using the equation
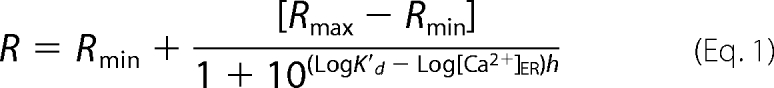 |
where Rmin and Rmax are the minimal and maximal ratio obtained at pCa >8 and <2, respectively, K′d is the apparent dissociation constant, and h is the Hill coefficient derived from the in situ Ca2+ titration of the D1ER probe in semipermeabilized cells, as described previously (36).
Mitochondrial and Cytosolic [ATP] Measurements
ATP imaging was performed on the epifluorescence system described above, with the genetically encoded ATP sensors ATeammito and ATeamcyto, for mitochondrial and cytosolic ATP, respectively. For the dual emission imaging of these two cameleon-based constructs, cells were excited and imaged as described previously for mitochondrial Ca2+ imaging. Images were acquired every 2 s. Fluorescence ratios were normalized on the minimum of fluorescence (Rmin), obtained after inhibition of glycolysis with 10 mm 2-deoxyglucose and in the presence of the inhibitor of mitochondrial ATP synthesis oligomycin A (10 μg/ml). Data were analyzed as described previously.
Permeabilized Cells
Cells were washed with high K+ intracellular buffer, containing (in mm) 110 KCl, 10 NaCl, 0.5 K2HPO4, 5 succinate, 10 mm HEPES (pH 7.0 at 37 °C), supplemented with 5 HEDTA or 1 EGTA. For permeabilization, 100 μm digitonin was added for 1 min, and the cells were then allowed to recover in intracellular buffer before the addition of CaCl2. Free [Ca2+] was calculated with the Maxchelator program.
Statistics
The significance of differences between means was established using the Student's t test for unpaired samples (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
RESULTS
Effect of UCP3 Depletion on Mitochondrial and Cytosolic Ca2+ Elevations
To clarify the role of UCPs in mitochondrial Ca2+ handling, we measured Ca2+ responses in the cytosol and in mitochondria in HeLa cells depleted or not of UCP3, the only novel UCP isoform expressed in this cell type (6). Consistent with earlier results (6), this treatment decreased UCP3 protein levels by 50% (supplemental Fig. S1). Cells were then transfected with a cameleon Ca2+ probe targeted to mitochondria (Fig. 1A) or to the cytosol (Fig. 1B) to enable [Ca2+]mit and [Ca2+]cyt recordings. As shown in Fig. 1A, UCP3 depletion strongly reduced the [Ca2+]mit elevations evoked by histamine, the peak [Ca2+]mit response being reduced by 57% and the integrated response being reduced by 62%. To exclude possible off-target effects linked to siRNA transfection, we overexpressed UCP3 protein and, in another set of experiments, attempted to rescue the defect caused by UCP3 siRNA by expressing another uncoupling protein, UCP2. As shown in supplemental Fig. S2A, UCP3 overexpression significantly increased the peak and integrated [Ca2+]mit responses evoked by histamine, mirroring the effects of UCP3 depletion, whereas expression of UCP2 restored normal [Ca2+]mit responses in cells depleted of UCP3 (supplemental Fig. S3A). These data indicate that the altered mitochondrial Ca2+ signals are indeed causally related to the changes in UCP3 levels. The UCP3-dependent Ca2+ defect was not restricted to mitochondria, however, because the cytosolic Ca2+ elevations were also reduced in cells depleted of UCP3, the peak and integrated [Ca2+]cyt responses being decreased by 25 and 44%, respectively (Fig. 1B). To confirm that the alteration in Ca2+ handling affected both compartments, we measured [Ca2+]cyt and [Ca2+]mit simultaneously instead of separately. In these conditions, both the cytosolic and the mitochondrial Ca2+ responses were blunted in cells depleted of UCP3 (supplemental Fig. S4). These data indicate that UCP3 knockdown has a global effect and not only blunts mitochondrial Ca2+ elevations but also reduces cytosolic Ca2+ elevations.
FIGURE 1.

Effect of UCP3 knockdown on mitochondrial and cytosolic Ca2+ elevations. A and B, HeLa cells were transiently co-transfected with the mitochondrial Ca2+ probe 4mtD3cpv (A) or the cytosolic Ca2+ probe YC3.6cyto (B) and with either scrambled siRNA (control siRNA) or UCP3-specific siRNA (UCP3 siRNA) for 48 h. A, left, averaged [Ca2+]mit recordings in HeLa cells stimulated with 100 μm histamine. Right, statistical evaluation of the UCP3 siRNA effects on the amplitude of the [Ca2+]mit signal evoked by histamine (upper panel) and on the integrated [Ca2+]mit response (lower panel). Bars are mean ± S.E. of 80 (n = 7) and 101 cells (n = 7) for Ctrl and UCP3 siRNA, respectively. AUC, area under the curve. B, left, averaged [Ca2+]cyto recordings in HeLa cells stimulated with 100 μm histamine. Right, statistical evaluation of the UCP3 siRNA effects on the amplitude of the [Ca2+]cyto signal evoked by histamine (upper panel) and on the integrated [Ca2+]cyto response (lower panel). Bars are mean ± S.E. of 40 (n = 3) and 37 cells (n = 3) for Ctrl and UCP3 siRNA, respectively. ***, p < 0.001.
Effect of UCP3 Depletion on Agonist-evoked Ca2+ Release and Influx
The novel UCPs have been recently proposed to specifically mediate the mitochondrial uptake of Ca2+ released from the ER, but not of the Ca2+ entering across SOCE channels (37). To verify this possibility, we separated the agonist-evoked Ca2+ response into its release and influx components. Cells were stimulated with histamine in Ca2+-free conditions to mobilize Ca2+ from stores, and Ca2+ was subsequently readmitted to promote Ca2+ entry. As shown in Fig. 2, the [Ca2+]cyt and [Ca2+]mit elevations caused by Ca2+ release from stores were slightly but significantly decreased. This inhibition, however, was much smaller than the inhibition observed in Ca2+-containing medium (Fig. 1). Unexpectedly, the [Ca2+]cyt elevations evoked by Ca2+ readmission were severely blunted in cells depleted of UCP3, the integrated [Ca2+]cyt response being reduced by 82% (Fig. 2A). An opposite effect was obtained by overexpressing UCP3, the [Ca2+]cyt responses increasing slightly (14%) during Ca2+ release from stores and markedly (75%) during Ca2+ influx (supplemental Fig. S2B). Moreover, UCP2 expression restored a normal Ca2+ influx in UCP3-depleted cells (supplemental Fig. S3B), confirming that this defect was due to the depletion of an uncoupling protein. As reported previously (38), the [Ca2+]mit elevations evoked by this Ca2+ readmission protocol were barely detectable and were not altered by UCP3 depletion (Fig. 2B).
FIGURE 2.

Effect of UCP3 knockdown on Ca2+ release and influx. A and B, HeLa cells were transiently co-transfected with the cytosolic Ca2+ probe YC3.6cyto (A) or the mitochondrial Ca2+ probe 4mtD3cpv (B) and with Ctrl (scramble) siRNA or the UCP3 siRNA for 48 h, washed, and stimulated with 100 μm histamine in Ca2+-free medium to deplete intracellular Ca2+ stores. Then Ca2+ was readmitted to monitor Ca2+ influx from plasma membrane. A, average of cytosolic Ca2+ responses (left) elicited by histamine and by Ca2+ readmission. Right, statistical evaluation of the UCP3 siRNA on the integrated [Ca2+]cyto response during release from ER and during Ca2+ readmission. Bars are mean ± S.E. of 84 (n = 9) and 86 cells (n = 8) for Ctrl and UCP3 siRNA, respectively. AUC, area under the curve. B, average of mitochondrial Ca2+responses elicited by histamine and Ca2+ readmission (left) and related statistical evaluation (right) on integrated [Ca2+]cyto response during Ca2+ release and influx. Bars are mean ± S.E. of 77 (n = 8) and 59 cells(n = 7) for Ctrl and UCP3 siRNA, respectively. *, p < 0.05; ***, p < 0.001. NS, not significant.
These observations indicate that UCP3 depletion slightly impairs Ca2+ release from stores and, surprisingly, strongly inhibits the cytosolic Ca2+ elevations caused by Ca2+ readmission to cells with depleted Ca2+ stores. The primary effect of UCP3 depletion thus appears to be a reduced entry of Ca2+ across SOCE channels.
Effect of UCP3 Depletion on ER Ca2+ Release and Refilling
SOCE channels are activated by the depletion of ER Ca2+ stores, which induces the translocation of the ER-resident Ca2+ sensor STIM1 to the plasma membrane, where it binds and activates the Orai and Transient Receptor Potential channels (39). The reduced Ca2+ entry of UCP3 knockdown cells suggested that the SOCE machinery was inhibited, prompting us to study the filling state of ER Ca2+ stores. We therefore measured [Ca2+]ER at rest and during store depletion with the ER-targeted cameleon Ca2+ indicator D1ER. As shown in Fig. 3A, D1ER basal ratio fluorescence levels were identical in control and UCP3-depleted cells, the basal [Ca2+]ER levels averaging 520 μm in both conditions (supplemental Fig. S5, A and B). Upon stimulation with histamine, however, [Ca2+]ER decreased much more slowly in cells depleted of UCP3, the half-time increasing from 72 to 269 s (Fig. 3B, inset). This four times slower [Ca2+]ER decrease was surprising considering that UCP3 knockdown cells released Ca2+ efficiently when stimulated with agonists (Fig. 2A). Reduced depletion of ER Ca2+ stores could reflect either a reduced passive permeability to Ca2+ or an increased Ca2+ pumping into the ER. To distinguish between these possibilities, we inhibited SERCA pumps with TG to reveal the intrinsic Ca2+ leak rates of the ER. As shown in Fig. 3C, the addition of TG elicited identical [Ca2+]ER decreases in control and UCP3 knockdown cells, indicating that the passive Ca2+ permeability of the ER was not affected by the depletion of UCP3. The addition of TG together with histamine accelerated the kinetics of [Ca2+]ER decrease by ∼2-fold in UCP3-depleted cells, but these cells still released Ca2+ more slowly than control cells (supplemental Fig. S5C). Because [Ca2+]ER decreased significantly ∼40 s after TG addition (supplemental Fig. S5D), we then added TG 40 s before histamine to ensure full inhibition of SERCA. In these conditions, the kinetics of [Ca2+]ER decrease evoked by histamine were similar in control and UCP3-depleted cells (Fig. 3D). These data indicate that the both the passive and the active (i.e. InsP3R-mediated) Ca2+ permeability of the ER are not affected by the depletion of UCP3. Similar effects were observed with the reversible SERCA inhibitor BHQ (not shown), confirming the involvement of SERCA. Of note, BHQ required more time than TG to completely inhibit SERCA pumps (supplemental Fig. S5E; see under “Discussion” to compare our results with recent data of the Graier group (37)). The restoration of normal ER Ca2+ release in cells depleted of UCP3 by SERCA inhibitors suggested that the activity of SERCA was increased in these cells. The expression levels of SERCA2b assessed by Western blot (Fig. 4A) and real-time quantitative PCR (not shown) were not influenced by UCP3 depletion. To directly estimate the activity of SERCA, intact cells were transiently treated with the reversible inhibitor BHQ to deplete ER Ca2+ stores, and Ca2+ was then readmitted to promote store refilling. As shown in Fig. 4B, the kinetics of ER refilling were faster in cells depleted of UCP3, the time required to reach half-maximal refilling being increased by 14 s, confirming that SERCA pumps were more active in these cells. This difference was not observed in permeabilized cells, however, the kinetics of ER refilling being identical in this condition (Fig. 4C). Our [Ca2+]ER measurements thus indicate that the activity of SERCA pumps is increased in intact cells depleted of UCP3.
FIGURE 3.

Effect of UCP3 knockdown on ER Ca2+ release. HeLa cells were transiently co-transfected with the ER Ca2+ probe D1ER and with the Ctrl (scramble) siRNA or the UCP3 siRNA for 48 h and washed, and Ca2+ responses were measured. A, resting D1ER ratio values of 73 (n = 8) and 77 (n = 8) cells, transfected with Ctrl and UCP3 siRNA, respectively. NS, not stimulated. B, averaged [Ca2+]ER recordings of HeLa cells stimulated with 100 μm histamine in Ca2+-free medium and related statistical evaluation (inset) of the UCP3 siRNA effects on the kinetics of Ca2+ release. For the latter statistics, the D1ER responses were fitted with a one-phase exponential decay function to extract the half-time. Bars are mean ± S.E. of 73 (n = 8) and 77 cells (n = 8) for Ctrl and UCP3 siRNA, respectively. ***, p < 0.001. C, the same as in B, but stimulating the cells with 1 μm TG. Bars are mean ± S.E. of 69 (n = 8) and 74 cells (n = 9) for Ctrl and UCP3 siRNA, respectively. D, the same as in B, but preincubating the cells with 1 μm TG 40 s before histamine stimulation. Bars are mean ± S.E. of 59 (n = 5) and 54 cells (n = 5) for Ctrl and UCP3 siRNA, respectively.
FIGURE 4.

Effect of UCP3 knockdown on ER Ca2+ refilling in intact and permeabilized cells. A, SERCA2 immunoblot of HeLa cells transfected with Ctrl or UCP3 siRNA for 48 h. 50 μg/lane of protein from cell extracts was analyzed, using actin as loading control. B, left, averaged [Ca2+]ER recordings of intact HeLa cells during ER Ca2+ refilling. After 15 μm BHQ induced Ca2+ release in Ca2+-free medium, cells were washed, and Ca2+ was then added to assess the kinetics of store refilling. Data were fitted with the sigmoidal equation to extract the EC50 (right), and they are mean ± S.E. of 40 (n = 5) and 30 cells (n = 4) for Ctrl and UCP3 siRNA, respectively. **, p < 0.01. C, left, averaged [Ca2+]ER recordings during ER Ca2+ release and refilling in permeabilized cells. The K+-rich intracellular buffer was supplemented with 1 mm Mg-ATP and 1 mm MgCl2 to allow ER Ca2+ refilling. Where indicated, 100 μm digitonin (dig), 15 μm BHQ, and 100 nm free Ca2+ were added. Right, statistical evaluation of the ER Ca2+ refilling kinetics. EC50 was determined as described for B. Data are mean ± S.E. of 47 (n = 3) and 33 cells (n = 3) for Ctrl and UCP3 siRNA, respectively. NS, not stimulated.
Effect of SERCA Inhibition on Mitochondrial Ca2+ Uptake in UCP3-depleted Cells
The effects of SERCA inhibitors on [Ca2+]ER handling prompted us to investigate whether SERCA inhibition could normalize mitochondrial Ca2+ uptake in cells depleted of UCP3. To test this possibility, we measured the [Ca2+]mit responses evoked by Ca2+ release and by Ca2+ influx in cells treated with TG. As shown in Fig. 5A, the [Ca2+]mit responses evoked by Ca2+ release from stores and by SOCE were identical in control and UCP3-depleted cells treated with the SERCA inhibitor. Parallel [Ca2+]cyto measurements confirmed that both the release and the influx components were normal in UCP3-depleted cells treated with TG (supplemental Fig. S6), whereas the SOCE component remained blunted after washout of the reversible SERCA inhibitor BHQ (supplemental Fig. S7). These results confirm that UCP3 modulates cellular Ca2+ signals by interfering with the activity of SERCA.
FIGURE 5.

Effect of SERCA inhibition or cell permeabilization on mitochondrial Ca2+ elevation. A, the same protocols and conditions as in Fig. 2 were used to deplete Ca2+ stores and to monitor Ca2+ release and then the influx component in mitochondria, but in the presence of the SERCA pump inhibitor 1 μm TG. Histamine was 100 μm. Left, average of [Ca2+]mit recordings from Ctrl (scramble) siRNA or the UCP3 siRNA cells. Right, statistical evaluation of the UCP3 knockdown on the integrated [Ca2+]mit responses evoked by histamine or after Ca2+ readmission, from data shown in the left panel. Bars are mean ± S.E. of 51 (n = 5) and 45 cells (n = 4) for Ctrl and UCP3 siRNA, respectively. NS, not stimulated. AUC, area under the curve. B, Ru360-sensitive mitochondrial Ca2+-uptake in permeabilized cells, in ATP-depleted medium. HeLa cells were transiently co-transfected with the mitochondrial calcium probe 4mtD3cpv and the indicated siRNAs. After permeabilization with digitonin, [Ca2+]mit was measured in intracellular buffer. Left, original [Ca2+]mit recordings of permeabilized HeLa cells during the addition of 3.5 μm free Ca2+ in the presence of or after wash-out of the mitochondrial Ca2+ uniporter inhibitor Ru360 (10 μm). Right, statistical evaluation of UCP3 siRNA effects on the slope of Ca2+ elevation. For the latter statistics, the Ca2+ responses for each trace were fitted with a linear function, and the Ru360-dependent slope was subtracted from the following one in the absence of the inhibitor. Bars are mean ± S.E. of 79 (n = 6) and 67 (n = 6) cells for Ctrl and UCP3 siRNA, respectively.
To verify that UCP3 depletion did not alter mitochondrial Ca2+ uptake, we measured [Ca2+]mit responses evoked by the addition of Ca2+ to permeabilized cells. As shown in Fig. 5B, the addition of 3.5 μm free Ca2+ to permeabilized cells evoked robust [Ca2+]mit elevations that were observed irrespective of the depletion of UCP3. The [Ca2+]mit elevations were prevented by the MCU-inhibitor Ru360 both in control and in UCP3-depleted cells, and the Ru360-sensitive mitochondrial Ca2+ uptake was not significantly different between the two conditions (Fig. 5B, right panel). These data indicate that UCP3 does not contribute to mitochondrial Ca2+ uptake.
Effect of UCP3 Depletion on Mitochondrial ATP Production
Because UCPs have been proposed to uncouple oxidative phosphorylation from ATP production, we postulated that mitochondria could generate more ATP at low UCP3 levels, thereby favoring the activity of nearby SERCA. Such a local control of SERCA pumps by mitochondrial ATP production was previously proposed (40), and the interaction between SERCA and mitochondria was postulated from the local competition between SERCA and mitochondria for the uptake of Ca2+ (41). To test whether mitochondria produced more ATP in UCP3-depleted cells, we prevented mitochondrial ATP production by inhibiting the ATP synthase with oligomycin or the respiratory chain with antimycin A and measured the [Ca2+]cyto influx component evoked by readmission of Ca2+ to cells stimulated with agonists, as in Fig. 2A. We used this experimental paradigm as agonist-evoked Ca2+ entry was the component most sensitive to UCP3 depletion. Unfortunately, inhibition of mitochondrial ATP production severely decreased agonist-evoked Ca2+ entry, an effect that had been previously reported (38). Interestingly, the effects of UCP3 depletion disappeared in the presence of the inhibitors (supplemental Fig. S8), consistent with a role of mitochondrial ATP production in the alterations in Ca2+ handling.
To confirm that the effects of UCP3 depletion are mediated by the availability of ATP, we measured changes in ATP levels occurring in individual living cells with the genetically encoded ATP-sensitive probes ATeam (33). These FRET-based probes were efficiently targeted to the cytosol and the mitochondrial matrix (Fig. 6B), enabling real-time monitoring of changes in [ATP]cyt and [ATP]mit by ratiometric measurements. As shown in Fig. 6A, basal [ATP]mit levels were slightly elevated in cells depleted of UCP3 and increased further following exposure to histamine (filled circles). In contrast, the addition of histamine had little effect on [ATP]mit in control cells (open circles). We then sequentially added oligomycin and 2-deoxyglucose to assess the contribution of the mitochondrial ATP synthase and of glycolysis to the [ATP]mit and[ATP]cyt signal. Oligomycin had a relatively modest effect on [ATP]cyt levels, indicating that most of the cytosolic ATP is of glycolytic origin in HeLa cells, consistent with the earlier ATeam study (33). In contrast, oligomycin significantly decreased [ATP]mit levels (Fig. 6A), indicating that the activity of the ATP synthase was detectable with the mitochondrial probe. Strikingly, the amplitude of the oligomycin-sensitive [ATP]mit component was nearly doubled in UCP3-depleted cells (Fig. 6B, bottom left panel). This effect was only observed with the mitochondria-targeted probe and not with the cytosolic probe (Fig. 6B, bottom right panel). The residual [ATP]mit levels of control and UCP3-depleted cells treated with oligomycin were similar and decreased with identical kinetics upon the addition of 2-deoxyglucose, indicating that the glycolytic activity was not affected by UCP3 depletion. The higher mitochondrial ATP concentration observed in UCP3-depleted cells stimulated with histamine therefore likely reflected the higher activity of the ATP synthase in these cells and indicates that mitochondria produce more ATP in cells depleted of UCP3. ATeam measurements in cells overexpressing UCP3 revealed that these cells produced less mitochondrial ATP (supplemental Fig. S2C), confirming the inverse correlation between UCP3 levels and mitochondrial ATP production.
FIGURE 6.
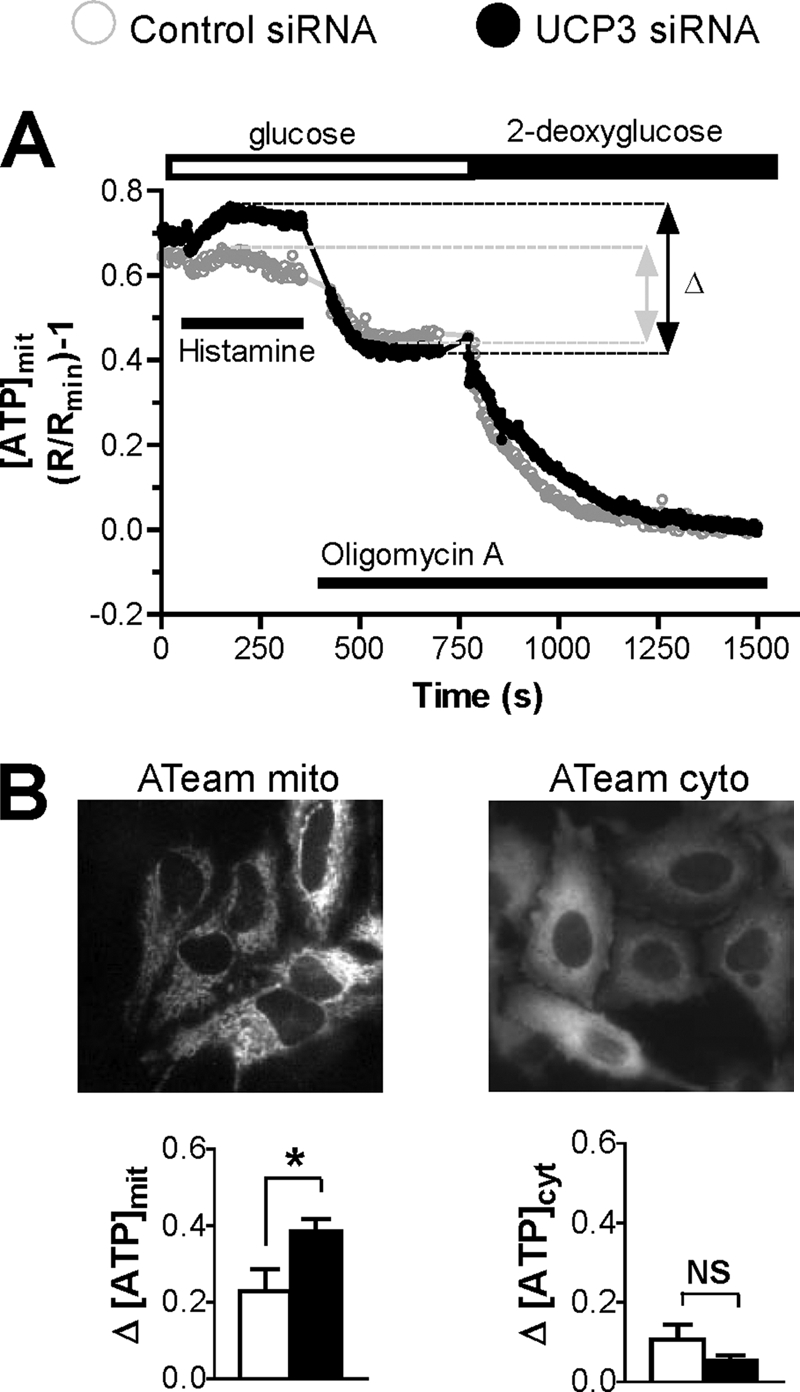
Effect of UCP3 knockdown on mitochondrial and cytosolic [ATP]. HeLa cells were transiently transfected with the mitochondrial or the cytosolic ATP probes ATeammito or ATeamcyto, respectively, together with the indicated siRNA for 48 h. A, averaged [ATP]mit changes elicited by 100 μm histamine and inhibition of mitochondrial ATP synthesis with oligomycin A (10 μg/ml) in cells perfused with 10 mm glucose as metabolic substrate and after inhibition of glycolysis with 10 mm 2-deoxyglucose. B, upper panels, mitochondrial (left) and cytosolic (right) ATeam signals recorded at 535 nm. Left lower panel, statistical evaluation of the histamine-induced, oligomycin A-sensitive [ATP]mit changes. Bars are mean ± S.E. of n = 4 (44 cells) and n = 5 (59 cells) for Ctrl and UCP3 siRNA, respectively. Right lower panel, statistical evaluation of the histamine-induced, oligomycin A-sensitive [ATP]cyt changes. Bars are mean ± S.E. of n = 5 (60 cells) and n = 5 (64 cells) for Ctrl and UCP3 siRNA, respectively. [ATP]cyt changes were recorded by applying the same protocol as in A. *, p < 0.05. NS, not stimulated.
DISCUSSION
There has been some confusion lately as to the identity of the long sought after Ca2+ uniporter of mitochondria because three distinct families of proteins were proposed to contribute to mitochondrial Ca2+ uptake: UCP2 and UCP3 (37), Letm1 (10), and MICU1 (12). MICU1 is a single-pass transmembrane protein unlikely to form a pore that has been proposed to modulate Ca2+ uniport activity (12). Letm1 was shown to catalyze electrogenic 1:1 Ca2+/H+ exchange, but this contradicts earlier studies showing that Letm1 drives electroneutral K+/H+ exchange (8, 42). The claim that UCP2 and UCP3 contribute to mitochondrial Ca2+ uptake was disputed early on because mice lacking the UCP2 and UCP3 isoforms have robust uniporter activity (7). In follow-up studies, UCPs were shown to facilitate the mitochondrial uptake of the Ca2+ released from the ER, but not of the Ca2+ entering across SOCE channels (37). Here, we show that UCP3 does not contribute to mitochondrial Ca2+ uptake but indirectly alters cellular Ca2+ homeostasis by modulating mitochondrial ATP production. Our data show that UCP3 silencing boosts mitochondrial ATP production and enhances the Ca2+ pumping activity of SERCA. The ensuing alteration of cellular Ca2+ handling mimics reduced mitochondrial Ca2+ uptake.
Consistent with the original finding of Trenker et al. (6), we observed that UCP3 depletion reduced [Ca2+]mit elevations evoked by histamine in intact cells, an effect most pronounced in Ca2+-containing medium (Figs. 1 and 2). However, this phenotype was not due to reduced uniport activity because UCP3 depletion did not reduce [Ca2+]mit signals in permeabilized cells and in cells treated with inhibitors of SERCA pumps or of mitochondrial ATP production (Figs. 5 and 6). Instead, UCP3 depletion was associated with decreased Ca2+ entry (Figs. 1B and 2A), blunted ER Ca2+ depletion (Fig. 3B), and increased mitochondrial ATP production (Fig. 6). The cytosolic and ER calcium defects also disappeared in cells treated with SERCA inhibitors (Figs. 3 and 5). These observations indicate that UCP3 does not mediate mitochondrial Ca2+ uptake but has a global effect on cellular Ca2+ homeostasis that requires functional SERCA and normal mitochondrial ATP production.
These data clarify the role of UCP3 in Ca2+ homeostasis and confirm several earlier observations, notably 1) the reduced [Ca2+]mit elevations in UCP3-depleted cells exposed to agonists (Fig. 2C of Ref. 43), Fig. 1C of Ref. 44, and Fig. 2A of Ref. 37); and 2) the lack of UCP3 effects on [Ca2+]mit elevations evoked by Ca2+ readmission to cells with inhibited SERCA (Figs. 2B and 3B of Ref. 37). These observations were interpreted as evidence that UCP3 is required for mitochondrial Ca2+ uptake during the rapid release of Ca2+ from ER stores, but not during the slow entry of Ca2+ across SOCE channels. Our simpler interpretation is that the effects of UCP3 depletion in intact cells are due to alterations in the rates of SERCA pumping because the differences disappear entirely in cells treated with SERCA inhibitors. Thus, in intact cells, the mitochondrial Ca2+ uptake rates indeed correlate with the expression levels of UCP2/3, as reported previously (6), but this effect is indirect and mediated by SERCA.
The requirement for active SERCA appears at odds with the blunted [Ca2+]mit responses reported in cells co-stimulated with histamine and the SERCA inhibitor BHQ (Figs. 2A, 3A, and 4A of Ref. 37). However, our data show that effective SERCA inhibition requires at least 30 s of preincubation with TG and that BHQ releases Ca2+ more slowly than TG (supplemental Fig. S3). Thus, SERCA pumps were likely not fully inhibited in these experiments, and the UCP3 effects could still reflect alterations in the rates of SERCA pumping. Our data from permeabilized cells also diverge from the data of Trenker et al. 6, who reported a reduced Ca2+ uptake in isolated liver mitochondria from UCP2−/− mice (Fig. 3 of Ref. 6). In our hands, the Ru360-sensitive component of mitochondrial Ca2+ uptake was not affected by UCP3 depletion in permeabilized HeLa cells (Fig. 5B). These data are consistent with the normal mitochondrial Ca2+ uptake reported by Brookes et al. (7) in purified heart and liver mitochondria treated with UCP inhibitors and in skeletal muscle mitochondria isolated from UCP2−/− and UCP3−/− mice (Figs. 1 and 2 of Ref. 44). To account for these discrepancies, Trenker et al. (44) argued that the failure of Brookes et al. (7) to detect UCP3 effects in permeabilized cells was due to the harsher conditions of their purification procedure that used differential centrifugation instead of density gradients. Size distribution analysis revealed that mitochondria isolated by density gradient had a larger diameter (0.75/1.00 μm versus 0.25/0.50 μm for differential centrifugation, supplemental Fig. S1 of Ref. 44). This increase in mitochondrial size could reflect a different amount of mitochondria-associated ER membranes, ER-derived structures that co-purify with mitochondria during isolation (45). Mitochondria-associated ER membranes are enriched in ERp57, a protein that directly interacts with SERCA2b and modulates its activity (46). The presence of functional SERCAs on mitochondria-associated ER membranes co-purified with mitochondria isolated by density gradient might explain the effects of UCP2 ablation in this mitochondrial preparation. Finally, another discrepancy is the normal rates of SERCA pumping reported in cells overexpressing UCP2 or UCP3 in the original study by Trenker et al. (6) (supplemental Fig. S1g of Ref. 9) and in a more recent publication (Fig. 2C of Ref. 37). In our study, increased SERCA pumping was evident in cells depleted of UCP3 (Fig. 4A), and the rates of [Ca2+]ER decrease were severely reduced during histamine stimulation (Fig. 3B). The increased activity of SERCA can account for the reduced influx of Ca2+ observed in cells depleted of UCP3 and treated with agonists (Fig. 2A), a phenotype that was not reported previously. Trenker et al. (6) did not attempt to measure [Ca2+]ER changes in cells depleted of UCP2 or UCP3; thus, our data cannot be readily compared with theirs. However, based on the effects of UCP3 depletion, we would predict that UCP3 overexpression would decrease SERCA pumping.
Several mechanisms could explain the sensitivity of SERCA to changes in UCP3 levels. First, mitochondria could produce more reactive oxygen species in response to UCP3 depletion because an increase in UCP3 levels is known to lower reactive oxygen species production (47). Because increased reactive oxygen species levels decrease SERCA activity, however (48, 49), UCP3 knockdown should decrease, rather than increase, SERCA activity. Alterations in reactive oxygen species levels are thus unlikely to account for the UCP3 effects that we report here. Second, UCP3 depletion could increase the number of ER-mitochondria contact sites or reduce the distance between mitochondria and the ER, enabling mitochondria to supply more ATP to nearby SERCA. Increased ER-mitochondria contact, however, should increase, rather than decrease, the efficiency of ER-mitochondria Ca2+ transmission. Finally, mitochondria could produce more ATP at low UCP3 levels, thereby fueling the activity of SERCA more efficiently. Our data indicate that this last mechanism is likely to occur because 1) the mitochondrial ATP production, measured with a genetically encoded ATP-sensitive indicator, was significantly increased following stimulation of cells with Ca2+-mobilizing agonists; and 2) the UCP3 effects disappeared in cells treated with inhibitors of mitochondrial respiration and of the ATP synthase. UCP2/3 are mitochondrial inner membrane proteins whose first postulated function is to uncouple oxidative phosphorylation from ATP production (20). Although this uncoupling function is also disputed (16), the simplest explanation for the increased production of mitochondrial ATP observed in UCP3-depleted cells is that the energy stored in the proton-motive force is used more efficiently by the ATP synthase. Thus, a mild uncoupling function of UCP3 could account for the whole phenotype that we report here in UCP3-depleted cells, including 1) the increased production of mitochondrial ATP, 2) increased activity of SERCA, 3) decreased store-operated Ca2+ entry (due to reduced store Ca2+ depletion), and finally, 4) reduced mitochondrial Ca2+ uptake due to sequestration of the released Ca2+ by SERCA and (predominantly) to the blunted SOCE.
Our data highlight the pitfalls of interpreting alterations in [Ca2+]mit signals occurring in intact cells. In intact cells, mitochondria are embedded in the ER, and the changes in matrix [Ca2+] are influenced by the activity of numerous Ca2+ transport proteins, including Ca2+ release and influx channels (InsP3R, Orai), Ca2+ pumps (SERCA, secretory pathway Ca2+-ATPase (SPCA), and plasma membrane Ca2+-ATPase (PMCA)), and Ca2+ buffering proteins, which are often overlooked. The simplistic interpretation that a reduced mitochondrial Ca2+ signal is due to reduced mitochondrial Ca2+ uptake disregards the complexity of the Ca2+ signaling circuitry in intact cells, where Ca2+ release and influx occur concomitantly with Ca2+ uptake catalyzed by SERCA and Ca2+ extrusion catalyzed by plasma membrane Ca2+-ATPase. Our data show that a phenotype typical of defective mitochondrial Ca2+ uptake can in fact be explained by a metabolic alteration that changes the activity of Ca2+ pumps.
Supplementary Material
Acknowledgments
We thank Drs. R. Y. Tsien and A. Palmer for providing the cameleon constructs, Drs. H. Imamura and H. Noji for providing the ATeam probes, Dr. Wolfgang Graier for UCPs constructs, and Ariane Widmer for expert technical assistance.
Note Added in Proof
A protein with all characteristic features of the Ca2+ uniporter of mitochondria was recently identified by two independent groups (50, 51). The purified MCU protein exhibited Ca2+ channel activity in lipid bilayers (50), confirming that UCPs are not essential for mitochondrial Ca2+ uptake. The effects of UCPs on mitochondrial Ca2+ handling that were previously reported therefore likely reflect the metabolic alterations described in this study.
This work was supported by the Swiss National Foundation Grant 31-068317 (to N. D.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S8.
- UCP
- uncoupling protein
- ER
- endoplasmic reticulum
- SERCA
- sarco/endoplasmic reticulum Ca2+ -ATPase
- MCU
- mitochondrial Ca2+ uniporter
- SOCE
- store-operated Ca2+ entry
- BHQ
- 2,5-di-t-butyl-1,4-benzohydroquinone
- [Ca2+]cyto
- cytosolic [Ca2+]
- [Ca2+]ER
- endoplasmic reticulum [Ca2+]
- [Ca2+]mit
- mitochondrial [Ca2+]
- TG
- thapsigargin
- YC
- yellow cameleon
- Ctrl
- control
- HEDTA
- N-(2-hydroxyethyl)ethylenediaminetriacetic acid.
REFERENCES
- 1. Szabadkai G., Duchen M. R. (2008) Physiology 23, 84–94 [DOI] [PubMed] [Google Scholar]
- 2. Rimessi A., Giorgi C., Pinton P., Rizzuto R. (2008) Biochim. Biophys. Acta 1777, 808–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Demaurex N., Poburko D., Frieden M. (2009) Biochim. Biophys. Acta 1787, 1383–1394 [DOI] [PubMed] [Google Scholar]
- 4. Bernardi P. (1999) Physiol. Rev. 79, 1127–1155 [DOI] [PubMed] [Google Scholar]
- 5. Kirichok Y., Krapivinsky G., Clapham D. E. (2004) Nature 427, 360–364 [DOI] [PubMed] [Google Scholar]
- 6. Trenker M., Malli R., Fertschai I., Levak-Frank S., Graier W. F. (2007) Nat. Cell Biol. 9, 445–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brookes P. S., Parker N., Buckingham J. A., Vidal-Puig A., Halestrap A. P., Gunter T. E., Nicholls D. G., Bernardi P., Lemasters J. J., Brand M. D. (2008) Nat. Cell Biol. 10, 1235–1237; author reply 1237–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nowikovsky K., Froschauer E. M., Zsurka G., Samaj J., Reipert S., Kolisek M., Wiesenberger G., Schweyen R. J. (2004) J. Biol. Chem. 279, 30307–30315 [DOI] [PubMed] [Google Scholar]
- 9. Dimmer K. S., Navoni F., Casarin A., Trevisson E., Endele S., Winterpacht A., Salviati L., Scorrano L. (2008) Hum. Mol. Genet. 17, 201–214 [DOI] [PubMed] [Google Scholar]
- 10. Jiang D., Zhao L., Clapham D. E. (2009) Science 326, 144–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Endele S., Fuhry M., Pak S. J., Zabel B. U., Winterpacht A. (1999) Genomics 60, 218–225 [DOI] [PubMed] [Google Scholar]
- 12. Perocchi F., Gohil V. M., Girgis H. S., Bao X. R., McCombs J. E., Palmer A. E., Mootha V. K. (2010) Nature 467, 291–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hajnóczky G., Csordás G. (2010) Curr. Biol. 20, R888–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Palty R., Silverman W. F., Hershfinkel M., Caporale T., Sensi S. L., Parnis J., Nolte C., Fishman D., Shoshan-Barmatz V., Herrmann S., Khananshvili D., Sekler I. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Da Cruz S., De Marchi U., Frieden M., Parone P. A., Martinou J. C., Demaurex N. (2010) Cell Calcium 47, 11–18 [DOI] [PubMed] [Google Scholar]
- 16. Azzu V., Brand M. D. (2010) Trends Biochem. Sci. 35, 298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cannon B., Nedergaard J. (2004) Physiol. Rev. 84, 277–359 [DOI] [PubMed] [Google Scholar]
- 18. Cadenas S., Echtay K. S., Harper J. A., Jekabsons M. B., Buckingham J. A., Grau E., Abuin A., Chapman H., Clapham J. C., Brand M. D. (2002) J. Biol. Chem. 277, 2773–2778 [DOI] [PubMed] [Google Scholar]
- 19. Couplan E., del Mar Gonzalez-Barroso M., Alves-Guerra M. C., Ricquier D., Goubern M., Bouillaud F. (2002) J. Biol. Chem. 277, 26268–26275 [DOI] [PubMed] [Google Scholar]
- 20. Krauss S., Zhang C. Y., Lowell B. B. (2005) Nat. Rev. Mol. Cell Biol. 6, 248–261 [DOI] [PubMed] [Google Scholar]
- 21. Chan C. B., MacDonald P. E., Saleh M. C., Johns D. C., Marbàn E., Wheeler M. B. (1999) Diabetes 48, 1482–1486 [DOI] [PubMed] [Google Scholar]
- 22. Himms-Hagen J., Harper M. E. (2001) Exp. Biol. Med. (Maywood) 226, 78–84 [DOI] [PubMed] [Google Scholar]
- 23. Goglia F., Skulachev V. P. (2003) FASEB J. 17, 1585–1591 [DOI] [PubMed] [Google Scholar]
- 24. Echtay K. S., Esteves T. C., Pakay J. L., Jekabsons M. B., Lambert A. J., Portero-Otín M., Pamplona R., Vidal-Puig A. J., Wang S., Roebuck S. J., Brand M. D. (2003) EMBO J. 22, 4103–4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brand M. D., Esteves T. C. (2005) Cell Metab. 2, 85–93 [DOI] [PubMed] [Google Scholar]
- 26. Cioffi F., Senese R., de Lange P., Goglia F., Lanni A., Lombardi A. (2009) Biofactors 35, 417–428 [DOI] [PubMed] [Google Scholar]
- 27. Bézaire V., Seifert E. L., Harper M. E. (2007) FASEB J. 21, 312–324 [DOI] [PubMed] [Google Scholar]
- 28. Arsenijevic D., Onuma H., Pecqueur C., Raimbault S., Manning B. S., Miroux B., Couplan E., Alves-Guerra M. C., Goubern M., Surwit R., Bouillaud F., Richard D., Collins S., Ricquier D. (2000) Nat. Genet. 26, 435–439 [DOI] [PubMed] [Google Scholar]
- 29. Brand M. D., Affourtit C., Esteves T. C., Green K., Lambert A. J., Miwa S., Pakay J. L., Parker N. (2004) Free Radic. Biol. Med. 37, 755–767 [DOI] [PubMed] [Google Scholar]
- 30. Nagai T., Yamada S., Tominaga T., Ichikawa M., Miyawaki A. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 10554–10559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Palmer A. E., Giacomello M., Kortemme T., Hires S. A., Lev-Ram V., Baker D., Tsien R. Y. (2006) Chem. Biol. 13, 521–530 [DOI] [PubMed] [Google Scholar]
- 32. Palmer A. E., Jin C., Reed J. C., Tsien R. Y. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 17404–17409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Imamura H., Nhat K. P., Togawa H., Saito K., Iino R., Kato-Yamada Y., Nagai T., Noji H. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 15651–15656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jousset H., Malli R., Girardin N., Graier W. F., Demaurex N., Frieden M. (2008) Cell Calcium 43, 83–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. De Marchi U., Campello S., Szabò I., Tombola F., Martinou J. C., Zoratti M. (2004) J. Biol. Chem. 279, 37415–37422 [DOI] [PubMed] [Google Scholar]
- 36. Poburko D., Liao C. H., van Breemen C., Demaurex N. (2009) Circ. Res. 104, 104–112 [DOI] [PubMed] [Google Scholar]
- 37. Waldeck-Weiermair M., Malli R., Naghdi S., Trenker M., Kahn M. J., Graier W. F. (2010) Cell Calcium 47, 433–440 [DOI] [PubMed] [Google Scholar]
- 38. Frieden M., James D., Castelbou C., Danckaert A., Martinou J. C., Demaurex N. (2004) J. Biol. Chem. 279, 22704–22714 [DOI] [PubMed] [Google Scholar]
- 39. Prakriya M. (2009) Immunol. Rev. 231, 88–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Landolfi B., Curci S., Debellis L., Pozzan T., Hofer A. M. (1998) J. Cell Biol. 142, 1235–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Csordás G., Hajnóczky G. (2001) Cell Calcium 29, 249–262 [DOI] [PubMed] [Google Scholar]
- 42. Zotova L., Aleschko M., Sponder G., Baumgartner R., Reipert S., Prinz M., Schweyen R. J., Nowikovsky K. (2010) J. Biol. Chem. 285, 14399–14414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Waldeck-Weiermair M., Duan X., Naghdi S., Khan M. J., Trenker M., Malli R., Graier W. F. (2010) Cell Calcium 48, 288–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Trenker M., Fertschai I., Malli R., Graier W. F. (2008) Nat. Cell Biol. 10, 1237–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hayashi T., Rizzuto R., Hajnoczky G., Su T. P. (2009) Trends Cell Biol. 19, 81–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li Y., Camacho P. (2004) J. Cell Biol. 164, 35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Toime L. J., Brand M. D. (2010) Free Radic. Biol. Med. 49, 606–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rosado J. A., Redondo P. C., Salido G. M., Pariente J. A. (2006) Mini Rev. Med. Chem. 6, 409–415 [DOI] [PubMed] [Google Scholar]
- 49. Ermak G., Davies K. J. (2002) Mol. Immunol. 38, 713–721 [DOI] [PubMed] [Google Scholar]
- 50. De Stefani D., Raffaello A., Teardo E., Szabo I., Rizzuto R. (2011) Nature, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Baughman J. M., Perocchi F., Girgis H. S., Plovanich M., Belcher-Timme C. A., Sancak Y., Bao X. R., Strittmatter L., Goldberger O., Bogorad R. L., Koteliansky V., Mootha V. K. (2011) Nature, in press [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.