

Abstract
β-Lactamases hydrolyze β-lactam antibiotics to provide drug resistance to bacteria. β-Lactamase inhibitory protein-II (BLIP-II) is a potent proteinaceous inhibitor that exhibits low picomolar affinity for class A β-lactamases. This study examines the driving forces for binding between BLIP-II and β-lactamases using a combination of presteady state kinetics, isothermal titration calorimetry, and x-ray crystallography. The measured dissociation rate constants for BLIP-II and various β-lactamases ranged from 10−4 to 10−7 s−1 and are comparable with those found in some of the tightest known protein-protein interactions. The crystal structures of BLIP-II alone and in complex with Bacillus anthracis Bla1 β-lactamase revealed no significant side-chain movement in BLIP-II in the complex versus the monomer. The structural rigidity of BLIP-II minimizes the loss of the entropy upon complex formation and, as indicated by thermodynamics experiments, may be a key determinant of the observed potent inhibition of β-lactamases.
Keywords: Enzyme Inhibitors, Enzyme Kinetics, Enzyme Structure, Protein Structure, Protein-Protein Interactions, Thermodynamics, Antibiotic Resistance, β-Lactamase, Molecular Recognition
Introduction
Protein-protein interactions govern many cellular processes, and an understanding of the determinants of molecular recognition would facilitate the rationale design of interactions for therapeutic purposes. Detailed examination of kinetic constants and thermodynamic driving forces, however, has been performed for relatively few protein-protein interaction complexes. Although significant progress has been made in the understanding and prediction of association rate constants, the determinants of dissociation rates remain poorly understood (1–5). The ability to predict the kinetic constants would be a significant contribution to the understanding of interaction networks in the systems biology era (6). Several model systems have been developed to analyze the principles of protein-protein interactions, and one such model is the interaction between β-lactamase enzymes and a set of β-lactamase inhibitory proteins (BLIPs)3 (7, 8).
β-Lactamases act by hydrolyzing the four-membered ring of β-lactam antibiotics, e.g. penicillins and cephalosporins, rendering the drugs inactive (9, 10). There are four classes of β-lactamases (A-D) based on primary amino acid sequences (11, 12). Class B β-lactamases are metalloenzymes that use a zinc-coordinated catalytic water to hydrolyze the β-lactam ring whereas classes A, C, and D are serine hydrolyases (13–15). Class A β-lactamases are widespread in both Gram-positive and Gram-negative bacteria and exhibit broad substrate hydrolysis profiles that include penicillins, cephalosporins, and for a few enzymes, carbapenems (12, 14).
The catalytic mechanism of serine β-lactamases is divided into two stages, acylation and deacylation (16). In class A enzymes, the catalytic Ser-70 residue nucleophilically attacks the carbonyl of the β-lactam and forms an acyl-intermediate (17, 18). An essential Glu-166 residue activates a highly coordinated water for deacylation (18, 19). Substitutions at Glu-166 result in an acylated, inactive enzyme (20–22).
Clinically available mechanism-based inhibitors are used in conjunction with β-lactam antibiotics to combat β-lactamase-mediated drug resistance (23). The emergence of β-lactamase mutants resistant to the action of mechanism-based inhibitors, however, has created the demand for new types of inhibitors (24, 25). The β-lactamase inhibitory proteins (such as BLIP, BLIP-I, and BLIP-II) have been found to be potent, competitive inhibitors of class A β-lactamases (26–28).
BLIP is an 18-kDa protein that is secreted by the soil bacterium Streptomyces clavuligerus (29). Although the physiological significance of BLIP is not fully understood, its ability to inhibit β-lactamases has been thoroughly investigated, with several site-directed mutagenesis, structural, biochemical, and thermodynamic studies performed (5, 8, 27, 30–40). BLIP uses a tandem domain to form a concave surface that primarily interacts with a conserved loop-helix region of β-lactamase adjacent to the active site. Two β-turns protrude into the β-lactamase active site, hindering substrate binding by interacting with residue that are required for binding β-lactam substrates (27).
BLIP-II is a 28-kDa secreted protein from Streptomyces exfoliatus that shares no sequence or structural homology with BLIP (41). Instead, BLIP-II forms an unusual seven-bladed β-propeller-fold with three β-strands and one α-helix per blade. It is believed to function in the S. exfoliatus sporulation process by potentially binding to peptidoglycan remodeling proteins such as penicillin-binding proteins (28). β-Propeller domains are ubiquitous among prokaryotes and eukaryotes and govern some of the most critical processes in the cell (42–44). For example, RCC-1 is a guanine nucleotide exchange factor for the Ran GTPase and is involved in nucleocytoplasmic transport (45). Interestingly, RCC-1 shares 21% sequence identity with BLIP-II and is the founding member of the RCC1-like repeat β-propellers (28).
β-Propellers use their β-turns and/or loops for protein-protein interactions and are capable of incorporating various secondary structures or domains within the β-propeller-fold (42). In the BLIP-II co-crystal structure with TEM-1, BLIP-II was shown to use these β-turns and loops to bind to the same loop-helix region of β-lactamase as that bound by BLIP. This interaction sterically blocks the active site (28). Previously, we demonstrated that BLIP-II inhibits all six of the class A β-lactamases we examined with low picomolar affinity, which is more potent than BLIP-mediated inhibition (46). In addition, BLIP exhibits a wide range of affinities (pm to μm Ki) for various class A β-lactamases, whereas BLIP-II inhibits every class A β-lactamase tested within a 10-fold affinity range (2.5–25 pm) (46).
Because of its high affinity for β-lactamases, BLIP-II could serve as a platform for the development of diagnostic agents to detect β-lactamases from clinical isolates as well as serve as a guide for inhibitor design. A detailed knowledge of the driving forces of the BLIP-β-lactamase interaction would facilitate the use of BLIP-II as a scaffold for diagnostic and inhibitor design as well as provide insights into the mechanisms behind the tight interactions of β-propeller proteins with their target molecules.
The high affinity of BLIP-II for class A β-lactamases made accurate determinations of the Ki values through traditional steady state kinetic methods difficult. Therefore, we expanded our understanding of the BLIP-II class A β-lactamase interactions using a combination of presteady state kinetics experiments and thermodynamics studies using isothermal titration calorimetry (ITC). The β-lactamases examined are clinically relevant enzymes found in pathogenic bacteria, including TEM-1 (the most common β-lactamase in Gram-negative bacteria) (47). In addition, crystals structures are presented of the BLIP-II-Bla1 complex (2.1 Å) and the monomeric form of BLIP-II (2.8 Å). Bla1 is the class A β-lactamase from Bacillus anthracis, the causative agent of anthrax (48). This analysis demonstrates that BLIP-II dissociates extremely slowly from β-lactamases, and the dissociation rate constants presented here are comparable with those found in the most stable protein-protein complexes (koff 10−4 to 10−7 s−1) known. Furthermore, crystallographic analysis reveals that BLIP-II is remarkably rigid, with no significant side chain movement in BLIP-II in the complex versus the monomer, which may minimize the entropic cost of association and contribute to tight binding.
EXPERIMENTAL PROCEDURES
Protein Purification
BLIP-II and the β-lactamases were purified as previously described (46, 49, 50). The purity of the BLIP-II and β-lactamase samples was determined to be >90% by SDS-PAGE. The protein concentrations were determined by a Bradford assay, the results of which were compared with a β-lactamase standard curve calibrated by quantitative amino acid analysis (40).
Stopped-flow Tryptophan Fluorescence Spectrometric Measurements of Association
The association rate constants of the BLIP-II-β-lactamase interactions were determined as previously described using a SLM 48000S fluorescence spectrometer with a MilliFlow stopped-flow reactor accessory to monitor the tryptophan intrinsic fluorescence over a time course (37). The proteins were dialyzed into 10 mm Tris, 150 mm NaCl at pH 7.0. Two injection syringes containing equal concentrations (5 μm) of both BLIP-II and either TEM-1, PC1, SHV-1, or Bla1 were loaded onto the instrument and used for mixing. A concentration of 1 mm β-mercaptoethanol was added during the dilutions to protect the proteins from UV damage (51). The excitation and emission wavelengths were 286 and 340 nm with bandwidths of 2 and 8 nm, respectively. The instrument collected data at 10-ms intervals with a 10-ms mixing dead time.
Multiple traces were collected at room temperature (∼23 °C) and averaged together. The averaged trace of the change in intrinsic fluorescence was then fitted with a second order kinetic time course (Equation 1) to determine the association constant using Graphpad Prism 5,
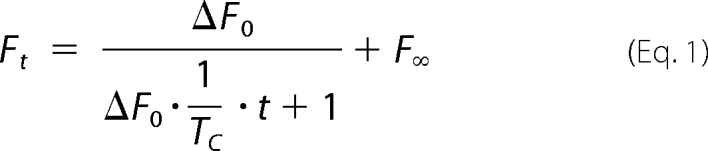 |
where Ft is the intrinsic fluorescence at time t, ΔF0 is the amplitude of the total change in the intrinsic fluorescence signal, and F∞ is the background intrinsic fluorescence after the association is complete. TC is the time parameter that is determined by fitting the data to the equation above. The association rate constant, kon, can then be calculated (Equation 2),
 |
where [m0] is the molar concentration of the unbound proteins.
Enzymatic Activity-based Measurements of Association
The association rate constants (kon) for the BLIP-II-β-lactamase interactions were determined by measuring the inhibition of β-lactamase activity over a given time period. The association rate constants were determined in conditions suitable for both second and pseudo-first order kinetics. For the determination of the on-rate constants by second order kinetics, BLIP-II was added at a 2-fold higher concentration than β-lactamase. Aliquots (0.3 ml) were taken out during the time course to measure the initial velocities of nitrocefin hydrolysis as indicated by the change in optical density at 482 nm. This activity is used as the readout of free enzyme. Excess concentrations of nitrocefin were used to yield the maximum initial velocity of each enzyme tested. The nitrocefin concentrations used in the experiment were 400, 200, 100, and 300 μm for TEM-1, Bla1, PC1, and SHV-1, respectively. The Km values of these enzymes for nitrocefin have previously been determined to be 84, 19, 1.5, and 21 μm for TEM-1, Bla1, PC1, and SHV-1, respectively (48, 49, 52, 53). The decrease in activity over the time course was fitted with the second order kinetic equation (Equation 3),
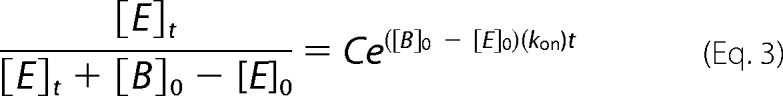 |
where [E]t is amount of free β-lactamase estimated by the enzymatic activity at time (t), [E]0 is the amount of free β-lactamase before the addition of BLIP-II, [B]0 is the initial BLIP-II concentration in the reaction, C is a fitting constant representing the background rate of nitrocefin hydrolysis, t is the time after mixing, and kon is the association rate constant of the interaction that is extrapolated from the data fitting.
BLIP-II was added in 5- and 10-fold excess to determine the association rate constants with pseudo first order kinetics (Equation 4),
where the terms of the equation are described above. However, we were unable to obtain sufficient data for the BLIP-II-PC1 association in this manner because the reaction was too fast to be accurately measured at the concentrations the assay required. The enzyme concentrations used were 0.5, 5, 5, and 1 nm for TEM-1, Bla1, PC1, and SHV-1, respectively. Attempts to lower the enzyme concentrations were unsuccessful because of the associated decease in observed nitrocefin hydrolysis. These experiments were performed at room temperature (∼23 °C) in 50 mm sodium phosphate, pH 7.0, supplemented with 1 mg/ml bovine serum albumin to minimize nonspecific interactions.
Enzymatic Activity-based Measurements of Dissociation
For the slow dissociation of the BLIP-II-β-lactamase complexes, the dissociation rate constant (koff) was determined by measuring the recovery of the wild-type β-lactamase activity by competitive displacement with an inactive TEM-1 mutant (E166A) (37). BLIP-II was incubated in a 2-fold excess with β-lactamase for 1 h. The BLIP-II-β-lactamase complex was then added to a solution containing a large excess of TEM-1 E166A. Aliquots were then used to measure initial velocities of nitrocefin hydrolysis over a time course. The TEM-1 E166A variant was used because this glutamate at position 166 is required for proper deacylation. Therefore, this mutant is functionally inactive after one round of acylation. Glu-166 is not involved in the BLIP-II-TEM-1 interface and was shown to not affect the BLIP-II association rate constant as determined by stopped-flow fluorescent spectrometric measurements. The final enzyme concentrations of TEM-1, Bla1, PC1, and SHV-1, after dilution into the TEM-1 E166A solution, were 2.5, 50, 150, and 10 nm, respectively. The concentrations of the competitor TEM-1 E166A protein were 10 μm for the PC1, Bla1, and SHV-1 dissociation reactions and 1.25 μm for the TEM-1 displacement. At these concentrations the signal created by nitrocefin hydrolysis of the active β-lactamases was significantly higher (at least 7-fold) than the background hydrolysis from the TEM-1 E166A enzyme and 1 mg/ml BSA in 50 mm sodium phosphate buffer, pH 7.0. An excess of nitrocefin was used to yield the maximal initial velocity for the β-lactamase enzymes. The concentrations of nitrocefin used in this experiment were 400, 200, 200, and 300 μm for TEM-1, Bla1, PC1, and SHV-1, respectively. The amount of active β-lactamase over the time course was fitted with first order kinetics to determine the kinetic parameters (Equation 5),
where [E]∞ is the amount of free β-lactamase when the dissociation had reached completion estimated by the enzymatic activity when uninhibited by BLIP-II, [E]t is the amount of free β-lactamase estimated by enzymatic activity at time (t), t is the time after mixing the BLIP-II-β-lactamase complex with the inactive TEM-1 E166A enzyme, C is the curve-fitting constant representing the background rate of nitrocefin hydrolysis (including the activity of the TEM-1 E166A enzyme), and koff is the dissociation rate constant extrapolated from the fitting the data. Due to the long duration of the experiment, positive and negative controls of the β-lactamase and inactive TEM-1 E166A alone, respectively, were used to assess the stability of the β-lactamases during the experiment. The activities of these enzymes did not significantly change during the time course of the experiment (>80% activity).
Isothermal Titration Calorimetry
The calorimetric experiments of binding between BLIP-II and the various class A β-lactamases were carried out using a VP-ITC calorimeter (GE Healthcare). The titrations were performed in up to five buffers that contain different ionization enthalpy quantities (ΔHion). The ionization enthalpy from the absorption or release of a proton from the buffer upon protein-protein binding affects the apparent enthalpy (ΔHapp) of the reaction. The different buffers were used to determine the proton uptake effect upon BLIP-II-β-lactamase binding (Equation 7). 10 mm concentrations of the buffers phosphate, Tris, ACES, PIPES or HEPES were supplemented with 150 mm NaCl. In each buffer condition, BLIP-II and the various class A β-lactamases were used both as the titrant and in the sample cell at concentrations of 40 and 4 μm, respectively. This procedure was carried out to validate the apparent enthalpy (ΔHapp) of the interaction at each temperature and buffer condition. Plotting the ΔHapp at a minimum of at least five temperatures aided in the determination of the ΔCp for each BLIP-II class A β-lactamase pair in phosphate-buffered saline. The ΔCp of each interaction in the different buffers did not vary significantly, with a S.D. less than 20%. Therefore, only the ΔCp determined in phosphate-buffered saline is presented. The temperature range was between 5 and 36 °C. The data were processed using the Origin 7.0 software package with an ITC add-on (OriginLab Corp., Northampton, MA). A list of the titrations performed is presented in supplemental Table 1. The ΔCp of the interaction was determined in phosphate-buffered saline using Equation 6,
 |
where ΔCp is the change in heat capacity, ΔHapp is the apparent change in enthalpy, and ΔT is the change in temperature (54, 55). The ΔHapp varied between buffers at identical temperatures, indicating that ΔHreal had to be determined by accounting for the enthalpy of buffer ionization (ΔHion) (Equation 7),
where n is the number of protons being exchanged with the buffer upon binding. If the slope of the line is positive, then the protons are absorbed when the complex is formed (55).
Crystallization and Structural Determination
Purified BLIP-II was obtained through batch purification with the TALON metal affinity resin, and the buffer was exchanged into 10 mm Tris, 150 mm NaCl, pH 7.0 (TBS), with a Sephadex S75 gel filtration column (GE Healthcare). Crystallization screening of the monomeric form of BLIP-II was set up on the TTP LabTech mosquito instrument. Diffraction quality crystals were formed in 2.0 m ammonium sulfate and were visible in approximately two months. X-ray diffraction data were collected at beamline 5.0.1 (Advanced Light Source, Berkeley, CA) and processed with HKL2000 software (56). The Phaser program from the CCP4 package was utilized for molecular replacement (57, 58). The BLIP-II molecule from the BLIP-II-TEM-1 structure (Protein Data Bank code 1JTD) was used as the reference molecule (28). After phasing, six molecules per asymmetric unit were found, and the model was fitted to the electron density using Coot (59). The initial refinement was performed by using simulated annealing and rigid body refinement in Phenix (60–62). This modified structure was further subjected to several cycles of refinement using Coot, Phenix, and Refmac5 (63). Ordered solvent was added using both Phenix and Coot (59, 60). The final refinement was performed in Refmac5 using TLS and noncrystallographic symmetry restraints. The TLS restraints were generated using the TLS motion determination server (64).
The BLIP-II-Bla1 complex (6 mg/ml in TBS) was crystallized in 0.1 m Bicine pH 8.5, 13% PEG 10,000. The crystals were visible in a few days and harvested a month later. Thirty percent glycerol and paratone-N were used as cryoprotectants. X-ray diffraction data were collected at beamline 5.0.1 (Advanced Light Source, Berkeley, CA) and processed into structure factors using HKL2000 (56). The symmetry of data was found to be C2 with one complex in the asymmetric unit. The initial model was obtained by molecular replacement (MolRep in CCP4 package) search using the BLIP-II model extracted from Protein Data Bank code 1JTD and a preliminary x-ray crystal structure of Bla1 (data not shown) (28, 65). Simulated annealing in Refmac5 was performed for the initial refinement, and the structure was then subjected to several rounds of refinement in the Coot program and Refmac5 using TLS restraints (59, 63). Water molecules were added using Coot (59). The final refinement of the modified structure was performed in Phenix (60–62). The analysis of the structures was done using Pymol and Coot (66). The Molprobity server was used to validate the structure (67). The buried surface area (ΔASA) was determined using the AREAIMOL program of the CCP4 package (58, 68). A list of data collection and refinement statistics for both structures is provided is supplemental Table 2.
Defining the Contact Residues of the BLIP-II Class A β-Lactamase Interfaces
The contact residues were assigned using the AquaProt server as previously described (33). This is an “all atom contact” method used to define the interactions between protein-protein interfaces. AquaProt is the sum of four modified, independent programs; REDUCE, PROBE, PARE, and NCI (32). REDUCE adds hydrogens through H-bond optimization (69). PROBE is used to analyze van der Waals interactions and H-bonds contacts (70). PARE and NCI define the electrostatic and aromatic interactions between the interacting proteins, respectively (5, 71). These programs allow for the identification of the individual atoms of the residues involved in the interaction. A two-dimensional representation of these interaction networks was made using the MAVisto program (72). For further details about these programs, refer to the individual publications.
RESULTS
BLIP-II Kinetics of Association with Class A β-Lactamases
To better understand the determinants of the high affinity of BLIP-II toward class A β-lactamases, the binding kinetics of association were determined using a stopped-flow fluorescence spectrophotometer. Equal concentrations of the interacting protein pairs were injected to a final concentration of 2.5 μm. The formation of the complex causes a quench of the intrinsic fluorescence signal decreasing the amplitude (Fig. 1). The resulting changes in fluorescence were used to approximate the changes in the amount of free unbound protein. This value was used to determine the association rate constant, kon. The association reaction during this time frame was treated as irreversible and fitted to the second order rate equation (see “Experimental Procedures,” Equations 1 and 2) (Fig. 1). The measurements of BLIP-II binding with TEM-1, PC1, SHV-1, and Bla1 revealed that the association rate constants were within a 10-fold range in the condition used (10 mm Tris, 150 mm NaCl, pH 7.0, at ∼23 °C). The association rate constants for the TEM-1, PC1, and SHV-1 interactions with BLIP-II were within a very narrow range (4.7–7.1 × 106 m−1s−1), whereas the BLIP-II-Bla1 interaction was ∼10-fold slower (6.8 × 105 m−1 s−1). These results are summarized in Table 1.
FIGURE 1.

Representative time-courses of the stopped-flow tryptophan fluorescence and enzymatic activity measurements to determine the association rate constants. A and B, shown is the average time course of at least 20 traces of the BLIP-II interactions with either the TEM-1 or Bla1 β-lactamase, respectively. The proteins were mixed at equal concentrations (5 μm) upon injection. The emission signal is represented as percent intrinsic fluorescence photon count (<Ieq>) of the equilibrium base line. Fluorescence emission scans are shown within the window. The spectra are labeled as TEM-1 and BLIP-II, complex, and sum. The complex is the normalized emission scan from the BLIP-II-β-lactamase, and the sum spectra are the addition of both the BLIP-II and β-lactamase spectra assuming the signal was not quenched by association. C and D, the time-course of the second order enzymatic activity-based measurement of association of the BLIP-II interactions is shown. Mobs is defined in Equation 3 as [E]t/([E]t + [B]o − [E]o) (see “Experimental Procedures”).
TABLE 1.
Kinetic constants of BLIP-II-β-lactamase interactions
| Molar ratio | TEM1 | PC1 | SHV-1 | Bla1 | |
|---|---|---|---|---|---|
| Experiment | |||||
| Stopped flowa, kon (105m−1 s−1) | 1:1 | 71 ± 21 | 47 ± 14 | 54 ± 16 | 6.8 ± 1.5 |
| Enzyme inhibitionb, kon (105m−1 s−1) | 10:1 | 36 ± 12 | NAc | 67 ± 23 | 7.3 ± 1.7 |
| 5:1 | 45 ± 11 | NAc | 87 ± 20 | 8.1 ± 2.0 | |
| 2:1 | 55 ± 20 | 63 ± 22 | 72 ± 18 | 8.9 ± 2.1 | |
| Kinetic parameters | |||||
| kon (105m−1 s−1)d | 54 ± 16 | 55 ± 18 | 70 ± 19 | 7.8 ± 1.8 | |
| koff (10−7 s−1)e | 43 ± 13 | 430 ± 97 | 2100 ± 530 | 8.3 ± 4.1 | |
| Calculated BLIP-II Kd (pm)f | 0.79 | 7.8 | 30 | 1.1 | |
| BLIP Kd (pm)g | 500 | 380,000 | 1,130,000 | 2,500 | |
a Stopped-flow tryptophan fluorescence spectrometry measurements at ambient temperature (23 °C).
b Activity-based determination of association at different molar ratios (BLIP-II-β-lactamase) at ambient temperature (23 °C). The on-rate constants were determined using the psuedo-first order rate equation for the 10:1 and 5:1 molar ratio assays. The second-order rate equation was used for the 2:1 molar ratio experiments (see “Experimental Procedures”).
c There were not enough data points to accurately determine the association rate constant using this method because of the rapid association due to the high concentrations of the PC1 β-lactamase enzyme needed for this assay.
d Average association rate constants from the different experimental methods.
e Activity-based dissociation experiments using inactive TEM-1 E166A-substituted enzyme in the competitive displacement assay. First-order reaction kinetics were used to determine the dissociation rate constants (see “Experimental Procedures”).
f The dissociation constant (Kd) was calculated from the dissociation rate constant and averaged association rate constant at ambient temperature (23 °C).
To validate the stopped-flow experiments and confirm that the association is accompanied by enzymatic inhibition, an enzyme activity-based measurement of the association rate was used. Decreases in enzyme activity upon the addition of BLIP-II were followed over a time course to determine the association rate constants. BLIP-II was added at either a 2:1, 5:1, or 10:1 molar ratio (BLIP-II-β-lactamase). The time course of enzymatic activity at the 2:1 molar ratio was fitted to the second order rate equation (see “Experimental Procedures,” Equation 3) (Fig. 1). The time course of the 10:1 and 5:1 molar ratios were fitted to the pseudo-first order rate equation (see “Experimental Procedures,” Equation 4). It was not possible to accurately determine the association rate constant for PC1 β-lactamase under pseudo-first order conditions because of the rapid complex formation and the high enzyme concentration needed to observe β-lactam hydrolysis. Overall, the association rate constants determined by either the activity-based assay at varying molar ratios or by stopped-flow fluorescence experiments were similar and fell within a less than a 2-fold range (Table 1) despite the different buffer conditions for the experiments (see “Experimental Procedures”). The association rate constants (Table 1) demonstrate that BLIP-II does not discriminate between the TEM-1, PC1, and SHV-1 β-lactamases via differences in association rates. The BLIP-II association rate constant for binding Bla1, however, is ∼7–10-fold slower than that observed for binding the other class A β-lactamases.
Measurements of BLIP-II Dissociation to Class A β-Lactamases
The dissociation rate constants were determined to gain further insight into binding of BLIP-II with class A β-lactamases. A time course of enzymatic activity recovery by competitive displacement was used to determine the dissociation rate constants. BLIP-II was added in 2-fold excess to class A β-lactamase and incubated for 1 h, and the BLIP-II-β-lactamase complex was then added to a large excess of inactive TEM-1 E166A. The crystal structure of the BLIP-II-TEM-1 complex reveals that Glu-166 does not interact with BLIP-II, and the association rate constant was determined to be 4.2 × 106 m−1 s−1 by stopped-flow fluorescence measurements. This result demonstrates that this substitution does not affect complex formation, whereas its poor enzymatic activity makes it a good candidate to use in excess in the activity-based dissociation measurements.
Examination of β-lactam hydrolysis in aliquots of the binding mixtures taken at various time points allowed for observation of the recovery of wild-type β-lactamase activity. The data were fitted to the first order rate equation because of the large excess of the E166A β-lactamase (see “Experimental Procedures,” Equation 5). The BLIP-II dissociation rate constants (koff) for the various β-lactamases fell within a 250-fold range of each other. BLIP-II dissociated from SHV-1 β-lactamase with the fastest rate constant of 2.1 × 10−4 s−1. The BLIP-II-Bla1 complex is the most stable with the slowest dissociation rate constant (8.3 × 10−7 s−1) and a half-life of ∼10 days (Fig. 2). The off-rate constants of the BLIP-II-PC1 and BLIP-II-TEM-1 complexes are 4.3 × 10−5 s−1 and 4.3 × 10−6 s−1, respectively. These results are summarized in Table 1. The 250-fold range of dissociation rate constants is significantly larger than the range observed for the association rates (10-fold). This indicates that the differences in binding affinity between BLIP-II and the various class A β-lactamases by BLIP-II is largely determined by differences in the dissociation rates of the complexes.
FIGURE 2.

Enzymatic activity-based measurements of dissociation. Plots of the dissociation between BLIP-II and either SHV-1 (A) or Bla1 (B) are shown. The BLIP-II-β-lactamase complex is bound and then added to a large excess of inactive, deacylation-deficient TEM-1 variant (E166A). The first order kinetics of this reaction is presented as the fraction activity recovery over time (see “Experimental Procedures,” Equation 5).
The previously reported BLIP-II equilibrium dissociation constants, Kd, for binding class A β-lactamases were determined using an enzyme inhibition assay (46). BLIP-II was added to β-lactamase at increasing concentrations and allowed to reach equilibrium after a 2-h incubation (46). However, the tight binding nature of BLIP-II made it difficult to accurately determine the Ki because this value is substantially lower than the β-lactamase concentration required to detect hydrolysis in the assay. For this reason, some values were given a less than or equal to value. For example, the Ki for the BLIP-II-PC1 and BLIP-II-Bla1 interactions were ≤16 and ≤25 pm, respectively (46). The Kd for each of the BLIP-II-β-lactamase interactions examined here were calculated using the individual association and dissociation rate constants (Table 1) and were found to be within ∼3-fold of the previously determined Ki values. The previously determined Ki for the BLIP-II-Bla1 interaction was less than or equal to 25 pm (46). However, when the individual on- and off-rate constants were used to calculate the Kd, it was determined to be 1.1 pm. Taken together, the results validate the interpretations of the previous data and the individual association and dissociation rate constants determined here (46).
Thermodynamic Comparison
To understand the driving forces of the BLIP-II-β-lactamase interactions, a detailed thermodynamic characterization was performed using isothermal titration calorimetry. The apparent enthalpies (ΔHapp) of the individual titrations for BLIP-II against the TEM-1, PC1, SHV-1, and Bla1 β-lactamases are listed in supplemental Table 1. It was not possible to determine affinity constants using ITC because these high affinity interactions resulted in too few data points along the transition of the binding curve (Fig. 3). The tight binding nature of these interactions, however, allowed for a clear transition to determine the ΔHapp for the binding reaction.
FIGURE 3.

ITC measurements of binding between BLIP-II and class A β-lactamases. A representative titration of the BLIP-II interactions is shown (A). In this particular titration, TEM-1 is injected into BLIP-II in phosphate-buffered saline at 30 °C. The steep transition region does not allow for an accurate measurement of affinity. B, the ΔCp of each interaction is calculated based on the slope of the line fitted to the apparent enthalpy (ΔHapp) shown at different temperatures (see “Experimental Procedures,” Equation 6). C, the protonation effect is shown as the ΔHapp varies depending on the buffer despite the same temperature (23 °C). The ΔHapp is plotted against the enthalpy of buffer ionization (ΔHion) to determine the number of protons absorbed upon complex formation (n) and the real binding enthalpy (ΔHreal) (see “Experimental Procedures,” Equation 7). Keys showing the colors corresponding to the respective β-lactamases are shown next to panels B and C.
The initial set of titrations was performed in phosphate-buffered saline at different temperatures to determine the ΔHapp and ΔCp of each interaction (Table 2). Each interaction was shown to be largely enthalpically driven in PBS at physiological temperatures (Fig. 3). The ΔCp for each interaction was determined by plotting the ΔHapp versus the temperature (see “Experimental Procedures,” Equation 6, Fig. 3). The ΔCp of the BLIP-II interactions revealed that the ΔCp values for binding TEM-1, PC1, and SHV-1 were striking similar (from −409 to −461 cal/mol × K), but the value for the BLIP-II-Bla1 interaction was significantly larger (−642 cal/mol × K) (Table 2). This trend is similar to the association rate constant determinations where each of these interactions had similar on-rate constants except for Bla1. The more negative ΔCp value of the Bla1-BLIP-II interaction suggests that either an increase in the buried surface area (ΔASA) or increased hydrophobic contacts are present in comparison to the other interactions. The ΔCp values presented here are less than that for the BLIP-TEM-1 interaction (−667 cal/mol × K).
TABLE 2.
Thermodynamics of the BLIP-II-β-lactamase interactions
| TEM-1 | PC1 | SHV-1 | Bla1 | |
|---|---|---|---|---|
| Kda (m) | 7.9 × 10−13 | 7.8 × 10−12 | 3.0 × 10−11 | 1.1 × 10−12 |
| ΔGb (kcal/mol) | −16.4 | −15.0 | −14.2 | −16.2 |
| ΔHc (kcal/mol) | −11.9 | −16.5 | −7.1 | −9.4 |
| ΔSd (cal/mol × K) | 15.0 | −4.9 | 24.0 | 22.9 |
| ΔCpe (cal/mol × K) | −457 | −409 | −461 | −642 |
| nf | 1.3 | 1.6 | 1.1 | 1.6 |
a Kd values were calculated from the individual rate constants in Table 1.
b Gibbs free energies are calculated from the Kd values for 23 °C.
c ΔH presented is the enthalpy of the interaction assuming the enthalpy of buffer ionization is 0 (Equation 7 and Fig. 3).
d Entropy, ΔS, was determined using the Gibbs free energy equation using both the calculated ΔG and the determined real ΔH from the ITC experiments.
e Change in heat capacity, ΔCp, was determined by plotting the ΔH as a function of temperature in phosphate-buffered saline.
f Number of protons absorbed upon binding.
The ΔHapp values for each interaction varied when the titrations were performed in a different buffer but at the same pH and temperature (Fig. 3, supplemental Table 1). This finding suggests that a proton is either being absorbed or released upon binding, and the difference in enthalpy observed is due to the ionization effect of the buffer. This protonation effect is not observed in either the BLIP-TEM-1 or barnase-barstar interactions (38, 73). Therefore, the ΔHapp observed in the different buffers is not the real enthalpy (ΔHreal) of the interaction. To correct for this effect, the titrations were carried out in multiple buffers (supplemental Table 1) with differing enthalpies of ionization (ΔHion) and at multiple temperatures. These titrations were used to determine the ΔHapp for each of the buffers at the temperature at which the kinetics assays were performed (see “Experimental Procedures,” Equation 7, Fig. 3). This experiment allowed for the real thermodynamics of the system to be determined at room temperature (23 °C). The linear relationship between the apparent and the buffer ionization enthalpies allowed for the determination of both the real enthalpy and the number of protons absorbed upon binding (see Equation 7). The number of protons (n) is extrapolated from the slope of the line. The real enthalpy is found when the effect of buffer ionization is zero (the y intercept). The number of protons absorbed in the BLIP-II interaction varied between β-lactamases from 1.1 for SHV-1 to 1.6 for PC1 and Bla1 (Table 2).
The Kd values determined by the presteady kinetics were used to calculate the Gibbs free energy of binding (ΔG). The determination of the ΔHreal (Equation 7) for each binding pair allowed for the calculation of the ΔS. These results are summarized in Table 2. Due to the tight binding nature of the BLIP-II interactions with β-lactamases, the ΔG values are very favorable (−14.2 to −16.4 kcal/mol). The ITC measurements showed that these reactions are largely enthalpically driven (−7.1 to −16.5 kcal/mol). The determined entropy of the systems was favorable for all of the β-lactamases except for PC1, for which binding must overcome an entropic cost of ∼1.5 kcal/mol (−TΔS).
Comparison of the BLIP-II Structures in Complex
Currently, there is only one x-ray structure of BLIP-II available, and it is in complex with TEM-1 β-lactamase (28). To aid in the understanding of the energetic terms described above, the structure of the monomeric form of BLIP-II was determined to 2.8 Å resolution and a complex of BLIP-II with Bla1 was solved to 2.1 Å (supplemental Table 2). The BLIP-II monomer structure packed into six molecules per asymmetric unit. This packing is not thought to be physiologically relevant because BLIP-II was shown to be a monomer by gel filtration (data not shown). The six molecules were very similar to each other with an root mean square deviation between 0.13 and 0.33 Å for the BLIP-II molecules (Fig. 4).
FIGURE 4.

Overview of the determined crystal structures. A, a schematic representation of the BLIP-II monomer is shown with six molecules per asymmetric unit. B, ribbon diagrams of each chain are superimposed with the blades of the β-propeller domain numbered. C, shown is superimposition of the schematic diagrams of the BLIP-II-Bla1 (red) structure and chain A of the BLIP-II monomer (purple).
The BLIP-II-Bla1 complex is interesting because of its unique on- and off-rate constants in comparison to the other β-lactamases examined. The association rate constant is ∼10-fold slower than the other β-lactamases, and it is the most stable complex with the slowest dissociation rate constant. Bla1 is a class A β-lactamase that is efficient at hydrolyzing penicillins and early generation cephalosporins, similar to TEM-1 and PC1, and has a similar overall fold (48). Bla1 shares ∼38% sequence identity with TEM-1 β-lactamase, and there is a 1.9 Å root mean square deviation between the two molecules. The affinity of BLIP-II toward TEM-1 and Bla1 is very similar (0.79 pm and 1.1 pm) despite the difference in presteady state kinetics, thermodynamics, and structure.
Cluster analysis has previously been used to define protein-protein interaction networks but has recently also been used to describe protein-protein interfaces (6, 74). Schreiber and co-workers (32, 33, 74, 75) have demonstrated that the interacting residues form a modular architecture. A module is defined as a cluster of strong interactions between the residues of the two proteins (74). The effect of mutating the residues within the module was shown to be cooperative, but mutating residues in different modules were shown to be additive (32). The conclusion was reached that hot spots, residues that decrease affinity by ≥10-fold upon mutation, can be defined by their connectively between interacting residues within the cluster or module (32). The AquaProt server is a tool to produce these interface contact maps and define the contact residues into clusters, defined as three or more connected residues (see “Experimental Procedures”) (33). This server was used to examine the differences between the BLIP-II-TEM-1 and BLIP-II-Bla1 structures (Figs. 5 and 6).
FIGURE 5.

Modular architecture of the BLIP-II interfaces. A, the crystal structure of the BLIP-II-TEM-1 (Protein Data Bank code 1JTD) complex (gray) is shown superimposed against the BLIP-II-Bla1 (green-red) structure (28). The modular architecture of the BLIP-II-TEM-1 (B) and BLIP-II-Bla1 (C) interfaces were determined using the AquaProt server (see “Experimental Procedures”). The blue circles and red boxes contain the BLIP-II and β-lactamase residue labels, respectively. The color of the edge represents van der Waals, hydrogen bonds, aromatic, and electrostatic interactions as black, blue, red, and green colors, respectively. An edge containing an arrowhead indicates a side-chain-backbone interaction, whereas the remaining edges are side-chain-side chain interactions. The diagrams were made using the motif analysis and visualization tool, MAVisto (72).
FIGURE 6.

Structure perspectives of the BLIP-II clusters. A cross-section view of Bla1 (red) superimposed against TEM-1 (gray) is shown where BLIP-II is removed from the illustration. Portions of the interactions are boxed, and a close-up view of the interaction is shown labeled with the corresponding cluster. BLIP-II (green) is only shown in the close-up views. The BLIP-II and β-lactamase residues are labeled in green and red, respectively.
Analysis of the BLIP-II-TEM-1 interface revealed a total of three clusters. Both C1 and C2 are large clusters containing a total of 11 residues and 15 residues, respectively (Fig. 5). C3 is considerably smaller, as it contains the fewest number of residues to be defined as a cluster with only Val-216 of TEM-1 forming van der Waals interactions with Trp-53 and Asp-52 of BLIP-II. The clusters of the BLIP-II-Bla1 interaction are very similar to the clusters of the BLIP-II-TEM-1 interaction (Figs. 5 and 6). The three clusters of the BLIP-II-TEM-1 interaction remain in the BLIP-II-Bla1 interaction, and the majority of contacts formed at the interface are similar with a few key exceptions. C2 and C3 are increased in size in the BLIP-II-Bla1 interaction. Connections by residues Lys-99, Glu-110, and Lys-111 of Bla1 are significantly increased, thereby increasing the elaborate network of C2 (Figs. 5 and 6). C3 was a minimal cluster in the BLIP-II-TEM-1 interaction. In the case of the BLIP-II-Bla1 interaction, more contacts are formed with an increase in van der Waals contacts and the addition of an electrostatic interaction between Arg244 in TEM-1 and Asp52 in BLIP-II (Figs. 5 and 6). This enrichment of hydrophobic and electrostatic interactions strengthens the contacts between the loop of BLIP-II that sterically hinders substrate binding to the active site.
Electrostatic Complementarity at the BLIP-II-Bla1 Interface
Cluster analysis showed that the BLIP-II-Bla1 interaction forms five salt bridges in contrast to the one formed in the BLIP-II-TEM-1 complex (Fig. 7). Lys-111 in TEM-1 forms the only salt bridge in the BLIP-II-TEM-1 interface with Asp-131 but is close to interacting with Asp-167, only 5 Å away (the cutoff in the cluster analysis is 4 Å) (Fig. 6). In the BLIP-II-Bla1 structure, Lys-111 has moved closer to Asp-167 (4 Å), where it forms two salt bridges with Asp-167 and Asp-131. Adjacent to these interactions, Gln-99 in the TEM-1 structure forms a hydrogen bond with Asp-206 and two van der Waals interactions with Tyr-208 and Phe-209 of BLIP-II. Instead, Lys-99 of Bla1 makes more contacts with BLIP-II (Fig. 6). The two van der Waals connections present in the BLIP-II-TEM-1 interaction are conserved in the BLIP-II-Bla1 structure. In addition, an aromatic interaction is formed between Lys-99 and Tyr-191 of BLIP-II, and two electrostatic interactions are formed between Lys-99 of Bla1 and two aspartate residues of BLIP-II (Asp-170 and Asp-206). Another salt bridge is formed between Arg-244 of Bla1 and Asp-52 of BLIP-II (Fig. 7). Although Arg-244 is present in both TEM-1 and Bla1, the arginine side chain in Bla1 has moved closer to BLIP-II by ∼1 Å, strengthening the interaction (Fig. 6).
FIGURE 7.

Electrostatic surface representations. The monomers of the BLIP-II-Bla1 complex are rotated ∼90° so the interface is shown. The active site serine (Ser-70) of the Bla1 enzyme and the residues involved in electrostatic interactions are labeled. The electrostatic surfaces of the SHV-1, TEM-1, and PC1 β-lactamases are also shown (PDB codes 1SHV, 1XPB, 3BLM) (92–94).
Rigidity of BLIP-II
The similarity between the six monomeric BLIP-II molecules and the two complexed BLIP-II molecules is remarkable. The root mean square deviation between each of the eight BLIP-II molecules is within a very narrow range with only 0.13–0.39 Å for the backbone atoms and 0.62–0.88 Å for all of the atoms in the molecule. These values were determined using the SuperPose server (76). Thus, the BLIP-II interface residues do not undergo significant conformation changes upon complex formation. There is very little side chain movement along the interface between the BLIP-II monomeric and complexed versions of BLIP-II (Fig. 8).
FIGURE 8.

Rigidity of the BLIP-II interface residues. A, the eight BLIP-II molecules, six monomers from the asymmetric unit and two from the complexes (TEM-1 and Bla1), are aligned and show very limited movements. The two contact residues that move significantly are Leu-91 and Tyr-208 (labeled). B, a close-up view of Tyr-208 where the residues are split between two different rotamers is shown. Three monomeric versions of BLIP-II are positioned uniquely in comparison to BLIP-II from the complexes and the remaining monomers (see “Results” and “Discussion”).
The only residues that show considerable movement are in the loop containing contact residue Leu-91 and the side chain of Tyr-208 (Fig. 8). Leu-91 in the BLIP-II-TEM-1 structure is moved away from the interface by ∼2.3 Å, preventing the residue from making contact. In the BLIP-II-Bla1 complex, this loop does not move upon binding but rather makes hydrophobic contacts with Ala-215 of Bla1, increasing the size of C3. The Tyr-208 side chain is shown to move when you compare the eight BLIP-II structures. Three of the monomeric BLIP-II molecules have Tyr-208 pointing in a different direction than the complexed state. However, a closer examination of the crystal packing shows that Thr-57 of a neighboring BLIP-II molecule sterically hinders that position of Tyr-208. The remaining three monomeric BLIP-II molecules are solvent-exposed and are positioned in the same direction as the complexed versions of the residue. This position of Tyr-208 in the complexed state is believed to be the native position of the rotamer, whereas the opposing rotamer positions shown are a crystallographic artifact. This speaks to the rigid nature of the β-propeller domain.
DISCUSSION
Association rate constants for protein-protein interactions vary from <103 to >109 m−1s−1 (4). Many interactions exhibit rate constants of ∼105-106 m−1s−1. Such interactions are hypothesized to be on the cusp of the rates at which protein-protein associations are diffusion-controlled (4). In this study we show that BLIP-II has a higher affinity for class A β-lactamases than BLIP and that the association rate constants are consistent with diffusion controlled rates (46). However, it is the exceptionally slow off-rates of BLIP-II that cause its overall affinity to be several orders of magnitude tighter than that observed for BLIP (34, 77).
The dissociation rate constants influence the overall Kd and, therefore, affect function (2). This point is illustrated by the linear correlation of the bioactivities of human growth hormone variants with the determined off-rate constants with its receptor (78). Similarly the wide-ranging dissociation rates of the BLIP-II-β-lactamase interactions dictate the specificity between β-lactamases and are responsible for the remarkably high affinity of BLIP-II for β-lactamase. The dissociation rate constants of BLIP-II are comparable with most femtomolar or even subfemtomolar interactions, which are some of the tightest interactions to be experimentally determined (79–82). For example, the association and dissociation rate constants of the well studied barnase-barstar interaction were determined to be 6 × 108 m−1s−1 and 8 × 10−6 s−1 (Kd = 1.3 × 10−14 m), respectively (82). The difference between the barnase-barstar interaction and BLIP-II affinities is represented primarily in the association rate constant where BLIP-II is ∼100-fold slower. However, the dissociation rate constants of some BLIP-II interactions were either equal to or slower than that of barnase-barstar. For example, the BLIP-II-Bla1 dissociation rate constant (8.3 × 10−7 s−1) is ∼10-fold slower than that of barnase-barstar, and the BLIP-II-TEM-1 dissociation is ∼2-fold slower (4.3 × 10−6 s−1). From our literature searches, the tightest BLIP-II interaction described here (BLIP-II-Bla1) has a dissociation rate constant only 18-fold faster than the slowest known for a protein-protein interaction, the trypsin-BPTI complex (koff 5 × 10−8 s−1) (79). The structure comparison between the two known BLIP-II-β-lactamase structures (TEM-1 and Bla1) reveals an increase in both electrostatic and hydrophobic interactions with Bla1 in comparison to TEM-1. We postulate that these close range contacts are responsible for stabilizing the interaction. The energetic contributions of the hydrophobic contacts are indicated by the more negative ΔCp value of the BLIP-II-Bla1 interaction in comparison to the other β-lactamases examined (Table 2).
The rate of association is often correlated with the degree of charge complementarity (1, 4). However, BLIP-II has the slowest association rate constant for Bla1 despite the increase in electrostatic interactions compared with the other β-lactamase complexes (Fig. 7). This observation suggests that something other than charge complementarity is a significant factor influencing the association rate constant. Perhaps the intermediate encounter complex/transition state or the rate of desolvation could be unfavorable in the BLIP-II-Bla1 complex as these characteristics have been shown to influence the association rate (3, 4, 83).
Although the physiological function of the interaction of various BLIPs with β-lactamases in vivo is unclear, it is likely that there is a selective pressure driving the evolution of the high affinities. Bla1 β-lactamase exhibits the slowest off rate with BLIP-II and is secreted by B. anthracis, the causative agent of anthrax (48). B. anthracis and Streptomyces (from which the BLIPs have been isolated) are soil dwelling bacteria and thus may exist in the same environment. Most of the tight binding complexes discussed here, such as barnase-barstar and trypsin-BPTI, are physiologically important, and the inhibition of the enzymes is necessary to prevent deleterious effects (79, 81, 82). Selective pressure then drives the formation of a faster and tighter complex. The tight interaction between BLIP-II and Bla1 may be the result of competition between bacteria to establish a niche where bacteria secrete β-lactams to kill competitors but also β-lactamases and β-lactamase inhibitors (such as BLIPs and clavulanic acid, a clinically available small molecule β-lactamase inhibitor) for self- preservation (29, 41).
The driving forces for binding mediated by β-propeller domains, such as those in the RCC1 superfamily including BLIP-II, are currently unknown because studies utilizing a combination of kinetic, thermodynamic, and structural approaches have not been performed. The available studies are often limited to either the apo-β-propeller structure or investigations of the interaction with a representative peptide of the interacting protein rather than the entire interface (84). It has been noted that the β-propeller protein motif forms a rigid structure (42). In this study, the picomolar affinity BLIP-II-β-lactamase interactions provide evidence that this rigidity is directly linked to the favorable enthalpy and entropy-driving forces of β-propeller-mediated protein assemblies.
In protein-protein interactions, the enthalpy driving force is related to the change in the entire noncovalent energy upon binding, whereas entropy is affected by conformational changes and water displacement from the interface (85, 86). Previous studies have shown that one-third of all interface residues are observed to undergo conformational changes (87, 88). These changes result in an entropic cost by being restricted to one rotamer conformation and an enthalpic cost by breaking the noncovalent intramolecular interactions. Polar hot spots of binding, residues that upon mutation to alanine result in a >10-fold decrease in affinity, demonstrate minimal movements to minimize the enthalpic and entropic cost of association (89). This mechanism is expanded to the entire BLIP-II interface in that a lack of side chain movement in the monomeric form versus the complex structures of BLIP-II was observed (42, 90). The lack of conformational changes upon binding provides a plausible explanation for why the BLIP-II interactions with β-lactamase are driven by both favorable enthalpy- and entropy-driving forces as indicated the ITC results. The rigidity minimizes the conformational entropy cost of binding and maximizes the favorable water displacement entropy common to protein-protein interactions (85). The favorable enthalpy is observed because the side chains only form additional noncovalent interactions upon association with β-lactamase but do not lose a majority of their intramolecular interactions. β-Propeller domains often serve as scaffolding proteins capable of interacting with several partners (84, 91). Therefore, if rigidity is conserved among β-propeller domains, these driving forces could be common among interactions involving proteins containing this motif.
The findings presented here are of interest with regard to the design of β-lactamase diagnostics. BLIP-II exhibits an extremely slow off-rate once bound to class A β-lactamases, with half-lives for complexes ranging from 1 h for SHV-1 to 9.7 days for Bla1 β-lactamase. These off-rates are conducive to the development of an ELISA-based detection system using BLIP-II to capture β-lactamase from clinical isolates. Ongoing alanine-scanning mutagenesis studies to compare the sequence requirements for binding various class A enzymes will provide insights into the basis of binding specificity as well as facilitate the engineering of BLIP-II variants with altered binding characteristics to recognize only certain class A enzymes and thereby provide more detailed diagnostic information.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants T90 DA 022885 (a training fellowship from the Keck Center Biomedical Discovery from the Large Scale Data Sets Training Program of the Gulf Coast Consortia) and AI32956 and AI092963 (to T. P.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables 1 and 2.
- BLIP
- β-lactamase inhibitory protein
- ITC
- isothermal titration calorimetry
- ACES
- 2-[(2-amino-2-oxoethyl)amino]ethanesulfonic acid.
REFERENCES
- 1. Shaul Y., Schreiber G. (2005) Proteins 60, 341–352 [DOI] [PubMed] [Google Scholar]
- 2. Bai H., Yang K., Yu D., Zhang C., Chen F., Lai L. (2011) Proteins 79, 720–734 [DOI] [PubMed] [Google Scholar]
- 3. Harel M., Cohen M., Schreiber G. (2007) J. Mol. Biol. 371, 180–196 [DOI] [PubMed] [Google Scholar]
- 4. Schreiber G., Haran G., Zhou H. X. (2009) Chem. Rev. 109, 839–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Selzer T., Albeck S., Schreiber G. (2000) Nat. Struct. Biol. 7, 537–541 [DOI] [PubMed] [Google Scholar]
- 6. Perkins J. R., Diboun I., Dessailly B. H., Lees J. G., Orengo C. (2010) Structure 18, 1233–1243 [DOI] [PubMed] [Google Scholar]
- 7. Schreiber G., Keating A. E. (2011) Curr. Opin. Struct. Biol. 21, 50–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yuan J., Huang W., Chow D. C., Palzkill T. (2009) J. Mol. Biol. 389, 401–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Page M. I. (1999) Curr. Pharm. Des. 5, 915–927 [PubMed] [Google Scholar]
- 10. Livermore D. M., Woodford N. (2006) Trends Microbiol. 14, 413–420 [DOI] [PubMed] [Google Scholar]
- 11. Frère J. M. (1995) Mol. Microbiol. 16, 385–395 [DOI] [PubMed] [Google Scholar]
- 12. Paterson D. L., Bonomo R. A. (2005) Clin. Microbiol. Rev. 18, 657–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Walsh T. R. (2008) Curr. Opin. Infect Dis. 21, 367–371 [DOI] [PubMed] [Google Scholar]
- 14. Walther-Rasmussen J., Høiby N. (2007) J. Antimicrob. Chemother. 60, 470–482 [DOI] [PubMed] [Google Scholar]
- 15. Bebrone C. (2007) Biochem. Pharmacol. 74, 1686–1701 [DOI] [PubMed] [Google Scholar]
- 16. Christensen H., Martin M. T., Waley S. G. (1990) Biochem. J. 266, 853–861 [PMC free article] [PubMed] [Google Scholar]
- 17. Chen C. C., Smith T. J., Kapadia G., Wäsch S., Zawadzke L. E., Coulson A., Herzberg O. (1996) Biochemistry 35, 12251–12258 [DOI] [PubMed] [Google Scholar]
- 18. Strynadka N. C., Adachi H., Jensen S. E., Johns K., Sielecki A., Betzel C., Sutoh K., James M. N. (1992) Nature 359, 700–705 [DOI] [PubMed] [Google Scholar]
- 19. Brown N. G., Shanker S., Prasad B. V., Palzkill T. (2009) J. Biol. Chem. 284, 33703–33712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adachi H., Ohta T., Matsuzawa H. (1991) J. Biol. Chem. 266, 3186–3191 [PubMed] [Google Scholar]
- 21. Escobar W. A., Tan A. K., Lewis E. R., Fink A. L. (1994) Biochemistry 33, 7619–7626 [DOI] [PubMed] [Google Scholar]
- 22. Guillaume G., Vanhove M., Lamotte-Brasseur J., Ledent P., Jamin M., Joris B., Frère J. M. (1997) J. Biol. Chem. 272, 5438–5444 [DOI] [PubMed] [Google Scholar]
- 23. Drawz S. M., Bonomo R. A. (2010) Clin. Microbiol. Rev. 23, 160–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chaïbi E. B., Sirot D., Paul G., Labia R. (1999) J. Antimicrob. Chemother. 43, 447–458 [DOI] [PubMed] [Google Scholar]
- 25. Drawz S. M., Bethel C. R., Hujer K. M., Hurless K. N., Distler A. M., Caselli E., Prati F., Bonomo R. A. (2009) Biochemistry 48, 4557–4566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gretes M., Lim D. C., de Castro L., Jensen S. E., Kang S. G., Lee K. J., Strynadka N. C. (2009) J. Mol. Biol. 389, 289–305 [DOI] [PubMed] [Google Scholar]
- 27. Strynadka N. C., Jensen S. E., Alzari P. M., James M. N. (1996) Nat. Struct. Biol. 3, 290–297 [DOI] [PubMed] [Google Scholar]
- 28. Lim D., Park H. U., De Castro L., Kang S. G., Lee H. S., Jensen S., Lee K. J., Strynadka N. C. (2001) Nat. Struct. Biol. 8, 848–852 [DOI] [PubMed] [Google Scholar]
- 29. Doran J. L., Leskiw B. K., Aippersbach S., Jensen S. E. (1990) J. Bacteriol. 172, 4909–4918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang W., Petrosino J., Palzkill T. (1998) Antimicrob. Agents Chemother. 42, 2893–2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang W., Zhang Z., Palzkill T. (2000) J. Biol. Chem. 275, 14964–14968 [DOI] [PubMed] [Google Scholar]
- 32. Reichmann D., Cohen M., Abramovich R., Dym O., Lim D., Strynadka N. C., Schreiber G. (2007) J. Mol. Biol. 365, 663–679 [DOI] [PubMed] [Google Scholar]
- 33. Reichmann D., Phillip Y., Carmi A., Schreiber G. (2008) Biochemistry 47, 1051–1060 [DOI] [PubMed] [Google Scholar]
- 34. Yuan J., Chow D. C., Huang W., Palzkill T. (2011) J. Mol. Biol. 406, 730–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hanes M. S., Jude K. M., Berger J. M., Bonomo R. A., Handel T. M. (2009) Biochemistry 48, 9185–9193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reynolds K. A., Hanes M. S., Thomson J. M., Antczak A. J., Berger J. M., Bonomo R. A., Kirsch J. F., Handel T. M. (2008) J. Mol. Biol. 382, 1265–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang J., Palzkill T., Chow D. C. (2009) J. Biol. Chem. 284, 595–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang J., Zhang Z., Palzkill T., Chow D. C. (2007) J. Biol. Chem. 282, 17676–17684 [DOI] [PubMed] [Google Scholar]
- 39. Zhang Z., Palzkill T. (2003) J. Biol. Chem. 278, 45706–45712 [DOI] [PubMed] [Google Scholar]
- 40. Zhang Z., Palzkill T. (2004) J. Biol. Chem. 279, 42860–42866 [DOI] [PubMed] [Google Scholar]
- 41. Park H. U., Lee K. J. (1998) Microbiology 144, 2161–2167 [DOI] [PubMed] [Google Scholar]
- 42. Fülöp V., Jones D. T. (1999) Curr. Opin. Struct. Biol. 9, 715–721 [DOI] [PubMed] [Google Scholar]
- 43. Paoli M. (2001) Prog. Biophys. Mol. Biol. 76, 103–130 [DOI] [PubMed] [Google Scholar]
- 44. Pons T., Gómez R., Chinea G., Valencia A. (2003) Curr. Med. Chem. 10, 505–524 [DOI] [PubMed] [Google Scholar]
- 45. Hadjebi O., Casas-Terradellas E., Garcia-Gonzalo F. R., Rosa J. L. (2008) Biochim. Biophys. Acta 1783, 1467–1479 [DOI] [PubMed] [Google Scholar]
- 46. Brown N. G., Palzkill T. (2010) Protein Eng. Des. Sel. 23, 469–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shah A. A., Hasan F., Ahmed S., Hameed A. (2004) Res. Microbiol. 155, 409–421 [DOI] [PubMed] [Google Scholar]
- 48. Materon I. C., Queenan A. M., Koehler T. M., Bush K., Palzkill T. (2003) Antimicrob. Agents Chemother. 47, 2040–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brown N. G., Pennington J. M., Huang W., Ayvaz T., Palzkill T. (2010) J. Mol. Biol. 404, 832–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zawadzke L. E., Smith T. J., Herzberg O. (1995) Protein Eng. 8, 1275–1285 [DOI] [PubMed] [Google Scholar]
- 51. Durchschlag H., Hefferle T., Zipper P. (2003) Radiat. Phys. Chem. 67, 479–486 [Google Scholar]
- 52. Bebrone C., Moali C., Mahy F., Rival S., Docquier J. D., Rossolini G. M., Fastrez J., Pratt R. F., Frère J. M., Galleni M. (2001) Antimicrob. Agents Chemother. 45, 1868–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thomson J. M., Distler A. M., Prati F., Bonomo R. A. (2006) J. Biol. Chem. 281, 26734–26744 [DOI] [PubMed] [Google Scholar]
- 54. Andújar-Sánchez M., Cámara-Artigas A., Jara-Pérez V. (2004) Biophys. Chem. 111, 183–189 [DOI] [PubMed] [Google Scholar]
- 55. Jelesarov I., Bosshard H. R. (1994) Biochemistry 33, 13321–13328 [DOI] [PubMed] [Google Scholar]
- 56. Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 57. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Potterton E., Briggs P., Turkenburg M., Dodson E. (2003) Acta Crystallogr. D. Biol. Crystallogr. 59, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 59. Emsley P., Cowtan K. (2004) Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 60. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) Acta Crystallogr. D. Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 61. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L. W., Read R. J., Adams P. D. (2008) Acta Crystallogr. D. Biol. Crystallogr. 64, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zwart P. H., Afonine P. V., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., McKee E., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Storoni L. C., Terwilliger T. C., Adams P. D. (2008) Methods Mol. Biol. 426, 419–435 [DOI] [PubMed] [Google Scholar]
- 63. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D. Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 64. Painter J., Merritt E. A. (2006) Acta Crystallogr. D. Biol. Crystallogr. 62, 439–450 [DOI] [PubMed] [Google Scholar]
- 65. Lebedev A. A., Vagin A. A., Murshudov G. N. (2008) Acta Crystallogr. D. Biol. Crystallogr. 64, 33–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Version 1.3, Schrodinger, LLC, New York [Google Scholar]
- 67. Davis I. W., Murray L. W., Richardson J. S., Richardson D. C. (2004) Nucleic Acids Res. 32, W615–W619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sevcik J., Dauter Z., Lamzin V. S., Wilson K. S. (1996) Acta Crystallogr. D. Biol. Crystallogr. 52, 327–344 [DOI] [PubMed] [Google Scholar]
- 69. Word J. M., Lovell S. C., Richardson J. S., Richardson D. C. (1999) J. Mol. Biol. 285, 1735–1747 [DOI] [PubMed] [Google Scholar]
- 70. Word J. M., Lovell S. C., LaBean T. H., Taylor H. C., Zalis M. E., Presley B. K., Richardson J. S., Richardson D. C. (1999) J. Mol. Biol. 285, 1711–1733 [DOI] [PubMed] [Google Scholar]
- 71. Babu M. M. (2003) Nucleic Acids Res. 31, 3345–3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schreiber F., Schwöbbermeyer H. (2005) Bioinformatics 21, 3572–3574 [DOI] [PubMed] [Google Scholar]
- 73. Frisch C., Schreiber G., Johnson C. M., Fersht A. R. (1997) J. Mol. Biol. 267, 696–706 [DOI] [PubMed] [Google Scholar]
- 74. Reichmann D., Rahat O., Albeck S., Meged R., Dym O., Schreiber G. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Reichmann D., Rahat O., Cohen M., Neuvirth H., Schreiber G. (2007) Curr. Opin. Struct. Biol. 17, 67–76 [DOI] [PubMed] [Google Scholar]
- 76. Maiti R., Van Domselaar G. H., Zhang H., Wishart D. S. (2004) Nucleic Acids Res. 32, W590–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Albeck S., Schreiber G. (1999) Biochemistry 38, 11–21 [DOI] [PubMed] [Google Scholar]
- 78. Pearce K. H., Jr., Cunningham B. C., Fuh G., Teeri T., Wells J. A. (1999) Biochemistry 38, 81–89 [DOI] [PubMed] [Google Scholar]
- 79. Castro M. J., Anderson S. (1996) Biochemistry 35, 11435–11446 [DOI] [PubMed] [Google Scholar]
- 80. Wallis R., Leung K. Y., Pommer A. J., Videler H., Moore G. R., James R., Kleanthous C. (1995) Biochemistry 34, 13751–13759 [DOI] [PubMed] [Google Scholar]
- 81. Wallis R., Moore G. R., James R., Kleanthous C. (1995) Biochemistry 34, 13743–13750 [DOI] [PubMed] [Google Scholar]
- 82. Schreiber G., Fersht A. R. (1993) Biochemistry 32, 5145–5150 [DOI] [PubMed] [Google Scholar]
- 83. Camacho C. J., Kimura S. R., DeLisi C., Vajda S. (2000) Biophys. J. 78, 1094–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Xu C., Min J. (2011) Protein Cell 2, 202–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Perozzo R., Folkers G., Scapozza L. (2004) J. Recept. Signal Transduct. Res. 24, 1–52 [DOI] [PubMed] [Google Scholar]
- 86. Grünberg R., Nilges M., Leckner J. (2006) Structure 14, 683–693 [DOI] [PubMed] [Google Scholar]
- 87. Guharoy M., Janin J., Robert C. H. (2010) Proteins 78, 3219–3225 [DOI] [PubMed] [Google Scholar]
- 88. Ruvinsky A. M., Kirys T., Tuzikov A. V., Vakser I. A. (2011) J. Mol. Biol. 408, 356–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ma B., Elkayam T., Wolfson H., Nussinov R. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 5772–5777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Takagi J., Yang Y., Liu J. H., Wang J. H., Springer T. A. (2003) Nature 424, 969–974 [DOI] [PubMed] [Google Scholar]
- 91. Noinaj N., Fairman J. W., Buchanan S. K. (2011) J. Mol. Biol. 407, 248–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Fonzé E., Charlier P., To'th Y., Vermeire M., Raquet X., Dubus A., Frère J. M. (1995) Acta Crystallogr. D. Biol. Crystallogr. 51, 682–694 [DOI] [PubMed] [Google Scholar]
- 93. Kuzin A. P., Nukaga M., Nukaga Y., Hujer A. M., Bonomo R. A., Knox J. R. (1999) Biochemistry 38, 5720–5727 [DOI] [PubMed] [Google Scholar]
- 94. Herzberg O. (1991) J. Mol. Biol. 217, 701–719 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.